

Abstract
Determining global proteome changes is important for advancing a systems biology view of cellular processes and for discovering biomarkers. Liquid chromatography, coupled to mass spectrometry, has been widely used as a proteomics technique for discovering differentially expressed proteins in biological samples. However, although a large number of high-throughput studies have identified differentially regulated proteins, only a small fraction of these results have been reproduced and independently verified. The use of different approaches to data processing and analyses is among the factors which contribute to inconsistent conclusions. This perspective provides a comprehensive and critical overview of bioinformatics methods for commonly used mass spectrometry-based quantitative proteomics, employing both stable isotope labeling and label-free approaches. We evaluate the challenges associated with current quantitative proteomics techniques, placing particular emphasis on data analyses. The complexity of processing and interpreting proteomics datasets has become a central issue as sensitivity, mass resolution, mass accuracy and throughput of mass spectrometers have improved. A number of computer programs are available to address these challenges, and are reviewed here. We focus on approaches for signal processing, noise reduction, and methods for protein abundance estimation.
Keywords: mass spectrometry, stable-isotope labeling, protein quantification, label-free quantification, isotope distribution, mass accuracy, mass resolution
1 INTRODUCTION
In recent years numerous efforts have been devoted to making mass spectrometry based proteomics an essential technique for quantitative analyses of proteomes[1-3]. Quantification of protein differences between two or more physiological states is critical for advancing a systems biology view of disease states[4], conducting biomarker discovery[5] and understanding signal transduction pathways[6]. Classical quantification methods using dye, fluorophore, or radioactive labeling exhibit good sensitivity, linearity, and dynamic range. However, these methods suffer from two important shortcomings: they require high-resolution protein separation (normally provided by 2D gels), and they do not permit protein identification. Thousands of peptides and proteins can be identified using mass spectrometry-based proteomics and platforms are being developed to reliably quantify these proteins. However, it should be mentioned the reliable quantification requires a much higher quality peptide signal than is required for peptide identification[7], and of the large number of studies conducted, only a few have resulted in a quantitative global description of a biological system.
In one popular approach, proteins in a biological sample are digested using site-specific enzymes, e.g., trypsin. The resulting peptides are separated, based on their physico-chemical properties, by multi-dimensional liquid chromatography. The separated peptides are positively ionized in an electro-spray ionization and subsequently enter the mass spectrometer. Here, the first mass analyzer performs full mass survey scans (full MS) and records elution times, mass-to-charge (m/z) ratios and ion abundances of the intact peptides (precursors). The intact peptides are then dissociated by such techniques as collision induced (CID)-, electron capture[8]- or electron transfer dissociations[9]. A second mass analyzer measures the masses of fragment (also called product) ions (also called MS/MS or tandem MS). Further fragmentation[10] of the product ions is used to improve the chances of high confidence peptide identifications or to locate post-translational modifications. Database search engines[11-16] then use the masses of the precursors and their fragment ions to identify peptides from protein sequence databases. This approach is termed bottom-up proteomics and is the focus of this review. In the alternative, top-down approach[16,17], intact proteins are ionized and fragmented. The fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. The proteins are identified from measured masses of fragments and protein sequence databases. Mass spectra of proteins allows assigning post-translational modifications of amino acids[18]. The technique is used in studies of biomarkers and is becoming throughput[19].
Note that there is another quantitative technique, named difference in gel electrophoresis (DIGE)[20]. It is a form of gel electrophoresis where up to three different protein samples can be labeled with fluorescent dyes prior to two-dimensional electrophoresis. After the gel electrophoresis, the gel is scanned with the excitation wavelength of each dye, one after the other. This technique is used to estimate changes in protein abundance. Our focus in this work is on the quantification methods using liquid chromatography-based separation.
