

Abstract
Preterm infants often experience hyperoxia while receiving supplemental oxygen. Prolonged exposure to hyperoxia during development is associated with pathologies such as bronchopulmonary dysplasia and retinopathy of prematurity. Over the last 25 years, however, experiments with animal models have revealed that moderate exposures to hyperoxia (e.g., 30–60% O2 for days to weeks) can also have profound effects on the developing respiratory control system that may lead to hypoventilation and diminished responses to acute hypoxia. This plasticity, which is generally inducible only during critical periods of development, has a complex time course that includes both transient and permanent respiratory deficits. Although the molecular mechanisms of hyperoxia-induced plasticity are only beginning to be elucidated, it is clear that many of the respiratory effects are linked to abnormal morphological and functional development of the carotid body, the principal site of arterial O2 chemoreception for respiratory control. Specifically, developmental hyperoxia reduces carotid body size, decreases the number of chemoafferent neurons, and (at least transiently) diminishes the O2 sensitivity of individual carotid body glomus cells. Recent evidence suggests that hyperoxia may also directly or indirectly impact development of central neural control of breathing. Collectively, these findings emphasize the vulnerability of the developing respiratory control system to environmental perturbations.
Keywords: developmental plasticity, control of breathing, hypoxic ventilatory response, hypoplasia, carotid body growth, O2 therapy
1. Introduction
The respiratory control system is under strong genetic regulation which guides its development and determines much of the adult respiratory phenotype (Han & Strohl, 2000; Strohl, 2003; Tankersley, 2003; Borday et al., 2004, 2005). Even so, the respiratory control system also retains substantial capacity for phenotypic plasticity (Carroll, 2003; Mitchell & Johnson, 2003; Bavis & Mitchell, 2008). Phenotypic plasticity describes the ability of a single genotype to produce a range of phenotypes in response to environmental variation. This capacity may be greatest during development, with specific windows of environmental sensitivity (i.e., critical periods) in which plasticity is inducible. Plasticity is often adaptive because it enables individuals to cope with changing demands throughout their lifetime; however, this environmental sensitivity may also yield maladaptive responses to disease, injury, or novel stimuli.
Environmental hyperoxia (inspired PO2 >160 mmHg) occurs rarely in nature except, for example, in certain isolated systems like tidal pools. On the other hand, supplemental O2 is a common therapeutic intervention in clinical settings which, in turn, leads to both acute and chronic hyperoxia in human and animal patients (e.g., Hagadorn et al., 2006; Claure and Bancalari, 2009; Finer and Leone, 2009). Moreover, it has been suggested that the earlier-than-normal rise in inspired O2 associated with preterm birth induces a state of “relative hyperoxia” with respect to normal gestational PO2 (Carroll, 2003). It is consequently important to understand the impact of hyperoxia (and relative hyperoxia) on critical homeostatic processes, such as regulation of blood gases.
The epithelium of the lungs and upper airways experience the highest PO2 during clinical O2 exposures and, consequently, may suffer direct oxidative injury through the overproduction of reactive oxygen species (ROS). The impact of hyperoxia may extend beyond these tissues due to elevated arterial PO2, however, and can include substantial morphological and functional plasticity in neural pathways critical to respiratory control. In this article, we review evidence that perinatal hyperoxia alters the development of the carotid body, the primary O2 chemoreceptor for the respiratory control system. Although the mechanism of hypoxia transduction within the carotid body is not completely understood, it appears to be initiated in the glomus (type I) cells, neuron-like secretory cells that synapse with neurons projecting to the brainstem via the carotid sinus nerve (CSN). Decreasing arterial PO2 depolarizes the glomus cells, increases intracellular calcium, and triggers the release of neurochemicals, ultimately enhancing afferent nerve activity and initiating the hypoxic ventilatory response (HVR) (Kumar, 2007; López-Barneo et al., 2008).
2. Ventilatory control after developmental hyperoxia
The first indication that chronic hyperoxia alters carotid body development came from studies of the HVR in cats and rats reared in moderate hyperoxia. Hanson et al. (1989) observed that the acute HVR was severely diminished in kittens reared in 30% O2 from birth. Later, Ling et al. (1996) demonstrated that the HVR remains blunted in rats reared in 60% O2 for the first postnatal month even after being returned to normoxia (21% O2) for several months; similar effects have been observed after only 1 or 2 weeks in 60% O2 (e.g., Fig. 1) (Bavis et al., 2003, 2007, 2008). Interestingly, rats exposed to a similar duration of 60% O2 as adults exhibit normal HVR (Ling et al., 1996, 1997a,c). Together, these data indicate that developmental hyperoxia induces a long-lasting reduction in the HVR and that this plasticity can only be elicited during development. Subsequent studies revealed that the critical period for long-lasting changes in the HVR is limited to the first two postnatal weeks in rats (Bavis et al., 2002). It is now known that developmental hyperoxia has similar effects on the HVR in several vertebrates species, including mice (Bavis et al., 2011a), quail (Bavis & Simons, 2008), chickens (Mortola, 2011), and zebrafish (Vulesevik and Perry, 2006); while fish do not have a carotid body per se, they have O2-sensitive chemoreceptors in their gills that are considered homologous to those in the mammalian and avian carotid body (Milsom and Burleson, 2007). No controlled experiments have been conducted in human infants, but there is correlative evidence suggesting that ventilatory control is affected by supplemental O2 in humans as well (Calder et al., 1994; Katz-Salamon and Lagercrantz, 1994; Katz-Salamon et al., 1996).
Fig. 1.

Representative effects of developmental hyperoxia on carotid body (CB) volume, CB hypoxic response (single-unit or whole-nerve CSN response), and hypoxic ventilatory response (HVR). Rats were exposed to 60% O2 for the first two postnatal weeks and studied (A) immediately (P13–14) or (B) >4 weeks (Adult) after return to room air. Note that the HVR for P13–14 rats represents the early phase of the response (i.e., first minute of hypoxia). Values (mean ± SEM) are expressed as a percentage of those measured for age-matched rats reared in room air (i.e., “Control” rats). All values are significantly reduced relative to Control (i.e., <100%; one-sample t-test, all P<0.001). Data were compiled from previously published studies (Donnelly et al., 2005; Bavis et al., 2007, 2008, 2010; Dmitrieff et al., 2012).
Although the eventual attenuation of the HVR is well documented in rats and other species exposed to developmental hyperoxia, recent studies indicate that changes to ventilatory control during the hyperoxic exposure are more complex than initially appreciated. The HVR is distinctly biphasic in newborn mammals, with an initial, carotid body-mediated increase in ventilation (early phase of the HVR) being followed by a secondary decline in ventilation linked to central neural inhibition (late phase of the HVR) (Eden and Hanson, 1987; Bissonnette, 2000; Teppema and Dahan, 2010). As individuals mature (over the first 7–14 postnatal days in rats), the overall magnitude of the HVR tends to increase and the biphasic response is gradually replaced with a sustained increase in ventilation. Bavis et al. (2010) assessed the ventilation of rats reared in 60% O2 from birth until studied at one of three postnatal ages: 4, 6–7, or 13–14 days of age (P4, P6–7, or P13–14, respectively). When acutely exposed to hypoxia (12.5% O2), the magnitude of the early phase of the HVR was similar to that of age-matched controls at P4, but became progressively diminished at P6–7 and P13–14 (Fig. 1A). Surprisingly, hyperoxia-treated rats exhibited a sustained increase in ventilation at P4 and P6–7 in contrast to the expected biphasic HVR observed in controls (for potential mechanisms, see Section 5 below). Thus, when expressed as a percentage increase from baseline, the HVR was actually enhanced during the later stages of hypoxia in hyperoxia-treated rats at these ages. The magnitude of the late HVR increased with postnatal maturation in control rats, however, and an overall reduction in the HVR of hyperoxia-treated rats emerged by P13–14 (Bavis et al., 2010).
In addition to changes in the HVR, Bavis et al. (2010) also noted changes to normoxic ventilation in neonatal rats exposed to developmental hyperoxia. Relative to age-matched control rats reared in normoxia, hyperoxia-treated rats exhibited substantially lower minute ventilation at P4 and P6–7 when acutely returned to 21% O2, but normoxic ventilation returned to normal in P14 rats despite their longer hyperoxic exposure. The reduced ventilation in hyperoxia-treated rats at P4 likely represents a true hypoventilation based on lower arterial O2 saturations in these pups (van Heerden and Bavis, 2011). Inasmuch as the carotid body contributes to normoxic ventilatory drive, lower resting ventilation is consistent with abnormal carotid body function in hyperoxia-treated neonates. This is supported by recent experiments showing that minute ventilation drops less in hyperoxia-treated rats during acute O2 inhalation (Dejour's test) than in age-matched controls at P4 and P6–7, but not at P13–14 (van Heerden et al., 2011). Moreover, normalization of normoxic ventilation by P13–14 could reflect a reduced contribution of carotid bodies to eupneic breathing with advancing age. Indeed, the ventilatory response to O2 inhalation was lower in control rats at P13–14 than at P4 (van Heerden et al., 2011).
3. Carotid body function after developmental hyperoxia
The carotid body responds to a decrease in arterial PO2 by increasing action potential activity on the carotid sinus nerve (CSN) which, in turn, stimulates breathing. Given the carotid body's primary role in initiating the HVR, it is a likely culprit in cases where the HVR is deemed abnormal. Indeed, direct measurements of CSN activity in cats and rats reared in developmental hyperoxia have revealed dramatic reductions in carotid body responses to hypoxia. Hanson et al. (1989) observed diminished carotid chemoafferent responses to hypoxia in single- and few-fiber CSN preparations in anesthetized cats that had been reared in 30% O2 from birth until studied at 12–23 days of age. Similarly, rats reared in 60% O2 for 1–4 weeks from birth exhibit greatly reduced (and in some cases absent) whole-nerve CSN responses to hypoxia, asphyxia, and cyanide administration when assessed 2–14 months after return to normoxia (Ling et al., 1997a; Fuller et al., 2002; Bisgard et al., 2003; Prieto-Lloret et al., 2004) (e.g., Fig. 1B). Collectively, these results point to abnormal development of the carotid body as a major, if not the sole, mechanism for long-lasting impairment of the HVR in hyperoxia-treated animals.
