

Summary
Heterologous prime-boost vaccination employing DNA-vaccinia virus (VACV) modality using the LACK (p36) antigen (Leishmania homologue of receptors for activated C kinase) has been shown to elicit protective immunity against both murine cutaneous and visceral leishmaniasis. However, DNA priming is known to have limited efficacy; therefore in the current studythe effect of NKT cell activation using α-galactosyl-ceramide (αGalCer) during intradermal DNAp36 priming was examined. Vaccinated mice receiving αGalCer+DNAp36 followed by a boost with VVp36 appeared to be resolving their lesions and had at 10 to 20-fold higher reductions in parasite burdens. NKT cell activation during αGalCer+DNAp36 priming resulted in higher numbers of antigen-reactive effector CD4+ and CD8+ T cells producing granzyme and IFNγ, with lower levels of IL-10. Although immunodepletion studies indicate both CD4 and CD8 T cells provide protection in the vaccinated mice, the contribution of CD4+ T cells was significantly increased in mice primed with DNAp36 together with αGalCer. Notably 5 months after boosting, mice vaccinated with DNAp36+αGalCer continued to show sustained and heightened T cell immune responses. Thus, heterologous prime-boost vaccination using αGalCer during priming is highly protective against murine cutaneous leishmaniasis, resulting in the heightened activation and development of CD4 and CD8 T cells (effectors and memory).
Keywords: Vaccine, Leishmania, NK-T, adjuvant, memory
Introduction
The leishmaniases are a group of vector-borne parasitic diseases caused by protozoa of the genus Leishmania. The disease is endemic in the tropics, subtropics and southern Europe [1]. The clinical forms range from the self-healing cutaneous leishmaniasis (CL) to visceral leishmaniasis (VL) in which the parasite spreads into the reticuloendothelial system with fatal outcome. Although drug treatment exists for leishmaniasis [2, 3], alternative approaches for the control of this disease (vector control, immunotherapeutic, chemotherapeutic and vaccine) are still needed [4-6]. Over the past decade, DNA vaccination has been shown to be a promising tool to combat intracellular infectious agents since it induces cell-mediated immunity [7] as well as humoral responses. LACK, a 36 kDa (p36) antigen from Leishmania, is an analogue of the receptors for activated C kinase. Immunofluorescence experiments indicate that LACK localizes to the cell cytoplasm (near the kinetoplast disc) [8], and is highly conserved among Leishmania species; LACK is expressed both by the promastigote and amastigote forms of the parasite [9]. Studies indicate that DNA encoding for the LACK antigen can provide protection against Leishmania major and that this is due to the induction of T cell responses and IFNγ production [10-14]. However, DNA vaccination with LACK has not proven to provide protection across Leishmania species causing both visceral and cutaneous forms of the disease [15-17], consequently new modalities have been sought.
The antigen delivery system is a critical component in determining antigenic efficacy. VACV and fowlpox vectors have been shown to be a good antigen delivery system for the control of infectious diseases in animal model studies [18] and, importantly, are known to provide long-term memory [19]. A DNA prime–poxvirus boost regime has been shown to induce protection against Plasmodium berghei malaria [18] and to induce immunological control of an immunodeficiency virus infection in monkeys [20]. A prime/boost regime using DNA and VACV expressing the leishmanial LACK antigen has been shown to be immunogenic and protective against murine L. major infection [21-23]. Further, the prime-boost modality is effective in inducing protection against visceral leishmaniasis caused by L. infantum, while DNA vaccination alone fails to protect [16, 24]. However, although significant reductions in parasite burdens were observed, protection was partial. Consequently, methods for enhancing the protection provided by the DNA-VACV prime boost modality have been sought. It is known that the potency of DNA priming is limited and this could potentially restrict the efficacy of boosting with recombinant VACV or other attenuated viral vectors. Consequently, adjuvants (cytokines, co-stimulatory molecules, delivery vehicles) with potential to enhance DNA vaccination have been evaluated [21, 25]. Both CD4 (Th1) and CD8 T cells have been demonstrated to be important for the control of Leishmania major infection [12, 13, 26-28]. NKT activation has been reported to lead to enhanced CD4 and CD8 T cell responses [29, 30] as well as induce dendritic cell maturation [31]; further, NK T cells have been shown to contribute to the control of L. major infection [32, 33]. Interestingly, TNFα (induced by various PAMPs/TLR) has been shown to be responsible for the recruitment of CD1d/NKT cells [34]. Further, CpG has been shown to enhance NKT cell activation through interplay between plasmacytoid and myeloid dendritic cells [35, 36]. Consequently, it might be anticipated that a vaccine employing DNA-priming (TLR9) together with NKT cell (αGalCer) activation would further amplify a developing Th1/TC1 immune response. However, αGalCer can drive both Th1 and Th2 responses [37, 38] and can induce the proliferation of Treg populations [39, 40]; further, the use of αGalCer as an adjuvant in vaccine studies has met with mixed results. Disease exacerbation and impairment of CD8 T cell function have been observed for systemic αGalCer-mediated NKT cell activation during DNA vaccination against Trypanosoma cruzi [41] infection. In contrast, studies have shown that the systemic activation of NKT cells (using the ligand αGalCer) during vaccination using viral vectors or irradiated sporozoites can lead to enhanced CD8 T cell responses and protection against murine malaria [42-44].
In this study, we have examined the effect of intradermal NKT activation during DNA priming immunization on vaccine efficacy in controlling murine leishmaniasis due to L. major. Intradermal priming with DNA-LACK together with αGalCer significantly enhances protection induced by a heterologous DNA-VACV prime-boost vaccination; this is due to increased levels of antigen-specific T cells (CD4 and CD8), in particular CD4+ T cells responses are increased. Further, the enhancement of T cell responses is sustained and is evident 5 months post-vaccination, indicating that NKT activation during DNA vaccination can contribute to the appropriate development of both T cell effector and memory populations.
Results
αGalactosylceramide enhances the protection induced by heterologous prime-boost (DNA-VACV) vaccination
We have previously demonstrated in a mouse model [22, 24, 45] that heterologous prime-boost DNA-VACV vaccination with each vector expressing the leishmanial LACK (Leishmania homologue of receptors for activated C kinase) antigen can provide protection against both cutaneous and visceral leishmaniasis. Although significant reductions in parasite burdens are achieved using this modality, protection observed was partial, overall resulting in a delay in onset of infection and less severe disease. Consequently, approaches were pursued for enhancing the protection provided by the DNA-VACV prime/boost modality.
NKT activation has been reported to lead to enhanced CD4 and CD8 T cell effector responses [29, 30, 46] as well as induce dendritic cell maturation [31] and hence might be expected to act to increase T cell responses during vaccination. As both CD4 (Th1) and CD8 T cell responses have been demonstrated to be important for the control of Leishmania major infection [12, 13, 26-28], we evaluated the efficacy of αGalCer as an adjuvant in DNA priming. Experimentally, mice were primed intradermally with p36 (LACK) plasmid DNA (DNAp36) together with or without α-galactosylceramide (αGalCer). Mice were then boosted two weeks later with VVp36 (1×107 PFU). Control groups of mice were inoculated with either DNA empty plasmid vector followed by 1×107 PFU WR-Luc (virus control) or received PBS alone. Three weeks after boosting, mice were infected in the right rear hind foot with 5 ×104 late-stationary phase L. major promastigotes. As shown in Figure 1A, mice that received αGalCer during DNA priming developed smaller lesions over time than mice receiving DNAp36 alone. In fact, the lesions of the mice in the αGalCer-DNAp36 group appeared to be resolving at 13 weeks post-infection. Evaluation of parasite burdens at 13 weeks post infection (Figure 1B) confirmed lesion development results and revealed that mice primed with αGalCer together with DNAp36 had 10-fold further reduction in their parasite burdens than those receiving DNAp36 alone.
Figure 1. The αGalCer Adjuvant during DNA Priming Enhances Protection against Leishmania major Infection.
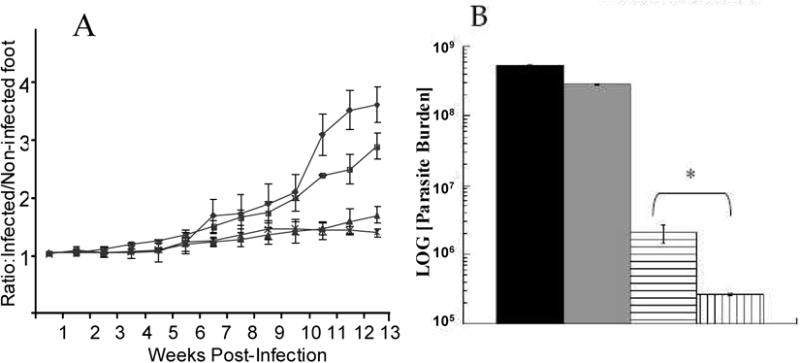
Mice were primed intradermally with p36 (LACK) plasmid DNA (DNAp36; 100 μg) with or without α-galactosylceramide (2μg αGalCer). Mice were boosted two weeks later with VVp36 (1×107 PFU). Control groups of mice were vaccinated with either control DNA followed by 1×107 PFU WR-Luc (vaccine controls) or received PBS alone (infection controls). Three weeks after boosting, mice were infected in the right rear hind foot with 5 ×104 late-stationary phase L. major promastigotes. A) Lesion development with time post-infection in mice receiving: -■-, PBS (control); -●- pCINeoDNA + WR-Luc (vector control); -▲-, DNAp36 + VVp36; -∗-, p36DNA+αGalCer + VVp36. (* - p36DNA versus p36DNA+ αGalCer p=0.03) B) Evaluation of parasite burdens at 13 weeks post-infection.
