

Abstract
Immunosuppression resulting from excessive post-trauma apoptosis of hyperactivated Tcells is controversial. TRAIL mediated Tcell apoptosis decreases highly activated Tcells’ responses. Caspase-10, a particular TRAIL target, was increased in trauma patients’ Tcells with concomitantly elevated plasma TRAIL levels. These patients’ Tcells developed anergy, implicating increased TRAIL-mediated Tcell apoptosis in post-trauma Tcell anergy. Control Tcells cultured with patients’ sera containing high TRAIL levels increased their Caspase-10 activity and apoptosis. Stimulated primary Tcells are TRAIL apoptosis resistant. Increased plasma Thrombospondin-1 and Tcell expression of CD47, a Thrombospondin-1 receptor, preceded patients’ Tcell anergy. CD47 triggering of Tcells increased their sensitivity to TRAIL-induced apoptosis. Augmentation of Tcell TRAIL-induced apoptosis was secondary to CD47 triggered activation of the Src homology-containing phosphatase-1(SHP-1) and was partially blocked by a SHP-1 inhibitor. We suggest that combined post-trauma CD47 triggering, SHP-1 mediated NFκB suppression, and elevated TRAIL levels increase patients’ CD47 expressing Tcell apoptosis, thus contributing to subsequent Tcell anergy.
Keywords: Human T cells, Trauma, TRAIL, Apoptosis, CD47
1. Introduction
Post-trauma development of T cell hyporesponsiveness has been linked to immunosuppression and patient pathology [1–5]. Suggested mechanisms causing post-trauma development of T cell hyporesponsiveness include depressed antigen presentation with decreased co-stimulatory receptor expression, activation of inhibitory signaling pathways, and regulatory T cell induction subsequent to excessive T cell apoptosis [2–6]. Only some severely injured trauma patients experience increased T lymphocyte apoptosis and T cell immunodepression. Consequently, identifying apoptosis inducing mechanism(s) in this patient subset may provide insight into development of post injury T cell hyporesponsiveness. Post-trauma increased T cell apoptosis can contribute to induction of patient pathology by depleting immunocompetent T cells with subsequent development of a hyporesponsive T cell population. Exposure to apoptotic T cells can also lead to induction of inhibitory APC function causing further immunosuppression [2, 4, 7]. Although post-trauma excessive T cell apoptosis is well documented, detection of Caspase-3 activation in ex vivo patient T cells can be missed, since this downstream effector Caspase is activated during terminal stages of apoptosis [8]. In this study, we found that increased activation of the human proximal upstream Caspase, Caspase-10, can be readily detected in the T cells of those trauma patients who later develop anergy and correlates to increased annexin-V binding indicating elevated apoptosis [8, 9]. T cell apoptosis can be mediated by several members of the TNF super-family as well as by Fas [1, 10]. The TNF related apoptosis inducing ligand (TRAIL) has been particularly implicated in triggering apoptosis of chronically and/or hyper-activated T cells such as occur in trauma patients [11, 12]. TRAIL also preferentially activates Caspase-10 in hyper-activated human T cells [10]. TRAIL mediated apoptosis has been linked to T cell depression in murine sepsis models [2]. Although activated primary human T cells express TRAIL receptors, they usually escape TRAIL-induced apoptosis by simultaneously activating NFκB [13]. Inhibition of NFκB using a synthetic inhibitor increases T cell TRAIL sensitivity suggesting that post injury in vivo mechanisms which decrease NFκB activation will increase TRAIL mediated T cell apoptosis [13]. Activation of T cell constitutively expressed CD47, by its ligand Thrombospondin-1 (TSP-1) is known to induce apoptosis in human T cells by a variety of mechanisms [14–16]. We have previously shown increased CD47 expression by trauma patients’ anergic T cells [17]. We and other investigators have also shown post injury increased TSP-1 levels in plasma, tissue and PBMC transcripts of trauma and septic patients [18, 19]. This increased TSP-1 can trigger the elevated CD47 receptors expressed by trauma patients’ T cells. Triggering of CD47 activates the Src homology domain containing phosphatase-1 (SHP-1) [20]. Activated SHP-1 can ultimately depress T cell NFκB activation and therefore CD47 triggered induction of SHP-1 could increase trauma patients’ T cell sensitivity to TRAIL mediated apoptosis [13]. We have previously reported that SHP-1 levels are increased in trauma patients’ anergic T cells [6]. TSP-1 triggering of CD47 is known to increase activated human T cells’ susceptibility to Fas-mediated apoptosis [14]. We postulated that elevated post injury TSP-1 levels interacting with augmented T cells’ CD47-receptor expression might also increase post injury T cell sensitivity to TRAIL mediated apoptosis.
To explore these possible activities, we assessed the timing of post-injury apoptosis in relationship to development of T cell anergy as an explanation for conflicting reported data on excessive post injury apoptosis. We investigated several mechanisms by which trauma could trigger increased T cell apoptosis particularly TRAIL mediated apoptosis.
2. Materials & methods
2.1 Study population
A total of 126 trauma patients admitted to the University of Rochester Medical Center were enrolled in our study. Patients’ samples were assessed approximately every 4 days post injury for 28 days or until release or demise. Patient leukocytes were always assayed in parallel with age, sex and ethnicity matched control samples. Data obtained from 113 patients (25 females & 88 males; Average age 43.09 ± 19.28 [Mean±SD]) were used for analysis. Data from 13 patients were excluded from analysis for at least one of the following reasons- i) unavailability of patients’ sample to assay at least three times because of discharge/demise, ii) patients were given immunosuppressive medications such as steroids during their ICU stay, or iii) patients’ T cell proliferation was between 50%–65% of parallel-processed control T cells and these patient T cells could not reliably be classified as either immunocompetent or hyporesponsive. All enrolled patients with trauma had APACHE scores ≥21. In addition to an APACHE score ≥21, patients with thermal trauma had total burned area at least 30% (or 15% after adjustment for age >55). Patients with severe brain injury, pregnancy, HIV-positive diagnosis or history of taking immunosuppressive drugs were excluded from the study. Representative volunteers from our institution served as healthy controls. The Institutional Review Board of University of Rochester approved the study. All subjects provided informed consent.
2.2 Antibodies
The functional grade azide-free low-endotoxin anti-human (αh) monoclonal antibodies αCD3 (clone- OKT3) (eBioscience, San Diego, CA); αCD47 (clone- B6H12) & its isotype control antibody (BD Pharmingen, San Diego, CA) were used for T cell stimulation. The antibodies used for phenotype studies, αCD2-PE-Cy7 (clone- L303.1), αCD47-FITC (clone- B6H12) and their isotype control antibodies were from BD Pharmingen. The anti programmed death receptor-1 (αPD-1) (clone J105) and its isotype control antibodies were purchased from eBioscience. The α-phospho-NFκB p65 (pS529) AF488 (clone- K10-895.12.50) antibody was from BD Phosflow (San Diego, CA), the α-phospho-SHP-1 (pY536) rabbit polyclonal antibody was purchased from ECM Biosciences (Versailles, KY), and the goat polyclonal anti rabbit-Ig Pacific Blue secondary antibody was from BioLegend (San Diego, CA).
