

Abstract
The impact of synthesis and solution formulation parameters on the swelling and mechanical properties of a novel class of thermally and chemically gelling hydrogels combining poly(N-isopropylacrylamide)-based thermogelling macromers containing pendant epoxy rings with polyamidoamine-based hydrophilic and degradable diamine crosslinking macromers was evaluated. Through variation of network hydrophilicity and capacity for chain rearrangement, the often problematic tendency of thermogelling hydrogels to undergo significant syneresis was addressed. The demonstrated ability to easily tune post-formation dimensional stability at both the synthesis and formulation stages represents a significant novel contribution towards efforts to utilize poly(N-isopropylacrylamide)-based polymers as injectable biomaterials. Furthermore, the cytocompatibility of the hydrogel system under relevant conditions was established, while demonstrating time- and dose-dependent cytotoxicity at high solution osmolality. Such injectable in situ forming degradable hydrogels with tunable water content are promising candidates for many tissue engineering applications, particularly for cell delivery to promote rapid tissue regeneration in non-load-bearing defects.
Keywords: poly(N-isopropylacrylamide), polyamidoamine, tissue engineering, injectable, hydrogel
Introduction
Injectable scaffolds and regeneration therapies are being pursued for many tissue engineering applications, since they are seen as being minimally invasive and capable of filling even irregular defects.1, 2 However, there are often inherent trade-offs involved in the development of materials solutions for such applications. Chief among these is the trade-off between the mechanical strength of the scaffold and its overall water content, which directly influences the ability of the scaffold to promote the sustained development of new tissue through the diffusion of nutrients. Beyond this, the overall injectable formulation must first promote the proliferation and differentiation of cells and then degrade in an appropriate time scale to make room for continued growth of new tissue.
Hydrogel-based injectable tissue engineering solutions possess high water content but generally low mechanical strength, and are often pursued as beneficial candidates for the co-delivery cells and/or drugs to a defect site.3, 4 Such solutions are typically restricted to non-load-bearing applications, such as many craniofacial defects caused by birth, trauma, or disease, or applications in soft tissues, where scaffold elasticity may also be a key design parameter. Though the mechanical integrity of hydrogel scaffolds can be improved through maximized gel fraction, high degrees of crosslinking, or the use of hard-setting yet highly porous composite systems, the time scale of degradation must also be taken into account, as these strategies will generally slow degradation and subsequently the potential rate of tissue regeneration.
One particular class of injectable in situ forming hydrogels that has received much attention over recent years is thermosensitive polymers, which have lower critical solution temperatures (LCSTs) below body temperature.5 Thereby, solutions of the polymer are liquid at room temperature or with refrigeration and undergo thermogellation upon temperature elevation to 37°C. Poly(N-isopropylacrylamide)-based (PNiPAAm) polymers have thus far been some of the most studied and most promising injectable candidates for tissue engineering applications due to the ease of polymer synthesis,6 copolymerization flexibility,6, 7 and a convenient LCST of 32°C in water, the exact location of which can be tuned up or down with appropriate copolymer compositional changes.6–9 However, thermosensitive in situ forming polymers generally undergo significant post-formation gel syneresis due to the hydrophilic to hydrophobic transition and associated coil to globule chain collapse.6, 10 This is worrisome, since a major challenge in tissue regeneration is promoting nutrient and cell exchange and ultimately tissue integration with surrounding host tissue. Significant scaffold syneresis, which can take hours to fully complete, complicates this effort through reduced efficacy of hydrogel contact with surrounding tissue. While this can be mitigated to an extent through concomitant chemical crosslinking,6, 11 more effective solutions are needed.
We have recently reported a novel, two-component hydrogel system, which by compartmentalizing the thermogellation and hydrophilicity of the hydrogel through combination of a PNiPAAm-based polymer with pendant epoxy rings and a hydrophilic and degradable polyamidoamine-based diamine crosslinker, has demonstrated the ability to decrease and the potential to completely eliminate hydrogel syneresis.7 The polyamidoamine macromer chemistry, a step-polymerization-formed structure with tunable hydrophilicity and degradation rate (through backbone amide linkages) and reported biocompatibility,12, 13 offers tremendous synthetic flexibility to complement that of PNiPAAm-based thermogelling macromers.14, 15 In addition, the two-component design offers added flexibility to tune ultimate hydrogel properties at the formulation stage. This system has been shown to offer a rapid and facile dual-gelation mechanism, with near-instantaneous thermogellation and concomitant chemical crosslinking largely completed within two hours, and a degradation time scale of approximately ten weeks in pH 7.4 PBS at 37°C.7
The objective of this study was to investigate the potential of creating injectable thermogelling hydrogels with tunable post-formation dimensional stability and to determine structure-property relationships inherent in this novel system through alteration of synthesis and formulation parameters, with the specific goal of eliminating post-formation syneresis. To this end, hydrogels with varying polyamidoamine molecular weights, compositional ratios, overall polymer contents, and formulation procedures were examined utilizing full and fractional factorial experimental designs. It was hypothesized that the use of factorial experimental designs would allow for examination of parameter interactions and provide additional insight into the general hydrogel behavior of such two-component in situ forming systems. Additionally, the in vitro cytocompatibility of the components, the hydrogel, and the degradation products was assessed.
Materials and Methods
Materials
N-isopropylacrylamide (NiPAAm), glycidyl methacrylate (GMA), 2,2′-azobis(2-methylpropionitrile) (azobisisobutyronitrile, AIBN), N,N′-methylenebisacrylamide (MBA), and piperazine (PiP) were purchased from Sigma-Aldrich (Sigma, St. Louis, MO) and used as received. The solvents; tetrahydrofuran (THF), dimethylformamide (DMF), diethyl ether, and acetone in analytical grade, and water, acetonitrile, chloroform, and methanol in HPLC-grade; were obtained from VWR (Radnor, PA) and used as received. Phosphate-buffered saline (PBS) solution was mixed from powder (pH 7.4, Gibco Life, Grand Island, NY), and ultrapure water was obtained from a Millipore Super-Q water system (Millipore, Billerica, MA).
