

Abstract
Rapid strides have been made in the field of hematology, and advances in immune thrombocytopenic purpura (ITP) management are no exception. From idiopathic to immune, the changed nomenclature is itself a testimonial to the growing awareness and improvements in the management of ITP. We discuss the pathophysiology, clinical presentation, and current management of this common pediatric disorder and summarize current guidelines for ITP treatment.
Keywords: Immune thrombocytopenia, purpura
INTRODUCTION
All pediatricians encounter cases of immune thrombocytopenia (ITP), especially in these days of Coulter counters and automated platelet counts. ITP is reported in approximately 5 per 100,000 children and 2 per 100,000 adults.1 ITP was called idiopathic thrombocytopenia until recently when several clinical and basic research studies unraveled the pathophysiology of the disease.
ITP follows a benign course in most children but has the potential to be life threatening. The risk of primary intracranial bleeding and soft tissue and mucosal bleeding secondary to trauma can cause morbidity and mortality. The lack of evidence-based management protocols is a potential cause for poor management of ITP because no evidence clearly demonstrates that treatment alters the final outcome in any way.2 We discuss the various presentations of ITP and management guidelines.
PATHOPHYSIOLOGY
At present, our understanding of the pathophysiology of ITP leads us to 2 central mechanisms: either immune-mediated increased destruction of platelets or decreased production of platelets that results in an overall decrease in circulating platelets. Harrington et al3 first highlighted the role of immunity in the destruction of platelets in ITP patients. In an unusual experiment, Harrington injected himself and other test subjects with blood from ITP patients. To everyone's surprise, he found a rapid decline in circulating platelet quantities in the test subjects.3 This experiment gave birth to the hypothesis of an antiplatelet factor, later confirmed to be an antibody against platelets.4 B and T cells are an integral part of the cascade involved in platelet destruction. Antiplatelet antibodies opsonize the platelets and then are attached to antigen-presenting cells with the help of Fcγ receptors. Opsonized platelets are finally phagocytosed by macrophages. T cells, at the same time, stimulate B cells to produce more antiplatelet antibodies, and new research shows that some cryptic epitopes from platelet antigens stimulate platelet-specific T cells.5
Reduced platelet production is another important mechanism that explains the pathophysiology of ITP in some patients. The recent discovery of thrombopoietin (TPO) and its role in thrombopoiesis helped us understand the role of reduced thrombopoiesis in ITP. An increase in platelet quantity after administering TPO mimetics in some study populations confirmed that TPO has a definitive role in ITP.6
Most ITP cases are self-limiting and require no treatment because most often the event responsible for antiplatelet antibody production is a viral illness. At present, most treatment protocols concentrate on the reduction of platelet destruction, and the drugs used are usually immunosuppressives. However, other drugs may be used in the near future if the TPO mimetic proves safe and effective in the various trials currently in progress.
CLINICAL PRESENTATION AND DIAGNOSIS
As depicted in Figure 1, the spectrum of disease fluctuates from an asymptomatic state to intracranial hemorrhage. The Intercontinental Childhood ITP Study (ICIS) group found the mean age of ITP presentation in a cohort of children to be 5.7 years.7 Boys younger than 10 years had a higher incidence of ITP.8-10 Physical examination is mostly positive for cutaneous manifestations as petechiae and bruising. One-fourth of children present with epistaxis, although hematuria is less frequent.11 Other atypical findings may include lymphadenopathy and hepatosplenomegaly.11,12
Figure 1.
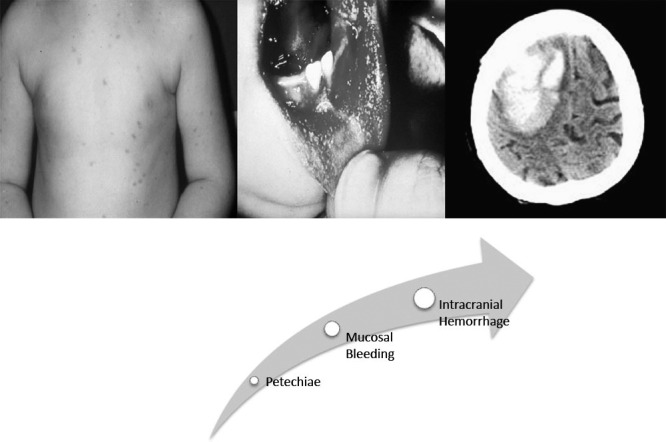
Clinical manifestations of immune thrombocytopenic purpura in increasing order of severity.