2.3 Isolation of T lymphocytes and monocytes from whole blood
Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation over Ficoll-Hypaque (GE Healthcare, Piscataway, NJ). T cells were isolated by sheep erythrocyte rosetting as described previously [17]. T cell purity was >95% by flowcytometry assay of CD2 staining. Monocytes were isolated from the non-rosette fraction of the PBMC by magnetic bead negative selection using αCD3, αCD19, αCD56 and αCD66b beads (Dynabeads, Invitrogen). Lymphocyte culture media was IMDM medium (Invitrogen-Gibco, Grand Island, NY) supplemented with 10% v/v FBS (Hyclone, Logan, UT), penicillin G (50IU/ml), gentamycin (50μg/ml), streptomycin (50μg/ml), fungizone (2.5μg/ml), L-glutamine (4mM), sodium pyruvate (1mM), MEM non-essential amino acids (1% v/v; CellGro, Manassas, VA), and 0.05mM β-mercaptoethanol (Sigma, St. Louis, MO). Polymyxin B ([100 IU/ml], Sigma) was added to all experiments. In the SHP-1 and NF κB assays, T cells were negatively isolated with RosetteSep T cell enrichment cocktail (StemCell Technologies) in vacutainer CPT (cell preparation tube) tubes following manufacturer’s protocol (BD Biosciences).
2.4 T cell stimulation and functional assessment
2×105 cells in 200μl complete IMDM medium were cultured for 72 hrs in a 96-well plate pre-coated with αCD3 ([1μg/well] eBioscience), pulsed with [3H]-thymidine ([1μCi/well], PerkinElmer, Wellesley, MA), then harvested after 18 hrs. T cell proliferation was expressed as counts per minute (CPM) in triplicate cultures. Patients’ T cell samples were defined as hyporesponsive (anergic) if their proliferation to direct TCR stimulation (immobilized αCD3) was <50% of that observed in parallel-processed age and sex matched healthy controls’ T cells.
2.5 Assessments of T cell apoptosis
T cells were stained for membrane Phosphatidyl Serine (PS) symmetry using an annexin-V PE and 7AAD kit (BD Biosciences) following manufacturer’s protocol. The patients’ and matched controls’ T cells were washed twice with PBS and incubated with annexin-V and 7AAD in 100 μl 1X Annexin binding buffer at room temperature, dark for 15 minutes. After incubation, reaction was stopped by adding 400 μl ice-cold buffer and the samples were acquired using a BD FACS Calibur flowcytometer immediately. To detect Caspase-10 activation, 1 × 105 T cells in complete IMDM medium were incubated with fluorescent FAM-AEVD-FMK peptide (Cell Technology, Mountain View, CA) for 30 minutes at 37°C. Caspase-10 activation was measured by assessing FAM-AEVD-FMK-peptide binding to active Caspase-10 by flowcytometry using FAM-peptide stained fresh T cells as control for background fluorescence.
2.6 Phenotypic analysis by flowcytometry
Freshly isolated T cells were stained with pre-titrated antibodies or appropriate isotype controls for 30 minutes at 4°C in the dark. After removal of excess antibodies, cells were analyzed in 9-color flowcytometry using a Cyan ADP flowcytometer (Beckman Coulter). CD2 was used as the T cell marker and always employed in T cell gating for analysis because CD3 is transiently down regulated in activated (e.g. in patient) T cells. Data are represented as percent positive cells and net mean fluorescence intensity (MFI) [as calculated by subtraction of the mean fluorescence intensity of the isotype control from that of the specific antibody]. The Cyan machine variability was controlled by weekly calibration.
2.7 Assessments of TRAIL and Thrombospondin-1 levels in patients’ plasma
The plasma levels of TRAIL and TSP-1 were determined using Quantikine human TRAIL and Quantikine human TSP-1 ELISA kits, (both from R&D Systems, Minneapolis, MN) following manufacturer’s protocol.
2.8 Serum extraction from patients’ plasma and determination of apoptosis in control T cells cultured with patient sera
Plasma samples from patients and matched controls were incubated with 2 Units/ml Thrombin (Sigma) and 10μl of 10% CaCl2 per ml plasma for 65 minutes at room temperature. After incubation, plasma samples were stored overnight at 4°C and then shaken vigorously using a vortex to dislodge the clots. Clotted samples were centrifuged at 3300 rpm for 30 min. The sera were collected over the clot-pellets and stored at −80°C until use.
To determine the effect of TRAIL present in patient sera on T lymphocytes, freshly isolated control T cells were stimulated with iαCD3 in culture as described above except FBS was replaced by 25% patient or control sera. To confirm that TRAIL was mediating triggering of Caspase-10 activation, either functional-grade αhTRAIL neutralizing antibody (clone-RIK2) or control IgG (both from eBioscience) was added to the culture 30 minutes before adding control T cells. Caspase-10 activation and T cell annexin-V binding were determined by flowcytometry.
2.9 Assessing if triggering of CD47 in control T cells sensitizes to TRAIL apoptosis
Freshly isolated control T cells (2 × 106 cells in 2 ml/well in complete IMDM medium) were cultured with 5 μg/well iαCD3, with or without 10 μg/ml αCD47 antibody and 1μg/ml rhTRAIL (PeproTech, Rocky Hill, NJ) to induce apoptosis. T cells cultured with iαCD3 alone, αCD47 alone, rhTRAIL alone or iαCD3+αCD47 served as controls. Cells were stimulated with iαCD3 plus anti-CD47 since CD47 activation occurs secondary to TCR engagement [15]. After 5 days, T cell apoptosis and Caspase-10 activation were measured as described above using IL-2 treated cultured T cells as control for background fluorescence to assess active Caspase-10 levels.
2.10 Assessment of SHP-1 and NFκB activation levels
Isolated control T cells were pre-incubated with αCD47 or isotype matched control Ig for 30 minutes, and then stimulated with plate-bound αCD3 (iαCD3) for 3 hours. After stimulation, cells were harvested, fixed with 1X Cytofix Buffer (BD Biosciences) for 10 min at 37°C, washed and then permeabilized with chilled Perm Buffer III (BD Phosflow) for 30 min over ice. After washing, cells were stained with α-phospho-NFκB p65 AF488 and rabbit α-phospho-SHP-1 antibodies. The anti-rabbit Ig Pacific Blue antibody was used to detect α-pSHP-1 antibody binding. Stained cells were acquired using a Cyan ADP flowcytometer. Unstimulated T cells served as controls. To determine NFκB nuclear localization, similarly treated cells were stained with α-NFκB p65 FITC (Santa Cruz Biotechnology) and nuclear stain DRAQ-5 (Invitrogen). Subcellular localizations of NFκB were determined using Amnis Imagestream X Imagecytometry (Amnis Inc, Seattle, WA).
