

Abstract
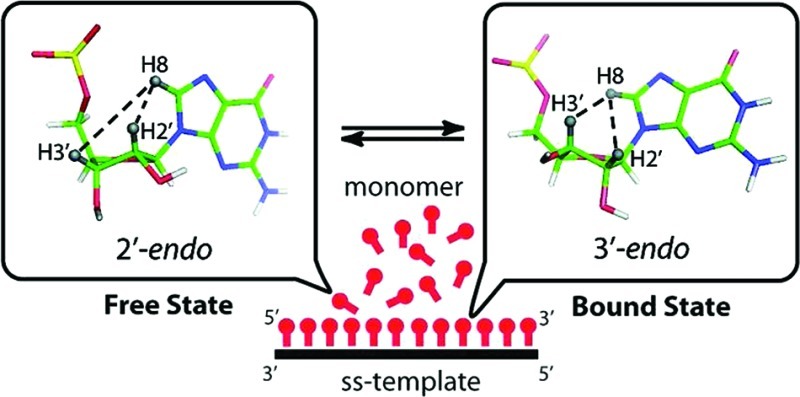
Template-directed polymerization of chemically activated ribonucleotide monomers, such as nucleotide 5′-phosphorimidazolides, has been studied as a model for nonenzymatic RNA replication during the origin of life. Kinetic studies of the polymerization of various nucleotide monomers on oligonucleotide templates have suggested that the A-form (C3′-endo sugar pucker) conformation is optimal for both monomers and templates for efficient copying. However, RNA monomers are predominantly in the C2′-endo conformation when free in solution, except for cytidine, which is approximately equally distributed between the C2′-endo and C3′-endo conformations. We hypothesized that ribonucleotides undergo a switch in sugar pucker upon binding to an A-type template and that this conformational switch allows or enhances subsequent polymerization. We used transferred nuclear Overhauser effect spectroscopy (TrNOESY), which can be used for specific detection of the bound conformation of small-molecule ligands with relatively weak affinity to receptors, to study the interactions between nucleotide 5′-phosphorimidazolides and single-stranded oligonucleotide templates. We found that the sugar pucker of activated ribonucleotides switches from C2′-endo in the free state to C3′-endo upon binding to an RNA template. This switch occurs only on RNA and not on DNA templates. Furthermore, activated 2′-deoxyribonucleotides maintain a C2′-endo sugar pucker in both the free and template-bound states. Our results provide a structural explanation for the observations that activated ribonucleotides are superior to activated deoxyribonucleotides and that RNA templates are superior to DNA templates in template-directed nonenzymatic primer-extension reactions.
Nonenzymatic template-directed polymerization of chemically activated mononucleotides has been studied extensively as a model of the chemical replication of nucleic acids during the origin of life.1−5 Nucleotide 5′-phosphorimidazolides (ImpdN for deoxynucleotides; ImpN for ribonucleotides) have commonly been used as activated monomers in such laboratory studies. Their polymerization on a single-stranded template involves monomer binding followed by nucleophilic attack of either the 2′- or 3′-hydroxyl of an RNA primer on the imidazole-activated 5′-phosphate of an adjacent bound monomer, with displacement of imidazole as the leaving group and subsequent formation of a 2′–5′ or 3′–5′ phosphodiester bond. The use of 2-methylimidazole in place of imidazole as the leaving group results in greater 3′–5′ regiospecificity of polymerization, for reasons that are unclear.6 Other leaving groups, such as 1-methyladenine and hydroxyazabenzotriazole, have also been used in studies of nucleotide polymerization.7,8 The efficiency of polymerization depends not only on the reactivity of the activated monomers but also on how tightly the monomers associate with the template and on the conformation adopted by the bound monomers. However, it has been difficult to study these effects independently and thus to arrive at a more complete understanding of the process of nonenzymatic template copying.