A diagnosis of ITP is confirmed by a finding of thrombocytopenia in a blood smear. More than 50% of acute ITP cases present with a platelet count less than 20 × 109/L. Chronic ITP is defined as platelet count less than 150 × 109/L for more than 6 months after the diagnosis.5 The presence of mild eosinophilia, along with a few megathrombocytes, is a common finding.
Peripheral Smear
Bone marrow examination is not routinely performed in children unless atypical clinical or laboratory findings are present. General pediatricians often obtain a marrow test to be interpreted by pathologists to exclude the possibility of acute leukemia, which could enter a temporary remission with steroid treatment, with disastrous results. Tests for retic count, erythrocyte sedimentation rate, antinuclear antibodies, blood group, Coombs, and Epstein-Barr virus may be needed in selected cases depending on associated symptoms. Close and continued monitoring of clinical status and hematologic status is the most important step in managing ITP.
Differential Diagnosis
Primary ITP is a diagnosis of exclusion by carefully ruling out causes of pseudothrombocytopenia, secondary ITP, and inherited ITP. Obtaining an accurate history combined with a complete blood count and a thorough evaluation of the blood smear is the first and most important step. Acute onset of bruising and petechiae in an otherwise healthy child—often with a recent history of viral infection—associated with isolated thrombocytopenia and large platelets in the smear suggests acute ITP. Prolonged fever, weight loss, bone pain, significant lymphadenopathy, and organomegaly are unusual and warrant careful examination to exclude leukemia, aplastic anemia, or other more serious illnesses.
Pseudothrombocytopenia is the result of platelets clumping in the presence of ethylenediaminetetraacetic acid. A smear must be evaluated to rule out a false count and to determine the morphology of red blood cells (RBCs), white blood cells (WBCs), and platelets themselves. Isolated thrombocytopenia with normal RBC morphology and larger-than-usual platelets but without any immature WBC series fits the classic description of ITP.13 Abnormalities in RBC or WBC series are unusual in standard ITP in children. Clinicians can exclude the common causes of secondary thrombocytopenia by investigating for Epstein-Barr virus, systemic lupus erythematosus, hepatitis C, and human immunodeficiency virus based on history and clinical examination findings.5 A detailed family history is essential to rule out inherited causes of thrombocytopenias such as Wiskott-Aldrich syndrome. The inherited thrombocytopenias are classified on the basis of platelet size and gene mutations.14 Other miscellaneous conditions to consider are thrombocytopenia secondary to drugs, infections (such as human immunodeficiency virus, Epstein-Barr virus, and cytomegalovirus), preeclampsia, HELLP syndrome, disseminated intravascular coagulation, large hemangiomas, aplastic anemia, metabolic disorders, and marrow infiltration.
TREATMENT
A sound understanding of pathophysiology is essential for the appropriate management of ITP in children. Rapid strides in pharmacotherapy have added to the preexisting predicament of when to treat and how to treat. Clinicians must clearly understand that no study has shown that any form of treatment decreases mortality or alters the risk of the disease process becoming chronic. Personalization of treatment based on platelet count, age, clinical picture, duration, lifestyle issues, economic considerations, and parental, patient, or physician concerns is the best approach. A standard treatment protocol for all ITPs would be a step backward.
Treatment of ITP can be divided into medical and surgical management. Medical management is further divided into first-line and second-line pharmacotherapy. Supportive therapy by a team experienced in the variable course and outcome of ITP is crucial. Education of the patient, teachers, siblings, parents, and primary care physician of the need for close monitoring for any acute bleed, especially intracranial or intraabdominal, can significantly decrease mortality and morbidity. The child and family also require psychosocial support and trauma prevention advice, including a strong warning about participating in contact sports.
Medical options for front-line drug therapy are corticosteroids, intravenous (IV) immunoglobulin (Ig), and IV Rh anti-D.