2.11 Statistical analysis
The statistical analyses were done using GraphPad Prism software (GraphPad Software Inc, La Jolla, CA). Analysis of variance (ANOVA), t test or Mann-Whitney test (as appropriate; indicated in the figure legend) was used to determine significant variance between each of the parameters. Data were expressed as median unless otherwise stated. Data were flagged significant when p-values were less than 0.05.
3. Results
3.1 Post-trauma increased T cell apoptosis precedes but does not coincide with T cell hyporesponsiveness
Freshly isolated hyporesponsive (anergic) and normal responsive T cells from trauma patients were simultaneously assessed for proliferation to direct TCR stimulation (using plate-bound immobilized anti CD3 [iαCD3] antibody) and for apoptosis by assessing annexin-V binding and Caspase-10 activation. Patients’ T cells were considered hyporesponsive if iαCD3-stimulated proliferation was <50% of parallel processed matched control T cells. T cells from patients were considered immunocompetent when their proliferation in response to iαCD3 was ≥65% of control T cell proliferation. Seventeen patients’ T cells were classified as hyporesponsive because at least one T cell assay was 50% during their clinical course. T cells from 96 patients were found to be immunocompetent at all assay points (Fig-1A). These patients’ T cells along with their matched control T cells were assessed for annexin-V binding and Caspase-10 activation by flowcytometry. T cells from patients who would later develop T cell hyporesponsiveness showed significantly elevated pre-anergy apoptosis (post-injury days 4–7) as determined by increased annexin-V binding and elevated Caspase-10 activation (Fig-1B). Pre-anergy increased T cell apoptosis was significantly elevated by linear regression analysis (p = 0.002, r2 =0.0627, plot not shown). T lymphocytes from patients whose T cells remained immunocompetent did not show significant elevation of apoptosis. However, at the point patients’ T cells were detected as anergic (post injury days 8–14) they no longer exhibited elevated levels of apoptosis (Fig-1C). Elevated terminal Caspase-3 levels were variably detected in the pre-anergic T cells when annexin-V and Caspase-10 were consistently elevated (data not shown). As well as being human apoptosis specific, Caspase-10 is activated early in the apoptotic cascade [8]. Consequently, its activation has a wider detection window in ex vivo activated patient T cells. In addition, engagement of TRAIL receptors on human T lymphocytes preferentially activates Caspase-10 [10]. Increased Caspase-10 mediated T cell apoptosis typified patient T cells that later became anergic, suggesting that excessive post-trauma T cell apoptosis might be triggered by elevated TRAIL levels. Plasma TRAIL levels were found to be significantly increased in patients experiencing elevated T cell apoptosis identified by increased Caspase-10 activation compared to T cells from immunocompetent patients and matched controls (Fig-1D).
Figure 1. Excessive post-injury T-cell apoptosis that presage the development of T-cell anergy is TRAIL-induced.
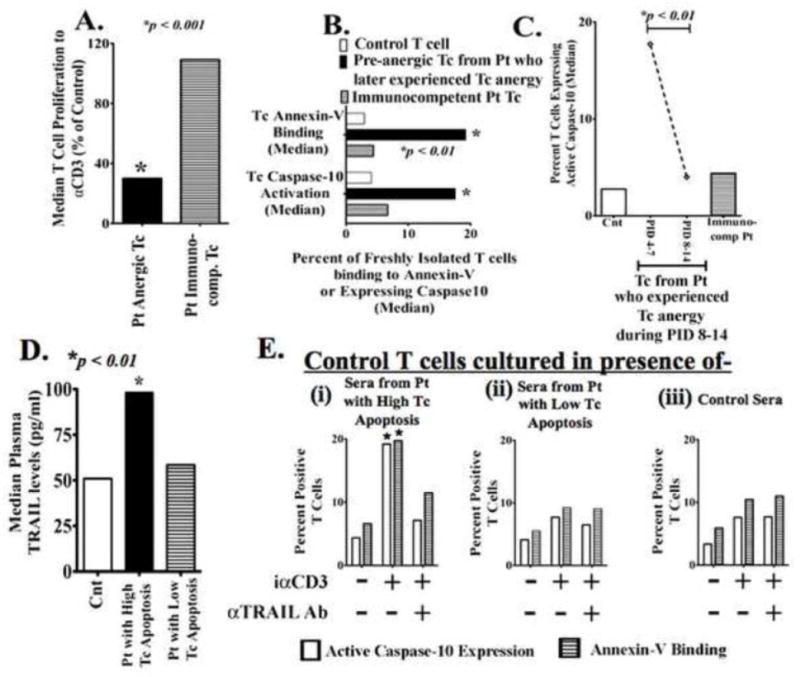
(A) Freshly isolated T-cells from trauma patients (Pt) and age, sex and ethnicity matched controls (Cnt) were stimulated with plate-bound αCD3 to assess T-cell (Tc) proliferation. Pt T-cells were considered anergic when proliferation to αCD3 was less than 50% of Cnt T-cell response. Data show the median value from 17 Pt with T-cell anergy and 96 immunocompetent Pt; p<0.001 by t test and ANOVA. (B) Trauma patients who experienced T-cell hyporesponsiveness showed earlier pre-anergy elevated T-cell apoptosis. Freshly isolated Patients’ (Pt) and matched Controls’ (Cnt) T-cells were incubated with annexin-V + 7AAD or FAM-AEVD-FMK peptides (binds specifically to human active Caspase-10) and assayed by flowcytometry. Data shows the median value. *p<0.01 by t test and ANOVA compared to controls’ and immunocompetent patients’ T-cells. (C) Only Pt with early (PID- 4–7) excessive T-cell apoptosis later (PID- 8–14) developed T-cell anergy. Pt actual anergic T-cell (PID- 8–14) did not show increased apoptosis. *p<0.01 by t test and ANOVA. (D) Patients experiencing increased T-cell apoptosis also had increased plasma TRAIL levels as determined by ELISA *p<0.01 by Mann-Whitney U test compared to matched controls. (E) Freshly isolated control T-cells were stimulated with iαCD3 in culture with IMDM medium plus (i) sera from Pt who experienced elevated T-cell apoptosis, (ii) sera from Pt with low T-cell apoptosis, or (iii) sera from matched healthy control volunteers. In the indicated experimental conditions, sera were pre-treated with αTRAIL blocking antibody to block existing TRAIL in the sera. After 5 days of culture, T-cell Caspase-10 activation and annexin-V binding were assessed n=3 for each Pt group; n=6 for Cnt *p < 0.05.