Optimal template-directed primer extension by activated monomers requires that all three components (primer, bound monomer, and template) adopt a conformation that positions the 3′-hydroxyl of the primer for in-line attack on the 5′-phosphate of the adjacent monomer.9 Several observations suggest that this optimal geometry is achieved when all three components adopt the A-form (C3′-endo sugar pucker) conformation typical of an RNA duplex. Markedly improved polymerization efficiency has been observed on A-form templates, such as RNA or hexitol nucleic acid (HNA), relative to B-form templates (e.g., DNA).10−12 Primers terminating in a 2′-deoxyribonucleotide are extended more slowly than primers ending in a ribonucleotide;13 in this case, however, the dominant effect may be the lower pKa of a ribonucleotide 3′-OH (pKa ∼ 12) versus that of a deoxyribonucleotide (pKa ∼ 16),14 making it difficult to assess the role of conformational differences. Finally, primers are extended more rapidly by activated ribonucleotides than by activated deoxyribonucleotides.12 Again, the role of potential conformational differences has not been directly examined.
Previous studies have shown that nonactivated nucleotide 5′-monophosphates, including RNA monomers (except cytidine), exhibit a C2′-endo-like sugar pucker when free in solution (see Table 1B of ref (15)).15 We have found that such nucleotides, when activated as 5′-phosphorimidazolides, retain the C2′-endo conformation in solution. This assessment is based on our measurements of the homonuclear 3JH1′–H2′ coupling constants for both activated and nonactivated monomers by one-dimensional 1H NMR [the coupling constants are listed in Table 1, and the spectra are shown in Figure S1 in the Supporting Information (SI)]. The homonuclear 3JH1′–H2′ coupling constant, which is sensitive to the dihedral angle, is the most direct determinant of the sugar pucker conformation. A typical C3′-endo (A-form) sugar pucker leads to a 3JH1′–H2′ value smaller than 2 Hz, while the C2′-endo (B-form) pucker gives a 3JH1′-H2′ of ∼8 Hz.16,17 In Table 1, all of the 3JH1′–H2′ values for various free monomers are ∼6 Hz. Because the magnitude of the 3JH1′–H2′ coupling constant is directly correlated with the C2′-endo/C3′-endo population ratio, we conclude that in the free state, the activated monomers ImpG, MeImpG, ImpdG, and MeImpdG as well as the nonactivated monomers GMP and dGMP all exist in a dynamic equilibrium between the C3′-endo and C2′-endo sugar conformations, with a preference for the latter. This equilibrium does not appear to be significantly altered by the presence of Mg2+ ions, as 3JH1′–H2′ for ImpG is 5.1 in the presence of either 10 or 100 mM Mg2+ versus 5.4 in the absence of Mg2+.
Table 1. Coupling Constants 3JH1′–H2′ for 5′-Mononucleotide Monomers.
| observed 3JH1′–H2′ (Hz)a |
||
|---|---|---|
| compound | 4 °C | 25 °C |
| ImpG | 5.4 | 5.3 |
| MeImpG | 5.3 | 5.2 |
| GMPb | 6.2 | 6.1 |
| ImpdG | 6.7 | 6.7 |
| MeImpdG | 6.7 | 6.7 |
| dGMP | 6.9 | 6.9 |
Data were collected in 20 mM phosphate buffer (pH 7.8).
A value of 5.8 Hz was previously reported for GMP in 50 mM Tris-HCl buffer (pH 7.5) at 30 °C (see ref (15)).
Previous studies have demonstrated that the sugars of nucleotides in a helical duplex are conformationally flexible and capable of switching between endo and exo conformations under certain circumstances, such as the presence of intercalators or mismatched base pairs.18,19 We therefore asked whether a sugar pucker switch occurs in nucleotide monomers upon binding to A-form templates. Unfortunately, the binding of nucleotide monomers to single-stranded templates is quite weak, with Kd values ranging from tens to hundreds of millimoles per liter.100 This weak binding makes it unfeasible to deduce the sugar pucker of template-bound nucleotides by the homonuclear J coupling constant method described above because of the poor sensitivity of this approach (for details, see the SI). Furthermore, line broadening due to the larger size of the monomer–template complex is likely to lead to overlap of the desired signals with signals derived from the template. In addition, the through-bond 3JH–H approach is unable to provide information about other important aspects of the nucleotide conformation (e.g., the glycosidic torsion angle) that could also influence nucleotide polymerization.