Corticosteroids
Sartorius15 in 1984 was the first to report the benefit of prednisolone in ITP. Another study by Buchanan and Holtkamp16 in the same year reaffirmed that prednisolone boosts platelet counts by day 7 of treatment. A well-established consensus exists regarding the initial benefit from oral prednisolone. Corticosteroids act by impairing the clearance of opsonized platelets in bone marrow and peripheral organs and reducing autoantibody levels in the body.17 Many prospective, randomized studies confirm that corticosteroids increase platelet levels more rapidly than no treatment.18 High-dose prednisone at approximately 4 mg/kg/d for 4 days or the shortest period possible can minimize side effects as well as maintain the therapeutic significance in treatment of ITP.19-23 Treatment duration and drug dose are determined by the response and side effects. Some of the common complications associated with corticosteroid treatment are avascular necrosis, diabetes, gastritis, ulcers, growth retardation, hypertension, insomnia, osteoporosis (in adults), personality changes, and opportunistic infections. Tapering the dose and terminating the drug on either stoppage of bleeding or on achieving a platelet count higher than 20 × 109/L are important. In fact, many doctors stop treatment after 2-3 weeks regardless of the response.
Intravenous Immunoglobulin G
Imbach et al24 were the first to propose the role of IV IgG in the reversal of thrombocytopenia. IV IgG acts by impairing the clearance of opsonized platelets,17 probably mediated through the FcγRIIb receptor.25 Some studies also suggest that IV IgG might cause increased clearance of antiplatelet antibodies via saturation of the neonatal Fc salvage receptor for IgG.26 Our present knowledge of IV IgG is mostly informed by 2 Canadian clinical trials.27,28 These studies concluded that IV IgG had a faster response rate compared to corticosteroids when the target platelet count was 50 × 109/L. Also, they found that single-dose IV IgG of 0.8 g/kg was as effective as and safer than the larger dose of 1 g/kg for 2 days. IV IgG worked better than IV Rh anti-D to achieve a 20 × 109/L platelet count. IV IgG, although more expensive than IV Rh anti-D, is definitely one of the safer options available and may shorten the length of the hospital stay because of the drug's rapid response, usually within 24-48 hours. Some common infusion-related side effects are headache, fever, chills, and nausea. Other worrisome but rare side effects include aseptic meningitis, renal impairment or failure, and thromboembolic events.
Intravenous Anti-D
Salama et al29 in 1983 reported an increase in platelet counts in ITP patients who were also positive for rhesus D antigen after the infusion of IV anti-D. Current wisdom favors the notion that IV anti-D coats the RBCs that are positive for D antigen, and these opsonized RBCs in turn compete with opsonized platelets in the spleen for sequestration.30 Reports suggest that a dose of 75 μg/kg over 3-5 minutes is more efficacious than a 50 μg/kg dose, although the side effects are more common with the higher dose. Some of the commonly encountered infusion-related side effects are fever, chills, nausea, and headache. Another important adverse effect is a fall in hemoglobin secondary to hemolysis. Usually the fall in hemoglobin is not more than 2 g/dL,31,32 but in rare cases, hemolysis can be severe and can lead to renal failure and disseminated intravascular coagulation.33,34
Second-Line Pharmacotherapy
Second-line pharmacotherapy mainly comprises immunosuppressants and rituximab. These drugs are used when first-line drugs have failed or patients have become intolerant. Immunosuppressants primarily act at the level of T cells. Azathioprine, cyclophosphamide, and cyclosporine are the main drugs in use. Dapsone, mycophenolate mofetil, danazol, and vinca alkaloids are a few other second-line drugs with unproven efficacy, but these agents are used rarely in children at the physician's discretion.