3.1.1 Patient sera with elevated TRAIL levels induce T cell apoptosis
To examine whether increased post-injury T cell apoptosis could be TRAIL mediated, control T cells from healthy donors were cultured with patients’ or control sera in the presence of either neutralizing αTRAIL or an isotype control antibody. Sera were used instead of EDTA-treated plasma to avoid clotting during addition of culture media. To neutralize TRAIL present in patients’ sera, specific TRAIL neutralizing antibody or its isotype control were added to the sera containing culture 30 minutes before the T cells were added. After five days of culture, T cell annexin-V binding and Caspase-10 activation were measured. Control iαCD3 stimulated T cells cultured with patients’ sera collected on the day when patients were experiencing elevated T cell apoptosis showed increased Caspase-10 activation and annexin-V binding compared to similarly treated control T cells cultured with control sera or sera from patients who did not experience elevated T cell death. Treating the patient sera with TRAIL-neutralizing antibodies reduced their apoptosis inducing activity in the control T cells (Fig-1E).
Patient anergic or normal responsive T cells were assessed for expression of receptors associated with apoptosis. We previously reported increased expression of both CD47 and PD-1 as associated with post-trauma anergic T cells [17]. Increased PD-1 expression is also characteristic of exhausted post apoptotic T cells while early triggering of CD47 has been reported to increase activated T cell apoptosis [14, 21]. Those trauma patients who experienced increased TRAIL mediated T cell apoptosis also had significantly increased CD47 expression concomitant to their depressed T cell activation and elevated apoptosis (Table 1). We consequently assessed whether post-injury increased T cell apoptosis correlates with elevation of a known trigger of CD47, TSP-1 [15]. Increased plasma TSP-1 was detected in the patients with hyporesponsive T cells concomitant to elevated patient T cell apoptosis (Fig-2A). Elevated TSP-1 mRNA levels have been repeatedly reported in microarrays of trauma and septic patients PBMC [18, 19]. The increased patients’ plasma TSP-1 levels were correlated to the patients’ elevation in T cell apoptosis (Fig-2B). Elevated T cell CD47 expression appeared before development of T cell anergy and decreased during anergy recovery (Fig-2C). In contrast, patients’ pre-anergic T cells had low PD-1 levels which increased as the patients developed then recovered from anergy (Fig-2D). These data suggest that early CD47 triggering contribute to elevated T cell apoptosis while PD-1 is indicative of an exhausted post apoptotic T cell.
Table 1.
Simultaneous increases in T cell CD47 expression and Caspase-10 activation precede post injury T cell anergy.
| Patients who Experienced T cell Anergy
|
Patients who NEVER Experienced T cell Anergy | |||
|---|---|---|---|---|
| Pre-Anergy | During Anergy | Post-Anergy | ||
| T cell Proliferation to immobilized αCD3 antibody (Percent of matched Control) | 99.3 | 31.0** | 93.7 | 115.5 |
| IL-2 secretion to αCD3 stimulation in 3 days culture (Percent of matched Control) | 38.57† | 11.42† | 77.67 | 71.79 |
| Caspase-10 Activation (Fold Change compared to matched Control) | 5.4* | 1.4 | 1.8 | 1.6 |
| CD47 Expression (Fold Change compared to matched Control) | 1.26## | 1.14# | 0.98 | 1.06 |
| PD-1 Expression (Fold Change compared to matched Control) | 1.14 | 1.57# | 2.7## | 1.2 |
All numbers represent median values. Fold changes in freshly isolated T cells’ expressions of active caspase-10 or PD-1 were determined as- (Percentage of patients’ T cells expressing active caspase-10 or PD-1 divided by percentage of parallel-processed control T cells expressing the same). Similarly fold changes in CD47 were determined as- [CD47 MFI in patients’ T cell/CD47 MFI in parallel-processed control T cells].
p < 0.001 vs. all other groups by Mann Whitney U test; n = 17 patients who experienced T cell anergy and 96 immunocompetent patients (who never experienced T cell anergy).
p < 0.05 compared to post-anergic T cells and T cells from immuno-competent patient group by t test; n = 3 for pre-anergy group, n = 11 for patients’ anergic T cell group, 3 for post anergic T cell group and 15 for immunocompetent T cell group.
p < 0.01 vs. all other groups by Mann Whitney U test; n = 6 for pre-anergy group, n = 16 for anergic T cell group, 13 for post anergic T cell group and 48 for immunocompetent T cell group.
p <0.01 and
p < 0.05 compared to post-anergic T cells and T cells from immuno-competent patient group by Mann Whitney U test; n= 8 patients who experienced T cell anergy and 92 immunocompetent patients.
Figure 2. Post trauma increased T cell apoptosis positively correlated with increased plasma Thrombospondin-1 levels.

(A) Patients experiencing elevated T cell apoptosis also had increased plasma Thrombospondin-1 levels. Elevated plasma TSP-1 levels in patients with increased T-cell apoptosis as determined by ELISA. T cell apoptosis was determined by assessing active Caspase-10 binding to FAM-AEVD-FMK peptide. *p<0.01 vs. control and immunocompetent Patients (Pt) T cells by Mann-Whitney U test compared to matched controls; n = 6 for each patient group. (B) Linear regression plot showing significant correlation between elevated plasma TSP-1 levels and T cell Caspase-10 activation. r2 = 0.734, **p < 0.001, n = 12. (C–D) Elevated T cell CD47 and PD-1 expression levels in patients who experienced T cell anergy. Freshly isolated T-cells from patients and matched controls were stained for CD47 (C) and PD-1 (D) by flowcytometry. Percent changes compared to parallely assessed T cells form matched controls. Dashed lines represent similar percent change values from immunocompetent patients’ T cells. All datapoints represent median value. *p<0.01 & #p < 0.05 by Mann Whitney U test, n = 8 for patients experiencing T cell anergy and 92 for patients’ immunocompetent T cells.
3.2 Control T cells from healthy donors become sensitive to TRAIL mediated apoptosis following CD47 engagement
Thrombospondin-1 triggering of its cell membrane receptor CD47 induces some human T cell apoptosis on its own [15, 22]. However, since TSP triggering of CD47 also increases T cell Fas sensitivity, we investigated whether T cell triggering by the combination of TRAIL and CD47 might increase control T cell apoptosis sensitivity to TRAIL similarly to what occurs with trauma patients’ T cells [14]. Control T cells from healthy donors were activated with iαCD3 and simultaneously treated with rhTRAIL and/or αCD47 for 5 days. T cells stimulated with iαCD3, αCD47 or TRAIL alone, served as controls. Although TRAIL alone treated, or αCD47 alone treated, stimulated T cells showed nearly identical low levels of apoptosis as stimulated T cells alone, stimulated T cells treated with both rhTRAIL and αCD47 antibody had significantly elevated levels of apoptosis as determined by Caspase-10 activation and annexin-V binding (Fig-3).
