

Abstract
Study of physiological angiogenesis and associated signalling mechanisms in adult heart has been limited by the lack of a robust animal model. We investigated thyroid hormone-induced sprouting angiogenesis and the underlying mechanism. Hypothyroidism was induced in C57BL/6J mice by feeding with propylthiouracil (PTU). One year of PTU treatment induced heart failure. Both 12 weeks- (young) and 1 year-PTU (middle age) treatment caused a remarkable capillary rarefaction observed in capillary density. Three-day Triiodothyronine (T3) treatment significantly induced cardiac capillary growth in hypothyroid mice. In cultured left ventricle (LV) tissues from PTU-treated mice, T3 also induced robust sprouting angiogenesis where pericyte-wrapped endothelial cells formed tubes. The in vitro T3 angiogenic response was similar in mice pre-treated with PTU for periods ranging from 1.5 to 12 months. Besides bFGF and VEGF164, PDGF-BB was the most robust angiogenic growth factor, which stimulated notable sprouting angiogenesis in cultured hypothyroid LV tissues with increasing potency, but had little effect on tissues from euthyroid mice. T3 treatment significantly increased PDGF receptor beta (PDGFR-β) protein levels in hypothyroid heart. PDGFR inhibitors blocked the action of T3 both on sprouting angiogenesis in cultured LV tissue and on capillary growth in vivo. In addition, activation of Akt signalling mediated in T3-induced angiogenesis was blocked by PDGFR inhibitor and neutralizing antibody. Our results suggest that hypothyroidism leads to cardiac microvascular impairment and rarefaction with increased sensitivity to angiogenic growth factors. T3-induced cardiac sprouting angiogenesis in adult hypothyroid mice was associated with PDGF-BB, PDGFR-β and downstream activation of Akt.
Keywords: thyroid hormone, sprouting angiogenesis, pericyte, PDGF, Akt
Introduction
Impaired blood flow and inadequate microvascular growth are commonly associated with cardiac diseases in adults [1–3]. Proper thyroid hormone (TH) levels are important in maintaining a healthy cardiovascular system. Importantly, low thyroid function increases the risk of cardiovascular diseases such as atherosclerosis [4], myocardial infarction (MI) [5] and heart failure [6]. Our previous study in rats showed that propylthiouracil (PTU)-induced hypothyroidism can eventually lead to heart failure with impaired myocardial blood flow and decreased myocardial arterioles [7]. This decrease in arterioles was consistently observed in rats with surgical thyroidectomy and was prevented by administration of thyroxin (T4) or the TH analogue, 3,5-diiodothyropropionic acid [8–10].
The pro-angiogenic effects of THs are both genomic and non-genomic [11]. Recent studies in mice [12] showed that T3-mediated capillary proliferation in the heart is mediated by TH receptor beta (TRβ), which mainly acts as a transcription factor. A rapid non-genomic mechanism of TH-induced angiogenesis in a chick chorioallantoic membrane model was also reported [13,14]. αvβ3 integrin was identified as a cell surface receptor for TH, which induced MAP kinase transduction and crosstalk with vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) [11,15]. Despite these advances, our understanding of the mechanisms by which THs induce angiogenesis in adult mammalian heart and the associated angiogenic growth factors are not well understood.
Akt regulates multiple aspects of vascular cell function such as cell growth, survival and metabolism [16–18]. The most important angiogenic growth factors, VEGF [19], angiopoietin-1 [20] and platelet-derived growth factor (PDGF) [21] are all known to activate Akt. Our recent studies showed that in vivo TH treatment caused activation of the Akt downstream signalling components GSK-3b, mTOR and S6 kinase in heart [22]. In rat heart after MI, short-term treatment (3 days) with T3 prevented cardiac myocyte apoptosis associated with phosphorylation of Akt [23]. At this time, the interaction of Akt and other angiogenic growth factors in TH-mediated cardiac angiogenesis is not clear. In this study, we investigated the induction of angiogenesis by T3 in adult heart from hypothyroid mice. We found that T3 activates the PDGF-Akt signalling pathway, which is essential for T3-induced sprouting angiogenesis.
Materials and methods
Animal model and study design
The use of animals in this study conformed to the Public Health Service Guide for Care and Use of Laboratory Animals and was approved by the Sanford Research/University of South Dakota Institutional Animal Care and Use Committee (Approval ID 35-05-08-11D). Female C57BL/6J mice were obtained from The Jackson Laboratory. Hypothyroidism was induced in ∼2-month-old mice by feeding with iodine-free chow supplemented with 0.15% PTU (catalog TD 97061; Harlan Teklan, Madison, WI, USA) for periods ranging between 6 weeks and 1 year. Age- and sex-matched mice fed normal food served as controls. For in vivo experiments, hypothyroid mice were randomly divided into the following experimental groups: (i) PTU; (ii) PTU + 3 days T3 (40 ng/g/day; Sigma-Aldrich, St. Louis, MO, USA); (iii) PTU + 3 days treatment with T3 and PDGF Ab (PDGF neutralizing antibody, 5 μg/g/day, i.p.; Millipore, Billerica, MA, USA); and (iv) PTU + 3 days treatment with T3 and Imatinib (PDGF receptor inhibitor, 100 μg/g/day, i.p.; Cayman Chemical, Ann Arbor, MI, USA). Body weight (BW), heart weight (HW), body temperature, heart rate (HR) and physiological parameters via echocardiogram were collected at terminal experiments. An inventory of adult PTU-treated hypothyroid mice was maintained for ongoing in vitro tissue culture experiments (minimum treatment period of 6 weeks).
Echocardiography
Echocardiographic measurements were performed under anaesthesia (3% isoflurane induction, 1% maintenance) using a Visual Sonics Vevo 660 High-Resolution Imaging System (Visual Sonics, Toronto, Ontario, Canada) with a 30-MHz linear array transducer (model RMV-707). Left ventricular dimensions and HR were measured from 2-D short-axis M-mode tracings at the level of the papillary muscle. Left ventricular mass and functional parameters were calculated using the above primary measurements.