 , PBS (control);
, PBS (control);
 , pCINeoDNA and WR-Luc (vector control);
, pCINeoDNA and WR-Luc (vector control);
 , DNAp36 + WRp36;
, DNAp36 + WRp36;
 , p36DNA+αGalCer + WRp36 (* - p36DNA versus p36DNA + αGalCer p=0.02).). The results presented are representative of 3 independent experiments.
, p36DNA+αGalCer + WRp36 (* - p36DNA versus p36DNA + αGalCer p=0.02).). The results presented are representative of 3 independent experiments.
An examination of the cellular immune responses of the mice either before infection (3 weeks after the final VVp36 boost) (Figure 2) or at 13 weeks post-infection (data not shown), were comparable and indicated that vaccinated mice receiving αGalCer together with DNAp36 produced higher levels of IFNγ and nitric oxide and reduced levels of IL-10 in response to leishmanial antigen (SLA or rLACK). In addition, the antibody responses of the vaccinated mice were evaluated at 13 weeks post-infection against both SLA (whole leishmanial lysate) and LACK antigen. As shown in Figure 3, mice primed with αGalCer together with DNAp36 in comparison to mice receiving DNAp36 alone, had elevated levels of IgG2a against LACK antigen, but comparable levels of IgG1 antibody. However, it should be noted that the overall antibody responses against soluble leishmanial antigen (which would be representative of the ongoing response to the parasite) were comparable for both groups of vaccinated mice as well as vaccine control mice (receiving pCINeo and VVLuc). Hence, overall the adjuvant αGalCer appeared to enhance a cellular Th1-like response in the vaccinated mice, which correlated with increased protection against L. major infection.
Figure 2. Immune Responses of Vaccinated Mice.

Mice were primed intradermally with p36 (LACK) plasmid DNA (DNAp36; 100 μg) with or without α-galactosylceramide (2 μg αGalCer) and were boosted two weeks later with WRp36 (1×107 PFU) (as indicated in Figure 1). Shown are the results from ELISA analyses of splenic cells stimulated with either recombinant LACK antigen (p36) or soluble leishmanial antigen (SLA), three weeks after the final VVp36 boost:
 , p36 antigen; □, soluble leishmanial antigen (SLA). These results are representative of 3 independent experiments.
, p36 antigen; □, soluble leishmanial antigen (SLA). These results are representative of 3 independent experiments.
Figure 3. Antibody Responses of Vaccinated Mice.

The antibody responses of the vaccinated and control mice were evaluated at 13 weeks post-infection against: A) LACK antigen (1:500) and B) SLA (whole leishmanial lysate; 1:1000). The levels of total IgG as well as specific IgG1 and IgG2 were determined as indicated in Materials and Methods Section. The antibody responses of mice receiving αGalCer+DNAp36 versus DNAp36were significantly different for: LACK: Total IgG (p=0.0001); IgG1 (p=0.04); IgG2a (p=0.00001); SLA: Total IgG (p=0.03); IgG2a (p=0.01); IgG1 (not significant). These results are representative of 3 independent experiments.
αGalactosylceramide enhanced responses during heterologous prime-boost (DNA-VACV) vaccination are long-lived
In order to evaluate the long-term effect of αGalCer on the overall immune response, several vaccinated mice (3/group) were left unchallenged. Five months after the final VVp36 boost, the immune response to leishmanial antigens and ConA were examined in these mice. As seen in Table 1, the IFNγ responses of the mice receiving DNAp36 together with αGalCer continued to be elevated in comparison to those of mice receiving DNAp36 alone. Further, the ratio of IFNγ/IL-10 remained higher (2-fold) in the mice receiving DNAp36 together with αGalCer. Interestingly and consistent with this observation, the mice receiving DNAp36 together with αGalCer also maintained higher levels of IgG2a LACK antibody (and higher IgG2a/IgG1) at 5 months post-boost than mice not receiving αGalCer (data not shown). The enhanced immunological responses of mice vaccinated with DNAp36 together with αGalCer were sustained. Therefore, NKT cell activation during DNA vaccination led not only to the development of higher numbers of T cell effectors and protection, but also to long-lived T cell immunological memory.
Table 1. T Cell Responses to LACK Antigen in Vaccinated Mice at Five Months after the VVp36 Boost.
| Vaccine Group | IFNγ (ng/ml) | IL-10 (pg/ml) | ||||
|---|---|---|---|---|---|---|
| ConA | SLA | LACK | ConA | SLA | LACK | |
| PBS | 12±4.1 | 15±0.5 | 15±0.1 | 152±2.8 | ND | 6.8±2.9 |
| Control DNA + WRLuc | 14±6.2 | 27±0.4 | 52±0.6 | 176.6±4.3 | ND | 127.2±4.6 |
| DNAp36 + VVp36 | 160±2.6 | 82±1.2 | 199±2.8 | 420±1.7 | ND | 634±15.0 |
| DNAp36,αGalCer + VVp36 | 190±4.1* | 111±4.0* | 272±6.0** | 46.8±4.9**** | ND | 459±15.6*** |
Cytokine Responses in Mice 5 Months after p36Vaccinia Virus Boost. Additional mice (3/group), vaccinated as part of the experiment shown in Figure 1, were allowed to rest for 5.5 months after the final VVp36 boost. Subsequently, splenic cells were isolated and stimulated with concanavalin A, rLACK antigen or soluble leishmanial antigen (SLA) and the levels of IFNγ and IL-10 determined. In addition, antibody responses were evaluated. These results indicate a sustained enhanced immunologic response in the mice receiving DNAp36 together with αGalCer. Comparison of cytokine levels between mice receiving DNAp36 + VVp36 to those vaccinated with DNAp36,αGalCer + VVp36:
p <0.05;
p <0.01;
p< 0.001;
p <0.0001;
ND= not determined.
NKT cells contribute to protection in mice vaccinated using αGalactosylceramide-during DNA priming
To verify the role of NKT cells in the αGalCer enhancement of protection induced during DNAp36 priming, groups of NKT deficient BALB/c mice were vaccinated and challenged with Leishmania major (Figure 4). NKT deficient mice were protectively vaccinated using the heterologous prime-boost modality. However, the lesion development over the 13 week period of observation did not show a reduction in lesion size for the group receiving αGalCer + DNAp36 in comparison to the group receiving DNAp36 alone; further, the parasite burdens were identical for these two groups of vaccinated mice. These results paralleled the immune responses (Table 2) found in these two groups of vaccinated mice. NKT deficient mice receiving DNAp36+ αGalCer or DNAp36alone displayed comparable IFNγ, granzyme, nitric oxide and IL-10 responses. It should be noted that this experiment was performed together with the experiment shown in Figure 5 (below). Hence an enhanced protection was observed for BALB/c mice vaccinated with p36DNA+αGalCer. Consequently, the effect of αGalCer on DNA priming was mediated through the activation of NKT cells.
Figure 4. NKT Cells are Required for Increased Protection Induced by αGalCer.
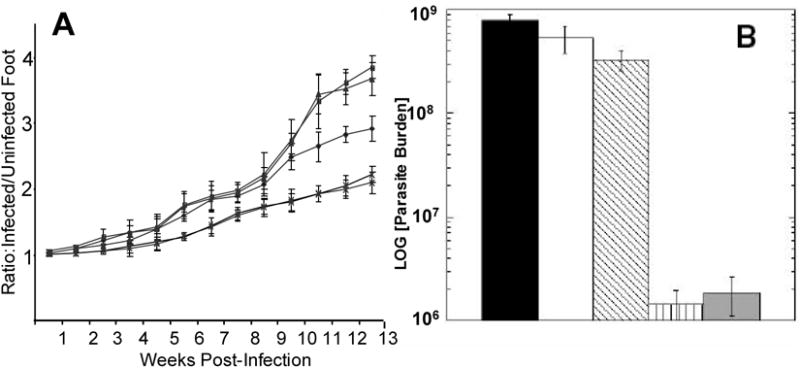
To verify the role of NKT cells in the αGalCer enhancement of protection induced during DNAp36 priming, groups of NKT deficient BALB/c mice were vaccinated and challenged with Leishmania major: -■-, PBS-NK-T (control) -●- pCINeoDNA and WR-Luc (vector control); -▲-, DNAp36 + VVp36; -∗-, p36DNA+αGalCer + VVp36; also shown is the course of infection for control -◆-, PBS WT-BALB/c mice; (DNAp36 versus DNAp36+ αGalCer p=0.45).
B) Evaluation of parasite burdens at 13 weeks post-infection.
, BALB/c (WT) PBS (control);
, NK-T PBS (Control);
 , NK-T pCINeoDNA and WR-Luc (vector control);
, NK-T pCINeoDNA and WR-Luc (vector control);
 , NK-T DNAp36 + VVp36;
, NK-T DNAp36+αGalCer + VVp36. (DNAp36 versus DNAp36+ αGalCer P=0.65) These results are representative of 2 independent experiments. It should be noted that this experiment was performed together with the experiment shown in Figure 5. Hence an enhanced protection was observed for BALB/c mice vaccinated with p36DNA+αGalCer.
, NK-T DNAp36 + VVp36;
, NK-T DNAp36+αGalCer + VVp36. (DNAp36 versus DNAp36+ αGalCer P=0.65) These results are representative of 2 independent experiments. It should be noted that this experiment was performed together with the experiment shown in Figure 5. Hence an enhanced protection was observed for BALB/c mice vaccinated with p36DNA+αGalCer.