Figure 3. Stimulated control T-cells expressed increased active Caspase-10 following combined treatment with rhTRAIL & αCD47 antibody.

Freshly isolated T-cells were cultured with (i) 10μg/ml αCD Ab only, or (ii) immobilized αCD3 antibody alone, or (iii) αCD3+ αCD Ab, or (iv) αCD3 + rhTRAIL [1 μg/ml], or (v) αCD3 + rhTRAIL + αCD Ab for five days. Cleaved Caspase-10 levels were determined as described previously. Annexin-V binding levels were determined by flowcytometry using an AnnexinV + 7AAD kit. (A) Histograms showing active Caspase-10 levels in T-cells. (B) Graphical representation & statistics of above experiments *p < 0.01 by t test and ANOVA (n=8).
3.3 CD47 induced activated SHP-1 reduces TCR-stimulated activation of NFκB phosphorylation resulting in increased T cell apoptosis sensitivity
TCR stimulation in primary human T cells is known to activate NFκB, protecting primary T cells from TRAIL-triggered and other TNF super family mediated apoptosis [13]. Engagement of T cell surface CD47 by its ligands can activate downstream Src homology containing phosphatase-1 (SHP-1) [17, 20]. Activated SHP-1 has a wide array of downstream inhibitory-dephosphorylation targets which will decrease final activation of NFκB [23]. Therefore, we assessed whether CD47 induced activation of T cell SHP-1 is mediating decreased activation of NFκB resulting in increased T cell sensitivity to TRAIL mediated apoptosis. T cells were pre-incubated with αCD47 antibody or its isotype control antibody for 30 minutes and then stimulated with αCD3 for 3 hours. Activation of SHP-1 was determined by assessing activating phosphorylation of Tyrosine 536 (pSHP-1) while NFκB activation levels were determined by assessing phosphorylation of S529 on the p65 subunit (pNFκB). Both T cell pSHP-1 and pNKκB were simultaneously measured at intracellular levels by flowcytometry. We detected significant elevation of NFκB phosphorylation in Control Ig treated T cells within 3 hours of αCD3 stimulation while SHP-1 activation levels remained similar to those of unstimulated T cells (Fig-4A). As expected, CD47-engagement resulted in increased phosphorylation of SHP-1 within 3 hours. Simultaneously, αCD47-treated T cells had decreased NFκB phosphorylation following TCR stimulation (Fig-4A). In fact, NFκB phosphorylation levels in αCD47-treated T cells were below the levels of unstimulated T cells (Fig-4B).
Figure 4. CD47 engagement in control T-cells inhibits TCR induced NFκB activation.
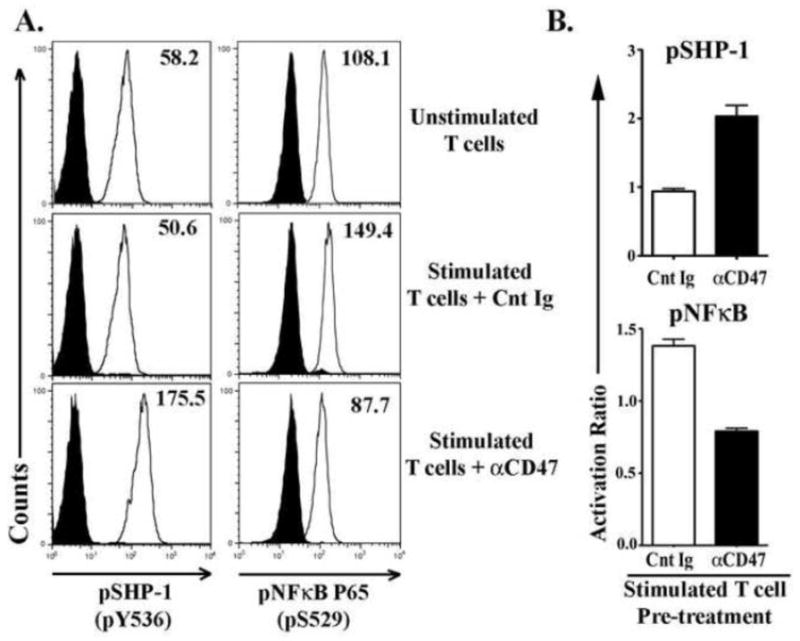
Freshly isolated T-cells from healthy control donors were pre-incubated with αCD47 antibody or an isotype matched nonspecific control immunoglobulin (Cnt Ig) for 30 minutes, then stimulated with plate-bound αCD3 Ab for 3 hours. Unstimulated T-cells served as controls for background phosphorylation levels. Cells were harvested and stained for intracellular pSHP-1 (pY536) and pNFκB p65 (pS529) and assessed by flowcytometry. (A) Histograms showing SHP-1 and NFκB phosphorylation levels in T-cells. (B) Graphs showing the activation ratio of SHP-1 and NFκB in αCD47 or Cnt Ig treated TCR-stimulated T-cells. Activation ratio has been calculated as- [Mean fluorescence intensity (MFI) in treated T-cells/MFI in unstimulated T-cells]. Data represented as Mean ± SEM, n=3.
In a final set of experiments, the SHP-1 inhibitor sodium stibogluconate (SSG) was added to anti CD47 triggered T cell cultures. The increased phosphorylated SHP-1 (pSHP-1) induced by αCD47 triggering of T cells was completely reversed in the presence of SSG (Fig-5A). In a second experimental design, we used the Amnis imaging flowcytometer to demonstrate that the reduced NFκB phosphorylation and depressed functional translocation to the T cell nucleus seen in αCD47 treated T cell cultures could also be reversed when the SHP-1 inhibitor SSG was also added to the cultures (Fig-5B). Most importantly, SSG addition to the αCD47 + TRAIL treated stimulated T cell cultures prevented the onset of TRAIL + αCD47 induced T cell apoptosis (Fig-5C).
Figure 5. Inhibition of SHP-1 by the pharmacological inhibitor, Sodium Stibogluconate (SSG), prevents CD47-induced NFκB deactivation and rescues T-cells from TRAIL-induced apoptosis.