As an alternative approach for determining the conformation of template-bound nucleotides, the nuclear Overhauser effect (NOE) can be exploited to obtain information about through-space distances between protons of interest. An especially attractive approach is transferred NOE spectroscopy (TrNOESY), which allows the conformation of bound ligands to be determined in cases where ligand–receptor affinity is weak (Kd values in the μM to mM range). This approach has been widely used in drug discovery and in the study of ligand–receptor complexes.20−22 Several aspects of NOE signals, including their magnitude, sign, and buildup rate, are related to the size of molecule. In general, smaller molecules yield small, positive NOEs that build up slowly and reach a maximum intensity at relatively long mixing times (>400 ms). In contrast, large molecules yield large, negative NOEs that build up rapidly and reach a maximum intensity at relatively short mixing times (100 ms).23 These differences in the NOE properties of small and large molecules form the basis of TrNOESY experiments. When a small ligand binds to a large target, it adopts the properties of the large complex. Fast chemical exchange (weak binding) allows the much stronger negative NOE corresponding to the bound state to develop in the monomer–template complex and then be transferred to the free-ligand state, where it can be measured from free-ligand resonances, which are much sharper and thus more easily detectable. We chose a short mixing time (100 ms) to ensure that the observed NOEs would come primarily from the bound state, meanwhile minimizing interference from spin diffusion.24
The NOE intensity is very sensitive to the distance between two protons. The ratio of 2′-endo to 3′-endo sugar puckering can be qualitatively determined from the pattern of NOE cross-peaks between base proton H8 (in purines) or H6 (in pyrimidines) and sugar protons H2′ versus H3′. In a C2′-endo nucleotide, whether free in solution or in a B-type helix, the intranucleotide distances between H8/H6 and H2′ are short (2.2 Å), resulting in a strong NOE, whereas the intranucleotide distances between H8/H6 and H3′ are relatively long (4.2 Å), resulting a weaker NOE (Figure 1B). This pattern is reversed in a C3′-endo nucleotide, where a relatively strong NOE between H8/H6 and H3′ (3.1 Å) and a weaker NOE between H8/H6 and H2′ (3.7 Å) are observed (Figure 1C).25
Figure 1.
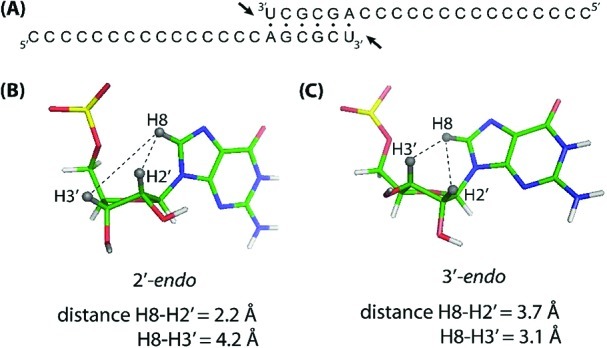
Primer–template complex and monomer conformations. (A) Dimerization of the 21-mer oligonucleotide forms a symmetrical primer–template complex, with two sites for monomer binding adjacent to each 3′-end. (B, C) Schematic illustrations showing the (B) 2′-endo and (C) 3′-endo sugar puckers of monomeric ImpG. The imidazole groups have been omitted for clarity. The characteristic distances between base and sugar protons are labeled.