Measurement of T4
Total Thyroxine (T4) levels were quantified using a Neo-Natal T4 ELISA kit (Monobind, Lake Forest, CA, USA). Procedures were performed as directed in the product insert. Whole blood was collected from the tail vein of mice and spotted onto #903 S&S filter paper, allowed to dry overnight at room temperature and stored at 4°C with desiccants. Absorbance was measured at 450 nm with a reference wavelength of 620 nm on a Thermomax microplate reader (Molecular Devices, Sunnyvale, CA, USA) and data collected using SoftMax (Molecular Devices) software for data analysis.
Analysis of capillaries in left ventricular myocardium
Capillary density was quantified from images of left ventricular tissues (six fields per slide, 40×, minimum sample area 0.5 mm2) stained with isolectin B4 conjugated to Fluorescein (IB4-FITC; Vector Laboratories, Burlingame, CA, USA). Data were collected only from fields containing circular capillary profiles and cross-sectioned myocytes identified by background fluorescence and/or Cy3-labelled wheat germ agglutinin staining (WGA-Cy3). The number of IB4-positive capillaries was normalized to tissue area to obtain numerical capillary density (number/mm2).
Real-time quantitative PCR for gene expression
mRNA expression was measured by the Genomic-Microarray/qPCR Core at the Sanford-Burnham Institute for Medical Research (La Jolla, CA, USA). Briefly, RNA from the left ventricle was extracted using the RNeasy Fibrous Tissue Mini Kit (Qiagen, Valencia, CA, USA). Oligo (dT) primed cDNA synthesis was performed with Superscript III (Invitrogen, Carlsbad, CA, USA). Expression of PDGFB, PDGFRB, VEGF, bFGF and GAPDH transcripts were measured using the TaqMan Gene Expression Assay kit using validated primers (Applied Biosystems, Foster City, CA, USA).
Western blot analysis
Tissue samples from the apex region in each animal were homogenized and incubated in Tissue Extraction Reagent I (Invitrogen) with protease inhibitors cocktail (Sigma-Aldrich) and 1mM PMSF (phenylmethylsulfonyl Fluoride) on ice for 15 min. The lysates were centrifuged at 13,000 g for 10 min. at 4°C. The supernatant was collected, aliquoted and stored at −80°C until time of use. Protein concentration was determined using Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA). Samples were mixed with Laemmli buffer containing 5% β-mercaptoethanol and were evenly loaded onto SDS-PAGE gels. Proteins were transferred to PVDF membranes. Membranes were incubated overnight at 4°C with primary antibodies, phospho-Akt-Ser473, Akt and PDGF receptor β (28E1; all from Cell Signaling Technology, Inc., Danvers, MA, USA) in 5% BSA. A horseradish peroxidase-conjugated secondary antibody was incubated for 1 hr at room temperature in 5% milk and processed for chemiluminescent detection using an ECL Advanced Western Blotting Kit (GE Healthcare, Piscataway, NJ, USA). Protein abundance on Western blots was quantified using densitometry with the Quantity One program from Bio-Rad.
In vitro LV tissue culture
The LV tissue culture method was based on a 3-D angiogenesis in vitro assay reported by Kiefer et al. [24]. Briefly, mice were anaesthetized with isofluorane and the chests cleaned with ethanol. Hearts were excised under sterile conditions, transferred to dishes with PBS, cut into small pieces (1 mm2), and imbedded between two layers of fibrin gel. After polymerization, gels were overlaid with standard DMEM containing the indicated amount of hormone-stripped foetal calf serum (FCS; gift from Paul Davis, Ordway Research Institute, Albany, NY, USA). Tissues were incubated under normoxic conditions (21% Oxygen). Media was changed every 2 days. After 7–10 days, tissue pieces with associated angiogenic sprouts were fixed overnight in 4% paraformaldehyde for immunofluorescent staining.
Sprouting angiogenesis was quantified from light microscope images of 10-day cultures. The tissue perimeter was outlined and measured using Image-Pro Plus (Media Cybernetics, Bethesda, MD, USA). Then using Image J, the radial grid plugin was employed to display 20 radii. This grid was positioned at the centre of the tissue. The grid radii were measured from tissue to furthest sprout intersection. As a result of the depth of focus provided by low-power 20× magnification and the use of finely sliced tissue, all sprouts perpendicular to the tissue surface were visible for quantification. Quantification values are shown as: angiogenic index = Sum of 20 radii lengths/perimeter.
Statistics
All values are presented as means ± S.E.M. Two groups were compared by unpaired Student's t-test. Three or more groups were analysed using one-way anova followed by a Student–Newman–Keuls post hoc test. Statistical analyses were performed with SigmaStat 3.5 (Aspire Software International, Ashburn, VA, USA) and P < 0.05 was considered significant.