Attenuation of single-unit chemoafferent responses to hypoxia in chronically hyperoxic kittens strongly suggests changes in cellular O2 sensitivity immediately after the hyperoxia treatment (Hanson et al., 1989). Whole-nerve CSN activity, on the other hand, is a function of the number of carotid chemoafferent neurons present, the number and strength of synapses between chemoreceptor cells and afferent neurons, and the activity of individual chemoreceptors. As such, whole-nerve recordings cannot distinguish between morphological changes to the carotid body and chemoafferent neurons (e.g., numbers of glomus cells, afferent neuron, and/or synapses) and functional changes (e.g., cellular O2 sensing and neurotransmission). In the following sections, we review evidence that hyperoxia influences both morphological and functional components of the carotid body and CSN. Importantly, these studies reveal that hyperoxia impacts multiple aspects of the carotid body and chemoafferent pathway.
3.1. Carotid body size
Rats and mice reared in 60% O2 for the first 1–4 postnatal weeks exhibit small carotid bodies as adults, often averaging only 25–40% of the size (i.e., volume or mass) of those collected from age-matched controls (e.g., Fuller et al., 2002; Bisgard et al., 2003; Prietol-Lloret et al., 2004; Bavis et al., 2011a) (e.g., Fig. 1); Erickson et al. (1998) mentioned that they observed similar effects in neonatal rats after one week of 30% O2, but these data have not been published. The reduction in carotid body size is evident after only four days in 60% O2 and becomes more prominent with longer durations of hyperoxia (Dmitrieff et al., 2012). Importantly, the change in carotid body volume should be functionally relevant given that it reflects a decrease in the volume occupied by the O2-sensitive glomus cells. Although the reduction in carotid body volume is partially explained by decreases in the volume occupied by non-glomic tissue (e.g., vasculature, sustentacular cells, connective tissue), there is a proportionate (Erickson et al., 1998) or slightly greater (Prieto-Loret et al., 2004) decrease in the volume occupied by glomus cells. The decrease in volume occupied by glomus cells reflects a reduced number of glomus cells (i.e., hypoplasia; see below); the size of individual cells has not been assessed quantitatively to our knowledge, but the glomus cells do not appear to be smaller (E.F. Dmitrieff and R.W. Bavis, personal observation).
Severe normobaric or hyperbaric hyperoxia (inspired PO2 of 745 – 3,800 mmHg) induces cellular injury and necrosis to the carotid body within 2–67 hours in juvenile and adult rats and cats, and this is correlated with a dose-dependent attenuation of carotid body O2 sensitivity and the HVR (Mokashi & Lahiri, 1991; Torbati et al., 1993, Di Giulio et al., 1998). It is questionable, however, whether ROS-mediated cell death contributes substantially to changes in carotid body volume during developmental exposures to 30–60% O2 (i.e., inspired PO2 ~230–460 mmHg; expected arterial PO2 ~130–250 mmHg; Penn et al., 1995). First, 60% O2 does not alter carotid body size in rats exposed to hyperoxia as adults (Dmitrieff et al., 2012); thus, unlike the severe hyperoxia discussed above, the morphological and functional effects of moderate hyperoxia appear to be limited to the developing carotid body (also see section 3.1.1, below). Second, treatment with two separate antioxidants (vitamin E and manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride; MnTMPyP) failed to prevent, or even reduce, changes to carotid body size during developmental hyperoxia (Bavis et al., 2008). Third, blood protein carbonyl concentrations (an indicator of oxidative damage) were unchanged in neonatal rats during chronic exposure to 60% O2 (Bavis et al., 2008). Finally, studies using TdT-mediated dUTP nick end labeling (TUNEL) to identify DNA fragmentation (often used as an indicator of cell death) have noted very low rates of TUNEL-positive cells in hyperoxic carotid bodies (Wang & Bisgard, 2005; Dmitrieff et al., 2012). Although Dmitrieff et al. (2012) detected a statistically significant increase in TUNEL-positive glomus cells in carotid bodies from neonatal rats (P2–P6) reared in 60% O2, this represented less than one percent of the glomus cells that were present (i.e., 0.7% of all glomus cells vs. 0.3% in age-matched controls). Moreover, the TUNEL assay also identifies cells with damaged DNA that will ultimately be repaired, so this assay likely overestimates cell death (O'Reilly, 2001). Collectively, these data suggest that ROS-mediated cell death makes a relatively small contribution to the overall change in carotid body size during developmental hyperoxia.
On the other hand, developmental hyperoxia appears to dramatically reduce cell proliferation within the carotid body (Wang & Bisgard, 2005; Dmitrieff et al., 2012). Postnatal growth of the carotid body, as indicated by changes in volume, appears to exhibit a biphasic pattern in rats: the carotid body initially decreases in size over the first four postnatal days (at least partially due to changes in the vascular compartment) and then grows rapidly for several weeks beyond that (Dmitrieff et al., 2012). When rats are reared in 60% O2, however, the carotid body exhibits the initial decrease in size but almost no growth after that. Consequently, the carotid body is significantly smaller than in age-matched controls by P4 and beyond (Dmitrieff et al., 2012). Treating rats with bromodeoxyuridine (BrdU) to label dividing cells revealed substantial decreases in the proliferation of carotid body cells during developmental hyperoxia (Wang & Bisgard, 2005; Dmitrieff et al., 2012), particularly at the earlier ages studied (P2 and P4 vs. P6). At P4, for example, only 9% of the glomus cells had undergone cell division in the preceding 24 hours, compared to 28% of the glomus cells in the age-matched control group (Dmitrieff et al., 2012). Hyperoxia appeared to inhibit cell proliferation of glomus cells and other cell types within the carotid body to a similar extent (Dmitrieff et al., 2012).
The mechanism by which hyperoxia inhibits cell proliferation in the developing carotid body is not yet known. Hyperoxia activates cell cycle “checkpoints” to arrest cell division in pulmonary epithelium, often through p53 and p21 signaling pathways (O'Reilly, 2001). However, no changes in the mRNA expression for p53 or p21, or protein expression for p21, were detected in the hyperoxic carotid body (Dmitrieff et al., 2012); p53 protein levels were not studied. Of course, these findings do not rule out the involvement of other signaling pathways that inhibit cell cycle progression or, alternatively, diminish the activity of mitogens. For example, carotid body glomus and sustentacular cells express numerous growth factors that stimulate cell proliferation and enhance cell survival in other tissues (Wang & Bisgard, 2005; Izal-Azcárate et al., 2008; Porzionato et al., 2008). Interestingly, brain-derived neurotrophic factor (BDNF) protein levels are reduced in the carotid bodies of hyperoxia-reared rats (Dmitrieff et al., 2011; Chavez-Valdez et al., 2012). The role of BDNF in proliferation of glomus cells has not been studied; however, glomus cells express TrkB receptors (Wang & Bisgard, 2005; Dmitrieff et al., 2011), consistent with an autocrine role for BDNF in carotid body growth and development.
3.1.1. Critical period for hyperoxia-induced changes in carotid body volume
As mentioned above, developmental hyperoxia reduces carotid body size while equivalent exposures (with respect to O2 level and duration) in adults have no such effect (Dmitrieff et al., 2012); in other words, there is a critical period for this plasticity during development. If hyperoxia primarily reduces carotid body size by inhibiting cell division, one might predict that the carotid body would be susceptible to morphological plasticity as long as it is actively growing. Alternatively, the period of cell proliferation may only be permissive for plasticity while the critical period is determined by another developmentally regulated process. To begin to distinguish between these possibilities, we constructed a growth curve for the carotid body and subsequently assessed the impact of hyperoxia at ages representing a range of apparent growth rates.
To prepare a carotid body growth curve, Sprague-Dawley rats (Charles River Laboratories, Portage, MI, USA) were reared in room air until their carotid bodies were collected at 2, 4, 6, 8, 10, and 12 weeks of age (n = 8–13 rats per age representing approximately equal numbers of males and females); several rats in each age group (n =3–5) were injected twice with BrdU (25 mg kg−1 per injection), 24 h and 12 h prior to collection, to label dividing cells. Following perfusion (4% paraformaldehyde in 0.1 M phosphate buffered saline, pH 7.4), carotid bodies were collected, frozen, sectioned, and processed with (1) hematoxylin and eosin staining or (2) immunohistochemistry for tyrosine hydroxylase (TH; marker for glomus cells) and BrdU; all methods were identical to those described by Dmitrieff et al. (2012). Carotid body volumes were computed by summing the area of the carotid body in each section multiplied by the section thickness (12 μm). For rats treated with BrdU, the number of cells double-labeled for TH and BrdU were counted on the two largest sections for each carotid body and averaged as an index of glomus cell division for that individual. As shown in Figure 2, the rat carotid body increased in size rapidly between 2 and 6 weeks of age but growth slowed thereafter. The slowing of carotid body growth corresponded to a strong decrease in the rate of glomus cell division with advancing age, such that very few BrdU-positive glomus cells were present at 10 or 12 weeks of age (Fig. 2); the number of BrdU-positive non-glomus cells also decreased with age (data not shown).
Fig 2.