Table 2. Immune Responses at 13 Weeks Post-Infection NK-T Deficient Mice.
| Vaccination Group | ||||
|---|---|---|---|---|
| Control (PBS) | pCINeo+GalCer + WRLuc | DNAp36 + VVp36 | DNAp36+αGalCer + VVp36 | |
| IFNγ (pg/ml) | 19.1 ± 6.4 | 56.6 ± 5.1 | 5153 ± 107* | 5958 ± 88* |
| IL-10 (pg/ml) | 835 ± 81 | 735 ± 70 | 202 ± 40* | 165 ± 50* |
| Granzyme (ng/ml) | 0.00 ± 0.00 | 0.03 ± 0.0 | 1.4 ± 0.7* | 1.26 ± 0.3* |
| Nitric Oxide (μM) | 0.06±0.01 | 0.12 ± 0.03 | 9.12 ± 0.83* | 8.02 ± 1.11* |
T Cell Responses of NKT Vaccinate Mice. Shown are the responses of splenic lymphocytes from NKT deficient mice vaccinated and then infected with Leishmania major as indicated in Materials and Methods Section. At 13 weeks post-infection, splenic lymphocytes were stimulated with SLA and the levels of IFNγ, IL-10, granzyme and nitric oxide measured. NKT deficient mice receiving DNAp36+ αGalCer or DNAp36 alone displayed comparable IFNγ, granzyme, nitric oxide and IL-10 responses. Consequently, the effect of αGalCer on DNA priming was mediated through the activation of NKT cells.
None of the responses found were signicantly different between the mice receiving DNAp36 + VVp36 in comparison to those vaccinated with DNAp36+αGalCer + VVp36; p>0.05. These results are representative of 2 independent experiments.
Figure 5. Immunodepletion of CD4 or CD8 T cells reverses protection in vaccinated mice.



Mice were vaccinated as indicated above and CD4 or CD8 T cells were depleted as previous described [62, 63] immediately prior to infection with L. major; lesion development was monitored (Figures 5 A and 5B), as described in Materials and Methods section.
At 13 weeks post-infection, the parasite levels in the cutaneous site of infection were enumerated (Figure 5 C).
, PBS (control);
, pCINeoDNA and WR-Luc (vector control);
, DNAp36 + VVp36;
, DNAp36+αGalCer + VVp36;
 , DNAp36 + VVp36- CD4 depleted;
, DNAp36 + VVp36- CD4 depleted;
 , DNAp36 + VVp36 – CD8 depleted;
, DNAp36 + VVp36 – CD8 depleted;
 , DNAp36+αGalCer + VVp36-CD4 depleted; and
, DNAp36+αGalCer + VVp36-CD4 depleted; and
 , DNAp36+αGalCer + VVp36 –CD8 depleted. These results represent the averaged values of 5 mice/group.
, DNAp36+αGalCer + VVp36 –CD8 depleted. These results represent the averaged values of 5 mice/group.
αGalactosylceramide-during DNA priming leads to enhanced CD4 and CD8 T cell IFNγ and granzyme responses
Both CD4 and CD8 T cells are known to contribute to protection against murine leishmaniasis in vaccinated mice (reviewed in [47]). It is well established that VACV can induce both CD8 and CD4 T cell responses; however, a generally dominant CD8 T cell response is observed [19]. Further, vaccination with DNAp36 alone or using a DNA-VACV prime-boost-modality has been shown primarily to lead to the development of a dominant CD8 T cell response [12, 13, 48]. However, activation of NKT cells has been shown to lead to dendritic cell maturation and CD4 T cell responses [49-52]. Consequently, given the enhanced protection observed, the impact of αGalCer on the development of CD4 and CD8 T cell responses was of interest.
To quantitatively evaluate the response of CD8 and CD4 T effector cell populations to the ongoing infection, the numbers of splenic CD4 and CD8 T cells responding in WT vaccinated and control mice after 13 weeks of infection were compared using FACS (Table 3). Proliferative responses (blast cells) together with the production of IFNγ or granzyme were employed as markers of activated T cells [53, 54]. Further, the effect of in vitro blocking of either the CD4 or CD8 T cell response on the overall ongoing response to SLA was determined for the vaccinated mice at 13 weeks post-infection (Table 4).
Table 3. Effect of NKT Activation During DNA36 Priming on the Development of CD4 and CD8 T cell Responses.
| PBS (2×) | pCIneo+Galcer+ WRluc | DNAp36+VVp36 | DNAp36+Galcer+ VVp36 | |
|---|---|---|---|---|
| % CD4 - IFNγ | 0.45±0.2 | 0.97±0.1 | 2.76±0.3* | 4.07±0.47* |
| % CD8 - IFNγ | 0.25±0.07 | 0.92±0.14 | 1.4±0.32** | 2.83±0.3** |
| % CD4 Granzyme B | 0.047±0.01 | 0.076±0.02 | 0.14±0.3*** | 0.83±0.1*** |
| % CD8 Granzyme B | 0.064±0.01 | 0.33±0.09 | 1.34±0.3** | 2.6±0.5*** |
FACS Analysis of Responding T cell Populations in Vaccinated and Control Mice at 13 Weeks Post-Infection. Shown are the effects of NKT activation during DNAp36 priming on the development of CD4 and CD8 T cell responses. The results of FACS analyses of spleen cells isolated from mice 13 weeks post-infection with L. major (PBS-Control, Vaccine Control (pCINeo+GalCer + WRLuc), DNAp36 +VVp36, and DNAp36+GalCer + VVp36) and stimulated for 3 days with SLA antigen are shown. Cell surface staining for (CD4 and/or CD8) and intracellular cytokine and granzyme staining were performed as indicated in the Materials and Methods Section. Cellular levels in mice vaccinated with DNAp36 followed by VVp36 in comparison to those receiving DNAp36+αGalCer and then boosted with VVp36 indicate significant differences with p values as follows:
p<0.05
p= 0.01
p=0.001.
Data represent the averaged values of three mice and are representative of 3 independent experiments.
Table 4. In vitro Inhibition of CD4 and CD8 T cell Responses at 13 weeks post-infection in BALB/c (WT) vaccinated mice.
| IFN((ng/ml) | IL-10 (pg/ml) | |||||||
|---|---|---|---|---|---|---|---|---|
| Group | SLA | SLA –anti-CD4 | SLA –anti-CD8 | SLA –anti-CD4 & CD8 | SLA | SLA –anti-CD4 | SLA –anti-CD8 | SLA –anti-CD4 & CD8 |
| PBS | 0±0 | 0±0 | 0±0 | 0±0 | 981±59 | 45.3±2.7 | 706±46 | 35.2±2.1 |
| Pcineo+Galcer+WRluc | 0±0 | 0±0 | 0±0 | 0±0 | 821±37 | 27.2± 3.4 | 626±18 | 20.8±1.3 |
| DNAp36+VVp36 | 66.2±1.2 | 2.7±0.04 | 47.6±3.1 | 0.7±0.1 | 281±15 | 11.7± 1.5 | 180±12 | 5.6±0.7 |
| DNAp36+Galcer+VVp36 | 85.5±0.9 | 3.3±0.2 | 73±4.2 | 1.05±0.03 | 164±8 | 8.3 ±0.7 | 122±20 | 2.5±0.9 |
Ongoing Immune Response to Infection of Vaccinated Mice Immunodepleted In Vitro of Either CD4 or CD8 T Cells. To further examine the effects of cellular depletion on the ongoing immune response and effect of the cells contributing to protection, cytokine responses of splenic lymphocytes were evaluated in vitro. Cells were treated or not with anti-CD4, anti-CD8 or both as indicated. Cells were then stimulated as indicated in the Materials and Methods Section for 72 hours and the cytokine levels determined.
FACS analyses indicate that NKT activation during DNA priming leads to higher numbers of antigen reactive CD4 and CD8 T cells, as the numbers of antigen-activated T cells (as determined by the production of either IFNγ or granzyme) increased (1.5 to 5.8 fold) in the mice receiving DNAp36+αGalCer in comparison to those receiving DNAp36 alone. The overall increases in DNAp36+αGalCer group in the responding CD4 or CD8 T cells producing IFNγ were comparable (1.5 and 2.0-fold, respectively), as were the corresponding mean intensities of fluorescence (data not shown). However, consistently (both LACK-vaccinated groups) significantly (2-fold) higher levels of CD4 than CD8 T cells were found to produce IFNγ, indicating that CD4 T cells are the predominant source of IFNγ. Overall the contribution of CD4 T cell IFNγ was higher in the mice receiving DNAp36+αGalCer. Further, the levels of cytolytic (producing granzyme) CD4 and CD8 T cells were also increased in the mice receiving DNAp36+αGalCer. Although the level of cytolytic CD4+ granzyme+ T cells increased 5.8-fold after vaccination with DNAp36+ αGalCer (compared to DNAp36 alone) in comparison to the increase in CD8+ granzyme+ T cells (2-fold), CD8+ T cells (2.6%) were the predominant granzyme-producing cells (Table 3).
FACS results were consistent with antibody blocking experiments (Table 4) of vaccinated mice that indicated a predominant contribution of CD4 T cells to the production of IFNγ. The level of inhibition of CD8 alone was partial, while blocking the response of CD4 T cells resulted in almost complete ablation of the IFNγ response. These results suggest that co-operation between CD4 and CD8 T cells are likely involved in an optimal secondary response, as has been observed in other systems [55-57] .