Results
T3 induced sprouting angiogenesis in cultured adult left ventricular tissue from hypothyroid mice
A tissue culture model was implemented to explore TH induction of angiogenesis. This model enabled culturing of mouse LV tissue pieces in a gel matrix and examination of outward sprouting from the tissue surface. Hypothyroidism was established by PTU treatment in mice for minimal 6 weeks up to 1 year. After 6 weeks treatment with PTU, T4 levels were undetectable compared with control mice with mean values of 39 ng/ml. Notably, in LV tissue from control mice, we did not see a T3-dependent angiogenic response. However, in LV tissue from PTU-induced hypothyroid mice, there was a remarkable induction of sprouting by T3 (Fig. 1). To minimize angiogenic effects from unknown growth factors, we first determined the minimal level of FCS needed to maintain cultures without inducing angiogenesis (Fig. 1A). In cultures with 1% FCS, there was no obvious sprouting response. With 2% FCS, there was minimal or no detectable sprouting in untreated cultures whereas T3 induced robust sprouting. With FCS concentrations of 3% and above, the T3 angiogenic effect was masked by a serum effect. Therefore, 2% hormone-stripped FCS was chosen for all subsequent experiments. At T3 concentrations ranging from 0.05 to 5 μM, 0.5 μM T3 induced the most robust sprouting (Fig. 1B) and was used in all subsequent experiments. The duration of PTU treatment, ranging from 1.5 to 12 months, had no effect on the in vitro T3-mediated angiogenic response (Fig. 1C). Consequently, an inventory of PTU-treated mice could be continuously maintained for culture experiments. T3-induced sprouting, after 7 days of T3 treatment is shown in cultured tissue pieces in Figure 1D. Sprouting was then examined by confocal microscopy (Fig. 1E–H). Sprouts were composed of endothelial cells (IB4, green) and pericytes (NG2, red). Pericytes were observed wrapping around endothelial cells (e.g. normal morphology) and, in some cases, preceding endothelial cells (Fig. 1E and F; 1G shows 3-d reconstruction). By 7 days of culture, lumena of individual sprouts (indicating maturation) were observed (Fig. 1H). Taken together, results showed a robust T3-induced sprouting response in hypothyroid mice with pericyte/endothelial tube formation.
Fig. 1.
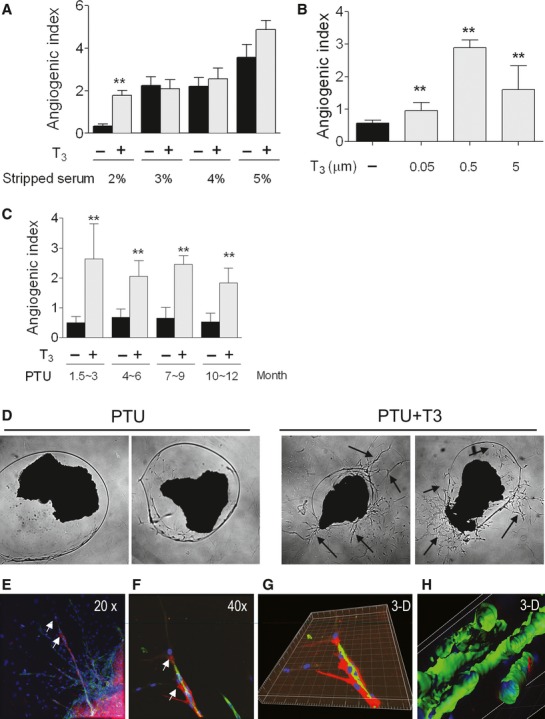
Thyroid hormone induced sprouting angiogenesis in cultured adult heart tissue from hypothyroid mice. (A) Cultured Left ventricular (LV) tissues were treated with different concentrations (2–5%) of thyroid hormone-stripped foetal calf serum (FCS) in the presence or absence of 0.5 μM T3. The optimal condition was determined as 2% FCS, with minimal serum effects and a distinct effect of T3. (B) Cultured LV tissues were treated with different concentration of T3 (0.05–5 μM). A dose of 0.5 μM T3 provided optimum sprouting. (C) Mice were fed PTU food for between 1.5 and 12 months. LV tissues were cultured with or without 0.5 μM T3 to determine if there was a difference in sprouting related to duration of PTU treatment. Values are expressed as mean ± SEM. Data represent 4–6 wells each group per experiment with at least three repeat experiments. **P < 0.01 versus control. (D) Representative image of progressive sprouting (arrows) induced by 7 days of T3 treatment in cultured LV tissues from PTU-fed mice. (E–H) Confocal images of T3-induced sprouting in cultured tissue. NG2 labelled pericytes (red), IB4-labelled endothelium (Green) and DAPI-labelled nuclei (blue) are shown. Images at 20× (E), 40× (F) and 3-D reconstructed image (G) show a view of pericytes wrapping around endothelial tubes and extending beyond endothelium (arrows mark the same vessel segment and point to pericytes). (H) 3-D reconstructed image of sprouts. Tubes, with clearly defined lumena, are readily observed and a pericyte process can be seen along the capillary on the right (red, NG2).
T3-induced myocardial capillary growth in vivo
From our in vitro data, we predicted that TH can induce capillary growth in vivo. To test our prediction, we used two models of cardiac capillary rarefaction in mice: early-onset hypothyroidism in young mice and chronic long-term hypothyroidism in middle-age mice. Mice were treated with PTU for 12 weeks starting from 3 weeks of age (Young) and for 1 year starting from 2 months of age (middle age). Comprehensive physical and functional data were collected from a subset of these mice. In both models, hypothyroidism led to a remarkable decrease in HW, BW and body temperature compared with controls. Three days injection of T3 significantly increased the HW and body temperature compared with the PTU group (Fig. 2A and B and data not shown). Echocardiographic data from young mice treated with PTU showed significantly decreased HR and ejection fraction (EF%). Middle-age mice with 1-year treatment of PTU showed severe cardiac dysfunction including decreased HR, ejection fraction and increased LV internal dimension, which confirmed our previous finding of dilated heart failure in rats [7]. Compared to the PTU group, 3 days of T3 treatment significantly increased HR and tended to increase ejection fraction and reduce LV internal dimension (Fig. 2C and D and data not shown). Most importantly, in both conditions (the early-onset PTU treatment in young mice and the chronic PTU treatment of middle-age mice), we observed capillary rarefaction or decrease in capillary density (Fig. 2E). T3 treatment of these mice induced an apparent capillary growth in cardiac tissue manifested as an increase in capillary density in young adult mice (Fig. 2E). Normalization of capillary density in older mice (Middle age, chronically hypothyroid) was not detected at 3 days after initiation of T3 treatment in this experiment (Fig. 2E).
Fig. 2.