Thermogelling Macromer (TGM) Synthesis
The thermogelling macromer, P(NiPAAm-co-GMA), was synthesized by free radical polymerization (Scheme 1), as previously reported.7 Ten grams of the comonomers, NiPAAm and GMA at 92.5 and 7.5 mol %, respectively, were dissolved in 100 mL of DMF and polymerized at 65°C under a nitrogen atmosphere. AIBN was added as a free radical initiator at 0.7 mol % of the total monomer content, and the reaction mixture was continuously stirred for 20 h. The product was concentrated by rotary evaporation, dissolved in THF, and twice precipitated in at least 10-times excess of cold diethyl ether to effectively remove the unreacted monomers and low molecular weight oligomers. The final filtrate was dried under vacuum at ambient temperature to yield a fine white powder. In addition, a similar procedure was followed to create a PNiPAAm homopolymer.
Scheme 1.

Synthesis of P(NiPAAm-co-GMA), PAMAM, chemical and thermal network gelation, and in situ PAMAM degradation.
Polyamidoamine (PAMAM) Synthesis
The polyamidoamine crosslinking macromers were synthesized by polyaddition of PiP with MBA (Scheme 1), following reported protocols.7, 14, 15 In a typical synthesis, 10.83 g of the comonomers were dissolved in 30 mL ultrapure water with a stoichiometric excess of PiP (r = [MBA]/[PiP] = 0.6, 0.78, or 0.85), stirred continuously under a nitrogen atmosphere at 30°C, and allowed to react for 48 h. The obtained viscous mixture was directly precipitated in 100 mL of acetone, filtered, and dried under vacuum at ambient temperature to yield a fine powder.
Proton Nuclear Magnetic Resonance Spectroscopy (1H NMR)
1H NMR spectra were obtained using a 400 MHz spectrometer (Bruker, Switzerland). Sample materials were dissolved in deuterium oxide (D2O; typical concentration of 20 mg/mL) that contained 0.75 wt % 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid, sodium salt (TSP) as internal shift reference (Sigma-Aldrich, St. Louis, MO). All post-acquisition data processing was performed with the MestRe-C NMR software package (Mestrelab Research S.L., Spain). The free induction decay (FID) was Fourier transformed, manually phased, referenced using the TSP signal, baseline corrected, and integrated.
MicroTOF Mass Spectroscopy
Molecular weight distributions of the synthesized PAMAM crosslinkers were analyzed using time-of-flight mass spectroscopy with positive-mode electrospray ionization on a Bruker microTOF ESI spectrometer (Bruker Daltonics, Billerica, MA) equipped with a 1200 series HPLC (Agilent Technologies, Santa Clara, CA) to deliver the mobile phase (50:50 HPLC-grade water and methanol). After complete PAMAM purification and drying, the final products were dissolved in a 50:50 mixture of water and acetonitrile (HPLC-grade; with 0.1% (v/v) formic acid added) at a concentration of 15 μM, and a 2 μL flow injection was delivered to the electrospray chamber. After data acquisition, all peaks (including degradation and secondary reaction products) were identified using microTOF Control software (Bruker), corrected for charge state (generally with H+ or Na+ and rarely K+ ions), and quantified for calculation of number- and weight-average molecular weights (Mn and Mw, respectively) and polydispersity index (PDI), following previously reported protocols.7
Rheological Characterization
A thermostatted, oscillating rheometer (Rheolyst AR1000, TA Instruments, New Castle, DE) equipped with a 6 cm steel cone (1 degree) with a gap size of 26 μm was used to evaluate the LCST of the synthesized thermogelling macromers. Solutions of 10 wt % TGM in pH 7.4 PBS were pipetted onto the rheometer, and the dynamic viscoelastic properties of the solutions, namely, the dynamic shear storage (G′) and loss (G″) moduli, complex viscosity (|η*|), and loss angle (δ), were recorded using the TA Rheology Advantage software (TA Instruments) as the solution temperature was increased at a rate of 1°C/min.
Hydrogel Formation and Swelling Properties
Individual solutions of the TGM and PAMAM macromers were prepared at twice the desired final solution concentrations, and 200 μL of each solution were combined in a glass vial at 4°C and mixed gently for ~30 s. After varied preparation times at 4°C, 90 μL injections were made into 37°C Teflon molds using cold pipette tips, and the hydrogels were allowed to chemically and thermally gel for 24 h, as illustrated in Scheme 1. Once ready, hydrogels were removed from the molds, weighed in air on an analytical balance, placed in an excess of pH 7.4 PBS, given an additional 24 h to equilibrate, reweighed, frozen at −80°C, lyophilized, and weighed a third time in the dry state. The hydrogels’ weight swelling ratios at formation, qf, and equilibrium, qeq, were calculated as the difference in swollen and dry weights divided by the dry weight. Additionally, hydrogels were similarly prepared with 250 μL each of TGM and PAMAM solutions in glass vials at 4°C, immediately immersed in a 37°C water bath, allowed to set for 24 h, and the hydrogels were collected for visual analysis.
Compressive Mechanical Testing
The mechanical integrity of hydrogel formulations was evaluated by determination of the compressive Young’s Modulus. Hydrogel cylinders of 6 mm in diameter and 15 mm in height were created by solution injection into 37°C Teflon molds and allowed to set for 24 h. The recovered cylinders were cut into multiple sections of nominally 4 mm in height, and two cylinders were used for each formulation tested, yielding 4–6 replicates. The cylindrical samples were then analyzed using a TA Instruments Thermomechanical Analyzer (TMA) 2940 (TA Instruments, Newcastle, DE) equipped with a wide compression probe (diameter of 6 mm). After pre-warming the oven and stage to 37°C, samples were placed on the stage, the sample height was determined by the instrument probe, the temperature was re-equilibrated at 37°C, the sample was compressed at a rate of 0.001 N/min, and the initial slope of the engineering stress versus engineering strain curve was used to determine the engineering unconfined compressive Young’s Modulus.16
Cell Culture
A rat fibroblast cell line (ATCC, CRL-1764) was cultured on T-75 flasks using Dulbecco’s modified Eagle medium (DMEM; Gibco Life, Grand Island, NY) supplemented with 10% (v/v) fetal bovine serum (FBS; Cambrex BioScience, Walkersville, MD) and 1% (v/v) antibiotics containing penicillin, streptomycin and amphotericin (Gibco Life). Cells were cultured in a humidified incubator at 37°C and 5% CO2. Cells of passage number 3 were used in this study.