Interestingly, blocking experiments indicated that CD4 T cells were also the predominant source of IL-10, as inhibition of CD4 cells extensively reduced the development of IL-10 responses. IL-10 CD4 Tr as well as CD4(+)CD25(−)Foxp3(−) Th1 populations have been reported to modulate leishmaniasis pathogenesis in non-vaccinated (naïve) mice [58-60] as well as to modulate protection elicited in response to prime-boost vaccination [61]. Although the precise CD4 T cell subpopulation(s) responsible for the IL-10 production remain to be determined, consistently in all groups of mice (control and vaccine), CD4 blockage led to lower levels of IL-10 produced, suggesting that CD4 T cells are generally the primary source of IL-10 during infection. As mice depleted of either CD4 or CD8 T cells (section below) before infection have levels of IL-10 comparable to those of non-vaccinated controls, vaccination importantly leads to overall reductions in IL-10 levels in response to infection; notably both T cell populations appear critical for this effect.
CD8 and CD4 T cells contribute to protection in mice vaccinated using αGalactosyl-ceramide-during DNA priming
To further evaluate the immunological consequences of NK T cell activation and evaluate the contributions of CD4 and CD8 T cells, we asked - which were the actual populations of effector cells induced by vaccination and did this significantly change in the presence of αGalCer during DNA priming. Cellular depletion (CD4 or CD8) was employed. Briefly, mice were vaccinated as indicated above and CD4 or CD8 T cells were depleted as previous described [62, 63] immediately prior to infection with L. major; depletion at this time in vaccinated mice would primarily target antigen-specific memory cells. Lesion development was monitored and immune responses to infection determined; at 13 weeks post-infection, the parasite levels at the cutaneous site of infection were enumerated.
As shown in Figure 5, lesion size development and parasite burdens were significantly increased in LACK-vaccinated mice depleted of either CD4 or CD8 T cells. In Figure 5A, although there was not significant difference between control mice (pCINeoDNA+WRLuc) and those vaccinated with DNAp36+VVp36 and depleted of CD8 T cells (p>0.05), vaccinated mice depleted of CD4 T cells were still significantly protected (p=0.01). Vaccinated mice depleted of either CD4 or CD8 T cells were significantly less protected than their non-depleted counterparts (p=0.001 and 0.0001, respectively). In vaccinated mice receiving αGalCer together with DNAp36(Figure 5B), mice depleted of either CD4 or CD8 T cells were significantly less protected than their non-depleted counterparts (p=0.01 and 0.001, respectively). Overall parasite burden analyses (Figure 5C) confirmed these results. Protection in mice receiving αGalCer together with DNAp36was better than mice vaccinated with DNAp36 alone (p=0.0001, ANOVA). Further, the parasite burdens in mice depleted of CD8 T cells in either group (αGalCer together with DNAp36 or DNAp36 alone) were significantly higher than mice depleted of CD4 T cells (p=0.01 and 0.001, respectively).
Overall these results reveal a larger contribution/role of CD8 T cells in protection induced by DNA-VACV prime-boost vaccination. In particular in the DNAp36 + VVp36 vaccinated group, the reversal of protection was more pronounced for mice depleted of CD8 T cells; parasite numbers at the site of infection in CD8-depleted mice were increased 65-fold, while in CD4 depleted mice parasite levels were increased only 2.4-fold. However for the mice receiving αGalCer during DNA priming, the effect of CD4 cell depletion was more substantial and approached that found after depletion of CD8 cells, with 156- (CD4) and 875-fold (CD8) increases in parasite levels at the site of infection. These data suggest that NKT cell activation increased the antigen-specific T cell responses (CD4 and CD8) during DNA priming. However, it appeared that NKT cell activation preferentially facilitated the development of antigen-specific CD4 T cells, leading to an enhanced contribution of CD4 T cells to protection.
To further examine the effects of cellular depletion on the ongoing immune response and effect of the cells contributing to protection, cytokine responses were evaluated in the T cell depleted mice at 14 weeks post-infection; splenic lymphocytes were stimulated using SLA and IFNγ, and granzyme, nitric oxide and IL-10 levels were determined. Overall the effect of CD4 or CD8 T cell depletion resulted in the ablation of IFNγ, nitric oxide and granzyme production in response to infection. In contrast, either CD4 or CD8 depletion just prior to infection led to increased IL-10 production, which approached that of control mice (Table 5). IL-10+CD4+ T cell populations have been reported to be critical for parasite persistence in murine leishmaniasis caused by L. major [58-61]. As mice depleted of either CD4 or CD8 T cells before infection have levels of IL-10 comparable to those of non-vaccinated controls, it can be concluded that vaccination importantly leads to overall reductions in IL-10 levels induced during infection and that interactions between CD4-CD8 T cells may be critical to the ablatement of the IL-10 response. . Given the decrease in IL-10 levels observed in the vaccinated mice receiving αGalCer during DNA priming, these results suggest that NKT activation in the context of TLR9 activation by CpG may prevent the known NKT enhancement of Tr cell populations [39, 40].
Table 5. Immune Responses of Vaccinated and CD4 and CD8 Deficient/Depleted BALB/c Mice in Response to Leishmania major Infection.
| Group | IFNγ (ng/ml±SD) | Granzyme (ng/ml±SD) | IL-10 (pg/ml+SD) | Nitric Oxide |
|---|---|---|---|---|
| PBS (2×) | 0±0 | 0±0 | 981±60 | 0.7±0.2 |
| pCIneo+Galcer+WRluc | 0±0 | 0±0 | 822±37 | 1.5±0.5 |
| DNAp36+VVp36 | 66.2±1.2 | 1.5±0.3 | 282±15 | 15.6±0.3 |
| DNAp36+Galcer+VVp36 | 85.5±1 | 3.5±0.6 | 164±8 | 24±0.5 |
| DNAp36+VVp36 CD4dpl | 5.7±0.7 | 0.8±0.2 | 470±21 | 6.3±0.1 |
| DNAp36+VVp36 CD8dpl | 2.1±0.3 | 0.4±0.1 | 650±25 | 2.6±0.1 |
| DNAp36+Galcer+VVp36 CD4dpl | 7±0.8 | 1+0.1 | 343±14 | 8.6±0.2 |
| DNAp36+Galcer+VVp36 CD8dpl | 1.3±0.7 | 0.5±0.2 | 640±60 | 4±0.6 |
To examine the effects of in vivo CD4 and CD8 T cell depletion on the ongoing immune response to infection, cytokine responses were evaluated in vitro of splenic lymphocytes. Cells were treated or not with anti-CD4, or anti-CD8 as indicated in the Materials and Methods Section. Cells were then stimulated for 72 hours and the cytokine levels determined.
Overall, experimental evidence indicates that NKT activation by αGalCer during intradermal DNA vaccination leads to enhanced curative Th1-like/TC1 CD4 and CD8 T cell responses which results in significant increases in protection against infection. Further, these studies appear to reveal that CD4↔CD8 interactions are critical/key to this protection during the memory/recall response, as the absence of either T cell subpopulation at the time of infection severely impacts on the development of the host immune response. Hence, CD4 and CD8 T cells are important as memory populations as well as their direct effector functions to the development of a protective response to infection.
Discussion
A heterologous DNA-vaccinia virus (VACV) prime-boost antigen delivery system has been shown to be efficacious for the control of infectious diseases in animal model studies and in human trials [18, 20, 64-73]. A prime/boost regime using DNA and vaccinia viruses expressing the leishmanial LACK antigen has been shown to be protective against murine L. major infection [21-23] and is effective in inducing protection against visceral leishmaniasis caused by L. infantum, while DNA vaccination alone fails to protect [16, 24]. However, the efficacy of DNA priming is known to be limited; consequently, adjuvants (cytokines, co-stimulatory molecules, delivery vehicles) with potential to enhance DNA priming have been sought [21, 25].
The use of recombinant protein cytokines or DNA encoding cytokines (IL-12, IL-18) have been demonstrated to enhance DNA priming, leading to increased protection against leishmanial infection [21]. However, in terms of vaccine development, the use of synthetic immunomodulatory compounds, such as αGalCer, offer similar advantages to DNA in terms of stability and production, while having direct immediate effects on the target cell population. Additionally, structural modification of the parent molecule has been shown to produce longer-lived NKT activation or the induction of specific cytokine profiles [51, 74]. Further, αGalCer has already been utilized in the treatment of human tumors [75, 76]. In terms of potential vaccine use, NKT activation has been reported to lead to enhanced CD4 and CD8 T cell effector responses [29, 30, 46] as well as induce dendritic cell maturation [31]. However, the precise response (Th1 versus Th2) that develops as a consequence to αGalcer activation of NKT cells is not always predictable [38]; for example, αGalCer provides protection in a murine model of malaria [42, 44], but disease exacerbation was found in Trypanosoma cruzi vaccine studies in mice [41]. However, CpG has been shown to enhance NKT cell activation through interplay between plasmacytoid and myeloid dendritic cells [35, 36], which may be in part dependent upon TNFα [35]. Recent studies indicate that activated NKT cells increase dendritic cell migration and enhance CD8 T cell responses in the skin [77]. Consequently, the interaction of adjuvants (and developing inflammatory milieu) in a vaccine employing intradermal DNA-priming (TLR9) together with NKT cell (αGalCer) activation could selectively amplify the developing Th1/TC1 immune response. Results from analyses of vaccination of mice indicate that this has occurred; mice vaccinated with αGalCer showed higher levels of both CD4 and CD8 T cells upon infection. These cells produced higher levels of IFNγ/granzyme and lower levels of IL-10 in response to infection. In addition, depletion experiments revealed that both populations (CD4 and CD8) were important for protection, while in mice not receiving αGalCer, a predominant role for CD8 T cells was apparent. Notably, these effects (elevated IFNγ/granzyme, lower IL-10 and increased protection) were not observed in vaccinated NKT deficient mice.