Postnatal growth of the carotid body in Sprague-Dawley rats in terms of carotid body volume (filled symbols, n=8–13 per age) and the number of glomus cells that underwent mitosis in the preceding 24 hours (i.e., number of BrdU-positive glomus cells) (open symbols, n=3–5 per age). Although this experiment focused on growth between 2 and 12 weeks of age, carotid body volumes for 24–26 week old rats from another experiment (i.e., Control rats from Fig. 3B) are plotted for comparison. No effect of sex was detected, so data for males and females have been pooled. All values are mean ±SEM.
Based on the carotid body growth curve, we investigated the effects of 7-day exposures to 60% O2 on carotid body size at P7–P14 (high cell proliferation), P28–P35 (moderate cell proliferation), and P70–P77 (low cell proliferation); age-matched control rats were reared in room air. Carotid bodies were harvested immediately at the end of the hyperoxic exposure (all ages) or after five months of recovery in room air (P7–P14 and P28–P35 groups only) (n = 6 per treatment group at each age). Carotid body volumes were determined as described above (see also Dmitrieff et al., 2012) and then compared between hyperoxia-treated and control rats at each age using unpaired t-tests; conclusions were identical when volumes were expressed in absolute units or normalized to body mass, so only mass-specific data are presented here. Rats reared in hyperoxia during P7–P14 had smaller carotid bodies than their age-matched controls at P14 (P<0.0001; Fig. 3A), and this effect was still evident five months later (P<0.0001; Fig. 3B). In contrast, hyperoxia had no effect on carotid body size when exposures occurred later in development (all P>0.05; Fig. 3). These results demonstrate that (1) hyperoxic exposures do not need to begin at birth in order to alter carotid body size, (2) changes to carotid body size, if present, persist into adulthood, and (3) the critical period for hyperoxia-induced plasticity includes the second postnatal week but ends by four weeks of age (if not sooner). The timing of this critical period is consistent with that previously reported for long-lasting impairment of the HVR (i.e., first two postnatal weeks; Bavis et al., 2002). Moreover, the critical period for morphological plasticity closes while the carotid body is actively growing: it is not defined solely by the period of cell division. The molecular pathways that regulate this age-sensitivity remain to be determined.
Fig 3.

Mass-specific carotid body (CB) volume in rats exposed to one week of 60% O2 at postnatal ages P7–P14, P28–P35, and P70–P77 (Hyperoxia) and in age-matched rats reared in room air (Control). Volumes were determined immediately following the hyperoxic exposure (panel A) or after a five month recovery in room air (panel B). All values are mean ±SEM; n=6 per treatment group at each age. * P<0.05 vs. age-matched Control.
3.2 Carotid body innervation
In addition to the morphological effects on the carotid body itself, developmental hyperoxia disrupts the afferent link between the carotid body and the central nervous system. Neurons in the petrosal ganglion innervate the carotid body via the carotid sinus nerve (CSN) and link carotid chemoreceptors to the nucleus tractus solitarii (NTS) in the brainstem. Although the CSN contains both myelinated and unmyelinated axons, electrophysiological studies indicate that carotid chemoafferent neurons generally have unmyelinated axons in neonatal rats (conduction velocities <1 m s−1; Donnelly, 2011). Importantly, Erickson et al. (1998) observed that rats reared in 60% O2 from birth through P28 exhibited a 41% decrease in the number of unmyelinated axons in the CSN; the number of Schwann cells was also reduced, but the total number of myelinated axons was comparable to that in age-matched controls. Hyperoxia also reduced the number of TH-positive neurons in the petrosal ganglion, but not in the nearby nodose ganglion, after only 7 days (Erickson et al., 1998). Since the subset of petrosal neurons that synapse with glomus cells are dopaminergic, Erickson and colleagues interpreted this latter result as evidence for rapid and selective degeneration of neurons in the carotid chemoafferent pathway during postnatal hyperoxia. More recently, Chavez-Valdez et al. (2012) retrogradely labeled chemoafferent neurons within the petrosal ganglion in hyperoxia-treated and control rats by applying rhodamine dextran to the carotid body ex vivo. This direct approach revealed a 55% decrease in the number of chemoafferent neurons after 14 days in 60% O2, and this deficit persisted for at least 7 days after return to normoxia (and is likely permanent). The number of chemoafferent neurons was not reduced after only 7 days in 60% O2, however. To reconcile the time courses observed by Erickson et al. (1998) and Chavez-Valdez et al. (2012), a reasonable interpretation is that reduced expression of TH is among the first signs of chemoafferent inactivity and/or neuron degeneration (Katz, 2005), and is thus apparent within 7 days, while axonal loss and/or cell death progresses more slowly (>7 days).
The degeneration of chemoafferent neurons may be secondary to carotid body hypoplasia. Petrosal ganglion neuron survival and functional maturation are dependent on carotid body-derived trophic support, and this interaction is mediated by neurotrophic factors such as brain-derived neurotrophic factor (BDNF) (Hertzberg et al., 1994; Brady et al., 1999) and glial cell line-derived neurotrophic factor (GDNF) (Erickson et al., 1996, 2001). Thus, given the reduced number of glomus cells and overall smaller size of the organ, the hyperoxic carotid body may not be able to support as large of a population of afferent neurons during postnatal development. This is likely exacerbated, however, by reduced BDNF expression in the remaining glomus cells. Carotid bodies from neonatal rats reared in 60% O2 exhibited a 70% reduction in BDNF protein after only 3 days (Dmitrieff et al., 2011), and a 93% reduction after 7 days (Chavez-Valdez et al., 2012); the expression of GDNF was unchanged (Dmitrieff et al., 2011; Chavez-Valdez et al., 2012). Importantly, this change in neurotrophin availability precedes the loss of chemoafferent neurons (Chavez-Valdez et al., 2012), as would be expected if the former causes the latter. Moreover, there was no change in the number of TH-positive neurons when rats were exposed to 60% O2 as adults (Erickson et al., 1998). This is significant because afferent neurons are no longer dependent on carotid body-derived trophic after the third postnatal week (i.e., carotid body removal no longer results in loss of TH-positive neurons) (Hertzberg et al., 1994).
In addition to changes in the number of afferent neurons, it is possible that developmental hyperoxia also alters functional properties of the remaining neurons. Action potentials evoked at the carotid body take longer to arrive at the petrosal ganglion in rats reared in 60% O2 than in age-matched controls (Donnelly et al., 2005, 2009). Although axon lengths were not determined in these studies, increased conduction time may reflect a lower conduction velocity and, therefore, may be symptomatic of altered membrane properties.
3.3. Glomus cell O2 sensitivity
Single-unit carotid chemoafferent responses to hypoxia are greatly diminished in chronically hyperoxic kittens (Hanson et al., 1989) and neonatal rats (Donnelly et al., 2005, 2009; Bavis et al., 2011b). The mechanism of hypoxia transduction within the carotid body is not fully understood, but hypoxia causes an increase in glomus cell calcium which, in turn, initiates the secretion of dense cored granules that modulate afferent neural activity (Gonzalez et al., 1994; Kumar, 2007; López-Barneo et al., 2008). Importantly, glomus cells isolated from neonatal rats reared in 60% O2 exhibit smaller intracellular Ca2+ responses to hypoxic challenges (Donnelly et al. 2009; Bavis et al., 2011b). Since the same glomus cells exhibited normal intracellular Ca2+ responses to high extracellular K+, it appears that hyperoxia specifically affects pathways linking hypoxia and membrane depolarization rather than the capacity for membrane depolarization itself. These data strongly indicate that diminished single-unit chemoafferent responses to hypoxia reflect changes in glomus cell O2 sensitivity, although additional plasticity at the synapse cannot be ruled out.
Glomus cell O2 sensitivity and corresponding single-unit chemoafferent responses to hypoxia are reduced after 4–5 days in 60% O2 and decrease further with continued hyperoxic exposure (Donnelly et al., 2009; Bavis et al., 2011b). The time course for these changes is similar regardless of whether hyperoxia is initiated at birth (Bavis et al., 2011b) or at 7 days of age (Donnelly et al., 2009), with one notable exception: when initiated at 7 days of age, glomus cell and single-unit chemoafferent responses to hypoxia are enhanced after one day in hyperoxia and become progressively smaller thereafter (Donnelly et al., 2009). Consistent with increased carotid body sensitivity, the HVR is also increased in neonatal rats after one day in 60% O2 at this age (Roeser et al., 2011). Whatever the mechanism for this transient enhancement, it appears to be developmentally regulated because no such increase in O2 sensitivity was observed in rats reared in hyperoxia from birth (Bavis et al., 2011b).
A critical period for hyperoxia-induced attenuation of glomus cell O2 sensitivity, if one exists, has not yet been defined. Donnelly et al. (2005) found that hyperoxia from P14–P28 decreased single-unit carotid chemoafferent response to hypoxia in rats to a similar extent as exposures during P0–P14. This differs from the critical period previously reported for long-lasting attenuation of the HVR, which is limited to the first two postnatal weeks (i.e., no lasting impairment of the HVR following exposures P14–21 or P21–P28; Bavis et al., 2002). The discordance between these observations suggests that the reduction in glomus cell O2 sensitivity measured immediately following chronic hyperoxia does not persist into adulthood (in contrast to the permanent morphological effects elicited by hyperoxia during P0–P14; see also Section 3.4).