We first acquired a NOESY spectrum of the free ribonucleotide 5′-phosphorimidazolide (ImpG) at a mixing time of 600 ms and examined the region of the spectrum covering the correlation between the base proton H8 and the sugar protons H2′ and H3′ (Figure 2A). All of the NOE cross peaks have the opposite sign as the diagonal peaks, as expected for the positive NOEs of a free monomer. The pattern of NOEs is characteristic of a primarily 2′-endo sugar pucker, with a stronger NOE between base proton H8 and sugar proton H2′ relative to that between H8 and sugar proton H3′. This result is consistent with the conclusion from the previous 3JH1′–H2′ analysis that the C2′-endo conformation is dominant in free monomers.
Figure 2.
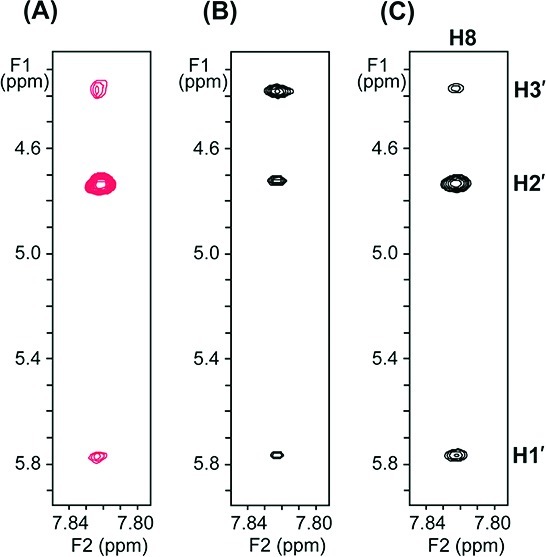
Expanded views of the spectral region covering NOEs between base proton H8 and sugar protons H1′, H2′, and H3′ of monomeric ImpG in D2O with 100 mM NaCl and 10 mM phosphate buffer (pH 7.8) at 4 °C. (A) NOESY spectrum (mixing time, 600 ms) of 10 mM ImpG in the absence of single-stranded template. (B) TrNOESY spectrum (mixing time, 100 ms) of 8 mM ImpG in the presence of an RNA single-stranded template (0.5 mM strand concentration). (C) TrNOESY spectrum (mixing time, 100 ms) of 9 mM ImpG in the presence of a DNA single-stranded template (0.6 mM strand concentration). The red cross-peaks in (A) correspond to positive NOEs having the opposite sign as the diagonal peaks; the black cross-peaks in (B) and (C) correspond to negative NOEs having the same sign as the diagonal peaks.
We next acquired a TrNOESY spectrum in the presence of a single-stranded RNA template at a mixing time of 100 ms (thus exclusively detecting bound monomers). The RNA template was a 21-mer oligonucleotide that dimerized to form a six-base-pair duplex stem flanked by 15-nucleotide 5′-overhanging single-stranded oligoC segments (Figure 1A). The large size of the dimeric primer–template complex enhances the distinction between the free and bound monomers and improves the quality of the TrNOESY spectra. In the presence of the RNA template (Figure 2B), we observed that all of the cross-peaks had the same sign as the diagonal peaks, indicating that the negative NOE from the bound ligand is dominant. Upon template binding, the NOE pattern corresponding to the sugar pucker clearly switches to that characteristic of a 3′-endo sugar pucker, as evidenced by a stronger NOE between base proton H8 and sugar proton H3′ relative to that between H8 and H2′. We observed qualitatively similar effects in parallel experiments with MeImpG, although the extent of the conformational shift was less dramatic than for ImpG (Figures S2 and S3). We were unable to measure the conformation of unactivated GMP in the bound state because of the much lower affinity of unactivated monomers for the template.
We then asked whether this sugar pucker switch depends on the nature of the template. In the TrNOESY spectrum of ImpG bound to a DNA version of the same template (Figure 2C), we observed that bound ImpG maintains the 2′-endo sugar pucker observed when it is free in solution. It therefore appears that an A-form (e.g., RNA) template is necessary to induce the C2′-endo to C3′-endo sugar pucker switch. Furthermore, this sugar pucker switch also depends on the nature of the bound monomer. The deoxyribonucleotide ImpdG showed no switch in sugar pucker when bound to either RNA or DNA templates (Figure 3).