CD4 and CD8 T cells producing INFγ have been demonstrated to be important for the control of cutaneous leishmaniasis caused by Leishmania major infection [12, 13, 26-28] as well as visceral disease [78-80]. In the current study, depletion experiments indicate that neither population is sufficient for protection, in that immunodepletion of either CD4 or CD8 T cells prior to infection for mice receiving αGalCer results in reversal of protection. Interestingly, αGalCer/NKT activation during DNAp36 priming leads to enhanced CD4 T cell contribution to protection in comparison to DNAp36-vaccination alone. These results suggest that the interactions between CD4-CD8 T cells are required for the activation and development of effector populations. Previous studies have demonstrated that the contributions of CD8 IFNγ was critical to vaccination induced by DNAp36 alone [12-14] and that this led to heightened CD4 T cell activation. Although somewhat controversial [81], the interaction between T cell subpopulations (and a requirement of both populations during chronic infection or secondary responses) has been documented in other systems and recently for L. major infection [55-57, 82]. It is unclear in the current study what the role of CD8 T cells might be in protection; however, it is clear that overall these cells are not the dominant source of IFNγ. Previous leishmaniasis vaccine studies have shown that CD8 IFNγ [13, 64] can be critical in the development of CD4 T cell responses and indeed, such mechanisms may also be involved in the prime-boost vaccinated mice. However, VACV vaccination is known to lead to the development of cytolytic CD8 T cells. The role of CTL CD8 T cells in vaccine-mediated protection has not been extensively studied. Only in the case of vaccine studies against L. amazonensis using a murine model and the P-8 proteoglycolipid complex antigen has a role for perforin and CD8 T cells been documented [63]. However, in mice primed with DNAp36 alone, little if any consequence of CD4 T cell depletion was found, suggesting that CD8 T cells alone can provide significant protection and the question arises as to the CD8-mechanisms pertinent to this protection. We found increased levels of granzyme B as a consequence of vaccination – in both CD4 and CD8 populations, suggesting that these CTL populations could contribute to disease control. Further studies are required to determine this point. However, these could point to additional effector targets for vaccine development.
The ideal vaccine against leishmaniasis would be one that protected against the various species and forms of disease (visceral, cutaneous and mucocutaneous). However, the literature suggests that the ability to provide cross-protection is elusive [16, 78, 83]. We have previously demonstrated that heterologous prime-boost vaccination (DNA-VACV) can provide protection against both cutaneous and visceral leishmaniasis. We have demonstrated herein that the development of protective memory and effector populations are enhanced through the activation of NKT populations during DNA priming. Consequently, a multiple-subunit vaccine employing this modality could provide for an effective protection across Leishmania species, and the development of a pan-leishmaniasis vaccine. However, work is required to determine if this modality represents a vaccine modality for humans; given the known effects of αGalCer in humans [29-31, 46, 75, 76] these results are encouraging.
Materials and Methods
Parasite strains and animals
L. major strain WR309 (MHOM/IL/79/LRC-L251) were grown as promastigotes at 24°C in Schneider's Drosophila medium (Sigma, Saint Louis, MO) supplemented with 15% heat inactivated fetal bovine serum and antibiotics, as previously described [84]. The virulence of the strain was preserved by periodic passage through BALB/c mice. All mice used in this study were BALB/c background (WT or NKT-deficient). Breeding pairs of BALB/c-NKT deficient mice were kindly provided by Dr. Malcolm Duthie (Infectious Disease Research Institute, Seattle, WA). BALB/c mice (4-6 weeks of age) were purchased from the NIH National Cancer Institute (Frederick, MD). All mice were housed in the Yale University School of Medicine, an American Association for Accreditation of Laboratory Animal Care-approved animal facility (Federal Animal Welfare Assurance number A3230-01). Sentinel mice were periodically checked for the presence of viruses in the colony.
Plasmids, recombinant p36 protein and recombinant vaccinia viruses
The gene encoding for L. infantum p36 protein was obtained as described [22] and inserted downstream of the cytomegalovirus (CMV) promoter into the SmaI site of the pCI-neo vector (Promega, Madison, WI). The empty plasmid pCI-neo (Promega) was used as control (DNA control). Plasmid DNA was purified using the Qiagen Endofree Plasmid Maxi Kit (Qiagen Inc., Valencia, CA) and eluted in pyrogen-free deionized water. The endotoxin level of the purified plasmid was tested using QCL-1000 Chromogenic LAL Test (Lonza Walkersville Inc., Walkersville, MD) before immunization. Preparations had a maximum of 0.1 ng LPS/100 μg DNA.
WR-LACK (WRp36) was derived from the WR strain and was prepared using standard methods previously described [22, 45, 85, 86].
Prime-boost vaccination and infection of mice with Leishmania major
Mice (7 to 10/group) were primed intradermally with p36 (LACK) plasmid DNA or control vector DNA (100 μg/mouse in 100 μl of phosphate buffered saline) together with or without α-galactosylceramide (αGalCer; 2 μg/mouse). Mice were boosted intraperitoneally two weeks later (14 d.p.i.) with WRp36 (1×107 PFU/mouse). Mice immunized earlier with the control DNA were boosted with 1×107 PFU of the control virus encoding for the luciferase gene (WR-Luc). Infection control mice received phosphate buffer saline (PBS) alone during the two immunizations. Three weeks after boosting, three of the mice from each group were sacrificed and spleen and draining lymph node were collected for immunological analyses. At the same time, other mice (minimally four per group) were infected in the right rear hind foot with 5 ×104 late-stationary phase L. major promastigotes. The course of infection was monitored by measuring the lesion development; using a dial gauge caliper (Starrett Thickness Gauge), the ratio of the infected compared with the (left) uninfected foot was determined. At the end of monitoring lesion development, the mice were sacrificed and the parasites were enumerated in the cutaneous tissue (site of infection) and draining lymph node by a limiting dilution assay, as reported previously [87].
Immunodepletion of CD4+ or CD8+ T cells
Anti-CD4 (L3T4) or anti-CD8 (Ly-2) monoclonal antibody obtained from eBioscience (San Diego, CA) was employed for cellular immunodepletion of vaccinated mice. For T cell depletion, mice were injected intraperitoneally with 100μg of anti-CD4 (clone GK1.5) or anti-CD8 (clone 53-6.7) antibody on days −3 and −1 of infection. Efficacy of depletion was monitored by flow cytometry and showed that more than 95% of the target cell population was depleted.
Cytokine Assays
Splenic lymphocytes and lymph node cells from control and immunized mice were harvested. Single cell preparations were made in RPMI medium supplemented with 10% fetal bovine serum, 2-mercaptoethanol, antibiotics and L-glutamine. Cells (5×106 per well) were plated in 24 well plates (Costar, Cambridge, MA) and stimulated at 37 °C with purified recombinant LACK (5 μg/ml) antigen, soluble leishmanial antigen (SLA; 25 :g/ml)), concanavalin A (1 :g/ml) or left unstimulated. For antibody blocking experiments, cells were cultured in the presence of 20 :g/ml of inhibitory anti-CD4 and/or anti-CD8 antibody (as indicated above for depletion experiments) or isotype control (eBiosciences, San Diego, CA). Preliminary experiments indicated that maximal blocking was achieved under these conditions. Supernatants were collected after 72 h of incubation and stored at –20°C until used. Cytokine levels were measured from culture supernatants by sandwich ELISA, according to the manufacturer's specifications, as previously described [84, 87]. Background cytokine levels were determined using supernatant from unstimulated cell populations.
FACS analyses were performed as previously described [63]. Cell surface staining for CD4 and/or CD8 was performed using specific FITC- or CyChrome antibody conjugates (BD Pharmingen, San Jose, CA). For intracellular cytokine staining, cells were treated with Golgi Plug (BD Pharmingen, San Jose, CA) for 4 hrs before harvesting, stained for surface markers (CD4 (L3T4 of H129.19 clone) and CD8 (Ly-2 of 53-6.7 clone)), and then fixed and permeabilized [63]. Cells were subsequently stained for either IFNγ (BD Pharmingen) or granzyme B (eBioSciences) using a PE-conjugated specific antibody. Cell fluorescence was measured using a FACScan (Beckton Dickinson); data were analyzed using Flow Jo software (Tree Star, Inc.).
Measurement of nitrite/nitric oxide
Nitric oxide (NO) production was measured as nitrite using the Greiss reaction as previously described [88]. Briefly, to each well 96-well flat-bottom microtiter plate (Nunc, USA) containing 50 :l of culture supernatant equal volumes of the Greiss reagents (1% sulfanilamide and 0.1% naphthylethylenediamine dichloride in 2.5% phosphoric acid) were added and incubated at room temperature for 10 minutes. Standards of NaNO2 using concentrations from 1.56 to 100 μM NaNO2 were employed with each assay. Absorbance was measured at 540 nm with the Bio-tek plate reader (Bio-tek Instruments, Winooski, VT). Background concentrations were assessed using supernatant from unstimulated splenocytes.
Antibody response
Antibody levels to LACK (p36) antigen and soluble leishmanial antigen (SLA) were assessed by ELISA. Total IgG as well as IgG1 and IgG2a isotype responses were evaluated. This was conducted on sera collected from each group of animals before challenge and at one month post-challenge. The ELISA was performed as previously described [21] with some modifications. Briefly, 96-well Maxisorp plates (Nunc, USA) were coated overnight at 4°C with 100μl of purified recombinant p36 protein (5 μg/ml). The following day, plates were washed four times with PBS-0.05% Tween 20 (PBS-T) and blocked with 3%BSA in PBS-T (blocking buffer) for two hours at 4°C. Sera were diluted in blocking buffer at 1:50 and 1:400 for pre- and post-challenge sera respectively, and incubated overnight at 4°C. Then, plates were washed and peroxidase-conjugated goat anti-mouse total IgG (H+L) (Pierce, Rockford, IL), IgG1 or IgG2a (Southern Biotechnology Associated, Birmingham, AL) was added. Reactions were developed using ATBS (Kirkegaard Perry Laboratories, Gaithersburg, MD). Absorbance was read at 405 nm with the Bio-tek plate reader (Bio-tek Instruments, Winooski, VT).