Indeed, hyperoxia-induced changes to glomus cell O2 sensitivity appear to be reversible upon return to normoxia. Single-unit carotid chemoafferent responses to hypoxia are severely reduced in P7 and P14 neonatal rats reared in 60% O2 continuously from birth (Donnelly et al., 2005; Bavis et al., 2011b). When hyperoxia-treated rats were returned to room air at P7, however, hypoxic responses were no different from those of age-matched controls by P14 (i.e., full recovery within 3–7 days) (Bavis et al., 2011b). Similarly, Prieto-Lloret et al. (2004) observed relatively normal chemoreceptor responses to hypoxia in rats exposed to 60% O2 for 28 days as neonates but subsequently reared in room air until studied at 3.5–4.5 months of age. Responses to hypoxia tended to be slower in hyperoxia-treated rats, but peak CSN responses (representing a mixture of single-unit, multi-unit, and whole-nerve recordings) and peak glomus cell calcium responses were not different than those observed in age-matched controls.
It should be noted, however, that Prieto-Lloret et al. (2004) had a relatively low success rate recording hypoxic responses from hyperoxia-treated rats (20% of CSN preparations vs. 80–85% in controls); when these “zero” responses were included in the data set, the average CSN responses were reduced in hyperoxia-treated rats. However, the significance of these “failed” preparations is difficult to interpret: failed preparations could reflect (1) difficulty isolating units given the lower density of chemoafferent axons (i.e., greater proportion of baroreceptors and other fiber types), (2) greater fragility of the neurons post-hyperoxia (i.e., more easily damaged during dissection), or (3) a subset of chemoreceptors in which chemosensitivity is completely abolished. Consistent with the latter hypothesis, Donnelly et al. (2005) encountered a greater frequency of “silent” chemoreceptor units in neonatal rats reared in hyperoxia compared to age-matched controls; these silent units were tentatively identified as chemoafferent fibers based on orthodromic activation via electrical stimulation of the carotid body, but they lacked spontaneous activity during hypoxia. Therefore, it is possible that some fraction of glomus cells and/or chemoafferent neurons is impaired by hyperoxia to such an extent that recovery is impossible while other cells (presumably those that retain some degree of hypoxic sensitivity immediately post-hyperoxia) recover rapidly upon return to normoxia. Prieto-Lloret et al. (2004) estimated that approximately 75% of surviving glomus cells are insensitive and/or unresponsive to hypoxia based on success rate with CSN recordings and reduced catecholamine secretion during hypoxia. The low frequency of “silent units” in single-unit chemoafferent recordings immediately post-hyperoxia suggests that this proportion may be much lower, at least after shorter hyperoxic exposures (i.e., 7–14 days vs. >28 days) (Donnelly et al., 2005; R.W. Bavis, personal observation).
At least two types of O2-sensitive channels are expressed by rat glomus cells, and both types are postulated to play a role in initiating the carotid body response to hypoxia. These include a calcium- and voltage-dependent K+ channel (BKCa) and a group of non-voltage-dependent `leak' channels (TASK) that are also selective for K+ (Gonzalez et al., 2009; Peers et al., 2010); both types are inhibited by hypoxia, thus depolarizing the glomus cell membrane. Developmental hyperoxia caused a >50% decrease in the mRNA expression for the TASK-1 channel within the carotid body after only 7 days (Bavis et al., 2011b); TASK-3 mRNA was also reduced (albeit over a slower time course), while BKCa mRNA expression actually increased. Since both nerve and calcium responses to hypoxia are reduced within 7 days of hyperoxia, this could indicate a role for TASK-1 (and/or TASK-1/3 heteromers) in hyperoxia-induced plasticity. This relationship is further supported by a correlated recovery of O2 sensitivity and TASK-1 mRNA expression upon return to room air (Bavis et al., 2011b). It remains to be determined whether these changes in mRNA expression produce a corresponding reduction in TASK channel density in the glomus cell membrane, and whether this diminishes O2 sensitivity. Indeed, reduced TASK channel density might be expected to increase glomus cell excitability, while normal responses to high extracellular K+ (Prieto-Lloret et al., 2004; Donnelly et al., 2009; Bavis et al., 2011b) suggests relatively normal K+ conductance overall. However, these apparent contradictions may be reconciled if decreases in O2-sensitive K+ channels were compensated by increased expression of O2-insensitive leak channels.
3.4. Potential mechanisms for carotid body plasticity during developmental hyperoxia
Figure 4 summarizes the (known) impact of postnatal hyperoxia on the developing carotid body, as described in the preceding sections. The net effect of these changes is to reduce carotid body input to the CNS, ultimately diminishing the HVR (and likely other carotid body-mediated hypoxic defenses such as arousal from sleep). Upon return to normoxia, there is a rapid recovery of O2 sensitivity in individual glomus cells, although a subset of cells may persist that is incapable of responding to hypoxia. Since changes to the numbers of glomus cells and afferent neurons appear permanent, this morphological plasticity likely explains much of the life-long attenuation of the whole-nerve CSN response to hypoxia and attenuation of the HVR (Fuller et al., 2002).
Fig. 4.

Developmental hyperoxia impairs the hypoxic ventilatory response (HVR) through its effects the carotid body, including carotid body hypoplasia, carotid sinus nerve (CSN) axon degeneration, and diminished glomus cell O2 sensitivity; changes to carotid body size and innervation persist into adulthood, while chemoreceptor O2 sensitivity may recover after return to normoxia. Current evidence suggests that CSN axon degeneration is secondary to carotid body hypoplasia (and associated loss of trophic support), but more direct effects of hyperoxia on chemoafferent neurons cannot be excluded (dotted line). Hypothesized pathways by which hyperoxia influences carotid body development are also described (text adjacent to arrows); although some pathways have been supported experimentally (e.g., changes in BDNF expression), question marks indicate that causality has not been established yet for any of these pathways.
The cellular pathways by which hyperoxia influences the structure and function of the carotid body are largely unknown (see arrows in Fig. 4), although altered expression of O2-sensitive K+ channels and neurotrophic factors such as BDNF have been implicated. Current evidence argues against direct oxidative damage to carotid body cells because the morphological effects are primarily localized to the carotid chemoafferent pathway (i.e., no effect on the nearby nodose ganglion and superior cervical ganglion; Erickson et al., 1998; Prieto-Lloret et al., 2004; Wang & Bisgard, 2005) and because antioxidants thus far have proven ineffective at preventing hyperoxia-induced plasticity (Bavis et al., 2008). It is possible that high O2 levels influence transcription of key genes, but no direct evidence is available to support or refute this hypothesis (reviewed in Bavis, 2005). Finally, it has been proposed that at least some of the effects of hyperoxia on respiratory control development are linked to carotid body inactivity (Ling et al., 1997a; Bavis, 2005; Chavez-Valdez et al., 2012). High arterial PO2 levels should decrease spontaneous carotid body depolarization, thereby altering activity-dependent gene expression and autocrine / paracrine signaling. For example, the transcription, translation, and release of BDNF are regulated in an activity-dependent manner in many neurons (Brady et al., 1999; Balkowiec & Katz, 2000; Cohen-Cory et al., 2010; Lau et al., 2010; Chavez-Valdez et al., 2012), and BDNF also modulates activity-dependent postnatal maturation of carotid chemoafferent neurons (Katz, 2005). Consistent with this “inactivity” hypothesis, concomitant exposure to intermittent hypercapnia (alternating hours of 0 and 7.5% CO2) during postnatal hyperoxia improved the HVR relative to rats reared in hyperoxia alone (Bavis et al., 2007). The therapeutic effect of intermittent hypercapnia may reflect carotid body stimulation, although this mechanism requires further study.
4. Induced recovery of carotid body function
The effects of developmental hyperoxia on carotid body size, whole-nerve CSN responses to hypoxia, and HVR persist into adulthood and appear to be permanent (Ling et al., 1997a; Fuller et al., 2002). The carotid body and other components of the respiratory control system retain the capacity for plasticity, however, which may provide a mechanism to enhance residual respiratory function. In adult rats that had been reared in 60% O2 for the first postnatal month, Fuller et al. (2001) found that one week of chronic sustained hypoxia (12% O2; CSH) or chronic intermittent hypoxia (cycling between 21% and 12% O2 at 5-min intervals, 12 h d−1; CIH) increased phrenic nerve responses to acute isocapnic hypoxia relative to hyperoxia-treated rats maintained in room air. The CSH rats also exhibited significantly larger carotid bodies, but their carotid bodies were still smaller than those of control rats reared in room air. The effects of CIH persisted for at least one week; longer recovery periods and the duration of CSH effects were not tested (Fuller et al., 2001). CSH applied during the neonatal period (i.e., P8–P14, following hyperoxia P0–P7) did not improve the CSN or phrenic nerve responses to hypoxia assessed in adulthood, suggesting that any improvement in respiratory function was short-lived (Bavis et al., 2004).
CSH and CIH both alter hypoxic responses in normal adult rats. CSH increases the HVR (i.e., ventilatory acclimatization) by enhancing both carotid body O2 sensitivity and the gain of the central neural integration of chemoafferent input (Powell et al., 2000; Powell, 2007). CSH also transiently increases carotid body volume (Kusakabe et al., 2004; Wang et al., 2008). Importantly, it appears that hyperoxia-treated rats are capable of qualitatively identical carotid body and ventilatory acclimatization in CSH (Wenninger et al., 2006). Likewise, CIH increases carotid body responses to hypoxia (Prabhakar et al., 2007) and enhances the gain of central neural integration of chemoafferent input (Ling et al., 2001), ultimately increasing respiratory responses to acute hypoxia. Thus, it is likely that CSH and CIH elicit transient improvement of respiratory function in hyperoxia-treated rats through the same mechanisms that CSH and CIH enhance hypoxic responses in normal rats rather than reversing the specific effects of developmental hyperoxia per se.