Cytocompatibility Assay
The cytocompatibility of the chemically and thermally gelled hydrogels was evaluated by exposure of cultured fibroblasts to dissolved PAMAM macromers, hydrogel leachables, and degradation products, according to established protocols.7, 17, 18 Cultured cells were harvested at 80–90% confluency with a Trypsin/EDTA solution (2 mL/flask), re-suspended at a density of 100,000 cells/mL, and seeded into 96-well tissue culture plates (100 μL cell suspension/well) for a seeding density of 10,000 cells/well. The plates were then incubated for 24–48 h before testing to achieve 80–90% confluency within the wells, and the test media were added to the cultured fibroblast cells in the 96-well plates (100 μL/well), replacing the culture media (n = 6). In addition, cells fed with identical media without the test diluents (DMEM supplemented with antibiotics and without serum) served as a positive (live) control, and cells exposed to 70% ethanol for 10 min served as a negative (dead) control (n=6). The plates were then incubated at 37°C, 95% relative humidity, and 5% CO2 for either 2 or 24 h.
Following incubation, media were removed, the cells were rinsed three times with pH 7.4 PBS, calcein AM and ethidium homodimer-1 in 2 μM and 4 μM concentrations in PBS, respectively (Live/Dead viability/cytotoxicity kit, Molecular Probes, Eugene, OR), were added, and the cells were incubated in the dark at room temperature for 30 min. Cell viability was then quantified using a fluorescence plate reader (Biotek Instrument FLx800, Winooski, VT) equipped with filter sets of 485/528 nm (excitation/emission) for calcein AM (live cells) and 528/620 nm (excitation/emission) for EthD-1 (dead cells). The fluorescence of the cell populations was recorded and the fractions of live and dead cells were calculated relative to the controls.
Cytocompatibility Test Media Preparation
PAMAM macromers of two molecular weights (1900 and 2600 Da) were first dissolved directly in media without the addition of serum at concentrations of 56.4 and 170 mg/mL or 92.4 and 277 mg/mL for P-1900 and P-2600, respectively. These concentrations were equivalent to what would be found in injectable solutions for 10 wt % TGM with a 1:1 amine:epoxy mol ratio and 20 wt % TGM with a 1.5:1 amine:epoxy mol ratio hydrogels, respectively. In addition, 10× and 100× dilutions of the highest PAMAM concentrations were tested for each macromer.
Next, P-1900 hydrogel discs with 10 wt % TGM, 30 min preparation time, and a 1:1 amine:epoxy mol ratio were formed in cell culture media (DMEM supplemented with antibiotics and without serum) by injection into 37°C Teflon molds (6 mm in diameter and 3 mm in height) and given 24 h to equilibrate. Hydrogels were then immersed in an excess of cell culture media without serum at a surface area to fluid volume ratio of 3 cm2/mL and incubated at 37°C for 24 h.18 The resulting hydrogel leachables solution was collected, sterile filtered, and prepared in 1, 10, and 100× dilutions.
Finally, a P-1900 hydrogel with 10 wt % TGM and a 1:1 amine:epoxy mol ratio was prepared in ultrapure water, thermogelled at 37°C for 24 h, and allowed to undergo accelerated degradation at 70°C. Upon complete degradation, the soluble degradation products were collected, isolated by lyophilization, re-dissolved in media without serum at the same concentration in which they were formed, sterile filtered, and prepared in 1, 10, and 100× dilutions.
pH and Osmolality Determination
The pH of the P-1900 and P-2600 PAMAM macromer solutions tested in the cytocompatibility assay was measured using a sympHony® pH meter (VWR, Radnor, PA) calibrated with standard buffers (pH 4, 7, and 10; VWR) using a sympHony® Posi-pHIo Epoxy pH electrode (VWR). In addition, the solution osmolality was determined by measurement with an Osmette A Automatic Osmometer (Precision Systems, Inc., Natick, MA) calibrated with 100 and 500 mOsm/kg H2O standard solutions (Precision Systems, Inc.) and run on the 0 – 2k mOsm range.
Statistics
The data are typically expressed as means ± standard deviation for three replicates, unless otherwise specified. Statistically significant differences were determined by Tukey’s post hoc test (p < 0.05) within time points for the cytocompatibility data or by a t-test (p < 0.05) in all other instances. Analysis of the main effects and parameter interactions in the 23 full factorial experimental designs was completed according to published methods.19
Results and Discussion
Macromer Synthesis
Synthesis and characterization of P(NiPAAm-co-GMA) have been previously reported to yield a number-average molecular weight, Mn, of ~9.2 kDa and a PDI of 3.1.7 In addition, the LCST was determined in this study by monitoring the rheological response of the injectable solution as the temperature was increased. The results (Figure 1) showed a decrease in the LCST of ~5°C compared to a PNiPAAm homopolymer resulting from the incorporation of 7.5 mol % GMA. The increase in shear moduli, attributed to physical thermogellation of the polymer network, was also accompanied by further evidence of a phase change with the transition of an initially transparent liquid to an opaque gel (e.g. a thermodynamic rather than simply kinetic transition occurred), a phenomenon which was observed in every formulation tested in this investigation. There is also often a thermal signal given off by the phase transition, which we have previously measured utilizing differential scanning calorimetry (DSC),7 however this method has its drawbacks, as different formulations may have different signal intensities and the thermal signal can even be completely eliminated in extreme cases. Finally, the rheometry data clearly demonstrate a 6-order of magnitude change in moduli consistent with gelation of a liquid which is sustained beyond the LCST. The ability to monitor changes in the system beyond the initial transition is a key benefit of rheometry in this application over other more commonly employed methods of LCST determination including DSC and light spectroscopy.