Evaluation of the parasite burden
The total number of parasites in the various infected tissues (cutaneous site, draining lymph node) was evaluated by limiting dilution analysis using Schneider's Drosophila medium, as previously described [87]. The individual parasite burdens from four mice per group were determined; results are expressed as the averaged value ± standard errors of the parasite burdens obtained from each group.
Statistical analysis of data
Data are presented as mean ± standard error. The Mann-Whitney Test was performed to assess the statistical significance between data acquired from mice primed with p36 (LACK) with and without alpha-galactosylceramide. For multiple group comparisons, Kruskal-Wallis one-way analysis of variance (ANOVA) was performed followed by a Dunn's post test. For all these statistical analyses, P values less than or equal to 0.05 were considered significant.
Acknowledgments
This work was supported by Grants from the NIH (AI 27811 and AI 06787 to DMc-P). Eva Perez-Jimenez is a recipient of predoctoral fellowship from the Ministerio de Educacion y Ciencia, Spain (BIO2004-03954 to M.E).
Abbreviations
- L
Leishmania
- VACV
vaccinia virus
- LACK
Leishmania homologue of receptors for activated C kinase
- αGalCer
α-galactosyl-ceramide
- CL and VL
cutaneous and visceral leishmaniasis
Footnotes
The authors have no financial conflicts of interest to the work reported herein.
References
- 1.Herwaldt BL. Leishmaniasis. Lancet. 1999;354:1191–1199. doi: 10.1016/S0140-6736(98)10178-2. [DOI] [PubMed] [Google Scholar]
- 2.Guerin PJ, Olliaro P, Sundar S, Boelaert M, Croft SL, Desjeux P, Wasunna MK, Bryceson AD. Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect Dis. 2002;2:494–501. doi: 10.1016/s1473-3099(02)00347-x. [DOI] [PubMed] [Google Scholar]
- 3.Sundar S, Jha TK, Thakur CP, Engel J, Sindermann H, Fischer C, Junge K, Bryceson A, Berman J. Oral miltefosine for Indian visceral leishmaniasis. N Engl J Med. 2002;347:1739–1746. doi: 10.1056/NEJMoa021556. [DOI] [PubMed] [Google Scholar]
- 4.Tesh RB. Control of zoonotic visceral leishmaniasis: is it time to change strategies? Am J Trop Med Hyg. 1995;52:287–292. doi: 10.4269/ajtmh.1995.52.287. [DOI] [PubMed] [Google Scholar]
- 5.Dye C. The logic of visceral leishmaniasis control. Am J Trop Med Hyg. 1996;55:125–130. doi: 10.4269/ajtmh.1996.55.125. [DOI] [PubMed] [Google Scholar]
- 6.Gramiccia M, Gradoni L. The current status of zoonotic leishmaniases and approaches to disease control. Int J Parasitol. 2005;35:1169–1180. doi: 10.1016/j.ijpara.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Gurunathan S, Klinman DM, Seder RA. DNA vaccines: immunology, application, and optimization*. Annu Rev Immunol. 2000;18:927–974. doi: 10.1146/annurev.immunol.18.1.927. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Aseguinolaza G, Taladriz S, Marquet A, Larraga V. Molecular cloning, cell localization and binding affinity to DNA replication proteins of the p36/LACK protective antigen from Leishmania infantum. Eur J Biochem. 1999;259:909–916. doi: 10.1046/j.1432-1327.1999.00122.x. [DOI] [PubMed] [Google Scholar]
- 9.Mougneau E, Altare F, Wakil AE, Zheng S, Coppola T, Wang ZE, Waldmann R, Locksley RM, Glaichenhaus N. Expression cloning of a protective Leishmania antigen. Science. 1995;268:563–566. doi: 10.1126/science.7725103. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed SB, Bahloul C, Robbana C, Askri S, Dellagi K. A comparative evaluation of different DNA vaccine candidates against experimental murine leishmaniasis due to L. major. Vaccine. 2004;22:1631–1639. doi: 10.1016/j.vaccine.2003.10.046. [DOI] [PubMed] [Google Scholar]
- 11.Bourreau E, Prevot G, Gardon J, Pradinaud R, Hasagewa H, Milon G, Launois P. LACK-specific CD4(+) T cells that induce gamma interferon production in patients with localized cutaneous leishmaniasis during an early stage of infection. Infect Immun. 2002;70:3122–3129. doi: 10.1128/IAI.70.6.3122-3129.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gurunathan S, Sacks DL, Brown DR, Reiner SL, Charest H, Glaichenhaus N, Seder RA. Vaccination with DNA encoding the immunodominant LACK parasite antigen confers protective immunity to mice infected with Leishmania major. J Exp Med. 1997;186:1137–1147. doi: 10.1084/jem.186.7.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurunathan S, Stobie L, Prussin C, Sacks DL, Glaichenhaus N, Iwasaki A, Fowell DJ, Locksley RM, Chang JT, Wu CY, Seder RA. Requirements for the maintenance of Th1 immunity in vivo following DNA vaccination: a potential immunoregulatory role for CD8+ T cells. J Immunol. 2000;165:915–924. doi: 10.4049/jimmunol.165.2.915. [DOI] [PubMed] [Google Scholar]
- 14.Gurunathan S, Prussin C, Sacks DL, Seder RA. Vaccine requirements for sustained cellular immunity to an intracellular parasitic infection. Nat Med. 1998;4:1409–1415. doi: 10.1038/4000. [DOI] [PubMed] [Google Scholar]
- 15.Dumonteil E, Maria Jesus RS, Javier EO, Maria del Rosario GM. DNA vaccines induce partial protection against Leishmania mexicana. Vaccine. 2003;21:2161–2168. doi: 10.1016/s0264-410x(02)00769-7. [DOI] [PubMed] [Google Scholar]
- 16.Melby PC, Yang J, Zhao W, Perez LE, Cheng J. Leishmania donovani p36(LACK) DNA vaccine is highly immunogenic but not protective against experimental visceral leishmaniasis. Infect Immun. 2001;69:4719–4725. doi: 10.1128/IAI.69.8.4719-4725.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coelho EA, Tavares CA, Carvalho FA, Chaves KF, Teixeira KN, Rodrigues RC, Charest H, Matlashewski G, Gazzinelli RT, Fernandes AP. Immune responses induced by the Leishmania (Leishmania) donovani A2 antigen, but not by the LACK antigen, are protective against experimental Leishmania (Leishmania) amazonensis infection. Infect Immun. 2003;71:3988–3994. doi: 10.1128/IAI.71.7.3988-3994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson RJ, Hannan CM, Gilbert SC, Laidlaw SM, Sheu EG, Korten S, Sinden R, Butcher GA, Skinner MA, Hill AV. Enhanced CD8+ T cell immune responses and protection elicited against Plasmodium berghei malaria by prime boost immunization regimens using a novel attenuated fowlpox virus. J Immunol. 2004;172:3094–3100. doi: 10.4049/jimmunol.172.5.3094. [DOI] [PubMed] [Google Scholar]
- 19.Amanna IJ, Slifka MK, Crotty S. Immunity and immunological memory following smallpox vaccination. Immunol Rev. 2006;211:320–337. doi: 10.1111/j.0105-2896.2006.00392.x. [DOI] [PubMed] [Google Scholar]
- 20.Ramsburg E, Rose NF, Marx PA, Mefford M, Nixon DF, Moretto WJ, Montefiori D, Earl P, Moss B, Rose JK. Highly effective control of an AIDS virus challenge in macaques by using vesicular stomatitis virus and modified vaccinia virus Ankara vaccine vectors in a single-boost protocol. J Virol. 2004;78:3930–3940. doi: 10.1128/JVI.78.8.3930-3940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tapia E, Perez-Jimenez E, Lopez-Fuertes L, Gonzalo R, Gherardi MM, Esteban M. The combination of DNA vectors expressing IL-12 + IL-18 elicits high protective immune response against cutaneous leishmaniasis after priming with DNA-p36/LACK and the cytokines, followed by a booster with a vaccinia virus recombinant expressing p36/LACK. Microbes Infect. 2003;5:73–84. doi: 10.1016/s1286-4579(02)00077-1. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalo RM, del Real G, Rodriguez JR, Rodriguez D, Heljasvaara R, Lucas P, Larraga V, Esteban M. A heterologous prime-boost regime using DNA and recombinant vaccinia virus expressing the Leishmania infantum P36/LACK antigen protects BALB/c mice from cutaneous leishmaniasis. Vaccine. 2002;20:1226–1231. doi: 10.1016/s0264-410x(01)00427-3. [DOI] [PubMed] [Google Scholar]
- 23.Lopez-Fuertes L, Perez-Jimenez E, Vila-Coro AJ, Sack F, Moreno S, Konig SA, Junghans C, Wittig B, Timon M, Esteban M. DNA vaccination with linear minimalistic (MIDGE) vectors confers protection against Leishmania major infection in mice. Vaccine. 2002;21:247–257. doi: 10.1016/s0264-410x(02)00450-4. [DOI] [PubMed] [Google Scholar]
- 24.Dondji B, Perez-Jimenez E, Goldsmith-Pestana K, Esteban M, McMahon-Pratt D. Heterologous prime-boost vaccination with the LACK antigen protects against murine visceral leishmaniasis. Infect Immun. 2005;73:5286–5289. doi: 10.1128/IAI.73.8.5286-5289.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Babiuk S, Babiuk LA, van Drunen Littel-van den Hurk S. DNA vaccination: a simple concept with challenges regarding implementation. Int Rev Immunol. 2006;25:51–81. doi: 10.1080/08830180600743008. [DOI] [PubMed] [Google Scholar]
