

 |
Malcolm Kohler (left) is a Senior Consultant at the Sleep Disorders Centre and Pulmonary Division of the University Hospital of Zurich, Switzerland. Awarded a European Respiratory Society Research Fellowship he spent 2 years at the Oxford Centre for Respiratory Medicine, where his research focused on vascular dysfunction in patients with obstructive sleep apnoea. Dr Kohler currently leads a research team, at the Centre for Integrative Human Physiology, University of Zurich, Switzerland, that is looking at the cardiovascular consequences of chronic breathing disorders such as obstructive sleep apnoea and chronic obstructive pulmonary disease. John Stradling (right) is a Professor of Respiratory Medicine at Oxford University and Director of the Respiratory Sleep Service. His doctorate (in 1981) was on sleep related breathing problems in chronic obstructive lung disease. After a period of research with Eliot Phillipson in Toronto 1984–85, he returned to Oxford as a Wellcome Senior Research Fellow for 7 years. His research is in the area of sleep related disorders of breathing, in particular epidemiology, cardiovascular consequences, simplified methods of diagnosis, and randomised controlled trials of therapy. He has published over 150 original publications in peer-reviewed journals.
Introduction
There is clearly an association between obstructive sleep apnoea (OSA) and vascular disease (Marin et al. 2005; Gottlieb et al. 2010). Much vascular disease remains unexplained even after allowing for well-known risk factors, such as hypertension and dyslipidaemia (Jackson et al. 2009) and, because OSA is common, it certainly could provide a significant extra contribution. In recent years several potential hypotheses have been advanced to link OSA with vascular disease, derived from the complex physiological changes that actually occur during OSA (Kohler & Stradling, 2010), and supported by animal models and laboratory-based studies; these include intermittent hypoxia, intermittent arousal from sleep and increased intrathoracic pressure swings. The link between these consequences of OSA and vascular disease are probably multifactorial, most likely augmented sympathetic activity, with less evidence for oxidative stress, systemic inflammation, and vibration damage to the carotid arteries. However, because of the close association between central obesity and both OSA (Davies et al. 1992) and vascular disease (Yusuf et al. 2005), it has been very difficult to disentangle the inter-relations and demonstrate that OSA is a truly independent risk factor for vascular disease. Although laboratory studies may support plausible hypotheses, this does not prove their relevance to clinical medicine, a painful lesson learnt in other areas of vascular risk (e.g. anti-oxidant treatment and hormone replacement therapy) where plausible hypotheses, based on laboratory experiments and cross-sectional studies, failed to be supported by randomised controlled trials (RCTs; Simon et al. 2001; Lawlor et al. 2004). Therefore, studies of robust design, allowing identification of true causality, are essential in this area. Each of the above-mentioned mechanisms that possibly underpin the association between OSA and vascular disease needs to be proven to be clinically relevant in RCTs. At present such data are most supportive for increased sympathetic nervous system activity, which appears more than adequate as a mechanism to explain the associations observed between OSA and vascular disease.
Mechanisms of sympathetic activation in OSA
Findings from both animal models and from human studies, have provided ample evidence that OSA is associated with augmented sympathetic activation, ascribed to recurrent arousals from sleep, intermittent hypoxia, and to a lesser extent intrathoracic pressure changes.
The exposure of rodents to intermittent hypoxia over 14–30 days increased catecholamines and substantially elevated blood pressure (BP; Lesske et al. 1997; Dematteis et al. 2008). Surgical denervation of peripheral chemoreceptors prevented the observed increase in BP, and adrenal demedullation and chemical denervation of the peripheral sympathetic nervous system prevented the elevation of BP (Lesske et al. 1997). In rats, chronic intermittent hypoxia has also been shown to augment chemoreflex-stimulated sympathetic outflow via the renin–angiotensin system and via modulation of O2-sensing in carotid bodies through activation of HIF-1 and downregulation of HIF-2 (La Rovere et al. 1998; Prabhakar et al. 2009). Furthermore, stimulation of carotid bodies by intermittent hypoxia has been shown to be associated with a reduction in baroreflex sensitivity in cats (Rey et al. 2004). In dogs, arousals from sleep have been shown to be associated with a marked increase in heart rate, which was abolished by sympathetic blockade (Horner et al. 1995).