Figure 1.

Complex shear storage (G′) and loss (G″) moduli for PNiPAAm and P(NiPAAm-co-GMA) with increasing temperature, as determined by rheology.
Synthesis of PAMAM diamine crosslinking macromers with varied stoichiometric feed ratios was first evaluated by 1H NMR, according to previously reported protocols.7 Table 1 lists the resulting Mn values determined for the three PAMAM crosslinkers. In addition, since the PAMAM synthesis reaction was run in water and the product is susceptible to hydrolytic degradation, further characterization of the product species was desired. Utilizing microTOF mass spectroscopy, all species in the reaction product, including the desired diamine heteroend variant, degradation products, and secondary reaction products, were identified and agreed with previously reported compositions (data not shown).7 The results of the microTOF analysis are presented in Table 1, and Figure 2 shows the molecular weight distributions of the three synthesized PAMAM macromers (both y-axes report the same value with differing scales for ease of distribution comparison). As can be seen, the reduced stoichiometric imbalance resulted in higher molecular weight crosslinkers, as expected by theory. Additionally, both the purity, expressed as mol and wt % of the total synthesis product that is the desired diamine, and distribution breadth of the synthesized diamine species increased with increasing molecular weight.
Table 1.
Molecular weights and purities of PAMAM crosslinkers determined by 1H NMR and microTOF mass spectroscopy for three stoichiometric reaction feed ratios, r.
| Feed | (NMR) | Desired Diamine Product (microTOF analysis) | All Species in Material | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| r | Mn | Mn | Mw | PDI | mol % of Total | wt % of Total | Mn | Mw | PDI | |
| P-800 | 0.6 | 675 | 820 | 920 | 1.12 | 82.8% | 86.0% | 790 | 890 | 1.12 |
| P-1900 | 0.78 | 1890 | 1,860 | 2,420 | 1.30 | 79.9% | 89.1% | 1,670 | 2,290 | 1.37 |
| P-2600 | 0.85 | 3000 | 2,580 | 3,370 | 1.31 | 83.9% | 93.2% | 2,320 | 3,230 | 1.39 |
Figure 2.

Molecular weight distributions of PAMAM crosslinkers determined by MicroTOF mass spectroscopy (arrows beside the legend indicate which set(s) of data are plotted on each y-axis).
Hydrogel Swelling Behavior
In order to evaluate the hypothesized potential to eliminate post-formation hydrogel syneresis and to establish structure-property relationships between the ultimate properties and the structural makeup and preparation conditions of the thermogelled and crosslinked injectable hydrogels, a number of parameters were varied: the content of TGM in the injectable solution (wt %); the molecular weight of the PAMAM crosslinker; the functionality ratio between amines available for reaction on the PAMAM crosslinker and epoxy rings on the TGM (the amine:epoxy mol ratio); and the preparation time, or the length of time the solution was held at 4°C after the TGM and PAMAM crosslinkers were mixed before temperature elevation to 37°C. Table 2 lists all values of each parameter that were considered.
Table 2.
All hydrogel formulation parameters tested.
| Parameter Level | 1 | 2 | 3 |
|---|---|---|---|
| TGM Content (wt %) | 10 | 15 | 20 |
| PAMAM MW (kDa) | 0.8 | 1.9 | 2.6 |
| Amine:Epoxy Mol Ratio | 0.5:1 | 1:1 | 1.5:1 |
| Preparation Time (min) | 10 | 20 | 30 |
In particular, the preparation time had a significant impact on the degree of post-formation syneresis or swelling of the injectable hydrogels. Figure 3 illustrates this effect as the preparation time at 4°C is increased from 0 to 30 min. As can be seen, the degree of syneresis was significantly reduced at the higher preparation times for P-800 hydrogels with 10 wt % TGM and a 0.5:1 amine:epoxy mol ratio; though, in this case even at 30 min preparation time, the hydrogel still expelled 60% of the initial liquid solution. Since the epoxy-amine reaction is quite favorable, crosslinking still occurs readily at 4°C, though at a reduced rate, and it will eventually result in a crosslinked but non-thermogelled hydrogel. This dependence on the initial degree of reaction before thermogellation was not initially expected to be such a significant factor; however it can be understood through consideration of two contributions.
Figure 3.
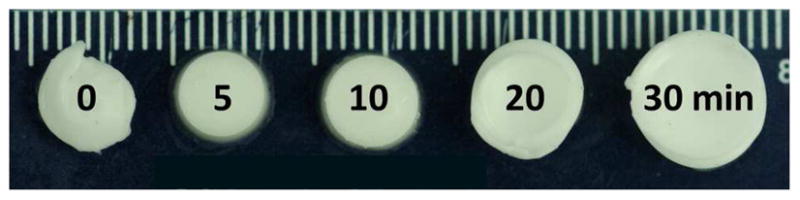
Images of P-800 hydrogels with 10 wt % TGM and a 0.5:1 amine:epoxy mol ratio at varying preparation times (0, 5, 10, 20, and 30 min).