Figure 3.
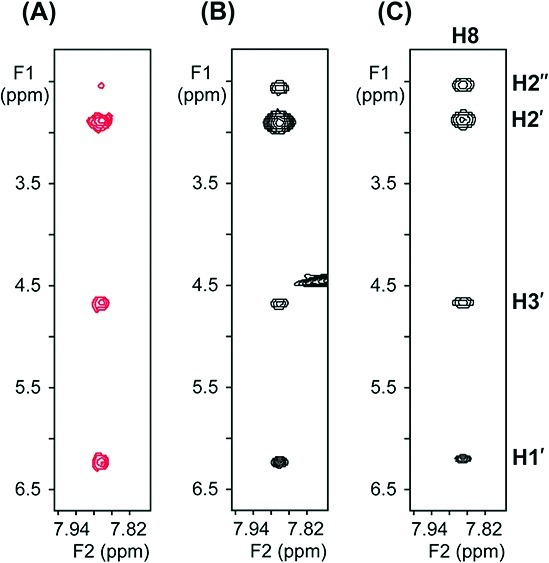
Expanded views of the spectral region covering NOEs between base proton H8 and sugar protons H1′, H2′/H2″, and H3′ of monomeric ImpdG in D2O with 100 mM NaCl and 10 mM phosphate buffer (pH 7.8) at 4 °C. (A) NOESY spectrum (mixing time, 600 ms) of 14 mM ImpdG in the absence of single-stranded template. (B) TrNOESY spectrum (mixing time, 100 ms) of 12 mM ImpdG in the presence of an RNA single-stranded template (0.7 mM strand concentration). (C) TrNOESY spectrum (mixing time, 100 ms) of 12 mM ImpdG in the presence of a DNA single-stranded template (0.8 mM strand concentration). The red cross-peaks in (A) correspond to positive NOEs having the opposite sign as the diagonal peaks; the black cross-peaks in (B) and (C) correspond to negative NOEs having the same sign as the diagonal peaks.
Another critical structural parameter is the glycosidic conformation, which is anti for antiparallel A- or B-form duplexes with canonical Watson–Crick base pairing. As long as Watson–Crick base pairing is maintained, the glycosidic conformation of bound monomers must be anti to allow the newly formed product strand to form an antiparallel duplex with the template strand. We therefore asked whether there is any detectable shift in the glycosidic angle of the monomer between the free and bound states. We were able to estimate anti or syn glycosidic conformations from the patterns of intraresidue NOEs between base proton H8 and the corresponding sugar protons. A syn conformation is associated with a short distance between base proton H8 and sugar proton H1′ (∼2.5 Å) and thus a stronger NOE, whereas the H2′ and H3′ protons point away from the base proton and therefore give weaker NOEs. On the other hand, in the anti conformation, base proton H8 is closer to protons H2′ and H3′ (stronger NOEs) than to proton H1′ (∼3.7 Å, weaker NOE).25 The observed NOE patterns for monomers in both the free and bound states (Figures 2 and 3) indicate that the base proton H8 points toward the sugar moiety, resulting in strong to moderate NOEs between proton H8 and sugar protons H2′ and H3′, along with a relatively weak NOE cross-peak between H8 and H1′. On the basis of these qualitative considerations, we therefore conclude that monomers remain in the anti conformation in both the free and bound states. The inclination of template-bound monomers to maintain the anti glycosidic conformation minimizes the chances of pyrophosphate bond formation between adjacent syn and anti monomers, which would terminate the polymerization reaction.