Quantitative proteomics is primarily applied in two contexts: targeted quantification and high-throughput quantification. In targeted quantification, experiments are set up to quantify specific proteins in a sample. Conversely in high-throughput quantification, the experiments are designed to quantify (in relative terms) all proteins in a sample. Datasets from targeted and high-throughput proteomics experiments are processed differently. A recent review[21] addresses the bioinformatics problems related to targeted quantification, as well as current approaches for their solutions. A contribution by Yingxin and colleagues in this theme issue discusses targeted quantification. In our review, we will focus on the bioinformatics methods for processing datasets from high-throughput quantitative proteomics[22]. Two main platforms have been developed and employed for these purposes: stable-isotope labeled[23-25] and label-free quantification[26-28], schematically shown in Figures 1A and, 1B respectively. In analyzing high-throughput datasets, both labeled and label-free techniques employ ratio estimation models that use statistical techniques to assign significance values to the computed ratios with the purpose of controlling error rates. However, there are some major differences between the bioinformatics solutions of the two approaches. These differences will be outlined in our discussion of ratio estimation techniques for each of the methods. We will present background information and a comparison of the bioinformatics methods. We will also emphasize several problems that still complicate routine application of quantitative proteomics methods.
Figure 1.


Quantification workflows.
A. A workflow for quantification using stable-isotope labeling. Note that in most cases, the processing step is specific to the labeling technique. For example, in SILAC experiments, labeled and unlabeled samples are mixed and the mixture is then digested by an enzyme. In enzymatic labeling experiments (18O-water labeling) the control and treatment samples are separately digested in 16O- and 18O-water, respectively. A common feature of all labeling techniques is that labeled and unlabeled species are analyzed in the same MS run. The spectrum shown in the figure is of a heavy and light peptide pair, where the heavy peptide is more abundant.
B. A workflow for label-free quantification. In the label-free approach, the control and treatment samples are processed separately, and run through LC-MS. The reconstructed ion chromatograms from different MS runs are then aligned (black and red are originals and red and blue are aligned RIC’s) and normalized before comparing peptide abundance levels in the two samples. This alignment was done using ChromAlign[87].
2 QUANTIFICATION USING STABLE-ISOTOPE LABELING
Stable-isotope labeling[29] relies on peptides incorporating specific stable heavy isotopes (deuterium, carbon-13, nitrogen-15 and oxygen-18). These isotopes are either incorporated metabolically by cells[30,31] or small organisms[32,33], by chemical[34,35] or enzymatic[36] reactions. For discussion of the advantages of various labeling methods, one is referred to other comprehensive reviews[7,29]. Stable-isotope labeling is commonly used in combination with liquid chromatography-mass spectrometry (LC-MS) to estimate differential expression of proteins in two or more samples[37]. In this method, two samples are taken and in one of the samples, the proteins/peptides are labeled using stable isotopes, Figure 1 A. Labeling changes the masses of the peptides but not their physico-chemical properties. The labeled and unlabeled samples (depending on the specifics of labeling platforms the samples could be cells, proteins or peptides) are then mixed in 1:1 ratio and the pairs of labeled and unlabeled peptides co-elute in a mass spectrometer. As long as the mass spectrometer has sufficient resolution power to separate light (unlabeled) and heavy (labeled) peptide ions or reporter product ions, relative protein ratios can be estimated from these isotopic profiles, Figure 1 A.
All stable-isotope labeling experiments share commonalities in workflow for processing datasets. Database search engines are used to identify peptides in the datasets, and quantification programs typically start with these results. Using peptide mass and elution times, these, programs generate reconstructed ion chromatograms (RIC) (also called extracted ion chromatogram, XIC), of the heavy and light peptide pairs. Areas under the reconstructed ion chromatograms, or alternatively peak heights, are used to estimate ratios of peptide pairs. Signal processing tools are used to smooth data, determine peak positions, and reduce noise. Due to the high throughput nature of data acquisition and the complexity of samples, RIC’s are affected by several artifacts. In complex samples with large numbers of peptides, some peptides co-elute and their mass profiles overlap. Noise signals and fluctuations in measurement by mass spectral analyzers can also affect ratio estimations. Some bioinformatics tools assign significance scores[38] to ratio estimations. Below, we describe specific labeling platforms and representative bioinformatics methods for analyzing their mass spectral data.