First, as the PAMAM crosslinkers begin to react with the TGM and create branched and crosslinked structures, the severity of the coil-globule chain collapse and expulsion of water can be reduced due to increased hydrophilicity of the average TGM chain. This occurs despite no appreciable change in the location of the hydrogel’s LCST between 0 and 30 min preparation time, as determined in this investigation by DSC (data not shown). This finding is consistent with published reports showing increased equilibrium hydrogel water content in PNiPAAm-based hydrogels containing increasing fractions of a super-water-absorbent comonomer without significantly altering the location or occurrence of the system’s LCST.20, 21
Second, the formation of branched and crosslinked polymer chains could impart greater diffusional restrictions to the syneresis process, which in turn allows more time for crosslinks to form prior to complete chain rearrangement. This in effect increases the degree of swelling of the network since a crosslinked structure will generally seek to maintain the configuration in which it was crosslinked. When taken with previous results showing completion of the crosslinking reaction within 2–3 hours despite near-instantaneous thermogellation and rapid equilibration of the hydrogels7 and the observation that increased preparation time did not noticeably affect the ease of solution injection into 37°C molds, the first effect is likely to be the more significant. Figure 4 shows quantitative equilibrium weight swelling ratios, q, of hydrogels with varied amounts of P-800 added to both 10 and 20 wt % TGM solutions. As can be seen, there were diminishing returns for increasing the preparation time beyond 20 min. Since one of the primary objectives of this investigation was to eliminate post-formation hydrogel syneresis, all future studies were performed at either 20 or 30 min preparation time at 4°C.
Figure 4.

Effect of preparation time on equilibrium swelling ratio, qeq, of hydrogels with varied mol fractions of P-800 and either 10 or 20 wt % TGM.
The swelling capacity, reported as the weight swelling ratio, q, of the thermogelling hydrogels was measured both after formation (subsequent to injection in a 37°C mold and allowing 24 h to equilibrate) and at equilibrium (after an additional 24 h in an excess of pH 7.4 PBS). The measurement of weight swelling both at gel formation and at equilibrium allowed for observation of post-formation hydrogel stability. Thus, whether hydrogels will undergo syneresis, remain dimensionally stable, or expand further after formation could be determined. Typical data are shown in Figure 5 for formulations with 20 min preparation time. The lower formation swelling ratios for 20 wt % TGM solutions can easily be understood since there is more hydrogel to hold the same initial volume of liquid. However, at equilibrium, the 20 wt % TGM hydrogels generally matched the 10 wt % hydrogels in terms of their weight swelling ratios. Some reduction in swelling was expected, since the 20 wt % TGM formulations were thought to provide a more tightly crosslinked network structure which might resist further expansion. It should also be mentioned that similarities in the formation weight swelling ratio as the PAMAM content was increased were largely a reflection of the limited supply of liquid at formation, thus indicating no syneresis of the hydrogel.
Figure 5.

Formation and equilibrium weight swelling ratios, q, for P-800 and P-2600 hydrogels with 20 min preparation time at 10 and 20 wt % TGM with varied amine:epoxy mol ratios.
Significantly, Figure 5 demonstrates the ability through parameter modification at both the synthesis and formulation stages to tune the swelling properties to create hydrogels that undergo syneresis, are dimensionally stable, or swell after initial formation. Together with the data presented in Figure 6, we can see that the primary contribution to the equilibrium degree of swelling of the hydrogels was the total content of the PAMAM crosslinker, or the overall hydrophilicity of the resulting hydrogel, which appeared largely insensitive to the degree of crosslinking or conditions under which the hydrogels were initially formed. The former effect is illustrated in Figure 6 first by continually increasing degrees of swelling as the degree of crosslinking is first increased and then decreased (after passing the theoretical maximum degree of crosslinking at equimolar amine and epoxy functional group contents) and second by the increased swelling at the same degree of crosslinking achieved through increasing PAMAM lengths. The latter effect is apparent in Figure 6 as trends were consistent with increasing PAMAM content despite differences in TGM content (10 vs. 20%). However, potential interactions and cross effects amongst the parameters are not accounted for in these experiments.
Figure 6.

Equilibrium weight swelling ratio, qeq, versus mol fraction PAMAM for P-800 and P-1900 hydrogels with 30 min preparation time at 10 and 20 wt % TGM.
In order to evaluate the effects and interactions of the three hydrogel composition parameters (TGM content, PAMAM MW, and amine:epoxy mol ratio) in a more formal manner, a 23 full factorial experimental design and analysis was completed with high/low values of each parameter, as detailed in Table 3. Also listed in Table 3 are the absolute weight swelling ratios at both formation and equilibrium with standard deviations for each group tested (n = 3). Data analysis was performed according to published protocols,19 and the results are presented in Figure 7, which shows the magnitude of the main effects of each individual parameter and the interactions amongst the parameters at both formation and equilibrium (the factor and interaction labels A, B, AB, etc. in Figure 7 and all subsequent figures are based upon group headings presented in Table 3). In the graph, the zero line is representative of the overall population mean at each effect (4.0 ± 1.4 and 4.4 ± 1.2 for formation and equilibrium swelling, respectively), and the changes associated with each parameter and parameter-pair interaction are shown with error bars representing the standard error of each effect population (formation and equilibrium swelling).
Table 3.
Hydrogel formulation parameters tested (i) and experimental weight swelling ratios at formation, qf, and equilibrium, qeq, and compressive Young’s Modulus, E, (ii; with standard deviations shown) for a 23 full factorial experimental design.
| (i) | TGM Content | PAMAM MW | Amine:Epoxy Mol Ratio | |||
|---|---|---|---|---|---|---|
| High (+) | 20 WT % | 2.6 KDA | 1:1 | |||
| Low (−) | 10 WT % | 0.8 KDA | 0.5:1 | |||
| (ii) Group | A | B | C | qf (formation) | qeq (equilibrium) | E (kPa) |
| (1) | − | − | − | 3.16 ± 0.37 | 3.12 ± 0.39 | 1.24 ± 0.22 |
| a | + | − | − | 2.28 ± 0.35 | 3.30 ± 0.14 | 3.03 ± 0.54 |
| b | − | + | − | 4.37 ± 0.30 | 3.27 ± 0.42 | 0.78 ± 0.08 |
| c | − | − | + | 5.23 ± 1.45 | 5.24 ± 0.48 | 1.42 ± 0.35 |
| ab | + | + | − | 4.49 ± 0.54 | 5.06 ± 0.72 | 3.51 ± 0.73 |
| ac | + | − | + | 2.31 ± 0.61 | 4.86 ± 0.08 | 2.47 ± 0.64 |
| bc | − | + | + | 5.55 ± 0.63 | 5.82 ± 0.50 | 1.18 ± 0.12 |
| abc | + | + | + | 3.62 ± 0.03 | 6.13 ± 0.33 | 4.26 ± 1.92 |
Figure 7.