In a RCT including 10 healthy males exposed to intermittent hypoxia or a sham procedure for 6 h per day for 4 days, mean blood pressure increased by 4 mmHg, and nitric oxide derivatives were reduced by 55%. This was associated with an increase in BP and cerebral vascular resistance response to a hypoxic challenge (Foster et al. 2009). In a similar study including 12 healthy subjects, 2 weeks of intermittent hypoxia exposure increased sympathetic outflow associated with an increase in daytime systolic BP of 8 mmHg; however, this was not accompanied by an increase in inflammatory markers (CRP, IL-1Ra, IL-8, TNF-α; Tamisier et al. 2011).
In a study including five healthy humans, increasing intensity of experimentally induced arousals caused progressively larger transient increases in BP and heart rate similar in size to those seen with the arousals that usually terminate an obstructive apnoea (Davies et al. 1993). In a larger study involving 14 healthy humans, arousal stimuli during sleep caused bursts of sympathetic nerve activity (as measured by microneurography of the peroneal nerve) and transient increases in BP (Somers et al. 1993). The increasing inspiratory effort against the collapsed pharynx during obstructive apnoeas is also likely to cause increased sympathetic nerve activity; in an experimental study of nine healthy humans, an intrathoracic pressure change induced by a Mueller manoeuvre generated an increase of more than 200% in postganglionic sympathetic nerve activity and a 14% increase in mean BP at the end of the apnoea (Somers et al. 1993).
Randomised controlled trials
At least five RCTs, with appropriate control subjects, have provided evidence that continuous positive airway pressure (CPAP) therapy in OSA reduces 24 h catecholamine excretion, consistent with a reduction in sympathetic nerve activity (Ziegler et al. 2001; Mills et al. 2006; Drager et al. 2007; Kohler et al. 2008, 2011). A study assessing 38 patients with obstructive sleep apnoea syndrome (OSAS) demonstrated a 26% reduction in 24 h urine norepinephrine (noradrenaline) levels after 7 days of CPAP treatment (Ziegler et al. 2001), and another trial involving 102 patients with moderate-to-severe OSAS reported a 26% reduction in urine normetanephrine levels and augmentation index (a measure of arterial stiffness) after 4 weeks of CPAP therapy; interestingly, however, no reduction in various markers of systemic inflammation (CRP, IL-6, INF-γ) was found (Kohler et al. 2008). In further studies, a reduction in urinary levels of norepinephrine of 38% during daytime, and of 22% during the night, was shown after 14 days of CPAP treatment in 50 patients (Mills et al. 2006), and in 24 patients, 4 months of CPAP therapy was associated with a 44% decrease of plasma norepinephrine levels (Drager et al. 2007). More recently, Kohler et al. 2011 showed in a trial, including 41 patients with OSA, that CPAP therapy withdrawal for 14 days increased urinary norepinephrine levels by 44%, associated with a substantial worsening of endothelial function, BP and heart rate, but no evidence of an increase in inflammatory markers (Fig. 1). Data from a RCT, comparing the effects of 14 days of CPAP with 14 days of overnight supplemental oxygen therapy on urinary norepinephrine levels in patients with OSAS, showed that the oxygen therapy also reduced urinary norepinephrine levels during the daytime (Mills et al. 2006; Norman et al. 2006).
Figure 1. Effects of 14 days of CPAP therapy withdrawal from patients with OSA.