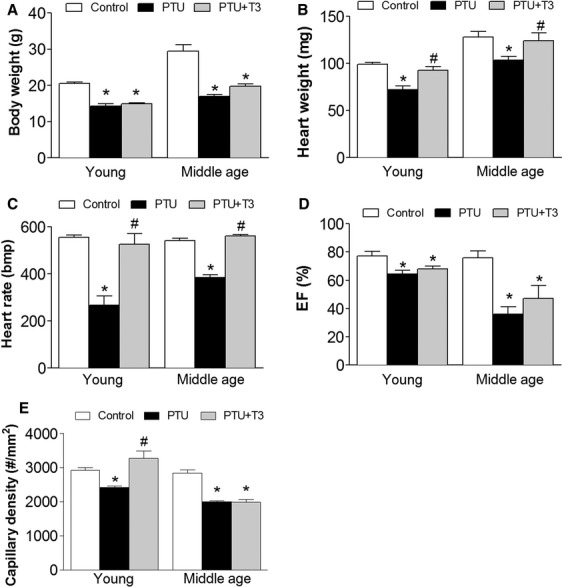
Thyroid hormone induced myocardial capillary growth in hypothyroid mice. Control mice were fed standard rodent chow. Mice were fed PTU food for 12 weeks starting from 3 weeks old (Young) or 1 year starting from 2 months old (Middle age) to induce hypothyroidism. PTU + T3, hypothyroid mice injected for three consecutive days with T3 (40 ng/g/day, i.p.). Changes in body weight (A), heart weight (B), heart rate (C), ejection fraction (EF%, (D) and capillary density (number/mm2, E) were measured and calculated. Bmp, beats per minute. Data are presented as means ± S.E.M. n = 6 for each group. *P < 0.05 versus control; #P < 0.05 versus PTU.
PDGFB/PDGFR-β signalling is involved in T3-induced angiogenesis
The ability of known angiogenic growth factors to induce sprouting angiogenesis in vitro was examined in LV tissues from untreated and PTU-treated mice. bFGF (10 ng/ml), VEGF164 (5 ng/ml), and PDGF-BB (10 ng/ml) were used. Optimum doses for growth factors were based on previous work by Kiefer [24]. LV tissues from control mice were not responsive to bFGF, VEGF164 or PDGF-BB stimulation. However, in LV tissues from hypothyroid mice, each tested growth factor induced significant angiogenic sprouting (Fig. 3A). Because PDGF-BB appeared to be the most robust inducer of angiogenesis in hypothyroid cardiac tissues in vitro, we examined the involvement of the PDGF pathway in T3-induced angiogenesis further. Inhibition of PDGF-BB signalling by either PDGFR tyrosine kinase inhibitor III or PDGFR inhibitor AG1296 prevented T3-induced angiogenic sprouting (Fig. 3B). Consistently, in in vivo experiments, PDGF neutralizing antibody (NAb) blocked T3-induced capillary growth in hypothyroid mice (Fig. 3C). Furthermore, 3-day T3 injection of PTU-treated mice did not alter mRNA and protein level of VEGF, bFGF or PDGF-BB whereas protein levels, but not mRNA level of PDGFR-β were increased by 1.7-fold compared with PTU treatment alone (Fig. 3D and F). Those observations indicate a post-translational regulation of PDGFR-β, which is involved in a non-genomic mechanism of the pro-angiogenic action of TH.
Fig. 3.
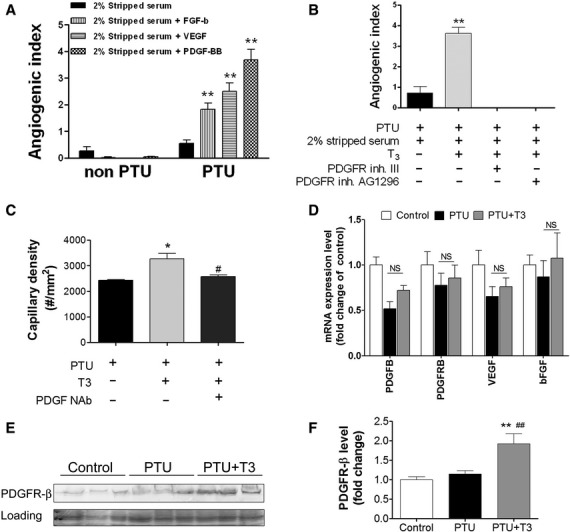
PDGFB-PDGFR-β signalling was involved in T3-induced angiogenesis in adult LV from hypothyroid mice. (A) LV tissues from PTU- and non-PTU-treated mice were cultured in DMEM with 2% stripped FCS. Cultures were treated with/without growth factors, bFGF (10 ng/ml), VEGF164 (5 ng/ml) and PDGF-BB (10 ng/ml) for 10 days. (B) LV tissues from PTU-treated mice were cultured with T3 (0.5 μM) in 2% stripped FCS. The angiogenic effects of PDGFR tyrosine kinase inhibitor III (100 nM) or PDGFR inhibitor AG1296 (1 μM) were examined in tissues treated with T3 for 10 days. Data were presented as mean ± S.E.M. A total of 4–6 wells each group with at least three independent experiments. **P < 0.01 versus control. (C) After 12 weeks of PTU treatment, mice were injected daily for 3 days with T3 (40 ng/g/day, i.p.) and anti-PDGF neutralizing antibody (5 mg/kg/day). Capillary density was measured and determined on LV tissue sections by confocal microscopy. (D–F) PTU-fed (1 year) mice were injected with T3 (40 ng/g/day, i.p.) for 3 days. mRNAs level of PDGFB, PDGFRB, VEGF, bFGF were determined using quantitative real-time PCR with Taqman probes (D). (E) Protein expression of PDGFR-β from LV tissues was determined by western blot. (F) Quantitative data in (E) are shown as mean ± S.E.M. n = 6 per group.**P < 0.01 versus control, ##P < 0.01 versus PTU, ns means P > 0.05 versus PTU.