Rituximab
Rituximab is a human-murine monoclonal antibody against the CD20 antigen on B lymphocytes that is used to treat lymphoma. It acts by reducing the number of B cells that produce autoantibodies. Results of a systemic review conducted by Arnold et al35 from 19 studies were promising and instill hope for the efficacy of rituximab. Severe side effects after rituximab therapy are fortunately rare but include the potential for neutropenia and the reactivation of chronic infections such as tuberculosis. Presently, rituximab seems to be the most promising drug for treatment of refractory ITP. The response to rituximab was complete, and the most frequently used dose was 375 mg/m2 for 4 weeks. Recent studies suggest a superior response rate if dexamethasone is given with rituximab. Also, some evidence favors rituximab being used before resorting to splenectomy in refractory ITP.36
Dapsone
Recently, some investigators have reported a reversal of thrombocytopenia in 40%-50% of patients taking dapsone.37 The dose recommended is 25 mg to 100 mg per day, and generally about a month passes before the response is apparent. The effects of the drug reportedly remain for a few months before relapse, which demands the continuation of therapy. Few people believe that dapsone in addition to prednisone should be recommended for patients who require low-dose steroids to maintain a high platelet count.
Surgical Management
Splenectomy is not a favored option for treating ITP in children, as reflected in various guidelines38 that prefer medical treatment over surgical management. The few indications that justify splenectomy are severe menorrhagia, life-threatening hemorrhage, and relentless lifestyle limitation. A major factor that deters most physicians from taking the surgical route is the risk of the patient developing overwhelming postsplenectomy sepsis and the lifetime risk of sepsis from encapsulated organisms. If splenectomy is selected, a laparoscopic approach is preferred with efforts made to identify and remove the accessory spleen. Also, physicians must immunize the child against Haemophilus influenzae type b, pneumococcus, and meningococcus. Some physicians recommend prophylactic penicillin in patients up to 5 years of age (or even later ages) because vaccines do not immunize against all pneumococcal serotypes. Education of the patient, parent, primary care physician, and emergency room physicians about the risk of sepsis is also essential.
In 80% of cases, acute ITP in children spontaneously resolves irrespective of any treatment. Of the 20% of patients who are truly chronic, 80% enter remission following splenectomy, but a few will relapse over a period of years. The explanation given for relapse is that in patients with significant antibodies, the liver or macrophages will continue to destroy sensitized platelets and the defective production is unable to compensate for the sustained destruction. Adolescents and older patients are more likely to have chronic disease, and physicians must remain alert for antibody-mediated destruction from collagen vascular diseases and/or bone marrow failure syndromes in adults.
Treatment Selection
The factors that influence the selection of a treatment regimen in a given patient are quality-of-life impact, adverse events, likelihood of response, bleeding risk, patient/parent anxiety, and economic issues. A detailed clinical history and thorough physical examination are vital. The clinical history has important implications not only in diagnosing the disease but also for selecting the treatment regimen. Based on the clinical history, patients can be subclassified into the following groups: emergent, acute responsive, acute refractory, chronic persistent, and chronic refractory.
Emergent Disease
The goal of treatment is immediate cessation of bleeding that can be achieved by either platelet-directed interventions or ancillary interventions. Platelet-directed interventions rely on IV corticosteroids, IV IgG, IV Rh anti-D, combination therapy, and platelet infusion. Platelet infusions by themselves are of no use because of immediate antibody-mediated destruction. Ancillary interventional options include the cessation of antiplatelet agents, antifibrinolytic therapy, and the use of recombinant factor VIIa. Emergency splenectomy under the cover of medical therapy may be very rarely indicated in extreme cases.
Acute ITP
To treat or not to treat is the most important and perhaps the most difficult question faced by a pediatric hematologist (Table, Figures 2 and 3).38-40 Many physicians believe in observation alone, while others prefer early initiation of therapy. Those who believe in observation justify their judgment by the fact that severe hemorrhage is rare and drug therapy may not prevent severe hemorrhage. Medical treatment may add to the financial burden and expose the child to severe drug-associated side effects. Proponents of the early initiation of therapy argue that platelet count increases T cells faster than without treatment, thus decreasing the likelihood of severe bleeding. Quality-of-life issues may decrease with a faster and better maintained response following drug treatment.
Table.
American Society of Hematology 2011 Guidelines for Immune Thrombocytopenic Purpura38

Figure 2.