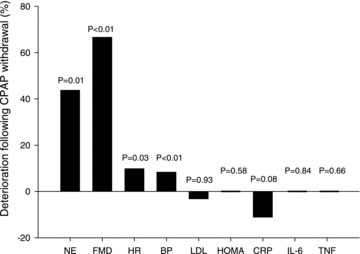
NE, urinary norepinephrine (noradrenaline); FMD, flow mediated dilatation (a measure of endothelial function); HR, heart rate; BP, diastolic blood pressure; LDL, low density lipoprotein; HOMA, homeostatic model assessment (a measure of insulin resistance); CRP, C-reactive protein; IL-6, interleukin 6; TNF, tumour necrosis factor α.
Baroreflex sensitivity, an established index of cardiac sympatho-vagal balance, is also depressed and, therefore, contributes to the development of hypertension and cardiovascular disease in patients with OSAS. Two RCTs have provided evidence for an independent link between OSAS and depressed daytime baroreflex sensitivity. In a study of 77 patients with OSA, 4 weeks of CPAP therapy increased baroreflex sensitivity by 24% (Kohler et al. 2008). Similarly, baroreflex sensitivity was increased with CPAP therapy in a trial involving 33 patients (Noda et al. 2007), consistent with a beneficial effect of OSAS treatment on the cardiac sympatho-vagal balance. The findings of these controlled trials provide evidence for a causal relationship between OSA, increased sympathetic nerve activity, and depressed baroreflex sensitivity.
Implications of sympathetic activation on vascular risk
There is extensive evidence that the above-described increase in sympathetic activity in patients with OSA, and its effect on heart rate, BP and vascular function, is of considerable clinical importance in determining vascular outcomes (Palatini & Julius, 2009; Fox & Ferrari, 2011). Furthermore, it has become increasingly clear that fluctuations in BP play a major additional role, over and above diurnal BP, in provoking vascular risk (Rothwell, 2010). With every obstructive apnoea there is a sympathetically induced rise in BP, as much as 80 mmHg, and this is likely to contribute to vascular risk, although this remains a hypothesis at present.
Conclusions
Thus, despite several plausible hypotheses to explain an increased vascular risk in OSA, only increased sympathetic activity has convincing supporting evidence from relevant clinical trials. Although there may be additional contributory factors, e.g. systemic inflammation, there is little robust evidence for these hypotheses and they are not needed to explain the adverse vascular outcomes associated with OSA.
Call for comments
Readers are invited to give their views on this and the accompanying CrossTalk articles in this issue by submitting a brief comment. Comments must not exceed 250 words, with a maximum of six references from peer reviewed publications only. To submit a comment, use the online form available in the centre panel on the HighWire site. If other responses have already been submitted, a ‘view comments’ link will be visible.
All comments will be moderated to avoid duplication of responses, and those deemed to add significantly to the discussion will be published online-only as footnotes to the articles. Comments may be posted up to 6 weeks after publication of the article, at which point the discussion will close and authors will be invited to submit a ‘final word’.
Questions about this call should be directed to Jerry Dempsey at jdempsey@wisc.edu.