PDGF-Akt axis mediated T3-induced angiogenesis in hypothyroid mice
Our previous report indicated that TH can activate the PI3K/Akt pathway. We next determine if the T3-activated PI3K/Akt pathway mediated T3-induced sprouting angiogenesis in hypothyroid mice. In LV tissues from PTU-induced hypothyroid mice, 3 days injection of T3 resulted in a 2.1-fold increase in phospho-Akt (Ser 473, Fig. 4A and B). T3 treatment also up-regulated phospho-Akt (Ser 473) expression (Red) in cultured LV tissues and sprouts. Untreated (PTU only) tissue had no sprouting and minimal Akt phosphorylation (Fig. 4C). Inhibition of the Akt pathway by either the PI3K inhibitor LY294002 (10 μM), Akt inhibitor IV (1 μM) or Akt inhibitor X (2 μM) prevented T3-induced angiogenic sprouting (Fig. 4D). Consistently, the T3-induced angiogenic response was observed in LV tissues in vivo from a PTU-treated wild-type mouse, but not in LV tissues from an Akt knockout mouse pre-treated with PTU for 8 weeks (Fig. 4E).
Fig. 4.
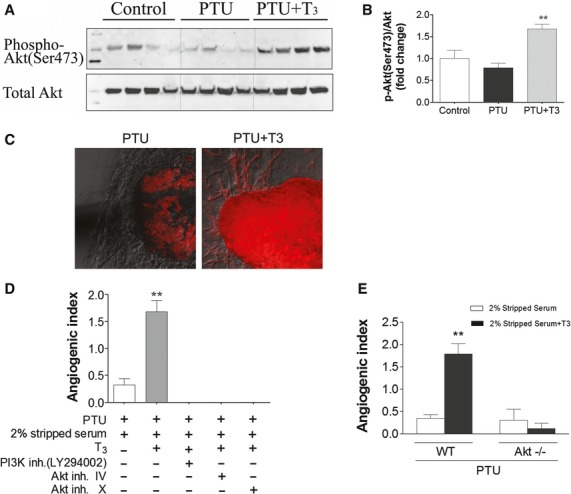
Akt signalling mediated T3-induced sprouting angiogenesis in adult LV from hypothyroid mice. (A, B) PTU-treated (1 year) mice were injected with T3 (40 ng/g/day, i.p.) for 3 days. Non-PTU-treated mice served as controls. Phosphorylation of Akt (Ser 473) and total Akt were measured by western blot as shown with representative blot (A) and quantification (B) (n = 6/group, **P < 0.01 versus PTU). (C) Confocal immunofluorescence staining of phospho- Akt (Ser 473) in cultured LV tissues from PTU-fed mice. T3 (0.5 μM) induced robust expression of phospho-Akt (Red) in tissue and sprouts, but no sprouting and reduced overall labelling in PTU treated only. (D) T3-induced angiogenic sprouts in cultured tissues were blocked by the PI3K inhibitor LY294002 (10 μM), Akt inhibitor IV (1 μM) and Akt inhibitor X (2 μM). **P < 0.01 versus PTU; 4–6 wells per group; at least three independent experiments. (E) Using standard conditions, LV tissues were cultured from a wild-type mouse (WT) and an Akt knockout mouse (Akt−/−) after 8 weeks PTU treatment. 0.5 μM T3 induced significant sprouting angiogenesis in WT mice, but not Akt knockout mouse.(**P < 0.01 versus PTU alone, n = 8 wells per group).
Because PDGF/PDGFR are critical for Akt activation [21,25], we further determined if blocking PDGF/PDGFR prevents activation of Akt signalling. In LV tissue from hypothyroid mice, T3-stimulated induction of phospho-Akt was reduced by PDGF neutralizing antibody and Imatinib (an inhibitor of the PDGF receptor tyrosine kinase, Fig. 5A and B). In addition, Akt is a major regulator of nitric oxide (NO) production, which has been demonstrated as a potent stimulator of angiogenesis [26,27]. To determine if NO production is downstream of Akt activation, we performed a rescue experiment in which we treated hypothyroid Akt-deficient cardiac tissue with T3 in the absence or in the presence of NO donor (S-Nitrosoglutathione, 1 μM). This experiment indicated that NO could at least partially restore T3-induced sprouting angiogenesis in cardiac tissue from Akt−/− mice (Fig. 5C). Together, these observations, as shown in schematic Figure 5D, indicate that T3-induced sprouting angiogenesis in LV from hypothyroid mice is mediated through PDGF-Akt signalling, both in vitro and in vivo.
Fig. 5.
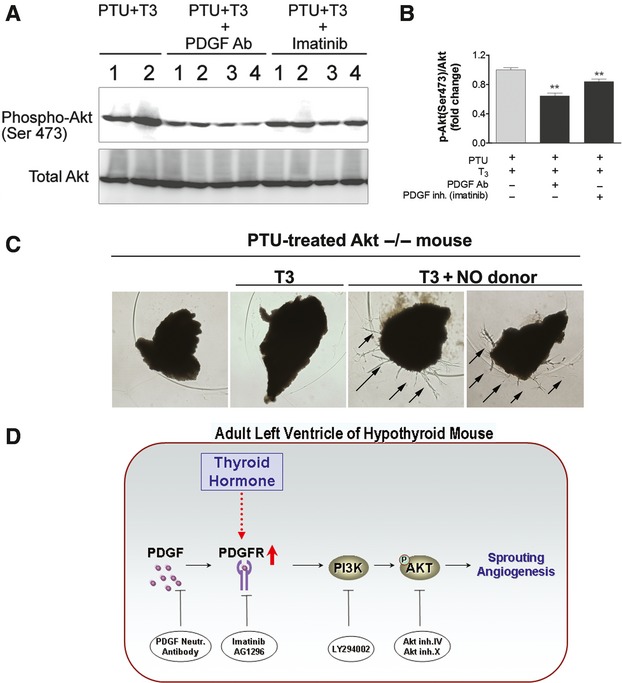
PDGFR-Akt axis was involved in T3 induced sprouting angiogenesis in adult LV from hypothyroid mice (A and B) After 1 year of PTU treatment, mice were injected daily for 3 days with T3 (40 ng/g/day, i.p.) and anti-PDGF neutralizing antibody (5 mg/kg/day), or with T3 and imatinib (100 mg/kg/day). Phospho-Akt expression was determined by western blot and quantified (**P < 0.01 versus PTU alone, n = 6 per group). (C) LV tissues were cultured from an Akt knockout mouse (Akt−/−) after 8 weeks PTU treatment. 1 μM S-Nitrosoglutathione (NO donor) treatment restored the sprouting angiogenesis in T3-treated Akt knockout mouse. (D) Schematic pathway of PDGFR-Akt axis in T3 induced sprouting angiogenesis in adult LV from hypothyroid mice.