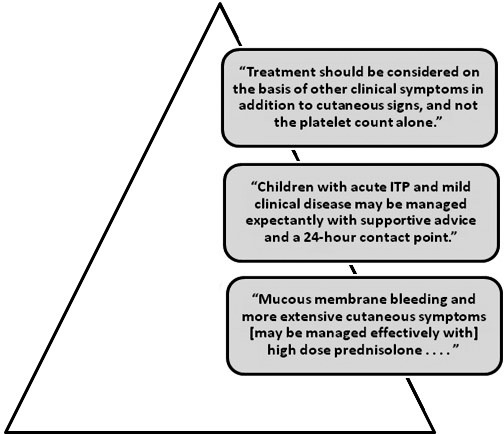
2003 British guidelines for investigating and managing idiopathic thrombocytopenic purpura (ITP).39
Figure 3.

2006 Indian Academy of Pediatrics Guidelines for diagnosing and managing immune thrombocytopenic purpura (ITP) in children.40
Chronic ITP
Chronic ITP includes all patients who have platelet counts less than 150 × 109/L persisting more than 6 months after diagnosis. Currently, there is no universally accepted regimen. For these patients, second-line pharmacotherapy can be considered a serious option. Treatment may be withheld in many cases if platelet counts are maintained above 20,000 × 109/L without any bleeding symptomatology. Azathioprine with or without prednisone, vincristine or vinblastine, cyclophosphamide, cyclosporin, dapsone, and combination chemotherapy are a few treatment options for chronic ITP. Patients with chronic ITP are also suitable candidates for splenectomy, as discussed before. A conservative approach is still the first choice, with observation alone or a combination of drugs used when indicated. Any plans for immunosuppression or splenectomy in a child with chronic ITP should be carefully reviewed with experienced pediatric hematologists and then discussed at length with parents and the child.
CONCLUSION
ITP is a common hematological problem that pediatricians face. The disease can be perplexing because no confirmatory tests exist to ensure that physicians are not missing a more sinister diagnosis. The treatment options vary from doing nothing to using immunosuppressants and chemotherapy. The individualization of care based on the medical, social, and other supporting evidence is the difficult but ideal choice.
Footnotes
The authors have no financial or proprietary interest in the subject matter of this article.
This article meets the Accreditation Council for Graduate Medical Education and the American Board of Medical Specialties Maintenance of Certification competencies for Patient Care and Medical Knowledge.
REFERENCES
- 1.Fogarty PF, Segal JB. The epidemiology of immune thrombocytopenic purpura. Curr Opin Hematol. 2007 Sep;14(5):515–519. doi: 10.1097/MOH.0b013e3282ab98c7. [DOI] [PubMed] [Google Scholar]
- 2.Cines DB, Bussel JB, McMillan RB, Zehnder JL. Congenital and acquired thrombocytopenia. Hematology Am Soc Hematol Educ Program. 2004. pp. 390–406. Erratum in: Hematology Am Soc Hematol Educ Program. 2005:543. [DOI] [PubMed]
- 3.Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med. 1951 Jul;38(1):1–10. [PubMed] [Google Scholar]
- 4.Shulman NR, Marder VJ, Weinrach RS. Similarities between known antiplatelet antibodies and the factor responsible for thrombocytopenia in idiopathic purpura. Physiologic, serologic and isotopic studies. Ann N Y Acad Sci. 1965 Jun 30;124(2):499–542. doi: 10.1111/j.1749-6632.1965.tb18984.x. [DOI] [PubMed] [Google Scholar]
- 5.Blanchette V, Bolton-Maggs P. Childhood immune thrombocytopenic purpura: diagnosis and management. Pediatr Clin North Am. 2008 Apr;55(2):393–420. doi: 10.1016/j.pcl.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 6.McMillan R. The pathogenesis of chronic immune thrombocytopenic purpura. Semin Hematol. 2007 Oct;44((4 Suppl 5)):S3–S11. doi: 10.1053/j.seminhematol.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Kühne T, Imbach P, Bolton-Maggs PH, Berchtold W, Blanchette V, Buchanan GR. Intercontinental Childhood ITP Study Group. Newly diagnosed idiopathic thrombocytopenic purpura in childhood: an observational study. Lancet. 2001 Dec 22-29;358(9299):2122–2125. doi: 10.1016/S0140-6736(01)07219-1. [DOI] [PubMed] [Google Scholar]