To submit a comment, go to http://jp.physoc.org/letters/submit/jphysiol;590/12/2813
Glossary
- BP
blood pressure
- CPAP
continuous positive airway pressure
- OSA(S)
obstructive sleep apnoea (syndrome)
- RCT
randomised controlled trial
References
- Davies RJ, Ali NJ, Stradling JR. Neck circumference and other clinical features in the diagnosis of the obstructive sleep apnoea syndrome. Thorax. 1992;47:101–105. doi: 10.1136/thx.47.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies RJ, Belt PJ, Roberts SJ, Ali NJ, Stradling JR. Arterial blood pressure responses to graded transient arousal from sleep in normal humans. J Appl Physiol. 1993;74:1123–1130. doi: 10.1152/jappl.1993.74.3.1123. [DOI] [PubMed] [Google Scholar]
- Dematteis M, Julien C, Guillermet C, Sturm N, Lantuejoul S, Mallaret M, Levy P, Gozal E. Intermittent hypoxia induces early functional cardiovascular remodeling in mice. Am J Respir Crit Care Med. 2008;177:227–235. doi: 10.1164/rccm.200702-238OC. [DOI] [PubMed] [Google Scholar]
- Drager LF, Bortolotto LA, Figueiredo AC, Krieger EM, Lorenzi-Filho G. Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med. 2007;176:706–712. doi: 10.1164/rccm.200703-500OC. [DOI] [PubMed] [Google Scholar]
- Foster GE, Brugniaux JV, Pialoux V, Duggan CT, Hanly PJ, Ahmed SB, Poulin MJ. Cardiovascular and cerebrovascular responses to acute hypoxia following exposure to intermittent hypoxia in healthy humans. J Physiol. 2009;587:5303–5304. doi: 10.1113/jphysiol.2009.171553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox KM, Ferrari R. Heart rate: a forgotten link in coronary artery disease? Nat Rev Cardiol. 2011;8:369–379. doi: 10.1038/nrcardio.2011.58. [DOI] [PubMed] [Google Scholar]
- Gottlieb DJ, Yenokyan G, Newman AB, O’Connor GT, Punjabi NM, Quan SF, Redline S, Resnick HE, Tong EK, Diener-West M, Shahar E. Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure. Circulation. 2010;122:352–360. doi: 10.1161/CIRCULATIONAHA.109.901801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner RL, Brooks D, Kozar LF, Tse S, Phillipson EA. Immediate effects of arousal from sleep on cardiac autonomic outflow in the absence of breathing in dogs. J Appl Physiol. 1995;79:151–162. doi: 10.1152/jappl.1995.79.1.151. [DOI] [PubMed] [Google Scholar]
- Jackson R, Marshall R, Kerr A, Riddell T, Wells S. QRISK or Framingham for predicting cardiovascular risk? BMJ. 2009;339:b2673. doi: 10.1136/bmj.b2673. [DOI] [PubMed] [Google Scholar]
- Kohler M, Pepperell JCT, Casadei B, Craig S, Crosthwaite N, Stradling JR, Davies RJO. CPAP and measures of cardiovascular risk in males with OSAS. Eur Respir J. 2008;32:1488–1496. doi: 10.1183/09031936.00026608. [DOI] [PubMed] [Google Scholar]
- Kohler M, Stoewhas AC, Ayers L, Senn O, Bloch KE, Russi EW, Stradling JR. The effects of CPAP therapy withdrawal in patients with obstructive sleep apnea: a randomized controlled trial. Am J Respir Crit Care Med. 2011;184:1192–1199. doi: 10.1164/rccm.201106-0964OC. [DOI] [PubMed] [Google Scholar]
- Kohler M, Stradling JR. Mechanisms of vascular damage in obstructive sleep apnea. Nat Rev Cardiol. 2010;7:677–685. doi: 10.1038/nrcardio.2010.145. [DOI] [PubMed] [Google Scholar]
- La Rovere MT, Bigger JT, Marcus FI, Mortara A, Schwartz PJ. Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. Lancet. 1998;351:478–484. doi: 10.1016/s0140-6736(97)11144-8. [DOI] [PubMed] [Google Scholar]
- Lawlor DA, Davey Smith G, Kundu D, Bruckdorfer KR, Ebrahim S. Those confounded vitamins: what can we learn from the differences between observational versus randomised trial evidence? Lancet. 2004;363:1724–1727. doi: 10.1016/S0140-6736(04)16260-0. [DOI] [PubMed] [Google Scholar]