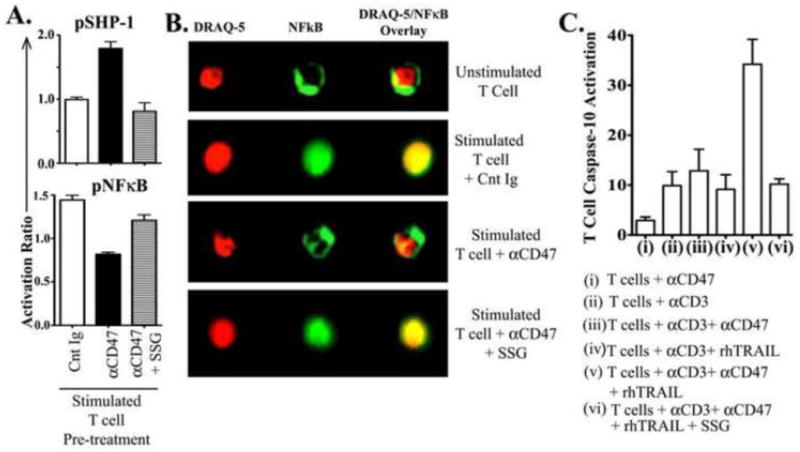
Freshly isolated T-cells from healthy control donors were pre-incubated either with a control immunoglobulin (Cnt Ig), or αCD47 antibody (Ab) or αCD47 AB + 15 μg/ml SSG for 30 minutes, then stimulated with plate-bound αCD3 Ab for 3 hours. (A) Activation ratio of intracellular pSHP-1 and pNFκB p65 were assessed as described above. Activation ratio has been calculated as [Mean fluorescence intensity (MFI) in treated T-cells/MFI in unstimulated T-cells]. Data represented as Mean±SEM, n=3. (B) Cells were stained for intracellular NFκB p65 (total protein) and nuclear dye DRAQ-5 to assess nuclear/cytoplasmic localization of NFκB by Amnis Image-cytometry. Data are representative of three experiments with similar results. (C) Freshly isolated T-cells were cultured with (i) 10μg/ml αCD47 Ab only, or (ii) immobilized αCD3 antibody alone, or (iii) αCD3 + αCD47 Ab, or (iv) αCD3 + rhTRAIL [1 μg/ml], or (v) αCD3 + rhTRAIL + αCD47 Ab, or (vi) αCD3 + rhTRAIL + αCD47 Ab + 15 μg/ml SSG for five days. Active Caspase-10 levels were determined as previously described. Data shown as Mean±SEM, n=3.
4. Discussion
Unlike post-sepsis induced T cell apoptosis, the importance of T cell apoptosis in post-trauma immunosuppression has been less clear. Similarly, the trauma induced triggers for elevated T cell apoptosis are ill defined. However, multiple investigators have described post-surgical and post-hemorrhage increased T cell apoptosis in murine models and patients with concomitant immune depression [1, 3–5, 18]. Hemorrhage and surgical intervention usually occur in severe trauma patients. However, some studies have identified no T cell apoptosis in post-injury patients’ cadaver spleens casting doubt on occurrence of post-injury T cell apoptosis [24]. Here we describe a changing time course of T cell apoptotic activity in T cells. Elevated T cell apoptosis occurred at less than day 7 post injury only in trauma patients who subsequently developed T cell unresponsiveness. However, there was no detectable increased apoptosis in these same patients’ anergic T cells at days >7 post injury. These hyporesponsive patient T cells appearing at >7 days post injury had elevated PD-1 levels similar to the anergic T cells remaining after apoptosis depletion of hyperactive T cells suggesting these post trauma anergic T cells are an exhausted, post apoptotic, residual population [11]. This variable time course for detection of T cell apoptosis may explain why some investigators’ examination of post-injury cadaver T cells shows little or no apoptosis elevation [24]. This study focused on the earliest detectible alteration in patients’ T cell responses as indicated by a depression in the anti CD3 direct T cell stimulated response. In previous studies we identified a more global form of anergy (irreversible by added exogenous IL-2 or CD28 stimulation) [25]. This global anergy occurred late in the post injury period and often overlapped septic or infectious episodes. Consequently, a secondary sepsis induced or reinforced immune suppression could not be eliminated. A subset of the present studies’ enrolled patients also suffered from sepsis. However, these post-trauma patients’ elevated T cell apoptosis preceded any development of T cell hyporesponsiveness representing an early post injury event that usually occurs within 1st week post-injury. Onset of sepsis was usually a later event. Although sepsis-linked alterations in leukocytes are well demonstrated, the detected post-trauma elevated peripheral blood T cell apoptosis occurred too early to be affected by later sepsis-related complications. Another reason for conflicting detection of post injury T cell apoptosis could be that measurements of terminal Caspase-3 activity in patients’ unstimulated T cells are variable because T cell apoptosis progresses quickly from appearance of terminal Caspase-3 activation to removal or lysis. This makes the window for Caspase-3 indicated apoptosis detection narrow [8]. Our measurement of Caspase-10 activation rather than Caspase-3 allows earlier apoptosis detection with a wider detection window. Caspase-10, a unique human expressed proximal Caspase, is also identified with alternative pathways of T cell apoptosis and unlike Caspase-8 is less associated with T cell activation [8–10]. We previously described increased post-injury mRNA transcripts for Caspase-10 but not Caspase-8 in trauma patient T cells with immunosuppression [18].