- 26.Stager S, Alexander J, Kirby AC, Botto M, Rooijen NV, Smith DF, Brombacher F, Kaye PM. Natural antibodies and complement are endogenous adjuvants for vaccine-induced CD8+ T-cell responses. Nat Med. 2003;9:1287–1292. doi: 10.1038/nm933. [DOI] [PubMed] [Google Scholar]
- 27.Mendez S, Gurunathan S, Kamhawi S, Belkaid Y, Moga MA, Skeiky YA, Campos-Neto A, Reed S, Seder RA, Sacks D. The potency and durability of DNA- and protein-based vaccines against Leishmania major evaluated using low-dose, intradermal challenge. J Immunol. 2001;166:5122–5128. doi: 10.4049/jimmunol.166.8.5122. [DOI] [PubMed] [Google Scholar]
- 28.Scott P. Immunologic memory in cutaneous leishmaniasis. Cell Microbiol. 2005;7:1707–1713. doi: 10.1111/j.1462-5822.2005.00626.x. [DOI] [PubMed] [Google Scholar]
- 29.Nishimura T, Kitamura H, Iwakabe K, Yahata T, Ohta A, Sato M, Takeda K, Okumura K, Van Kaer L, Kawano T, Taniguchi M, Nakui M, Sekimoto M, Koda T. The interface between innate and acquired immunity: glycolipid antigen presentation by CD1d-expressing dendritic cells to NKT cells induces the differentiation of antigen-specific cytotoxic T lymphocytes. Int Immunol. 2000;12:987–994. doi: 10.1093/intimm/12.7.987. [DOI] [PubMed] [Google Scholar]
- 30.Eberl G, MacDonald HR. Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur J Immunol. 2000;30:985–992. doi: 10.1002/(SICI)1521-4141(200004)30:4<985::AID-IMMU985>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 31.Silk JD, Hermans IF, Gileadi U, Chong TW, Shepherd D, Salio M, Mathew B, Schmidt RR, Lunt SJ, Williams KJ, Stratford IJ, Harris AL, Cerundolo V. Utilizing the adjuvant properties of CD1d-dependent NK T cells in T cell-mediated immunotherapy. J Clin Invest. 2004;114:1800–1811. doi: 10.1172/JCI22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishikawa H, Hisaeda H, Taniguchi M, Nakayama T, Sakai T, Maekawa Y, Nakano Y, Zhang M, Zhang T, Nishitani M, Takashima M, Himeno K. CD4(+) v(alpha)14 NKT cells play a crucial role in an early stage of protective immunity against infection with Leishmania major. Int Immunol. 2000;12:1267–1274. doi: 10.1093/intimm/12.9.1267. [DOI] [PubMed] [Google Scholar]
- 33.Mattner J, Donhauser N, Werner-Felmayer G, Bogdan C. NKT cells mediate organ-specific resistance against Leishmania major infection. Microbes Infect. 2006;8:354–362. doi: 10.1016/j.micinf.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Mempel M, Ronet C, Suarez F, Gilleron M, Puzo G, Van Kaer L, Lehuen A, Kourilsky P, Gachelin G. Natural killer T cells restricted by the monomorphic MHC class 1b CD1d1 molecules behave like inflammatory cells. J Immunol. 2002;168:365–371. doi: 10.4049/jimmunol.168.1.365. [DOI] [PubMed] [Google Scholar]
- 35.Montoya CJ, Jie HB, Al-Harthi L, Mulder C, Patino PJ, Rugeles MT, Krieg AM, Landay AL, Wilson SB. Activation of plasmacytoid dendritic cells with TLR9 agonists initiates invariant NKT cell-mediated cross-talk with myeloid dendritic cells. J Immunol. 2006;177:1028–1039. doi: 10.4049/jimmunol.177.2.1028. [DOI] [PubMed] [Google Scholar]
- 36.Marschner A, Rothenfusser S, Hornung V, Prell D, Krug A, Kerkmann M, Wellisch D, Poeck H, Greinacher A, Giese T, Endres S, Hartmann G. CpG ODN enhance antigen-specific NKT cell activation via plasmacytoid dendritic cells. Eur J Immunol. 2005;35:2347–2357. doi: 10.1002/eji.200425721. [DOI] [PubMed] [Google Scholar]
- 37.Brutkiewicz RR. CD1d ligands: the good, the bad, and the ugly. J Immunol. 2006;177:769–775. doi: 10.4049/jimmunol.177.2.769. [DOI] [PubMed] [Google Scholar]
- 38.Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol. 2005;23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742. [DOI] [PubMed] [Google Scholar]
- 39.La Cava A, Van Kaer L, Fu Dong S. CD4+CD25+ Tregs and NKT cells: regulators regulating regulators. Trends Immunol. 2006;27:322–327. doi: 10.1016/j.it.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 40.Jiang S, Game DS, Davies D, Lombardi G, Lechler RI. Activated CD1d-restricted natural killer T cells secrete IL-2: innate help for CD4+CD25+ regulatory T cells? Eur J Immunol. 2005;35:1193–1200. doi: 10.1002/eji.200425899. [DOI] [PubMed] [Google Scholar]
- 41.Miyahira Y, Katae M, Takeda K, Yagita H, Okumura K, Kobayashi S, Takeuchi T, Kamiyama T, Fukuchi Y, Aoki T. Activation of natural killer T cells by alpha-galactosylceramide impairs DNA vaccine-induced protective immunity against Trypanosoma cruzi. Infect Immun. 2003;71:1234–1241. doi: 10.1128/IAI.71.3.1234-1241.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez-Aseguinolaza G, de Oliveira C, Tomaska M, Hong S, Bruna-Romero O, Nakayama T, Taniguchi M, Bendelac A, Van Kaer L, Koezuka Y, Tsuji M. alpha -galactosylceramide-activated Valpha 14 natural killer T cells mediate protection against murine malaria. Proc Natl Acad Sci U S A. 2000;97:8461–8466. doi: 10.1073/pnas.97.15.8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmieg J, Yang G, Franck RW, Tsuji M. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand alpha-Galactosylceramide. J Exp Med. 2003;198:1631–1641. doi: 10.1084/jem.20031192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gonzalez-Aseguinolaza G, Van Kaer L, Bergmann CC, Wilson JM, Schmieg J, Kronenberg M, Nakayama T, Taniguchi M, Koezuka Y, Tsuji M. Natural killer T cell ligand alpha-galactosylceramide enhances protective immunity induced by malaria vaccines. J Exp Med. 2002;195:617–624. doi: 10.1084/jem.20011889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalo RM, Rodriguez JR, Rodriguez D, Gonzalez-Aseguinolaza G, Larraga V, Esteban M. Protective immune response against cutaneous leishmaniasis by prime/booster immunization regimens with vaccinia virus recombinants expressing Leishmania infantum p36/LACK and IL-12 in combination with purified p36. Microbes Infect. 2001;3:701–711. doi: 10.1016/s1286-4579(01)01426-5. [DOI] [PubMed] [Google Scholar]
- 46.Eberl G, Brawand P, MacDonald HR. Selective bystander proliferation of memory CD4+ and CD8+ T cells upon NK T or T cell activation. J Immunol. 2000;165:4305–4311. doi: 10.4049/jimmunol.165.8.4305. [DOI] [PubMed] [Google Scholar]
- 47.Kedzierski L, Zhu Y, Handman E. Leishmania vaccines: progress and problems. Parasitology. 2006;133(Suppl):S87–112. doi: 10.1017/S0031182006001831. [DOI] [PubMed] [Google Scholar]
- 48.Perez-Jimenez E, Kochan G, Gherardi MM, Esteban M. MVA-LACK as a safe and efficient vector for vaccination against leishmaniasis. Microbes Infect. 2006;8:810–822. doi: 10.1016/j.micinf.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Munz C, Steinman RM, Fujii S. Dendritic cell maturation by innate lymphocytes: coordinated stimulation of innate and adaptive immunity. J Exp Med. 2005;202:203–207. doi: 10.1084/jem.20050810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. 2003;171:5140–5147. doi: 10.4049/jimmunol.171.10.5140. [DOI] [PubMed] [Google Scholar]
- 51.Fujii S, Shimizu K, Hemmi H, Fukui M, Bonito AJ, Chen G, Franck RW, Tsuji M, Steinman RM. Glycolipid alpha-C-galactosylceramide is a distinct inducer of dendritic cell function during innate and adaptive immune responses of mice. Proc Natl Acad Sci U S A. 2006;103:11252–11257. doi: 10.1073/pnas.0604812103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen YG, Choisy-Rossi CM, Holl TM, Chapman HD, Besra GS, Porcelli SA, Shaffer DJ, Roopenian D, Wilson SB, Serreze DV. Activated NKT cells inhibit autoimmune diabetes through tolerogenic recruitment of dendritic cells to pancreatic lymph nodes. J Immunol. 2005;174:1196–1204. doi: 10.4049/jimmunol.174.3.1196. [DOI] [PubMed] [Google Scholar]
- 53.Jabbari A, Harty JT. Simultaneous assessment of antigen-stimulated cytokine production and memory subset composition of memory CD8 T cells. J Immunol Methods. 2006;313:161–168. doi: 10.1016/j.jim.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 54.Masopust D, Murali-Krishna K, Ahmed R. Quantitating the magnitude of the lymphocytic choriomeningitis virus-specific CD8 T-cell response: it is even bigger than we thought. J Virol. 2007;81:2002–2011. doi: 10.1128/JVI.01459-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lutjen S, Soltek S, Virna S, Deckert M, Schluter D. Organ- and disease-stage-specific regulation of Toxoplasma gondii-specific CD8-T-cell responses by CD4 T cells. Infect Immun. 2006;74:5790–5801. doi: 10.1128/IAI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu HM, Winter H, Urba WJ, Fox BA. Divergent roles for CD4+ T cells in the priming and effector/memory phases of adoptive immunotherapy. J Immunol. 2000;165:4246–4253. doi: 10.4049/jimmunol.165.8.4246. [DOI] [PubMed] [Google Scholar]