- 8.Bolton-Maggs PH, Moon I. Assessment of UK practice for management of acute childhood idiopathic thrombocytopenic purpura against published guidelines. Lancet. 1997 Aug 30;350(9078):620–623. doi: 10.1016/s0140-6736(97)04143-3. [DOI] [PubMed] [Google Scholar]
- 9.Sutor AH, Harms A, Kaufmehl K. Acute immune thrombocytopenia (ITP) in childhood: retrospective and prospective survey in Germany. Semin Thromb Hemost. 2001 Jun;27(3):253–267. doi: 10.1055/s-2001-15255. [DOI] [PubMed] [Google Scholar]
- 10.Rosthøj S, Hedlund-Treutiger I, Rajantie J, et al. NOPHO ITP Working Group. Duration and morbidity of newly diagnosed idiopathic thrombocytopenic purpura in children: a prospective Nordic study of an unselected cohort. J Pediatr. 2003 Sep;143(3):302–307. doi: 10.1067/s0022-3476(03)00245-2. [DOI] [PubMed] [Google Scholar]
- 11.Choi SI, McClure PD. Idiopathic thrombocytopenic purpura in childhood. Can Med Assoc J. 1967 Sep 9;97(11):562–568. [PMC free article] [PubMed] [Google Scholar]
- 12.Lusher JM, Zuelzer WW. Idiopathic thrombocytopenic purpura in childhood. J Pediatr. 1966 Jun;68(6):971–979. doi: 10.1016/s0022-3476(66)80218-4. [DOI] [PubMed] [Google Scholar]
- 13.Payne BA, Pierre RV. Pseudothrombocytopenia: a laboratory artifact with potentially serious consequences. Mayo Clin Proc. 1984 Feb;59(2):123–125. doi: 10.1016/s0025-6196(12)60247-x. [DOI] [PubMed] [Google Scholar]
- 14.Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood. 2004 Jan 15;103(2):390–398. doi: 10.1182/blood-2003-05-1742. Epub 2003 Sep 22. [DOI] [PubMed] [Google Scholar]
- 15.Sartorius JA. Steroid treatment of idiopathic thrombocytopenic purpura in children. Preliminary results of a randomized cooperative study. Am J Pediatr Hematol Oncol. 1984 Summer;6(2):165–169. doi: 10.1097/00043426-198406020-00008. [DOI] [PubMed] [Google Scholar]
- 16.Buchanan GR, Holtkamp CA. Prednisone therapy for children with newly diagnosed idiopathic thrombocytopenic purpura. A randomized clinical trial. Am J Pediatr Hematol Oncol. 1984 Winter;6(4):355–361. doi: 10.1097/00043426-198424000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Sandler SG, Tutuncuoglu SO. Immune thrombocytopenic purpura—current management practices. Expert Opin Pharmacother. 2004 Dec;5(12):2515–2527. doi: 10.1517/14656566.5.12.2515. [DOI] [PubMed] [Google Scholar]
- 18.George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996 Jul 1;88(1):3–40. [PubMed] [Google Scholar]
- 19.Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993 Oct-Dec;10(4):317–321. doi: 10.3109/08880019309029508. [DOI] [PubMed] [Google Scholar]
- 20.Suarez CR, Rademaker D, Hasson A, Mangogna L. High-dose steroids in childhood acute idiopathic thrombocytopenia purpura. Am J Pediatr Hematol Oncol. 1986 Summer;8(2):111–115. [PubMed] [Google Scholar]
- 21.van Hoff J, Ritchey AK. Pulse methylprednisolone therapy for acute childhood idiopathic thrombocytopenic purpura. J Pediatr. 1988 Sep;113(3):563–566. doi: 10.1016/s0022-3476(88)80654-1. [DOI] [PubMed] [Google Scholar]
- 22.Jayabose S, Patel P, Inamdar S, Brilliant R, Mamtani R. Use of intravenous methylprednisolone in acute idiopathic thrombocytopenic purpura. Am J Pediatr Hematol Oncol. 1987 Summer;9(2):133–135. doi: 10.1097/00043426-198722000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Carcao MD, Zipursky A, Butchart S, Leaker M, Blanchette VS. Short-course oral prednisone therapy in children presenting with acute immune thrombocytopenic purpura (ITP) Acta Paediatr Suppl. 1998 Jun;424:71–74. doi: 10.1111/j.1651-2227.1998.tb01239.x. [DOI] [PubMed] [Google Scholar]