Caspase-10 is also a particular target of the TNF superfamily member TRAIL [10]. TRAIL has also recently been implicated in sepsis induced apoptosis and immune suppression [2]. We demonstrated that TRAIL levels were increased simultaneously in sera of patients with early elevated T cell apoptosis and subsequent T cell anergy. These same patients’ sera induced increased apoptosis in primary stimulated control T cells through a TRAIL mediated mechanism (inhibited by anti TRAIL antibody). A circulating sera factor(s) has been recently described as increasing septic patients’ T cell apoptosis [26]. This septic sera factor was not identified but our data suggest elevated plasma TRAIL can increase T cell apoptosis when TSP-1 is concomitantly present. Since TRAIL alone is unable to mediate apoptosis in primary stimulated T cells, an additional serum mediator needs to be involved in mediating the control T cell apoptosis induced by trauma patients’ sera [13, 27, 28]. TSP-1 is increased post-injury and can trigger CD47 (its receptor) on T cells to induce apoptosis [15, 22]. We showed that TSP-1 was also increased concomitant to TRAIL in the plasma of trauma patients who were experiencing elevated apoptosis. CD47 triggering by TSP-1 is known to increase Fas mediated apoptosis and we now demonstrate that CD47 triggering also increases TRAIL mediated apoptosis [14]. Although CD47 has other inhibitory ligands such as SIRPα (signal regulatory protein), only TSP-1 – CD47 interactions have been associated with increased T cell apoptosis [15]. The onset of T cell hyporesponsiveness after excessive apoptosis is well documented and has previously been suggested as a mechanism for post trauma immunosuppression [29–31]. Increased TRAIL release by hyperactivated T cells (as occurs after severe injury) as well as tolerance induction is well described [21, 32]. Although we cannot rule out that elevated post injury steroids might also increase T cell apoptosis sensitivity, our data link increased TRAIL mediated apoptosis to the simultaneous elevation of TSP-1 and its receptor CD47 on T cells. The post-injury T cell anergy developing subsequent to TSP-1 triggering of CD47 could also result from other CD47 triggered inhibitory functions [33]. However, we have now demonstrated CD47 expression to be increased on patients’ T cells concomitant to elevated sera levels of TSP-1 and increased T cell apoptosis but preceding T cell anergy development. We have also shown that CD47 triggering induces the phosphatase SHP-1, which inhibits multiple T cell activation pathways [34]. We previously reported SHP-1 levels as increased in trauma patients’ hyporesponsive T cells [17]. TSP-1 mediated CD47 triggering can increase TRAIL mediated primary stimulated T cell apoptosis by diminishing several of these T cell activation pathways. Most prominently, T cell resistance to TRAIL mediated apoptosis is pivotally dependent on TCR triggering of NFκB, and NFκB activation is also critical to sepsis survival where T cell apoptosis is known to play a major immunosuppressive role [13, 35]. Consequently, our demonstration that CD47 triggering depresses NFκB phosphorylation through a SHP-1 dependent mechanism and that a SHP-inhibitor reverses TRAIL + αCD47 augmented apoptosis of primary stimulated T cells illustrates one mechanism for increased post injury T cell apoptosis. Our data further suggest that TSP-1 triggering of CD47 induction of SHP-1 activation can critically contribute to TRAIL triggered post injury apoptosis by depressing NFκB.
5. Conclusions
Our data support a role for excessive T cell apoptosis in post injury immunosuppression, reconcile previous contradictory reports indicating both no or increased post trauma T cell apoptosis, and demonstrate TRAIL involvement in this post injury T cell apoptosis. Our demonstration that sera from patients with excessive T cell apoptosis have concomitantly increased TSP-1 levels and that these patients’ T cells have increased expression of the TSP-1 receptor, CD47, imply that elevated TSP-1 can also contribute to post injury T cell apoptosis through triggering CD47. The demonstration that control T cells are sensitive, not resistant, to TRAIL mediated apoptosis by these patients’ TRAIL and TSP-1 containing sera suggested that TSP-1 triggering of CD47 was augmenting TRAIL mediated apoptosis. Finally, our data showed that CD47 triggering, by elevating SHP-1 phosphatase and depressing NFκB activation, could induce increased sensitivity of control T cells to TRAIL mediated apoptosis. We suggest that post injury elevated TRAIL levels combined with injury released TSP-1 are major contributors to the excessive post injury T cell apoptosis that results in T cell elimination and a residual anergic T cell population.
Highlights.
Trauma patients’ Tcells with elevated apoptosis & CD47 expression become anergic.
Only patients with high plasma TRAIL&TSP-1 developed Tcell apoptosis then anergy.
TRAIL & TSP-1 containing patient plasma induced TRAIL apoptosis in control Tcells.
Control Tcells were sensitive to TRAIL mediated apoptosis after CD47 triggering.
CD47 triggered SHP-1’s depression of NFκB to increase Tcells’ TRAIL apoptosis.
Acknowledgments
We thank Sanjukta Bandyopadhyay and Leanne Staples for excellent technical support. This study was supported by National Institutes of Health grants R01-GM065237 and R01-GM036214.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hsieh YC, Athar M, Chaudry IH. When apoptosis meets autophagy: deciding cell fate after trauma and sepsis. Trends Mol Med. 2009;15:129–138. doi: 10.1016/j.molmed.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unsinger J, Kazama H, McDonough JS, Griffith TS, Hotchkiss RS, Ferguson TA. Sepsis-induced apoptosis leads to active suppression of delayed-type hypersensitivity by CD8+ regulatory T cells through a TRAIL-dependent mechanism. J Immunol. 2010;184:6766–6772. doi: 10.4049/jimmunol.0904054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hostmann A, Jasse K, Schulze-Tanzil G, Robinson Y, Oberholzer A, Ertel W, et al. Biphasic onset of splenic apoptosis following hemorrhagic shock: critical implications for Bax, Bcl-2, and Mcl-1 proteins. Crit Care. 2008;12:R8. doi: 10.1186/cc6772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wesche DE, Lomas-Neira JL, Perl M, Chung C-S, Ayala A. Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol. 2005;78:325–337. doi: 10.1189/jlb.0105017. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Liang L, Wu W, Gao Y, Chen ZB, Liang ZY, et al. Resuscitation with hydroxyethyl starch solution prevents CD4+ T-lymphocyte apoptosis and modulates the balance of T helper type 1 and T helper type 2 responses in the rat with traumatic virgule/shill hemorrhagic shock. Shock. 2008;30:692–698. doi: 10.1097/SHK.0b013e31816f260d. [DOI] [PubMed] [Google Scholar]