Main effects and interactions for weight swelling ratios at formation and equilibrium from the 23 full factorial experimental design.
The most significant factor at formation was the TGM content of the hydrogel, however as previously indicated from Figure 5, this effect is shown to completely disappear at equilibrium swelling. All other conditions being equivalent, changing the PAMAM MW from low to high (factor B) resulted in a theoretical three-fold increase in the total mass content of PAMAM in the polymer, while changing the amine:epoxy ratio (factor C) from low to high resulted in a 50% theoretical increase. While both factors had a significant positive impact on swelling at formation, with the more significant increase in hydrophilicity (factor B) trending towards a greater impact in degree of swelling, at equilibrium this trend was reversed, and the lesser expected increase in hydrophilicity resulted in a statistically greater increase in the water content of the hydrogels. To understand this phenomenon, it is important here to note that, with the increase of PAMAM content through the amine:epoxy mol ratio, the high value represents the maximum theoretical degree of crosslinking with sufficient amines to react all epoxy rings without creating significant quantities of branched structures, whereas the low ratio represents the capacity to consume only 50% of the available epoxy rings. Thus, while factor B was initially hypothesized to have a greater impact on equilibrium swelling than factor C, it is likely that the presence versus absence of epoxy rings in the network contributed significantly to the unexpectedly higher impact of increasing factor C. This assertion is supported by the decrease in LCST associated with introduction of GMA into the PNiPAAm backbone (Figure 1) which indicates the creation of a more hydrophobic thermogelling polymer.
It can also be seen from Figure 7 that the various parameters tested exhibited interdependence upon one another through the AB, AC, and BC interactions. These cross effects are further illustrated for the equilibrium weight swelling data in Figure 8. For both the AC and BC interactions, we see that the increase in equilibrium swelling associated with increasing the content of the PAMAM crosslinker was enhanced by decreasing either the TGM content or the PAMAM MW. The reduced increase in equilibrium swelling due to increased hydrophilic content when moving to a higher initial TGM content (AC interaction) was likely a consequence of initially forming the crosslinked network in a more compact/dense state; thereby leading to a reduced hydrogel capacity for further expansion after formation.
Figure 8.

The interaction effects between wt % TGM, PAMAM MW, and amine:epoxy mol ratio on the weight swelling ratio at equilibrium, qeq, of crosslinked hydrogels from the 23 full factorial experimental design.
Similarly, the BC interaction can be understood as the consequence of forming a tighter network structure due to shorter interchain bridge lengths, thereby imparting a reduced capacity for elastic expansion of the hydrogel. This conclusion is supported by recent observations on the effects of crosslinker interchain bridge length on dynamic swelling of polymer networks.22 This same trend is evident once again in the AB interaction, where despite leading to a decrease in swelling upon increase in TGM content from 10 to 20 wt % for the 0.8 kDa hydrogels, the 2.6 kDa PAMAM hydrogels displayed increased swelling. Essentially, a more loosely crosslinked network structure due to longer interchain bridge lengths allowed the hydrogel to respond more readily to changes in other properties through increased swelling. Thereby, the decrease in weight swelling ratio for the 0.8 kDa hydrogel at 20 wt % TGM was considered a reflection of increased initial polymer mass. Parameter interactions on formation weight swelling ratios mimicked those at equilibrium swelling (data not shown).
The results of the full factorial experimental swelling designs were quite interesting and offered several insights into the hydrogel network dynamics involved in this and similar systems that were not initially anticipated or evident in studies evaluating the effects of changing each parameter individually. Chief among these was that increasing the number of crosslink points had only a very minimal impact on equilibrium degrees of hydrogel swelling, due most likely to the intertwined nature of several variables. For example, decreasing the crosslinking degree would normally increase swelling; however this effect was mitigated in this system by increased network hydrophobicity due to more unreacted pendant epoxide rings. However, the overall initial network configuration did still have significant impacts as a result of altering the ability of the hydrogel to respond to access to additional liquid going from formation to equilibrium swelling conditions. Both increasing the chain density (and hence the crosslinking density on a per-unit-volume basis) at formation through increased TGM content and reducing the interchain crosslinker bridge length through reduced PAMAM molecular weight led to a reduced capacity for subsequent hydrogel expansion due to a more tightly crosslinked network. These observations are directly relevant to this and similar injectable in situ hardening polymeric systems where post-formation network expansion is possible.
Hydrogel Mechanical Behavior
In addition to swelling behavior, the effects of the varied network composition parameters on the unconfined compressive Young’s Modulus were evaluated in a 23 full factorial experimental design. The groups were identical to those in the swelling study with the exception of a 30 min preparation time for all samples, and the groups, absolute values of the unconfined compressive Young’s Modulus, and standard deviations (n = 4–6) are given in Table 3. The results at gel formation were analyzed as before, and the main effects and interactions are presented in Figure 9, with the zero line representing the population mean of 2.3 ± 1.4 kPa. Whereas increasing the PAMAM molecular weight (factor B) resulted in a significant increase in the compressive modulus, increasing the PAMAM molar content and the associated degree of crosslinking (factor C) did not. Since the data were collected on gels after formation, the former effect is likely a reflection of increased polymer content with the much longer crosslinker. As expected, increasing the TGM content (factor A) led to significant increases in modulus.
Figure 9.

Main effects and interactions for compressive Young’s Modulus, E, at formation from the 23 full factorial experimental design.