Discussion
Thyroid hormones have been shown to promote coronary angiogenesis in hyperthyroidism [28,29] and after MI [30]. An important question is whether THs induce cardiac angiogenesis directly or as a result of increased tissue metabolism and myocyte hypertrophy. Work by Tomanek et al. [28] suggested that T4 induced capillary proliferation within 24 hrs after injection and before development of cardiac hypertrophy. However, this did not rule out capillary growth as a result of increased metabolism. Recent work by Makino et al. [12] showed that T3 stimulated increased capillary networks in cultured mouse coronary endothelial cells in a TRβ-dependent manner. In vitro data reported here also support direct stimulation of angiogenesis by T3. Heron demonstrated a modest proliferation of endothelial cells in hyperthyroid rats, but much bigger increase after T3 treatment in hypothyroid rats [31]. A goal of this study was to create an in vitro model with maximum vascular growth potential by first sensitizing the tissue to angiogenic stimulation by inducing a low thyroid condition. Ideally, such an in vitro model would work in adult myocardium and allow the use of gene-targeted mouse models for future exploration of molecular mechanisms.
Previously, we reported that long-term PTU-induction of hypothyroidism in rats led to cardiac atrophy, increased chamber diameter/wall thickness ratio, severe cardiac dysfunction, series addition of sarcomeres in myocytes without transverse growth (a hallmark of dilated heart failure) and impaired myocardial blood flow with dramatic loss of arterioles [7]. In this study, we confirmed similar cardiac functional impairment and remodelling suggestive of heart failure in mice after 1 year PTU treatment. PTU-treated mice exhibited cardiac capillary rarefaction by a ∼20% reduction (young mice, 12 weeks PTU treatment) and ∼30% reduction (middle-age mice, 1 year PTU treatment) in capillary density. Consistently, cultured heart tissues from PTU-treated mice were similarly responsive to T3-induced angiogenesis regardless of the duration of hypothyroidism or age of hypothyroid tissue donors. As noted previously by Keifer et al. [24], it is difficult to induce angiogenesis in tissues from adult mouse heart under normal culture conditions. Their work showed increased angiogenic sensitivity to hypoxia whereas we showed that hypothyroid tissues have increased sensitivity to angiogenic stimuli, including growth factors and T3. This study demonstrates that hypothyroidism, induced by PTU treatment in mice, also sensitizes LV tissue for angiogenic stimuli delivered by T3 in culture. Our in vivo data suggested similar pro-angiogenic cardiac effects of T3 treatment of young adult hypothyroid mice manifested as an increase in LV capillary density after only 3 days of T3 treatment. This combined in vitro/in vivo approach to study T3-induced cardiac angiogenesis in adult mice should provide a useful tool for dissection of angiogenic mechanisms, particularly as the model can be easily superimposed on gene-targeted mouse models and genetic lineage tracing systems.
As noted above, we used the in vitro angiogenesis model described by Kiefer et al. [24] using heart tissues cultured in a three-dimensional fibrin gel matrix to directly visualize vessel sprouting. Kiefer found that hypoxia and serum were required for angiogenesis to occur in culture of adult mouse heart tissues. They were able to induce sprouting only under hypoxia and higher FCS concentrations (5% or more), but not under normoxia, bFGF, VEGF164 or PDGF-BB stimulation. With the increased sensitivity of hypothyroid tissues to angiogenic stimulation, we were able to consistently observe robust T3-induced sprouting in tissues under normoxic conditions with only 2% FCS whereas sprouting was rare and sporadic in non-T3-treated control cultures. In addition, bFGF, VEGF164 and PDGF-BB induced a robust angiogenic response under these culture conditions whereas there was no response when using tissues from euthyroid mice. In addition to the study by Kiefer et al. [24], other in vivo and in vitro studies have demonstrated an age-related reduction in angiogenic potential [32–35]. Intriguingly, this in vitro hypothyroid model does not show such a reduction in angiogenic potential, at least up to about 14 months of age (e.g. middle age).
A growing body of literature suggests important interactions of THs and other important angiogenic inducers, especially bFGF [11,13,28]. Tomanek indicated, in a T4-induced cardiac hypertrophy model, capillary growth was accompanied by up-regulation of bFGF mRNA and protein [28]. Using a chick chorioallantoic model, Paul Davis’ laboratory identified integrin αvβ3 as a TH receptor on cell membranes. Integrin αvβ3 non-genomically initiate pro-angiogenic action of TH by activation of MAP kinases and increased transcription of bFGF and VEGF, and additionally engages in crosstalk with the VEGF and bFGF receptors [11,36]. In this study, we found that PDGF signalling robustly induces sprouting angiogenesis and is required for T3-generated sprouts in cultured adult heart tissues from hypothyroid mice. This finding implies that TH-induced angiogenesis interplays with growth factors, which depend on tissue TH levels. Although we did not observe in vivo changes in mRNA and protein expression of VEGF, bFGF or PDGF-BB in heart tissue at the 3-day T3 treatment time point, the up-regulation of PDGFR-β protein expression, but not mRNA level indicate a post-translational regulation of TH on pro-angiogenesis through a non-genomical crosstalk between TH and PDGF receptor. It would be interesting to identify new direct ligands in the future study for TH to engage the crosstalk with PDGF signalling pathway in a non-genomical way. Furthermore, inhibition of PDGFR blocked T3-induced sprouting angiogenesis and T3-induced Akt activation. In addition, Akt has been shown to regulate NO synthesis, which is also a potent stimulator stimulated angiogenesis and arteriogenesis. Our data indicate that NO donor could rescue impaired sprouting angiogenesis in cardiac tissue from Akt−/− mice. Therefore, in this hypothyroid model, it appears that T3-induced sprouting angiogenesis primarily involves PDGF-PDGFR-β and downstream activation of the Akt-NO pathway.