- 24.Imbach P, Barandun S, d'Apuzzo V, et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet. 1981 Jun 6;1(8232):1228–1231. doi: 10.1016/s0140-6736(81)92400-4. [DOI] [PubMed] [Google Scholar]
- 25.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001 Jan 19;291(5503):484–486. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 26.Hansen RJ, Balthasar JP. Effects of intravenous immunoglobulin on platelet count and antiplatelet antibody disposition in a rat model of immune thrombocytopenia. Blood. 2002 Sep 15;100(6):2087–2093. [PubMed] [Google Scholar]
- 27.Blanchette VS, Luke B, Andrew M, et al. A prospective, randomized trial of high-dose intravenous immune globulin G therapy, oral prednisone therapy, and no therapy in childhood acute immune thrombocytopenic purpura. J Pediatr. 1993 Dec;123(6):989–995. doi: 10.1016/s0022-3476(05)80400-7. [DOI] [PubMed] [Google Scholar]
- 28.Blanchette V, Imbach P, Andrew M, et al. Randomised trial of intravenous immunoglobulin G, intravenous anti-D, and oral prednisone in childhood acute immune thrombocytopenic purpura. Lancet. 1994 Sep 10;344(8924):703–707. doi: 10.1016/s0140-6736(94)92205-5. [DOI] [PubMed] [Google Scholar]
- 29.Salama A, Mueller-Eckhardt C, Kiefel V. Effect of intravenous immunoglobulin in immune thrombocytopenia. Lancet. 1983 Jul 23;2(8343):193–195. doi: 10.1016/s0140-6736(83)90175-7. [DOI] [PubMed] [Google Scholar]
- 30.Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc. 2004 Apr;79(4):504–522. doi: 10.4065/79.4.504. [DOI] [PubMed] [Google Scholar]
- 31.Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med. 2002 Mar 28;346(13):995–1008. doi: 10.1056/NEJMra010501. [DOI] [PubMed] [Google Scholar]
- 32.Scaradavou A, Woo B, Woloski BM, et al. Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Blood. 1997 Apr 15;89(8):2689–2700. [PubMed] [Google Scholar]
- 33.Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following Rh(0)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood. 2005 Sep 1;106(5):1532–1537. doi: 10.1182/blood-2004-11-4303. Epub 2005 May 5. [DOI] [PubMed] [Google Scholar]
- 34.Gaines AR. Acute onset hemoglobinemia and/or hemoglobinuria and sequelae following Rh(o)(D) immune globulin intravenous administration in immune thrombocytopenic purpura patients. Blood. 2000 Apr 15;95(8):2523–2529. [PubMed] [Google Scholar]
- 35.Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med. 2007 Jan 2;146(1):25–33. doi: 10.7326/0003-4819-146-1-200701020-00006. [DOI] [PubMed] [Google Scholar]
- 36.Zaja F, Baccarani M, Mazza P, et al. 50th ASH Annual Meeting and Exposition. American Society of Hematology. San Francisco, CA: 2008. A prospective randomized study comparing rituximab and dexamethasone vs dexamethasone alone in ITP: results of final analysis and long term follow up. December 7. https://ash.confex.com/ash/2008/webprogram/Paper8341.html. Accessed June 22, 2012. [Google Scholar]
- 37.Vancine-Califani SM, De Paula EV, Ozelo MC, Orsi FL, Fabri DR, Annichino-Bizzacchi JM. Efficacy and safety of dapsone as a second-line treatment in non-splenectomized adults with immune thrombocytopenic purpura. Platelets. 2008 Nov;19(7):489–495. doi: 10.1080/09537100802315110. [DOI] [PubMed] [Google Scholar]
- 38.Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Jr, Crowther MA. American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011 Apr 21;117(16):4190–4207. doi: 10.1182/blood-2010-08-302984. Epub 2011 Feb 16. [DOI] [PubMed] [Google Scholar]
- 39.British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol. 2003 Feb;120(4):574–596. doi: 10.1046/j.1365-2141.2003.04131.x. [DOI] [PubMed] [Google Scholar]
- 40.Indian Academy of Pediatrics. IAP Guidelines 2006 on Diagnosis. Management of ITP in Children. 2006. [PubMed]