- Lesske J, Fletcher EC, Bao G, Unger T. Hypertension caused by chronic intermittent hypoxia – influence of chemoreceptors and sympathetic nervous system. J Hypertens. 1997;15:1593–1603. doi: 10.1097/00004872-199715120-00060. [DOI] [PubMed] [Google Scholar]
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet. 2005;365:1046–1053. doi: 10.1016/S0140-6736(05)71141-7. [DOI] [PubMed] [Google Scholar]
- Mills PJ, Kennedy BP, Loredo JS, Dimsdale JE, Ziegler MG. Effects of nasal continuous positive airway pressure and oxygen supplementation on norepinephrine kinetics and cardiovascular responses in obstructive sleep apnea. J Appl Physiol. 2006;100:343–348. doi: 10.1152/japplphysiol.00494.2005. [DOI] [PubMed] [Google Scholar]
- Noda A, Nakata S, Koike Y, Miyata S, Kitaichi K, Nishizawa T, Nagata K, Yasuma F, Murohara T, Yokota M. Continuous positive airway pressure improves daytime baroreflex sensitivity and nitric oxide production in patients with moderate to severe obstructive sleep apnea syndrome. Hypertens Res. 2007;30:669–676. doi: 10.1291/hypres.30.669. [DOI] [PubMed] [Google Scholar]
- Norman D, Loredo JS, Nelesen RA, Ancoli-Israel S, Mills PJ, Ziegler MG, Dimsdale JE. Effects of continuous positive airway pressure versus supplemental oxygen on 24-hour ambulatory blood pressure. Hypertension. 2006;47:840–845. doi: 10.1161/01.HYP.0000217128.41284.78. [DOI] [PubMed] [Google Scholar]
- Palatini P, Julius S. The role of cardiac autonomic function in hypertension and cardiovascular disease. Curr Hypertens Rep. 2009;11:199–205. doi: 10.1007/s11906-009-0035-4. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK, Nanduri J. Intermittent hypoxia-mediated plasticity of acute O2 sensing requires altered red-ox regulation by HIF-1 and HIF-2. Ann N Y Acad Sci. 2009;1177:162–168. doi: 10.1111/j.1749-6632.2009.05034.x. [DOI] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560:577–586. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PM. Limitations of the usual blood-pressure hypothesis and importance of variability, instability, and episodic hypertension. Lancet. 2010;375:938–948. doi: 10.1016/S0140-6736(10)60309-1. [DOI] [PubMed] [Google Scholar]
- Simon JA, Hsia J, Cauley JA, Richards C, Harris F, Fong J, Barrett-Connor E, Hulley SB. Postmenopausal hormone therapy and risk of stroke: The Heart and Estrogen-progestin Replacement Study (HERS) Circulation. 2001;103:638–642. doi: 10.1161/01.cir.103.5.638. [DOI] [PubMed] [Google Scholar]
- Somers VK, Dyken ME, Mark AL, Abboud FM. Sympathetic-nerve activity during sleep in normal subjects. N Engl J Med. 1993;328:303–307. doi: 10.1056/NEJM199302043280502. [DOI] [PubMed] [Google Scholar]
- Somers VK, Dyken ME, Skinner JL. Autonomic and hemodynamic responses and interactions during the Mueller maneuver in humans. J Auton Nerv Syst. 1993;44:253–259. doi: 10.1016/0165-1838(93)90038-v. [DOI] [PubMed] [Google Scholar]
- Tamisier R, Pepin JL, Remy J, Baguet JP, Taylor JA, Weiss JW, Levy P. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J. 2011;37:119–128. doi: 10.1183/09031936.00204209. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Hawken S, Ounpuu S, Bautista L, Franzosi MG, Commerford P, Lang CC, Rumboidt Z, Onen CL, Lisheng L, Tanomsup S, Wangai P, Razak F, Sharma AM, Anand SS INTERHEART study investigators. Obesity and the risk of myocardial infarction in 27,000 participants from 52 countries: a case-control study. Lancet. 2005;366:1640–1649. doi: 10.1016/S0140-6736(05)67663-5. [DOI] [PubMed] [Google Scholar]
- Ziegler MG, Mills PJ, Loredo JS, Ancoli-Israel S, Dimsdale JE. Effect of continuous positive airway pressure and placebo treatment on sympathetic nervous activity in patients with obstructive sleep apnea. Chest. 2001;120:887–893. doi: 10.1378/chest.120.3.887. [DOI] [PubMed] [Google Scholar]