5. Does developmental hyperoxia elicit plasticity outside the carotid body?
Thus far we have attributed reduced HVR (and reduced normoxic ventilation in neonates) entirely to impairment of the carotid body and its first order afferent neurons. Several observations have been used to argue against possible contributions of other components of the respiratory system to diminished HVR in hyperoxia-treated rats. First, developmental hyperoxia has no lasting effect on the hypercapnic ventilatory response in rats (Ling et al., 1996; Prieto-Lloret et al., 2004) or mice (Dauger et al., 2003), suggesting that there is no generalized impairment of ventilatory control. Similarly, rats and mice reared in hyperoxia can elicit near normal maximal responses to acute hypoxia, although they require a stronger hypoxic stimulus to achieve this response (i.e., blunting reflects a leftward shift in the ventilation–PO2 relationship) (Dauger et al., 2003; Prieto-Lloet et al., 2004; Bavis et al., 2011a). Finally, there currently is no evidence that decreased function of the central nervous system contributes to the attenuated HVR. Ling et al. (1997b) and Fuller et al. (2002) studied the central integration of carotid chemoafferent inputs in anesthetized rats by electrically stimulating the CSN and monitoring the frequency and amplitude of integrated respiratory bursts along the phrenic nerve. Over a range of stimulation frequencies, both studies found phrenic responses to CSN stimulation to be virtually identical between hyperoxia-treated rats (reared in 60% O2 through P28) and control rats.
A normal input-output relationship for electrical CSN stimulation is somewhat surprising, however, since the absolute number of afferent neurons available to be stimulated is smaller in hyperoxia-treated rats (Erickson et al., 1998). Moreover, Chavez-Valdez et al. (2012) recently reported an increase in pro-apoptotic markers and a reduction in TH expression in the NTS following 7–14 days in 60% O2, which could indicate a loss of both first and second order neurons during developmental hyperoxia. One interpretation of the normal input-output relationship is that there is redundancy in the chemoafferent pathway such that activation of a subset of axons can produce responses equivalent to those produced by activation of the full population. Indeed, several studies have reported that unilateral carotid body denervation has minimal effects on the HVR while bilateral denervation abolishes, or nearly abolishes, the HVR (Busch et al., 1983; Cragg and Khrisanapant, 1994; but see Fatemian et al., 2003). Alternatively, central neural integration of carotid chemoafferent input might actually be enhanced in adult rats raised in hyperoxia, perhaps compensating for a reduction in tonic synaptic activation (i.e., homeostatic plasticity; Turrigiano and Nelson, 2004; Kline, 2008).
The time course for changes in the HVR of neonatal rats provides additional, albeit indirect, evidence for central neural plasticity during developmental hyperoxia. As described above, carotid body size and chemoreceptor O2 sensitivity are already noticeably reduced after four days in 60% O2, but the early, carotid body-mediated phase of the HVR is normal (if not somewhat enhanced; 0.05<P<0.10) at the same age (Fig. 5). This pattern suggests increased excitability of the central limb of the hypoxic chemoreflex. Consistent with this hypothesis, hyperoxia-treated rats exhibit a sustained increase in ventilation during hypoxia (versus a biphasic HVR) at an earlier age (≤P4) than rats reared in room air (>P7) (Bavis et al., 2010). Ventilatory decline during the late phase of the biphasic HVR is not fully understood (Bissonnette, 2000; Teppema and Dahan, 2010), but it is usually attributed to inhibitory processes in the CNS: central hypoxia may directly depress CNS function and/or inhibitory neurons may modulate excitatory input from the peripheral chemoreceptors. The latter process appears particularly influential since hypoxic ventilatory decline was no longer observed in neonatal rabbits following carotid body denervation (Schramm and Grunstein, 1987). If ventilatory decline is dependent on carotid body input to the CNS, the lack of a biphasic HVR in hyperoxia-treated neonatal rats could reflect diminished chemoafferent activity. This is unlikely to explain the complete lack of a biphasic HVR, however, since hyperoxia-treated rats continue to show modest increases in chemoafferent activity and ventilation in hypoxia (which are lacking in denervated preparations). Instead, the lack of the biphasic HVR in hyperoxia-treated rats may indicate an earlier shift (i.e., developmental plasticity) in the balance between inhibitory and excitatory neuromodulation during hypoxia in the developing NTS and/or other brainstem regions (Bissonnette, 2000; Simakajornboon and Kuptanon, 2005). Presumably the effects of diminished carotid body function on the HVR become more prominent with advancing age due to (1) further impairment of carotid body size and O2 sensitivity in hyperoxia-treated rats and (2) increased HVR in untreated control rats associated with normal postnatal maturation, ultimately yielding an attenuated HVR in the hyperoxia-treated animals (Fig. 5).
Fig. 5.
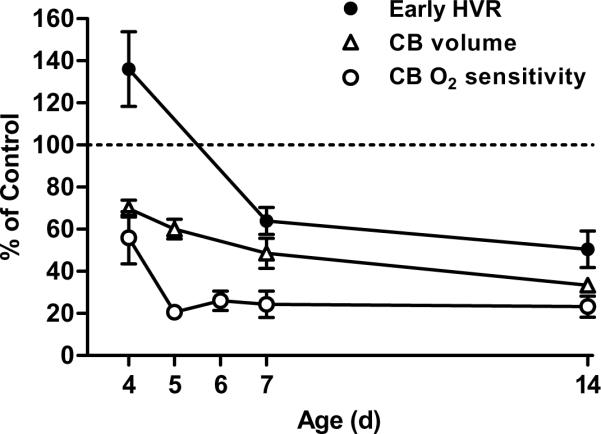
Time course for changes in carotid body (CB) volume, CB O2 sensitivity, and the hypoxic ventilatory response (HVR) in neonatal rats reared from birth in 60% O2. Carotid body O2 sensitivity is based on the peak single-unit chemoreceptor activity during severe hypoxia (0% O2), while HVR represents the early phase of the response to 12% O2 (i.e., first minute of hypoxia). Values (mean ± SEM) are expressed as a percentage of those measured for age-matched rats reared in room air (i.e., “Control” rats); n=6 per age for CB volume, n= 6–16 per age for CB O2 sensitivity, and n=15–17 per age for HVR. Data were compiled from previously published studies (Donnelly et al., 2005; Bavis et al., 2010, 2011b; Dmitrieff et al., 2012).
Although aortic bodies and other O2-sensitive peripheral chemoreceptors outside the carotid body normally contribute little to the HVR in adults animals (Martin-Body et al., 1985), the activity of these extracarotid chemoreceptors increases following bilateral carotid body denervation and may restore the HVR (Forster, 2003). However, there currently is no evidence that extracarotid chemoreceptors compensate for reduced carotid body function after developmental hyperoxia. Indeed, vagotomy (which severs afferents from the aortic bodies and abdominal chemoreceptors) does not alter residual hypoxic sensitivity in hyperoxia-treated rats (Ling et al., 1997c). In contrast, hypoxic sensitivity is abolished following bilateral section of the CSN (Ling et al., 1997c), suggesting that the carotid body is the principal determinant of the residual HVR. It is possible that the remaining carotid body function is sufficient to inhibit compensatory upregulation of extracarotid chemoreceptors (Serra et al., 2002). Alternatively, developmental hyperoxia may impair the function of all O2-sensitive peripheral chemoreceptors, not just those in the carotid body.
6. Prenatal hyperoxia and carotid body development
In placental mammals, birth is associated with a rapid rise in arterial O2. This “relative hyperoxia” is recognized as an important stimulus for physiological changes that mark the transition from fetal to neonatal life, including postnatal maturation of glomus cell O2 sensitivity (Carroll, 2003). The carotid body normally exhibits low O2 sensitivity at birth, but this sensitivity increases progressively over the next few days to weeks (Carroll and Kim, 2005; Donnelly, 2005). Blanco et al. (1988) ventilated fetal lambs with relatively hyperoxic gas mixtures in utero for 24–31 hours (raising mean arterial PO2 from 26 to 180 mmHg). Carotid body O2 sensitivity (single- and few-fiber CSN recordings) was substantially increased following this protocol when compared to sheep maintained at normal fetal PO2, suggesting premature resetting of O2 sensitivity (Blanco et al., 1988). Conversely, postnatal hypoxia (mimicking PO2 in utero) delays resetting of carotid body O2 sensitivity at the level of the glomus cell in rats (Sterni et al., 1999). Thus, brief periods (~one day) of prenatal hyperoxia can alter the timing and/or rate of carotid body development (i.e., heterokairy; cf. Spicer & Rundle, 2007).
Relatively little is known about the effects of chronic prenatal hyperoxia on the control of breathing in mammals. This is partly explained by the experimental challenges posed by prenatal manipulations in placental mammals: mothers may buffer developing offspring from experimental treatments and/or introduce confounding maternal effects. Moreover, the elegant experimental design used by Blanco et al. (1988) to ventilate fetal lambs in utero may not be practical for studies of longer duration. Egg-laying species have been used to circumvent these obstacles since environmental O2 levels of developing embryos can be controlled in incubators (i.e., prenatal = pre-hatching). Like mammals, quail (Bavis and Simons, 2008) and chickens (Mortola, 2011) exhibited blunted HVR following developmental hyperoxia, and these effects may persist into adulthood (Bavis and Simons, 2008); hypercapnic ventilatory responses were unaffected (Mortola, 2011). It is likely that the diminished HVR in these birds is explained by abnormal carotid body function, although this has not been tested directly. Importantly, chicken embryos were exposed to hyperoxia (40 or 60% O2) for the last week of incubation through hatching, and quail were exposed to hyperoxia (60% O2) for 2–4 weeks spanning both the prenatal and postnatal periods or only during the prenatal period; quail exposed to hyperoxia primarily during postnatal development exhibited normal HVR (Bavis and Simons, 2008). Thus, these findings suggest that the critical period for hyperoxia-induced plasticity extends prenatally in these precocial bird species. Moreover, this critical period is consistent with the timing of embryonic carotid body development (Fontaine, 1973) and the prenatal maturation of carotid body O2 sensitivity in birds (Menna and Mortola, 2003; Mortola, 2004).