Angiogenesis primarily involves endothelial cells (ECs), pericytes and smooth muscle cells (SMCs). SMCs are associated with arteries and veins with multiple concentric layers in larger arteries, whereas pericytes are associated with the smallest vessels, primarily capillaries and venules. Pericytes and their interaction with ECs are involved in pivotal processes of angiogenesis and have become putative targets in tumour therapy [37]. In addition to NG2 [38], PDGFR-β has been shown to be a promising pericyte marker [39]. PDGF-PDGFR-β signalling involves pericyte recruitment for angiogenesis [37,39]. In this study, we showed that T3-induced sprouts are composed of endothelial tubes (IB4 positive) wrapped by pericytes (NG2 positive), which, occasionally precede ECs. This is consistent with previous observations from Ozerdem and Stallcup [40] who observed EC-free (pericyte) segments of growing spouts in the microvasculature of normally developing tissue (mouse retina) and various tumour models. Up-regulation of PDGFR-β expression in LV tissues of hypothyroid mice treated with T3 further suggests an active role for pericyte participation in T3-induced sprouting angiogenesis.
Our recent studies indicate that the milieu of ‘pro-arteriogenic’ factors induced by TH in the heart is complex and indeed leads to arteriogenesis [10,41]. In addition to the PDGF system, TH appear to activate an Angiopoietin response as well [41]. In this study, we specifically show the direct effect of PDGFBB in sprouting angiogenesis in hypothyroid mouse heart tissue. A future study employing genetic models of PDGF and Angiopoietin deficiencies is necessary to delineate their relative contributions to arteriogenesis during the restoration of a euthyroid state in the heart.
The study of physiological angiogenesis and its mechanisms in adult organs, especially in adult heart, has been rather limited because of the lack of a suitable animal model. T3 is known to stimulate angiogenesis. Akt is known to play a major role in angiogenesis. Until now, the mechanistic link between T3 and Akt in angiogenesis was not clear. In this study, we used a PTU-induced hypothyroid model and showed global, rapid and robust induction of angiogenesis in adult heart with T3 treatment both in vivo and in vitro. PDGF-PDGFR-β and downstream activation of Akt played a key role. These in vivo and in vitro models, coupled with the coronary EC culture model used by Dillmann's group [42], provide important tools to further examine angiogenic signalling in adult heart.
Acknowledgments
This study was supported by the National Center for Research Resources (NCRR) Grant P20RR017662-06A1 and RO1 HL093160-01A1 (AMG) from the National Institutes of Health (NIH). We would like to thank Ms. Rebecca Redetzke of the Sanford Research Physiology Core and Ms. Kelly Graber of the Sanford Research Imaging Core for their assistance with the project.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Tomanek RJ, Torry RJ. Growth of the coronary vasculature in hypertrophy: mechanisms and model dependence. Cell Mol Biol Res. 1994;40:129–36. [PubMed] [Google Scholar]
- 2.Shiojima I, Sato K, Izumiya Y, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–18. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoon YS, Uchida S, Masuo O, et al. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation. 2005;111:2073–85. doi: 10.1161/01.CIR.0000162472.52990.36. [DOI] [PubMed] [Google Scholar]
- 4.Auer J, Berent R, Weber T, et al. Thyroid function is associated with presence and severity of coronary atherosclerosis. Clin Cardiol. 2003;26:569–73. doi: 10.1002/clc.4960261205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franklyn JA, Gammage MD, Ramsden DB, et al. Thyroid status in patients after acute myocardial infarction. Clin Sci (Lond) 1984;67:585–90. doi: 10.1042/cs0670585. [DOI] [PubMed] [Google Scholar]
- 6.Ascheim DD, Hryniewicz K. Thyroid hormone metabolism in patients with congestive heart failure: the low triiodothyronine state. Thyroid. 2002;12:511–5. doi: 10.1089/105072502760143908. [DOI] [PubMed] [Google Scholar]
- 7.Tang YD, Kuzman JA, Said S, et al. Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation. 2005;112:3122–30. doi: 10.1161/CIRCULATIONAHA.105.572883. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Redetzke RA, Said S, et al. Serum thyroid hormone levels may not accurately reflect thyroid tissue levels and cardiac function in mild hypothyroidism. Am J Physiol Heart Circ Physiol. 2008;294:H2137–43. doi: 10.1152/ajpheart.01379.2007. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Sherer BA, Redetzke RA, et al. Regulation of arteriolar density in adult myocardium during low thyroid conditions. Vascul Pharmacol. 2010;52:146–50. doi: 10.1016/j.vph.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Wang D, Redetzke RA, et al. Thyroid hormone analog 3,5-diiodothyropropionic acid promotes healthy vasculature in the adult myocardium independent of thyroid effects on cardiac function. Am J Physiol Heart Circ Physiol. 2009;296:H1551–7. doi: 10.1152/ajpheart.01293.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luidens MK, Mousa SA, Davis FB, et al. Thyroid hormone and angiogenesis. Vascul Pharmacol. 2010;52:142–5. doi: 10.1016/j.vph.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Makino A, Suarez J, Wang H, et al. Thyroid hormone receptor-beta is associated with coronary angiogenesis during pathological cardiac hypertrophy. Endocrinology. 2009;150:2008–15. doi: 10.1210/en.2008-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis FB, Mousa SA, O'Connor L, et al. Proangiogenic action of thyroid hormone is fibroblast growth factor-dependent and is initiated at the cell surface. Circ Res. 2004;94:1500–6. doi: 10.1161/01.RES.0000130784.90237.4a. [DOI] [PubMed] [Google Scholar]