2.1 18O-WATER LABELING
The trypsin-mediated 18O-water labeling employs the protease-catalyzed exchange of 18O into the C-terminal carboxylic acid group as method for stable-isotope incorporation[36,39]. The incorporation of two 18O atoms produces a 4 Da mass shift in the peptides. Experimentally, control sample is subjected to trypsin-mediated exchange in ordinary 16O water while the test sample is subjected to trypsin-mediated exchange in 18O-water. The peptides from the two samples are then pooled and analyzed simultaneously by LC-MS. The ratios of unlabeled to labeled peptides are then determined from abundance levels of monoisotopic (no 18O atoms) and the corresponding labeled (4 Da mass shift) species.
If we denote the abundances of the unlabeled species as A, singly 18O labeled species as B1 and doubly 18O labeled species as B2, then they are related to the experimentally observed ion abundance levels via the following relationships[40]:
| (1) |
where I0, I2 and I4 are the abundance levels of the monoisotopic, second (overlapping with the monoisotopic peak of the singly 18O labeled peptide) and fourth (overlapping with the monoisotopic peak of the doubly 18O labeled peptide) isotopic peaks; and M0, M2 and M4 are the theoretical relative abundance levels of the monoisotopic, second and fourth naturally occurring isotopic peaks, respectively. The ratio of peptide pairs, R, is determined from the solutions of the above equations for A, B1 and B2:
| (2) |
The above formulae assume that the portion of the second isotopic peak not accounted for by the monoisotopic peak of the unlabeled peptide is due to a single 18O-labeled peptide.
Several algorithmic approaches for processing data sets from 18O-labeling experiments have been reported in the literature[41,42]. Publicly available ZoomQuant[43,44] software uses ratio expressions similar to those reported by Yao et al.[36,45] and Johnson et al.[46] These ratios are obtained based on the assumption that all peptides in the labeled sample have incorporated at least one 18O atom. ZoomQuant works with mass spectral data from linear ion trap mass spectrometers, LTQ, and processes search results from the database search engine, SEQUEST[11]. MassXplorer[47], too, works with data sets from low resolution ion trap mass spectrometers. It uses power spectrum analysis to filter out high-frequency noise and band-filter contaminant peaks from co-eluting peptides with differing charge states when analyzing zoom scans from linear ion traps. The filtered spectrum is back-transformed into the mass domain and used in a correlation analysis to locate the monoisotopic peak position of the unlabeled peptides. After fixing the peak position, peak shapes and peak heights are obtained by a fit using the Levenberg-Marquardt method. The ratios are computed from monoisotopic peak heights, Eq. (2).
Ramos-Fernandez et al.[48] have developed a kinetic model for 18O incorporation which uses a binomial model to describe 18O incorporation and estimates labeling efficiency from the parametric fit of mixed Gaussians and other exponential forms to zoom scans. Mason et al.[49] have used a similar binomial model to describe 18O incorporation. However, they used a multivariate regression approach which concurrently models the joint distribution of the labeled and unlabeled samples. Multivariate regression was first used in a similar problem by Mirgorodskaya et al.[50]. Their approach was developed for high mass accuracy instruments and does not require a prior knowledge of peptide sequence. An average amino acid (averagine[51]) model has also been used to approximate chemical composition from molecular mass, with labeling efficiency directly estimated from the regression model. This approach has been implemented in the program, RAAMS[49,52] which works with high mass accuracy data. A STEM algorithm to interpret proteomics experimental results has been developed by Shinkawa et al.[53]. The algorithm implements a qualitative approach for protein ratio calculations; no statistical or signal processing technique is employed.
2.2 BIOINFORMATICS METHODS FOR CHEMICAL LABELING
2.2 A Isotope Coded Affinity Tag (ICAT)
In the isotope coded affinity tag (ICAT)[34] technique, stable isotope signatures are introduced into peptides via chemical tagging of cysteins. Labeled peptides, which differ in mass by 8 Da or 9 Da are mixed and co-elute in the mass spectrometer. Li et al.[23] have developed an algorithm for automated statistical analysis of protein abundance ratios (ASAPRatio) in LC-MS/MS experiments using ICAT. Along with the abundance ratio, the algorithm also provides statistical significance (p-value) for assessing protein abundance change between two samples. The algorithm has the following basic steps.