7. Clinical significance
Developmental hyperoxia is a useful experimental model to explore fundamental questions of O2 chemotransduction and perhaps, more generally, to study activity-dependent development of sensory systems. Whether the effects of hyperoxia on the developing respiratory control system are also clinically significant remains an important question. Supplemental O2 is among the most common therapies for preterm and very low birth weight infants, but there is much uncertainty as to what O2 levels are “normal” and/or safe for these infants (Tin and Gupta, 2007; Finer and Leone, 2009). Fetal development is programmed to occur normally at relatively low O2 levels in utero, and mechanisms for dealing with oxidative stress are poorly developed in preterm infants. Thus, while O2 therapy may be critical to the survival of infants in respiratory distress, higher arterial PO2 are linked to conditions such as bronchopulmonary dysplasia (BPD) and retinopathy of prematurity (ROP) (Tin and Gupta, 2007; Sola et al., 2008; Finer and Leone, 2009). To minimize these adverse outcomes, target pulse oximeter O2 saturations (SpO2) of 85–93 or 95% are commonly used in neonatal intensive care units. Despite awareness of the potential dangers of hyperoxia, preterm infants are hyperoxic relative to their intended SpO2 range for 30–40% of the time that they are receiving O2 therapy (Hagadorn et al., 2006; Claure and Bancalari, 2009; Finer and Leone, 2009), with the measured SpO2 fluctuating around the target values. When sampled directly, arterial PO2 values are frequently greater than 80 mmHg (often considered the acceptable limit for neonatal PO2) when SpO2>93% (e.g., mean PO2=107 mmHg, range 34–316 mmHg; Castillo et al., 2008). Taken together, these observations indicate that many preterm infants experience chronic intermittent hyperoxia while on supplemental O2. Importantly, initial studies in rats reared in chronic intermittent hyperoxia indicate deficits in HVR qualitatively similar to those produced by sustained hyperoxia (Bavis et al., 2007; Tobin and Bavis, 2012); whether this plasticity reflects abnormal carotid body function has not been confirmed yet.
Preterm infants that have received supplemental O2 often exhibit blunted ventilatory responses to changes in inspired O2 (Calder et al., 1994; Katz-Salamon and Lagercrantz, 1994; Katz-Salamon et al., 1996), and this is related to the duration of their O2 treatment (Katz-Salamon and Lagercrantz, 1994). Moreover, recent studies indicate that these infants continue to express abnormal HVR into adulthood (Beshish et al., 2012). It is not possible to attribute this impaired chemosensitivity directly to O2 therapy (and associated hyperoxia) versus other underlying factors in these uncontrolled studies, but the experimental evidence from animal models reviewed above is fully consistent with a causal relationship. At a minimum, these data indicate a need to assess the potential impact of developmental hyperoxia on carotid body function in infants receiving supplemental oxygen.
Acknowledgments
The new experiments described in Section 3.1.1 (Critical period for hyperoxia-induced changes in carotid body volume) and Figs. 2 and 3 were supported by the Bates College Faculty Development Fund and by grants from the National Center for Research Resources (P20 RR-016463-12) and the National Institute of General Medical Sciences (P20 GM-103423) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J. Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW. Developmental plasticity of the hypoxic ventilatory response after perinatal hyperoxia and hypoxia. Respir. Physiol. Neurobiol. 2005;149:287–299. doi: 10.1016/j.resp.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Bavis RW, Mitchell GS. Long-term effects of the perinatal environment on respiratory control. J. Appl. Physiol. 2008;104:1220–1229. doi: 10.1152/japplphysiol.01086.2007. [DOI] [PubMed] [Google Scholar]
- Bavis RW, Simons JC. Developmental hyperoxia attenuates the hypoxic ventilatory response in Japanese quail (Coturnix japonica) Respir. Physiol. Neurobiol. 2008;164:411–418. doi: 10.1016/j.resp.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Olson EB, Jr., Mitchell GS. Critical developmental period for hyperoxia-induced blunting of hypoxic phrenic responses in rats. J. Appl. Physiol. 2002;92:1013–1018. doi: 10.1152/japplphysiol.00859.2001. [DOI] [PubMed] [Google Scholar]
- Bavis RW, Olson EB, Jr., Vidruk EH, Bisgard GE, Mitchell GS. Level and duration of developmental hyperoxia influence impairment of hypoxic phrenic responses in rats. J. Appl. Physiol. 2003;95:1550–1559. doi: 10.1152/japplphysiol.01043.2002. [DOI] [PubMed] [Google Scholar]
- Bavis RW, Olson EB, Jr., Mitchell GS, Bisgard GE. Can developmental hypoxia prevent impairment of the adult hypoxic ventilatory response after developmental hyperoxia in rats? FASEB J. 2004;18:A336. Abstract. [Google Scholar]
- Bavis RW, Russell KER, Simons JC, Otis JP. Hypoxic ventilatory responses in rats after hypercapnic hyperoxia and intermittent hyperoxia. Respir. Physiol. Neurobiol. 2007;155:193–202. doi: 10.1016/j.resp.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Bavis RW, Wenninger JM, Miller BM, Dmitrieff EK, Olson EB, Jr., Mitchell GS, Bisgard GE. Respiratory plasticity after perinatal hyperoxia is not prevented by antioxidant supplementation. Respir. Physiol. Neurobiol. 2008;160:301–312. doi: 10.1016/j.resp.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Young KM, Barry KJ, Boller MR, Kim E, Klein PM, Ovrutsky AR, Rampersad DA. Chronic hyperoxia alters the early and late phases of the hypoxic ventilatory response in neonatal rats. J. Appl. Physiol. 2010;109:796–803. doi: 10.1152/japplphysiol.00510.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Dmitrieff EF, Young KM, Piro SE. Hypoxic ventilatory response of adult rats and mice after developmental hyperoxia. Respir. Physiol. Neurobiol. 2011a;177:342–346. doi: 10.1016/j.resp.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Kim I, Pradhan N, Nawreen N, Dmitrieff EF, Carroll JL, Donnelly DF. Recovery of carotid body O2 sensitivity following chronic postnatal hyperoxia in rats. Respir. Physiol. Neurobiol. 2011b;177:47–55. doi: 10.1016/j.resp.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshish A, Bates ML, Farrell ET, Pegelow DF, Eldridge MW. Blunted hypoxic ventilatory drive in adult humans with a history of premature birth. FASEB J. 2012;26:1090.11. Abstract. [Google Scholar]
- Bisgard GE, Olson EB, Jr., Wang Z-Y, Bavis RW, Fuller DD, Mitchell GS. Adult carotid chemoafferent responses to hypoxia after 1, 2, and 4 wk of postnatal hyperoxia. J. Appl. Physiol. 2003;95:946–952. doi: 10.1152/japplphysiol.00985.2002. [DOI] [PubMed] [Google Scholar]
- Blanco CE, Hanson MA, McCooke HB. Effects on carotid chemoreceptor resetting of pulmonary ventilation in the fetal lamb in utero. J. Dev. Physiol. 1988;10:167–174. [PubMed] [Google Scholar]
- Bissonnette JM. Mechanisms regulating hypoxic respiratory depression during fetal and postnatal life. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000;278:R1391–R1400. doi: 10.1152/ajpregu.2000.278.6.R1391. [DOI] [PubMed] [Google Scholar]
- Borday C, Wrobel L, Fortin G, Champagnat J, Thaëron-Antôno C, Thoby-Brisson M. Developmental gene control of brainstem function: views from the embryo. Prog. Biophys. Mol. Biol. 2004;84:89–106. doi: 10.1016/j.pbiomolbio.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Borday C, Chatonnet F, Thoby-Brisson M, Champagnat J, Fortin G. Neural tube patterning by Krox20 and emergence of a respiratory control. Respir. Physiol. Neurobiol. 2005;149:63–72. doi: 10.1016/j.resp.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Brady R, Zaidi SI, Mayer C, Katz DM. BDNF is a target-derived survival factor for arterial baroreceptor and chemoafferent primary sensory neurons. J. Neurosci. 1999;19:2131–2142. doi: 10.1523/JNEUROSCI.19-06-02131.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch MA, Bisgard GE, Mesina JE, Forster HV. The effects of unilateral carotid body excision on ventilatory control in goats. Respir. Physiol. 1983;54:353–361. doi: 10.1016/0034-5687(83)90078-6. [DOI] [PubMed] [Google Scholar]
- Calder NA, Williams BA, Smyth J, Boon AW, Kumar P, Hanson MA. Absence of ventilatory response to alternating breaths of mild hypoxia and air in infants who have had bronchopulmonary dysplasia: implications for the risk of sudden infant death. Pediatr. Res. 1994;35:677–681. doi: 10.1203/00006450-199406000-00011. [DOI] [PubMed] [Google Scholar]
- Carroll JL. Developmental plasticity in respiratory control. J. Appl. Physiol. 2003;94:375–389. doi: 10.1152/japplphysiol.00809.2002. [DOI] [PubMed] [Google Scholar]