In conclusion, our TrNOESY experiments have shown that the sugar pucker of activated ribonucleotides switches upon binding to an RNA template, from a more C2′-endo-like pucker in the free state to a C3′-endo pucker in the bound state. This switch is specific to RNA as opposed to DNA templates. Our observations are consistent with the hypothesis that the 3′-endo sugar pucker is the productive monomer conformation for polymerization on an RNA template,9 with the caveat that our observations reflect the dominant equilibrium conformations, whereas a minor conformer may be critical to this kinetically controlled reaction. The observed conformational switch can be rationalized in terms of the assembly of adjacent monomers on the template strand to form a pseudoduplex structure in which one strand is discontinuous and consists of noncovalently linked monomers. Such a prepolymerization structure cannot form a B-type duplex because of the steric clash between the monomer 2′-hydroxyls and the phosphodiester backbone (see Figure 4 of ref (26)). The switch of the ribonucleotide monomers in the bound state to a 3′-endo sugar pucker is necessary for the discontinuous strand to form the sterically favored A-type duplex.26 The change in monomer conformation upon template binding suggests that monomers that are preorganized in the 3′-endo conformation should bind more tightly and polymerize more efficiently. Thus, 2-thio-UMP, which exists in a 3′-endo conformation when free in solution,27 should be a better substrate for nonenzymatic template-directed polymerization than standard UMP. Our observations may also explain the poor polymerization of ribonucleotide monomers on DNA templates as a consequence of the energetic penalty required for transformation of the DNA template strand to an A-type (3′-endo) conformation following the assembly of the prepolymerization discontinuous RNA strand.
What factors might induce the conformational change observed in monomers upon template binding? This process must be largely mediated by both the adjacent primer–monomer and bound monomer–monomer interactions when they are aligned on a template by base pairing, since the basic duplex structure keeps the sugar–phosphate leaving group segments of the incoming monomers away from the template strand. One possibility for an interaction that would stabilize a monomer 3′-endo conformation is hydrogen bonding between the 2′-hydroxyl of a bound monomer and O4′ of an adjacent monomer, as observed in an RNA duplex. A direct role for the monomer 2′-hydroxyl would help to explain why ribonucleotides, but not deoxyribonucleotides, exhibit the observed conformational shift. Another possibility is that water of hydration, which plays a critical role in stabilizing the A-form conformation of RNA,28 could influence the monomer conformation. In an A-form duplex but not a B-form duplex, a water molecule can bridge the free phosphate oxygens of adjacent nucleotides (see Figure 1 of ref (28)). As a result, fewer water molecules are needed to hydrate an A-form duplex than a B-form duplex, decreasing the entropic cost of hydration.28 In a monomer–template complex, such bridging water molecules could stabilize the pseudostrand of bound monomers before the formation of covalent phosphodiester bonds during polymerization. Finally, as noted above, the steric clash between the 2′-OH and the backbone could prevent RNA monomers from forming a B-type discontinuous strand, thereby favoring an A-type helix and 3′-endo sugar pucker. Further structural studies of the transient complex formed between activated ribonucleotides and an RNA template will no doubt lead to a greater understanding of the mechanism of nonenzymatic template-directed RNA copying chemistry.
Acknowledgments
We are grateful to Itay Budin, Ting Zhu, and Aaron Engelhart for helpful discussions and comments on the manuscript. This research was funded in part by Grant CHE-0809413 from the NSF. J.W.S. is an Investigator of the Howard Hughes Medical Institute.