- 6.Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Ivashkiv LB. IFN-gamma abrogates endotoxin tolerance by facilitating Toll-like receptor-induced chromatin remodeling. Proc Natl Acad Sci U S A. 2010;107:19438–19443. doi: 10.1073/pnas.1007816107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wachmann K, Pop C, van Raam BJ, Drag M, Mace PD, Snipas SJ, et al. Activation and specificity of human caspase-10. Biochemistry. 2010;49:8307–8315. doi: 10.1021/bi100968m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lafont E, Milhas D, Teissie J, Therville N, Andrieu-Abadie N, Levade T, et al. Caspase-10-dependent cell death in Fas/CD95 signalling is not abrogated by caspase inhibitor zVAD-fmk. PLoS One. 2010;5:e13638. doi: 10.1371/journal.pone.0013638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engels IH, Totzke G, Fischer U, Schulze-Osthoff K, Janicke RU. Caspase-10 sensitizes breast carcinoma cells to TRAIL-induced but not tumor necrosis factor-induced apoptosis in a caspase-3-dependent manner. Mol Cell Biol. 2005;25:2808–2818. doi: 10.1128/MCB.25.7.2808-2818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oh S, Perera LP, Terabe M, Ni L, Waldmann TA, Berzofsky JA. IL-15 as a mediator of CD4+ help for CD8+ T cell longevity and avoidance of TRAIL-mediated apoptosis. Proc Natl Acad Sci U S A. 2008;105:5201–5206. doi: 10.1073/pnas.0801003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herbeuval JP, Grivel JC, Boasso A, Hardy AW, Chougnet C, Dolan MJ, et al. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood. 2005;106:3524–3531. doi: 10.1182/blood-2005-03-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morales JC, Ruiz-Magana MJ, Ruiz-Ruiz C. Regulation of the resistance to TRAIL-induced apoptosis in human primary T lymphocytes: role of NF-kappaB inhibition. Mol Immunol. 2007;44:2587–2597. doi: 10.1016/j.molimm.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 14.Manna PP, Dimitry J, Oldenborg P-A, Frazier WA. CD47 Augments Fas/CD95-mediated Apoptosis. J Biol Chem. 2005;280:29637–29644. doi: 10.1074/jbc.M500922200. [DOI] [PubMed] [Google Scholar]
- 15.Lamy L, Foussat A, Brown EJ, Bornstein P, Ticchioni M, Bernard A. Interactions between CD47 and thrombospondin reduce inflammation. J Immunol. 2007;178:5930–5939. doi: 10.4049/jimmunol.178.9.5930. [DOI] [PubMed] [Google Scholar]
- 16.Kaur S, Kuznetsova SA, Pendrak ML, Sipes JM, Romeo MJ, Li Z, et al. Heparan sulfate modification of the transmembrane receptor CD47 is necessary for inhibition of T cell receptor signaling by thrombospondin-1. J Biol Chem. 2011;286:14991–15002. doi: 10.1074/jbc.M110.179663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bandyopadhyay G, De A, Laudanski K, Li F, Lentz C, Bankey P, et al. Negative signaling contributes to T-cell anergy in trauma patients. Crit Care Med. 2007;35:794–801. doi: 10.1097/01.CCM.0000256847.61085.A5. [DOI] [PubMed] [Google Scholar]
- 18.Laudanski K, Miller-Graziano C, Xiao W, Mindrinos MN, Richards DR, De A, et al. Cell-specific expression and pathway analyses reveal alterations in trauma-related human T cell and monocyte pathways. Proc Natl Acad Sci U S A. 2006;103:15564–15569. doi: 10.1073/pnas.0607028103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornell TT, Wynn J, Shanley TP, Wheeler DS, Wong HR. Mechanisms and regulation of the gene-expression response to sepsis. Pediatrics. 2010;125:1248–1258. doi: 10.1542/peds.2009-3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, et al. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol. 2005;174:2004–2011. doi: 10.4049/jimmunol.174.4.2004. [DOI] [PubMed] [Google Scholar]
- 21.Griffith TS, Kazama H, Vanoosten RL, Earle JK, Jr, Herndon JM, Green DR, et al. Apoptotic Cells Induce Tolerance by Generating Helpless CD8+ T Cells That Produce TRAIL. J Immunol. 2007;178:2679–2687. doi: 10.4049/jimmunol.178.5.2679. [DOI] [PubMed] [Google Scholar]
- 22.Isenberg JS, Annis DS, Pendrak ML, Ptaszynska M, Frazier WA, Mosher DF, et al. Differential interactions of thrombospondin-1, -2, and -4 with CD47 and effects on cGMP signaling and ischemic injury responses. J Biol Chem. 2009;284:1116–1125. doi: 10.1074/jbc.M804860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neznanov N, Neznanova L, Kondratov RV, O’Rourke DM, Ullrich A, Gudkov AV. The ability of protein tyrosine phosphatase SHP-1 to suppress NFkappaB can be inhibited by dominant negative mutant of SIRPalpha. DNA Cell Biol. 2004;23:175–182. doi: 10.1089/104454904322964779. [DOI] [PubMed] [Google Scholar]
- 24.Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Jr, Hui JJ, Chang KC, et al. Sepsis-Induced Apoptosis Causes Progressive Profound Depletion of B and CD4+ T Lymphocytes in Humans. J Immunol. 2001;166:6952–6963. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 25.De AK, Kodys KM, Pellegrini J, Yeh B, Furse RK, Bankey P, Miller-Graziano CL. Induction of global anergy rather than inhibitory Th2 lymphokines mediates posttrauma T cell immunodepression. Clin Immunol. 2000;96:52–66. doi: 10.1006/clim.2000.4879. [DOI] [PubMed] [Google Scholar]
- 26.Vaki I, Kranidioti H, Karagianni V, Spyridaki A, Kotsaki A, Routsi C, et al. An early circulating factor in severe sepsis modulates apoptosis of monocytes and lymphocytes. J Leukoc Biol. 2011;89 doi: 10.1189/jlb.0410232. [DOI] [PubMed] [Google Scholar]
- 27.Jin CH, Chae SY, Kim TH, Yang HK, Lee EY, Song YW, et al. Effect of tumor necrosis factor-related apoptosis-inducing ligand on the reduction of joint inflammation in experimental rheumatoid arthritis. J Pharmacol Exp Ther. 2010;332:858–865. doi: 10.1124/jpet.109.159517. [DOI] [PubMed] [Google Scholar]
- 28.Bosque A, Aguilo JI, del Rey M, Paz-Artal E, Allende LM, Naval J, et al. Cell cycle regulation by FasL and Apo2L/TRAIL in human T-cell blasts. Implications for autoimmune lymphoproliferative syndromes. J Leukoc Biol. 2008;84:488–498. doi: 10.1189/jlb.0108043. [DOI] [PubMed] [Google Scholar]
- 29.Griffith TS, Brincks EL, Gurung P, Kucaba TA, Ferguson TA. Systemic Immunological Tolerance to Ocular Antigens Is Mediated by TRAIL-Expressing CD8+ T Cells. J Immunol. 2011;186:791–798. doi: 10.4049/jimmunol.1002678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nat Med. 2009;15:496–497. doi: 10.1038/nm0509-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolkers MC, Gerlach C, Arens R, Janssen EM, Fitzgerald P, Schumacher TN, et al. Nab2 regulates secondary CD8+ T-cell responses through control of TRAIL expression. Blood. 2012;119:798–804. doi: 10.1182/blood-2011-08-373910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van VQ, Darwiche J, Raymond M, Lesage S, Bouguermouh S, Rubio M, et al. Cutting edge: CD47 controls the in vivo proliferation and homeostasis of peripheral CD4+ CD25+ Foxp3+ regulatory T cells that express CD103. J Immunol. 2008;181:5204–5208. doi: 10.4049/jimmunol.181.8.5204. [DOI] [PubMed] [Google Scholar]
- 34.Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol. 2003;4:248–254. doi: 10.1038/ni895. [DOI] [PubMed] [Google Scholar]
- 35.Courtine E, Pene F, Cagnard N, Toubiana J, Fitting C, Brocheton J, et al. Critical role of cRel subunit of NF-{kappa}B in sepsis survival. Infect Immun. 2011 doi: 10.1128/IAI.00021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]