- 57.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 58.Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4(+)CD25(-)Foxp3(-) Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med. 2007;204:285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, Sacks DL. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med. 2001;194:1497–1506. doi: 10.1084/jem.194.10.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 61.Stober CB, Lange UG, Roberts MT, Alcami A, Blackwell JM. IL-10 from regulatory T cells determines vaccine efficacy in murine Leishmania major infection. J Immunol. 2005;175:2517–2524. doi: 10.4049/jimmunol.175.4.2517. [DOI] [PubMed] [Google Scholar]
- 62.Kar S, Metz C, McMahon-Pratt D. CD4+ T cells play a dominant role in protection against New World leishmaniasis induced by vaccination with the P-4 amastigote antigen. Infect Immun. 2005;73:3823–3827. doi: 10.1128/IAI.73.6.3823-3827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Colmenares M, Kima PE, Samoff E, Soong L, McMahon-Pratt D. Perforin and gamma interferon are critical CD8+ T-cell-mediated responses in vaccine-induced immunity against Leishmania amazonensis infection. Infect Immun. 2003;71:3172–3182. doi: 10.1128/IAI.71.6.3172-3182.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gurunathan S, Wu CY, Freidag BL, Seder RA. DNA vaccines: a key for inducing long-term cellular immunity. Curr Opin Immunol. 2000;12:442–447. doi: 10.1016/s0952-7915(00)00118-7. [DOI] [PubMed] [Google Scholar]
- 65.Kirman JR, Seder RA. DNA vaccination: the answer to stable, protective T-cell memory? Curr Opin Immunol. 2003;15:471–476. doi: 10.1016/s0952-7915(03)00068-2. [DOI] [PubMed] [Google Scholar]
- 66.Melby PC. Recent developments in leishmaniasis. Curr Opin Infect Dis. 2002;15:485–490. doi: 10.1097/00001432-200210000-00005. [DOI] [PubMed] [Google Scholar]
- 67.Melby PC. Vaccination against cutaneous leishmaniasis: current status. Am J Clin Dermatol. 2002;3:557–570. doi: 10.2165/00128071-200203080-00006. [DOI] [PubMed] [Google Scholar]
- 68.van Drunen Littel-van den Hurk S, Babiuk SL, Babiuk LA. Strategies for improved formulation and delivery of DNA vaccines to veterinary target species. Immunol Rev. 2004;199:113–125. doi: 10.1111/j.0105-2896.2004.00140.x. [DOI] [PubMed] [Google Scholar]
- 69.Hill AV, Reece W, Gothard P, Moorthy V, Roberts M, Flanagan K, Plebanski M, Hannan C, Hu JT, Anderson R, Degano P, Schneider J, Prieur E, Sheu E, Gilbert SC. DNA-based vaccines for malaria: a heterologous prime-boost immunisation strategy. Dev Biol (Basel) 2000;104:171–179. [PubMed] [Google Scholar]
- 70.Huygen K. Plasmid DNA vaccination. Microbes Infect. 2005;7:932–938. doi: 10.1016/j.micinf.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 71.Gherardi MM, Ramirez JC, Esteban M. Towards a new generation of vaccines: the cytokine IL-12 as an adjuvant to enhance cellular immune responses to pathogens during prime-booster vaccination regimens. Histol Histopathol. 2001;16:655–667. doi: 10.14670/HH-16.655. [DOI] [PubMed] [Google Scholar]
- 72.Hanke T, McMichael AJ, Dorrell L. Clinical experience with plasmid DNA- and modified vaccinia virus Ankara-vectored human immunodeficiency virus type 1 clade A vaccine focusing on T-cell induction. J Gen Virol. 2007;88:1–12. doi: 10.1099/vir.0.82493-0. [DOI] [PubMed] [Google Scholar]
- 73.Gilbert SC, Moorthy VS, Andrews L, Pathan AA, McConkey SJ, Vuola JM, Keating SM, Berthoud T, Webster D, McShane H, Hill AV. Synergistic DNA-MVA prime-boost vaccination regimes for malaria and tuberculosis. Vaccine. 2006;24:4554–4561. doi: 10.1016/j.vaccine.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 74.Parekh VV, Singh AK, Wilson MT, Olivares-Villagomez D, Bezbradica JS, Inazawa H, Ehara H, Sakai T, Serizawa I, Wu L, Wang CR, Joyce S, Van Kaer L. Quantitative and qualitative differences in the in vivo response of NKT cells to distinct alpha- and beta-anomeric glycolipids. J Immunol. 2004;173:3693–3706. doi: 10.4049/jimmunol.173.6.3693. [DOI] [PubMed] [Google Scholar]
- 75.Okai M, Nieda M, Tazbirkova A, Horley D, Kikuchi A, Durrant S, Takahashi T, Boyd A, Abraham R, Yagita H, Juji T, Nicol A. Human peripheral blood Valpha24+ Vbeta11+ NKT cells expand following administration of alpha-galactosylceramide-pulsed dendritic cells. Vox Sang. 2002;83:250–253. doi: 10.1046/j.1423-0410.2002.00217.x. [DOI] [PubMed] [Google Scholar]
- 76.Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, von Blomberg BM, Scheper RJ, van der Vliet HJ, van den Eertwegh AJ, Roelvink M, Beijnen J, Zwierzina H, Pinedo HM. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res. 2002;8:3702–3709. [PubMed] [Google Scholar]
- 77.Gorbachev AV, Fairchild RL. Activated NKT cells increase dendritic cell migration and enhance CD8+ T cell responses in the skin. Eur J Immunol. 2006;36:2494–2503. doi: 10.1002/eji.200636075. [DOI] [PubMed] [Google Scholar]
- 78.Rachamim N, Jaffe CL. Pure protein from Leishmania donovani protects mice against both cutaneous and visceral leishmaniasis. J Immunol. 1993;150:2322–2331. [PubMed] [Google Scholar]
- 79.Stager S, Smith DF, Kaye PM. Immunization with a recombinant stage-regulated surface protein from Leishmania donovani induces protection against visceral leishmaniasis. J Immunol. 2000;165:7064–7071. doi: 10.4049/jimmunol.165.12.7064. [DOI] [PubMed] [Google Scholar]
- 80.Squires KE, Schreiber RD, McElrath MJ, Rubin BY, Anderson SL, Murray HW. Experimental visceral leishmaniasis: role of endogenous IFN-gamma in host defense and tissue granulomatous response. J Immunol. 1989;143:4244–4249. [PubMed] [Google Scholar]
- 81.Castellino F, Germain RN. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu Rev Immunol. 2006;24:519–540. doi: 10.1146/annurev.immunol.23.021704.115825. [DOI] [PubMed] [Google Scholar]
- 82.Herath S, Kropf P, Muller I. Cross-talk between CD8(+) and CD4(+) T cells in experimental cutaneous leishmaniasis: CD8(+) T cells are required for optimal IFN-gamma production by CD4(+) T cells. Parasite Immunol. 2003;25:559–567. doi: 10.1111/j.0141-9838.2004.00668.x. [DOI] [PubMed] [Google Scholar]
- 83.Dumonteil E, Andrade-Narvarez F, Escobedo-Ortegon J, Ramirez-Sierra MJ, Valencia-Pacheco G, Flores-Serrano A, Canto-Lara S, Arjona-Torres A. Comparative study of DNA vaccines encoding various antigens against Leishmania mexicana. Dev Biol (Basel) 2000;104:135–141. [PubMed] [Google Scholar]
- 84.Ahmed S, Colmenares M, Soong L, Goldsmith-Pestana K, Munstermann L, Molina R, McMahon-Pratt D. Intradermal infection model for pathogenesis and vaccine studies of murine visceral leishmaniasis. Infect Immun. 2003;71:401–410. doi: 10.1128/IAI.71.1.401-410.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chakrabarti S, Brechling K, Moss B. Vaccinia virus expression vector: coexpression of beta-galactosidase provides visual screening of recombinant virus plaques. Mol Cell Biol. 1985;5:3403–3409. doi: 10.1128/mcb.5.12.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gomez CE, Rodriguez D, Rodriguez JR, Abaitua F, Duarte C, Esteban M. Enhanced CD8+ T cell immune response against a V3 loop multi-epitope polypeptide (TAB13) of HIV-1 Env after priming with purified fusion protein and booster with modified vaccinia virus Ankara (MVA-TAB) recombinant: a comparison of humoral and cellular immune responses with the vaccinia virus Western Reserve (WR) vector. Vaccine. 2001;20:961–971. doi: 10.1016/s0264-410x(01)00389-9. [DOI] [PubMed] [Google Scholar]
- 87.Soong L, Duboise SM, Kima P, McMahon-Pratt D. Leishmania pifanoi amastigote antigens protect mice against cutaneous leishmaniasis. Infect Immun. 1995;63:3559–3566. doi: 10.1128/iai.63.9.3559-3566.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Green SJ, Aniagolu J, Raney JJ. Oxidative metabolism of murine macrophages. Greene Pub. Associates and Wiley-Interscience; New York: 1994. [Google Scholar]