Additionally, the only particularly significant cross effect (AB; shown in Figure 10) demonstrated the cooperative impact of increasing the total polymer content through increasing either or both the TGM and PAMAM content. Finally, Figure 11 shows typical absolute values of the compressive Young’s Modulus for P-1900 hydrogels at 30 min preparation time with increasing TGM and PAMAM content with statistically significant differences (as determined by a t-test; p < 0.05) shown. As can be seen, first increasing the degree of crosslinking (going from an amine:epoxy mol ratio of 0.5:1 to 1:1) and subsequently decreasing the degree of crosslinking through further incorporation of PAMAM (from 1:1 to 1.5:1) resulted in similar increases in the unconfined compressive Young’s modulus. Taken with the factorial design results, we conclude that in these systems the degree of crosslinking had only a minimal impact on the compressive properties, with the overall polymer content accounting for nearly all changes. This was initially unexpected, but is likely a reflection of the compressive dynamics in networks formed at very high solvent contents (e.g. the standard conceptualization of a network as a homogeneous ‘mesh’ may not be appropriate) and the fact that unconfined compression allows for the expulsion of liquid during compression. The degree of crosslinking would still be expected to more significantly affect both tensile and shear moduli.
Figure 10.

The interaction effect between wt % TGM and PAMAM MW on the compressive Young’s Modulus, E, of crosslinked hydrogels.
Figure 11.

Compressive Young’s Modulus, E, for P-1900 hydrogels at 30 min prep time with varying wt % TGM at a 1:1 amine:epoxy mol ratio and varying amine:epoxy mol ratios at 10 wt % TGM (* indicates a statistical difference determined by a t-test; p < 0.05).
In Vitro Cytocompatibility Assessment
Cytocompatibility of the novel hydrogel system was evaluated in vitro by treating live cells with media with either P-1900 or P-2600 PAMAM macromers dissolved at varying concentrations, with leachables from P-1900 hydrogels, or with degradation products from a P-1900 hydrogel and incubating at 37°C for 2 or 24 h (Figure 12). Statistically significant differences were determined by a Tukey’s test within time points and a t-test across time points (p < 0.05). As can be seen, the short-term leachables from the P-1900 hydrogel system (c) were cytocompatible, which agrees with a previously reported leachables study with hydrogels made from P-2200 PAMAM crosslinkers.7 Leachables from hydrogels with 10 wt % TGM and no added PAMAM were likewise cytocompatible at all conditions tested (data not shown). The dissolved PAMAM crosslinkers and soluble degradation products displayed a time- and dose-dependent response.
Figure 12.

Viability of rat fibroblasts exposed to media with varying dissolved concentrations of P-1900 (a) and P-2600 (b) PAMAM crosslinkers; with 1, 10, and 100× dilutions of leachables (c) from P-1900 hydrogels with a 1:1 amine:epoxy ratio, 10 wt % TGM, and 30 min preparation time; and with 1, 10, and 100× dilutions of degradation products (d) from P-1900 hydrogels with a 1:1 amine:epoxy ratio, 10 wt % TGM, and 30 min preparation time over 2 and 24 h relative to the live controls, as determined by fluorescence intensity from Live/Dead staining, with live and dead controls shown (# and * indicate statistical differences within and between time points, respectively).
When dissolved directly in media without modification or available reaction pathways, the P-1900 and P-2600 PAMAM crosslinkers were cytocompatible at low concentrations and cytotoxic at higher concentrations. To understand the cause behind this response, we must consider the nascent effects caused by high concentrations of the ionizable PAMAM diamine species. Figure 13 demonstrates the effect of increased content of P-1900 and P-2600 macromers dissolved in media without serum on the pH and osmolality of the solutions. Whereas the increase in pH is initially very significant, there is a diminishing return on continued addition of PAMAM. In contrast, the osmolality scaled linearly with the mass concentration of the PAMAM species over two orders of magnitude. It is not surprising that at such high values of pH and osmolality the dissolved PAMAM macromers exhibited a cytotoxic response.
Figure 13.

pH and osmolality of media without the addition of serum as a function of concentration (mg/mL) of polyamidoamine crosslinkers P-1900 and P-2600.
However, despite these unfavorable conditions, only the very highest concentrations ever tested in this investigation (170 and 277 mg/mL for P-1900 and P-2600, respectively, which correspond to hydrogels with 20 wt % TGM and a 1.5:1 amine:epoxy mol ratio) exhibited significant cytotoxicity over a 2 h exposure period. Since the amine-epoxy reaction has been shown previously to be largely completed within ~110 min,7 the higher cytotoxicity at 24 h is not relevant to planned usage of the materials but illustrates the time- and dose-dependent response of the cells to the soluble PAMAM macromers. The additional treatment concentrations correspond to 10× and 100× dilutions from the maximum concentrations tested, as well as a more relevant working concentration representing hydrogels with 10 wt % TGM and a 1:1 amine:epoxy mol ratio (56 and 92 mg/mL for P-1900 and P-2600, respectively).
Finally, a P-1900 hydrogel with 10 wt % TGM and a 1:1 amine:epoxy mol ratio formed in ultrapure water (as opposed to PBS) was degraded under accelerated conditions at 70°C, and the soluble degradation products were isolated, dissolved in media without serum, and their cytocompatibility was evaluated (Figure 12(d)). The use of ultrapure water and elevated temperature for accelerated degradation, as opposed to PBS or sodium hydroxide solutions, allowed for isolation by lyophilization of the pure concentrated degradation products. Thus, the time- and dose-dependent response shown is a consequence only of the degradation products, without additional mass and ion concentrations. The 1× dilution represents the full, concentrated dose of the degradation products, and was cytotoxic.
This outcome was anticipated from examination of the response of cells to the PAMAM macromers alone (Figure 12(a)). In that case, cytotoxicity at high concentrations was attributed primarily to the high increases in solution osmolality (Figure 13). At complete degradation, we have taken those PAMAM molecules and cleaved them into roughly 15 times the number of ionizable molecules. With complete long-term degradation of the PAMAM macromers having been reported at approximately ten weeks7 and a reasonable assumption that release of degradation products will proceed at an essentially linear rate, the anticipated exposure of cells to these degradation products over an average 24 h period is expected to lie close to the 100× dilution, particularly considering the high water content and associated favorable dilution and diffusion conditions inherent in the current hydrogel design.