- Carroll JL, Kim I. Postnatal development of carotid body glomus cell O2 sensitivity. Respir. Physiol. Neurobiol. 2005;149:201–215. doi: 10.1016/j.resp.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Castillo A, Sola A, Baquero H, Neira F, Alvis R, Deulofeut R, Critz A. Pulse oxygen saturation levels and arterial oxygen tension values in newborns receiving oxygen therapy in the neonatal intensive care unit: is 85% to 93% an acceptable range? Pediatrics. 2008;121:882–889. doi: 10.1542/peds.2007-0117. [DOI] [PubMed] [Google Scholar]
- Chavez-Valdez R, Mason A, Nunes AR, Northington FJ, Tankersley C, Ahlawat R, Johnson SM, Gauda EB. Effect of hyperoxic exposure during early development on neurotrophin expression in the carotid body and nucleus tractus solitarii. J. Appl. Physiol. 2012;112:1762–1772. doi: 10.1152/japplphysiol.01609.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claure N, Bancalari E. Automated respiratory support in newborn infants. Semin. Fetal Neonatal. Med. 2009;14:35–41. doi: 10.1016/j.siny.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Kidane AH, Shirkey NJ, Marshak S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 2010;70:271–288. doi: 10.1002/dneu.20774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg PA, Khrisanapant W. Is the second carotid body redundant? Adv. Exp. Med. Biol. 1994;360:297–299. doi: 10.1007/978-1-4615-2572-1_51. [DOI] [PubMed] [Google Scholar]
- Dauger S, Ferkdadji L, Saumon G, Vardon G, Peuchmaur M, Gaultier C, Gallego J. Neonatal exposure to 65% oxygen durably impairs lung architecture and breathing pattern in adult mice. Chest. 2003;123:530–538. doi: 10.1378/chest.123.2.530. [DOI] [PubMed] [Google Scholar]
- Di Giulio C, Di Muzio M, Sabatino G, Spoletini L, Amicarelli F, Di Ilio C, Modesti A. Effect of chronic hyperoxia on young and old rat carotid body ultrastructure. Exp. Gerontol. 1998;33:319–329. doi: 10.1016/s0531-5565(97)00097-1. [DOI] [PubMed] [Google Scholar]
- Dmitrieff EF, Wilson JT, Dunmire KB, Bavis RW. Chronic hyperoxia alters the expression of neurotrophic factors in the carotid body of neonatal rats. Respir. Physiol. Neurobiol. 2011;175:220–227. doi: 10.1016/j.resp.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitrieff EF, Piro SE, Broge TA, Jr., Dunmire KB, Bavis RW. Carotid body growth during chronic postnatal hyperoxia. Respir. Physiol. Neurobiol. 2012;180:193–203. doi: 10.1016/j.resp.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DF. Development of carotid body/petrosal ganglion response to hypoxia. Respir. Physiol. Neurobiol. 2005;149:191–199. doi: 10.1016/j.resp.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Donnellr DF. Developmental changes in the magnitude and activation characteristics of Na+ currents of petrosal neurons projecting to the carotid body. Respir. Physiol. Neurobiol. 2011;177:284–293. doi: 10.1016/j.resp.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Donnelly DF, Kim I, Carle C, Carroll JL. Perinatal hyperoxia for 14 days increases nerve conduction time and the acute unitary response to hypoxia of rat carotid body chemoreceptors. J. Appl. Physiol. 2005;99:114–119. doi: 10.1152/japplphysiol.01009.2004. [DOI] [PubMed] [Google Scholar]
- Donnelly DF, Bavis RW, Kim I, Dbouk HA, Carroll JL. Time-course of alterations in pre- and post-synaptic chemoreceptor function during developmental hyperoxia. Respir. Physiol. Neurobiol. 2009;168:189–197. doi: 10.1016/j.resp.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden GJ, Hanson MA. Maturation of the respiratory response to acute hypoxia in the newborn rat. J. Physiol. 1987;392:1–9. doi: 10.1113/jphysiol.1987.sp016765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, Yancopoulos G, Katz DM. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. J. Neurosci. 1996;16:5361–5371. doi: 10.1523/JNEUROSCI.16-17-05361.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JT, Mayer C, Jawa A, Ling L, Olson EB, Jr., Vidruk EH, Mitchell GS, Katz DM. Chemoafferent degeneration and carotid body hypoplasia following chronic hyperoxia in newborn rats. J. Physiol. 1998;509:519–526. doi: 10.1111/j.1469-7793.1998.519bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JT, Brosenitsch TA, Katz DM. Brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor are required simultaneously for survival of dopaminergic primary sensory neurons in vivo. J. Neurosci. 2001;21:581–589. doi: 10.1523/JNEUROSCI.21-02-00581.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemian M, Nieuwenhuijs DJ, Teppema LJ, Meinesz S, van der Mey AG, Dahan A, Robbins PA. The respiratory response to carbon dioxide in humans with unilateral and bilateral resections of the carotid bodies. J. Physiol. 2003;549:965–973. doi: 10.1113/jphysiol.2003.042259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finer N, Leone T. Oxygen saturation monitoring for the preterm infant: the evidence basis for current practice. Pediatr. Res. 2009;65:375–380. doi: 10.1203/PDR.0b013e318199386a. [DOI] [PubMed] [Google Scholar]
- Fontaine J. Development of the carotid body and the ultimobranchial body in birds. Monoamine content and L-dopa uptake capacity in glomic and calcitonin cells during embryonic development. Arch. Anat. Microsc. Morphol. Exp. 1973;62:89–100. [PubMed] [Google Scholar]
- Forster HV. Plasticity in the control of breathing following sensory denervation. J. Appl. Physiol. 2003;94:784–794. doi: 10.1152/japplphysiol.00602.2002. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Wang Z-Y, Ling L, Olson EB, Jr., Bisgard GE, Mitchell GS. Induced recovery of hypoxic phrenic responses in adult rats exposed to hyperoxia for the first month of life. J. Physiol. 2001;536:917–926. doi: 10.1111/j.1469-7793.2001.00917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Bavis RW, Vidruk EH, Wang Z-Y, Olson EB, Jr., Bisgard GE, Mitchell GS. Life-long impairment of hypoxic phrenic responses in rats following 1 month of developmental hyperoxia. J. Physiol. 2002;538:947–955. doi: 10.1113/jphysiol.2001.012908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol. Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Vaquero LM, López-López JR, Pérez-García MT. Oxygen-sensitive potassium channels in chemoreceptor cell physiology: making a virtue of necessity. Ann. N.Y. Acad. Sci. 2009;1177:82–88. doi: 10.1111/j.1749-6632.2009.05037.x. [DOI] [PubMed] [Google Scholar]
- Hagadorn JI, Furey AM, Nghiem TH, Schmid CH, Phelps DL, Pillers DA, Cole CH, AVIOx Study Group Achieved versus intended pulse oximeter saturation in infants born less than 28 weeks' gestation: the AVIOx study. Pediatrics. 2006;118:1574–1582. doi: 10.1542/peds.2005-0413. [DOI] [PubMed] [Google Scholar]
- Han F, Strohl KP. Inheritance of ventilatory behavior in rodent models. Respir. Physiol. 2000;121:247–256. doi: 10.1016/s0034-5687(00)00132-8. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Eden GJ, Nijhuis JG, Moore PJ. Peripheral chemoreceptors and other oxygen sensors in the fetus and newborn. In: Lahiri S, Forster RE, Davies RO, Pack AI, editors. Chemoreceptors and Reflexes in Breathing: Cellular and Molecular Aspects. Oxford University Press; New York: 1989. pp. 113–120. [Google Scholar]
- Hertzberg T, Fan G, Finley JCW, Erickson JT, Katz DM. BDNF supports mammalian chemoafferent neurons in vitro and following peripheral target removal in vivo. Dev. Biol. 1994;166:801–811. doi: 10.1006/dbio.1994.1358. [DOI] [PubMed] [Google Scholar]
- Izal-Azcárate A, Belzunegui S, San Sebastián W, Garrido-Gil P, Vázquez-Claverie M, López B, Marcilla I, Luquin MA. Immunohistochemical characterization of the rat carotid body. Respir. Physiol. Neurobiol. 2008;161:95–99. doi: 10.1016/j.resp.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Katz DM. Regulation of respiratory neuron development by neurotrophic and transcriptional signaling mechanisms. Respir. Physiol. Neurobiol. 2005;149:99–109. doi: 10.1016/j.resp.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Katz-Salamon M, Lagercrantz H. Hypoxic ventilatory defense in very preterm infants: attenuation after long term oxygen treatment. Arch. Dis. Child. Fetal Neonatal. Ed. 1994;70:F-90–95. doi: 10.1136/fn.70.2.f90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz-Salamon M, Eriksson M, Jonnson B. Development of chemoreceptor function in infants with chronic lung disease (CLD) with initially lacking hyperoxic response. Arch. Dis. Child. 1996;75:4–9. doi: 10.1136/fn.75.1.f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD. Plasticity in glutamatergic NTS neurotransmission. Respir. Physiol. Neurobiol. 2008;164:105–111. doi: 10.1016/j.resp.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. Sensing hypoxia in the carotid body: from stimulus to response. Essays Biochem. 2007;43:43–60. doi: 10.1042/BSE0430043. [DOI] [PubMed] [Google Scholar]
- Kusakabe T, Hirakawa H, Oikawa S, Matsuda H, Kawakami T, Takenaka T, Hayashida Y. Morphological changes in the rat carotid body 1, 2, 4, and 8 weeks after the termination of chronically hypocapnic hypoxia. Histol. Histopathol. 2004;19:1133–1140. doi: 10.14670/HH-19.1133. [DOI] [PubMed] [Google Scholar]