- 14.Bergh JJ, Lin HY, Lansing L, et al. Integrin alphaVbeta3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology. 2005;146:2864–71. doi: 10.1210/en.2005-0102. [DOI] [PubMed] [Google Scholar]
- 15.Mousa SA, Bergh JJ, Dier E, et al. Tetraiodothyroacetic acid, a small molecule integrin ligand, blocks angiogenesis induced by vascular endothelial growth factor and basic fibroblast growth factor. Angiogenesis. 2008;11:183–90. doi: 10.1007/s10456-007-9088-7. [DOI] [PubMed] [Google Scholar]
- 16.Somanath PR, Razorenova OV, Chen J, et al. Akt1 in endothelial cell and angiogenesis. Cell Cycle. 2006;5:512–8. doi: 10.4161/cc.5.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–50. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 18.Dimmeler S, Zeiher AM. Akt takes center stage in angiogenesis signaling. Circ Res. 2000;86:4–5. doi: 10.1161/01.res.86.1.4. [DOI] [PubMed] [Google Scholar]
- 19.Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–43. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 20.Fukuhara S, Sako K, Noda K, et al. Tie2 is tied at the cell-cell contacts and to extracellular matrix by angiopoietin-1. Exp Mol Med. 2009;41:133–9. doi: 10.3858/emm.2009.41.3.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuzman JA, Vogelsang KA, Thomas TA, et al. L-Thyroxine activates Akt signaling in the heart. J Mol Cell Cardiol. 2005;39:251–8. doi: 10.1016/j.yjmcc.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 23.Chen YF, Kobayashi S, Chen J, et al. Short term triiodo-L-thyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J Mol Cell Cardiol. 2008;44:180–7. doi: 10.1016/j.yjmcc.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiefer FN, Munk VC, Humar R, et al. A versatile in vitro assay for investigating angiogenesis of the heart. Exp Cell Res. 2004;300:272–82. doi: 10.1016/j.yexcr.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Bajraszewski N, Wu E, et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–8. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murohara T, Asahara T, Silver M, et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–78. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duda DG, Fukumura D, Jain RK. Role of eNOS in neovascularization: NO for endothelial progenitor cells. Trends Mol Med. 2004;10:143–5. doi: 10.1016/j.molmed.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Tomanek RJ, Doty MK, Sandra A. Early coronary angiogenesis in response to thyroxine: growth characteristics and upregulation of basic fibroblast growth factor. Circ Res. 1998;82:587–93. doi: 10.1161/01.res.82.5.587. [DOI] [PubMed] [Google Scholar]
- 29.Breisch EA, White FC, Hammond HK, et al. Myocardial characteristics of thyroxine stimulated hypertrophy. A structural and functional study. Basic Res Cardiol. 1989;84:345–58. doi: 10.1007/BF02650869. [DOI] [PubMed] [Google Scholar]
- 30.Tomanek RJ, Zimmerman MB, Suvarna PR, et al. A thyroid hormone analog stimulates angiogenesis in the post-infarcted rat heart. J Mol Cell Cardiol. 1998;30:923–32. doi: 10.1006/jmcc.1998.0671. [DOI] [PubMed] [Google Scholar]
- 31.Heron MI, Rakusan K. Proliferating cell nuclear antigen (PCNA) detection of cellular proliferation in hypothyroid and hyperthyroid rat hearts. J Mol Cell Cardiol. 1995;27:1393–403. doi: 10.1006/jmcc.1995.0132. [DOI] [PubMed] [Google Scholar]
- 32.Rivard A, Fabre JE, Silver M, et al. Age-dependent impairment of angiogenesis. Circulation. 1999;99:111–20. doi: 10.1161/01.cir.99.1.111. [DOI] [PubMed] [Google Scholar]
- 33.Swift ME, Kleinman HK, DiPietro LA. Impaired wound repair and delayed angiogenesis in aged mice. Lab Invest. 1999;79:1479–87. [PubMed] [Google Scholar]
- 34.Arthur WT, Vernon RB, Sage EH, et al. Growth factors reverse the impaired sprouting of microvessels from aged mice. Microvasc Res. 1998;55:260–70. doi: 10.1006/mvre.1998.2078. [DOI] [PubMed] [Google Scholar]
- 35.Rivard A, Berthou-Soulie L, Principe N, et al. Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J Biol Chem. 2000;275:29643–7. doi: 10.1074/jbc.M001029200. [DOI] [PubMed] [Google Scholar]
- 36.Luidens MK, Mousa SA, Davis FB, et al. Thyroid hormone and angiogenesis. Vascul Pharmacol. 2010;52:142–5. doi: 10.1016/j.vph.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 37.Gaengel K, Genove G, Armulik A, et al. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–8. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 38.Virgintino D, Girolamo F, Errede M, et al. An intimate interplay between precocious, migrating pericytes and endothelial cells governs human fetal brain angiogenesis. Angiogenesis. 2007;10:35–45. doi: 10.1007/s10456-006-9061-x. [DOI] [PubMed] [Google Scholar]
- 39.Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23. doi: 10.1007/s00441-003-0745-x. [DOI] [PubMed] [Google Scholar]
- 40.Ozerdem U, Stallcup WB. Early contribution of pericytes to angiogenic sprouting and tube formation. Angiogenesis. 2003;6:241–9. doi: 10.1023/B:AGEN.0000021401.58039.a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Savinova OV, Liu Y, Aasen GA, et al. Thyroid hormone promotes remodeling of coronary resistance vessels. PLoS One. 2011;6:e25054. doi: 10.1371/journal.pone.0025054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makino A, Platoshyn O, Suarez J, et al. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am J Physiol Cell Physiol. 2008;295:C221–30. doi: 10.1152/ajpcell.00433.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]