Conclusions
The effects of synthesis and formulation parameters on the swelling and mechanical behavior of a recently developed in situ dual-gelling class of injectable hydrogels combining PNiPAAm-based thermogelling macromers with PAMAM-based crosslinking macromers were investigated. Hydrogel swelling behavior was shown to depend primarily on the total hydrophilic content of the network and secondly on the ability of the network to undergo structural rearrangement, while the compressive mechanical response was dependent primarily on the total hydrogel polymer content, with all other parameters having a relatively negligible impact. Through alteration of formulation parameters, the advantageous and novel ability to fully tune post-formation swelling behavior as desired, from significant syneresis to dimensional stability to significant expansion, was demonstrated. The hydrogel system was further shown to be cytocompatible for relevant macromer and degradation product concentrations and relevant time scales, while displaying a time- and dose-dependent cytotoxicity response influenced primarily by solution osmolality.
Injectable in situ forming hydrogels with high water content have many potential biomaterials applications, particularly in drug delivery and tissue engineering. The hydrogel system vetted in this investigation harnesses the benefits of thermogelling systems, chiefly the near-instantaneous initial gelation upon injection, while addressing the often problematic issue of significant post-formation syneresis through incorporation of hydrophilic yet degradable polyamidoamine crosslinks. The documented synthetic and formulation flexibility to tune hydrogel properties as needed makes this a promising two-component system worthy of further investigation, particularly concerning the co-delivery and encapsulation of cells in injectable tissue engineering therapies.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01 DE17441) and Baylor’s Scientific Training Program for Dental Academic Research (T32 DE018380).
References
- 1.Gutowska A, Jeong B, Jasionowski M. Anat Rec. 2001;263:342–349. doi: 10.1002/ar.1115. [DOI] [PubMed] [Google Scholar]
- 2.Temenoff JS, Mikos AG. Biomaterials. 2000;21:2405–2412. doi: 10.1016/s0142-9612(00)00108-3. [DOI] [PubMed] [Google Scholar]
- 3.Kretlow JD, Klouda L, Mikos AG. Adv Drug Del Rev. 2007;59:263–273. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Kretlow JD, Young S, Klouda L, Wong M, Mikos AG. Adv Mater. 2009;21:3368–3393. doi: 10.1002/adma.200802009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klouda L, Mikos AG. Eur J Pharm Biopharm. 2008;68:34–45. doi: 10.1016/j.ejpb.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hacker MC, Klouda L, Ma BB, Kretlow JD, Mikos AG. Biomacromolecules. 2008;9:1558–1570. doi: 10.1021/bm8000414. [DOI] [PubMed] [Google Scholar]
- 7.Ekenseair AK, Boere KWM, Tzouanas SN, Vo TN, Kasper FK, Mikos AG. Biomacromolecules. 2012;13:1908–1915. doi: 10.1021/bm300429e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui ZW, Lee BH, Pauken C, Vernon BL. J Biomater Sci, Polym Ed. 2010;21:913–926. doi: 10.1163/156856209X451323. [DOI] [PubMed] [Google Scholar]
- 9.Cui ZW, Lee BH, Pauken C, Vernon BL. J Biomed Mater Res, Part A. 2011;98A:159–166. doi: 10.1002/jbm.a.33093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robb SA, Lee BH, McLemore R, Vernon BL. Biomacromolecules. 2007;8:2294–2300. doi: 10.1021/bm070267r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kretlow JD, Hacker MC, Klouda L, Ma BB, Mikos AG. Biomacromolecules. 2010;11:797–805. doi: 10.1021/bm9014182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Magnaghi V, Conte V, Procacci P, Pivato G, Cortese P, Cavalli E, Pajardi G, Ranucci E, Fenili F, Manfredi A, Ferruti P. J Biomed Mater Res, Part A. 2011;98A:19–30. doi: 10.1002/jbm.a.33091. [DOI] [PubMed] [Google Scholar]
- 13.Navath RS, Menjoge AR, Dai H, Romero R, Kannan S, Kannan RM. Mol Pharm. 2011;8:1209–1223. doi: 10.1021/mp200027z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dey RK, Ray AR. J Macromol Sci, Part A: Pure Appl Chem. 2005;A42:351–364. [Google Scholar]
- 15.Ferruti P, Marchisio MA, Duncan R. Macromol Rapid Commun. 2002;23:332–355. [Google Scholar]
- 16.Meyers MA, Chawla KK. Mechanical Behavior of Materials. Prentice-Hall, Inc; Upper Saddle River, NJ: 1999. [Google Scholar]
- 17.Klouda L, Hacker MC, Kretlow JD, Mikos AG. Biomaterials. 2009;30:4558–4566. doi: 10.1016/j.biomaterials.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Timmer MD, Shin H, Horch RA, Ambrose CG, Mikos AG. Biomacromolecules. 2003;4:1026–1033. doi: 10.1021/bm0300150. [DOI] [PubMed] [Google Scholar]
- 19.Montgomery DC. Design and Analysis of Experiments. 3. John Wiley & Sons; New York: 1991. [Google Scholar]
- 20.Scognamillo S, Alzari V, Nuvoli D, Mariani A. J Polym Sci, Part A: Polym Chem. 2010;48:2486–2490. [Google Scholar]
- 21.Scognamillo S, Alzari V, Nuvoli D, Illescas J, Marceddu S, Mariani A. J Polym Sci, Part A: Polym Chem. 2011;49:1228–1234. [Google Scholar]
- 22.Ekenseair AE, Seidel RN, Peppas NA. Polymer. 2012 doi: 10.1016/j.polymer.2012.06.036.. [DOI] [Google Scholar]