- Lau AG, Irier HA, Gu J, Tian D, Ku L, Liu G, Xia M, Fritsch B, Zheng JQ, Dingledine R, Xu B, Lu B, Feng Y. Distinct 3'UTRs differentially regulate activity-dependent translation of brain-derived neurotrophic factor (BDNF) Proc. Natl. Acad. Sci. USA. 2010;107:15945–15950. doi: 10.1073/pnas.1002929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr., Vidruk EH, Mitchell GS. Attenuation of the hypoxic ventilatory response in adult rats following one month of perinatal hyperoxia. J. Physiol. 1996;495:561–571. doi: 10.1113/jphysiol.1996.sp021616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr., Vidruk EH, Mitchell GS. Developmental plasticity of the hypoxic ventilatory response. Respir. Physiol. 1997a;110:261–268. doi: 10.1016/s0034-5687(97)00091-1. [DOI] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr., Vidruk EH, Mitchell GS. Integrated phrenic responses to carotid afferent stimulation in adult rats following perinatal hyperoxia. J. Physiol. 1997b;500:787–796. doi: 10.1113/jphysiol.1997.sp022058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr., Vidruk EH, Mitchell GS. Phrenic responses to isocapnic hypoxia in adult rats following perinatal hyperoxia. Respir. Physiol. 1997c;109:107–116. doi: 10.1016/s0034-5687(97)00045-5. [DOI] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J. Neurosci. 2001;21:5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barneo J, Ortega-Sáenz P, Pardal R, Pascual A, Piruat JI. Carotid body oxygen sensing. Eur. Respir. J. 2008;32:1386–1398. doi: 10.1183/09031936.00056408. [DOI] [PubMed] [Google Scholar]
- Martin-Body RL, Robson GJ, Sinclair JD. Respiratory effects of sectioning the carotid sinus glossopharyngeal and abdominal vagal nerves in the awake rat. J. Physiol. 1985;361:35–45. doi: 10.1113/jphysiol.1985.sp015631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menna TM, Mortola JP. Ventilatory chemosensitivity in the chick embryo. Respir. Physiol. Neurobiol. 2003;137:69–79. doi: 10.1016/s1569-9048(03)00109-5. [DOI] [PubMed] [Google Scholar]
- Milsom WK, Burleson ML. Peripheral arterial chemoreceptors and the evolution of the carotid body. Respir. Physiol. Neurobiol. 2007;157:4–11. doi: 10.1016/j.resp.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J. Appl. Physiol. 2003;94:358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Mokashi A, Lahiri S. Aortic and carotid body chemoreception in prolonged hyperoxia in the cat. Respir. Physiol. 1991;86:233–243. doi: 10.1016/0034-5687(91)90083-u. [DOI] [PubMed] [Google Scholar]
- Mortola JP. Ventilatory response to hyperoxia in the chick embryo. Comp. Biochem. Physiol. A. 2004;137:723–730. doi: 10.1016/j.cbpb.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Mortola JP. Prenatal hyperoxia blunts the hypoxic ventilatory chemosensitivity of the 1-day old chicken hatchling. Respir. Physiol. Neurobiol. 2011;178:352–356. doi: 10.1016/j.resp.2011.06.015. [DOI] [PubMed] [Google Scholar]
- O'Reilly MA. DNA damage and cell cycle checkpoints in hyperoxic lung injury: braking to facilitate repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;281:L291–L305. doi: 10.1152/ajplung.2001.281.2.L291. [DOI] [PubMed] [Google Scholar]
- Peers C, Wyatt CN, Evans AM. Mechanisms for acute oxygen sensing in the carotid body. Respir. Physiol. Neurobiol. 2010;174:292–298. doi: 10.1016/j.resp.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Penn JS, Henry MM, Wall PT, Tolman BL. The range of PaO2 variation determines the severity of oxygen-induced retinopathy in newborn rats. Invest. Ophthalmol. Vis. Sci. 1995;36:2063–2070. [PubMed] [Google Scholar]
- Porzionato A, Macchi V, Parenti A, De Caro R. Trophic factors in the carotid body. Int. Rev. Cell. Mol. Biol. 2008;269:1–58. doi: 10.1016/S1937-6448(08)01001-0. [DOI] [PubMed] [Google Scholar]
- Powell FL. The influence of chronic hypoxia upon chemoreception. Respir. Physiol. Neurobiol. 2007;157:154–161. doi: 10.1016/j.resp.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell FL, Huey KA, Dwinell MR. Central nervous system mechanisms of ventilatory acclimatization to hypoxia. Respir. Physiol. 2000;121:223–236. doi: 10.1016/s0034-5687(00)00130-4. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Kumar GK, Pawar A. Altered carotid body function by intermittent hypoxia in neonates and adults: relevance to recurrent apneas. Respir. Physiol. Neurobiol. 2007;157:148–153. doi: 10.1016/j.resp.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Prieto-Lloret J, Caceres AI, Obeso A, Rocher A, Rigual R, Agapito MT, Bustamante R, Castañeda J, Perez-Garcia MT, López-López JR, González C. Ventilatory responses and carotid body function in adult rats perinatally exposed to hyperoxia. J. Physiol. 2004;554:126–144. doi: 10.1113/jphysiol.2003.049445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeser JC, Brackett DG, van Heerden ES, Young KM, Bavis RW. Potentiation of the hypoxic ventilatory response by one day of hyperoxia in neonatal rats. Respir. Physiol. Neurobiol. 2011;176:50–56. doi: 10.1016/j.resp.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm CM, Grunstein MM. Respiratory influence of peripheral chemoreceptor stimulation in maturing rabbits. J. Appl. Physiol. 1987;63:1671–1680. doi: 10.1152/jappl.1987.63.4.1671. [DOI] [PubMed] [Google Scholar]
- Serra A, Brozoski D, Hodges M, Roethle S, Franciosi R, Forster HV. Effects of carotid and aortic chemoreceptor denervation in newborn piglets. J. Appl. Physiol. 2002;92:893–900. doi: 10.1152/japplphysiol.00819.2001. [DOI] [PubMed] [Google Scholar]
- Simakajornboon N, Kuptanon T. Maturational changes in neuromodulation of central pathways underlying hypoxic ventilatory response. Respir. Physiol. Neurobiol. 2005;149:273–286. doi: 10.1016/j.resp.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Sola A, Saldeño YP, Favareto V. Clinical practices in neonatal oxygenation: where have we failed? What can we do? J. Perinatol. 2008;28(Suppl 1):S28–S34. doi: 10.1038/jp.2008.47. [DOI] [PubMed] [Google Scholar]
- Spicer JI, Rundle SD. Plasticity in the timing of physiological development: physiological heterokairy – what is it, how frequent is it, and does it matter? Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007;148:712–719. doi: 10.1016/j.cbpa.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Sterni LM, Bamford OS, Wasicko MJ, Carroll JL. Chronic hypoxia abolished the postnatal increase in carotid body type I cell sensitivity to hypoxia. Am. J. Physiol. 1999;277:L645–L652. doi: 10.1152/ajplung.1999.277.3.L645. [DOI] [PubMed] [Google Scholar]
- Strohl KP. Periodic breathing and genetics. Respir. Physiol. Neurobiol. 2003;135:179–185. doi: 10.1016/s1569-9048(03)00036-3. [DOI] [PubMed] [Google Scholar]
- Tankersley CG. Genetic aspects of breathing: on interactions between hypercapnia and hypoxia. Respir. Physiol. Neurobiol. 2003;135:167–178. doi: 10.1016/s1569-9048(03)00035-1. [DOI] [PubMed] [Google Scholar]
- Teppema LJ, Dahan A. The ventilatory response to hypoxia in mammals: mechanisms, measurement, and analysis. Physiol. Rev. 2010;90:675–754. doi: 10.1152/physrev.00012.2009. [DOI] [PubMed] [Google Scholar]
- Tin W, Gupta S. Optimum oxygen therapy in preterm babies. Arch. Dis. Child. Fetal Neonatal. Ed. 2007;92:F143–147. doi: 10.1136/adc.2005.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin KE, Bavis RW. Intermittent hyperoxia attenuates the hypoxic ventilatory response in neonatal rats. FASEB J. 2012;26:LB819. doi: 10.1016/j.resp.2015.09.015. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbati D, Sherpa AK, Lahiri S, Mokashi A, Albertine KH, DiGiulio C. Hyperbaric oxygenation alters carotid body ultrastructure and function. Respir. Physiol. 1993;92:183–196. doi: 10.1016/0034-5687(93)90037-b. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- van Heerden ES, Bavis RW. Cardiorespiratory effects of chronic hyperoxia in neonatal rats. FASEB J. 2011;25:LB631. Abstract. [Google Scholar]
- van Heerden ES, Brackett DG, Leso JI, Dmitrieff EF, Bavis RW. Chronic hyperoxia alters the hyperoxic ventilatory response and diaphragm muscle fiber composition in neonatal rats. FASEB J. 2011;25:1111.5. Abstract. [Google Scholar]
- Vulesevik B, Perry SF. Developmental plasticity of ventilatory control in zebrafish, Danio rerio. Respir. Physiol. Neurobiol. 2006;154:396–405. doi: 10.1016/j.resp.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Wang Z-Y, Bisgard GE. Postnatal growth of the carotid body. Respir. Physiol. Neurobiol. 2005;149:181–190. doi: 10.1016/j.resp.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Wang Z-Y, Olson EB, Jr., Bjorling DE, Mitchell GS, Bisgard GE. Sustained hypoxia-induced proliferation of carotid body type I cells in rats. J. Appl. Physiol. 2008;104:803–808. doi: 10.1152/japplphysiol.00393.2007. [DOI] [PubMed] [Google Scholar]
- Wenninger JM, Olson EB, Wang Z, Keith IM, Mitchell GS, Bisgard GE. Carotid sinus nerve responses and ventilatory acclimatization to hypoxia in adult rats following 2 weeks of postnatal hyperoxia. Respir. Physiol. Neurobiol. 2006;150:155–164. doi: 10.1016/j.resp.2005.05.014. [DOI] [PubMed] [Google Scholar]