Supporting Information Available
Experimental procedures and supporting text and figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Orgel L. E. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99. [DOI] [PubMed] [Google Scholar]
- Wu T.; Orgel L. E. J. Am. Chem. Soc. 1992, 114, 5496. [DOI] [PubMed] [Google Scholar]
- Wu T.; Orgel L. E. J. Am. Chem. Soc. 1992, 114, 7963. [DOI] [PubMed] [Google Scholar]
- Mansy S. S.; Schrum J. P.; Krishnamurthy M.; Tobé S.; Treco D. A.; Szostak J. W. Nature 2008, 454, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervio E.; Hochgesand A.; Steiner U. E.; Richert C. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T.; Orgel L. E. J. Mol. Biol. 1982, 162, 201. [DOI] [PubMed] [Google Scholar]
- Prabahar K. J.; Ferris J. P. J. Am. Chem. Soc. 1997, 119, 4330. [DOI] [PubMed] [Google Scholar]
- Vogel S. R.; Deck C.; Richert C. Chem. Commun. 2005, 4922. [DOI] [PubMed] [Google Scholar]
- Kozlov I. A.; Orgel L. E. Mol. Biol. 2000, 34, 781. [PubMed] [Google Scholar]
- Kurz M.; Gobel K.; Hartel C.; Gobel M. W. Helv. Chim. Acta 1998, 81, 1156. [Google Scholar]
- Kurz M.; Gobel K.; Hartel C.; Gobel M. W. Angew. Chem., Int. Ed. Engl. 1997, 36, 842. [Google Scholar]
- Kozlov I. A.; Politis P. K.; Van Aerschot A.; Busson R.; Herdewijn P.; Orgel L. E. J. Am. Chem. Soc. 1999, 121, 2653. [DOI] [PubMed] [Google Scholar]
- Wu T.; Orgel L. E. J. Am. Chem. Soc. 1992, 114, 317. [DOI] [PubMed] [Google Scholar]
- Birnbaum G. I.; Giziewicz J.; Huber C. P.; Shugar D. J. Am. Chem. Soc. 1976, 98, 4640. [DOI] [PubMed] [Google Scholar]
- Ts’o P. O.; Kondo N. S.; Schweizer M. P.; Hollis D. P. Biochemistry 1969, 8, 997. [DOI] [PubMed] [Google Scholar]
- Altona C.; Sundaralingam M. J. Am. Chem. Soc. 1973, 95, 2333. [DOI] [PubMed] [Google Scholar]
- Bae S. H.; Cheong H. K.; Lee J. H.; Cheong C.; Kainosho M.; Choi B. S. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X. L.; Patel D. J. Q. Rev. Biophys. 1989, 22, 93. [DOI] [PubMed] [Google Scholar]
- Yi-Brunozzi H. Y.; Brabazon D. M.; Lener D.; Le Grice S. F.; Marino J. P. J. Am. Chem. Soc. 2005, 127, 16344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Szostak J. W. In preparation.
- Ray B. D.; Jarori G. K.; Raghunathan V.; Yan H.; Rao B. D. Biochemistry 2005, 44, 13762. [DOI] [PubMed] [Google Scholar]
- Martin J. N.; Muñoz E. M.; Schwergold C.; Souard F.; Asensio J. L.; Jiménez-Barbero J.; Cañada J.; Vicent C. J. Am. Chem. Soc. 2005, 127, 9518. [DOI] [PubMed] [Google Scholar]
- Johnson E. C.; Feher V. A.; Peng J. W.; Moore J. M.; Williamson J. R. J. Am. Chem. Soc. 2003, 125, 15724. [DOI] [PubMed] [Google Scholar]
- Neuhaus D.; Williamson M. P.. The Nuclear Overhauser Effect in Structural and Conformational Analysis, 2nd ed.; Wiley-VCH: New York, 2000. [Google Scholar]
- Clore G. M.; Gronenborn A. M. J. Magn. Reson. 1982, 48, 402. [Google Scholar]
- Wüthrich K.NMR of Proteins and Nucleic Acids; Wiley: New York, 1986. [Google Scholar]
- Sundaralingam M.; Rao S. T. Int. J. Quantum Chem. 1983, 24(Suppl. S10), 301. [Google Scholar]
- Yokoyama S.; Yamaizumi Z.; Nishimura S.; Miyazawa T. Nucleic Acids Res. 1979, 6, 2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenger W.; Hunter W. N.; Kennard O. Nature 1986, 324, 385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.