Evaluation of abundance ratio for each identified peptide. The abundance of unlabeled and labeled peptide is estimated by the area under its RIC. The latter is checked for sufficient signal-to-noise ratio and its error is calculated to be used in the estimation of the error of the abundance ratio. Abundance ratios are calculated in this way for all present charge states of a peptide and then aggregated into a single ratio in a weighted fashion.
Evaluation of unique peptide ratio for multiple identifications of a single peptide. First, abundance ratios of peptides identified during the same elution peak are averaged to calculate an abundance ratio for the peak. Second, abundance ratios of different peaks are averaged to calculate the unique peptide ration. On each of these two steps, averaging is done with weights equal to the largest area under corresponding chromatograms.
Evaluation of protein abundance ratio for each identified protein. The unique peptide ratios are averaged with weights proportional to their errors. A graphical user interface is available for users to verify protein abundance ratios and their errors. Protein abundance ratios are assigned to all proteins having at least one identified and quantified peptide.
Evaluation of the significance of abundance change for each identified protein. This is done under assumption that a large number of identified proteins have the same abundance in the samples being analyzed. The distribution of the logarithm of all unique peptide ratios is fitted with normal distribution centered at point r0, where r0 is the most likely abundance ratio of a protein of unchanged abundance. The fitted distribution is then used to derive the probability of a protein not to change its calculated abundance in the samples being analyzed.
For each of the steps described above, Dixon’s test is used to identify and exclude outlying values whenever aggregating of three or more values is required. In step one, the Savitzky-Golay[54] smoothing filter is used in chromatogram reconstruction. The performance of the ASAPRatio program has been evaluated in two experiments[23]. In the first, five mixtures of bovine serum albumin protein were used with protein abundance ratios ranging from 1:1 to 1:100. It was shown that ASAPRatio accurately determined abundance ratios over the full dynamic range of the mass spectrometer and produced reasonable results even for more than 10-fold differences in protein abundance. In the second experiment, RNA Pol II transcription complex data was used. The abundance ratios were used to identify proteins that were enriched in the sample containing the functional complex compared to the control sample containing proteins nonspecifically binding to the DNA template. For this data, results produced by ASAPRatio have been compared to results produced by the XPRESS[55] tool. This work showed that ASAPRatio was more effective for processing and analyzing data to distinguish proteins with significant changes in abundance from those of unchanged abundance.
2.2 B Isobaric Tag for Relative and Absolute Quantification (iTRAQ)
i-Tracker[56] is a program developed for quantitative analysis of tandem MS data obtained using the isobaric tag for relative and absolute quantification, iTRAQ[35], technique. iTRAQ is a method for chemical labeling of N-terminus and side chain amines and allows for simultaneous quantification of eight different samples. After fractionation and CID, eight different reporter groups one per reagent, are observed in MS/MS at 113, 114, 115, 116, 117, 118, 119 and 121 Thompsons (Th). Quantification is based on MS/MS-dependent workflow. Only peptides that have been successfully identified from the database search are used in quantification.
i-Tracker performs relative quantification using the same peak areas of reporter ions utilized by iTRAQ. Each reporter ion peak is assumed to be comprised of a series of ions within the user-specified window of its m/z value. The area of a reporter ion peak is calculated as a sum of areas of trapezoids formed between captured peaks within that m/z window. The purity of iTRAQ reagents supplied by ABI is routinely characterized by the percentages of the reporter ions differing by -2, -1, 1, 2 Da from the nominal mass values due to isotopes. This information can be used by the i-Tracker algorithm to correct reporter ion peak areas. A user can specify thresholds for minimum acceptable reporter ion peak intensity and ion count to be used by the algorithm. i-Tracker accepts input data in dta (SEQUEST) and mgf (MASCOT) formats and generates output in comma-separated values (CSV) format. In a comparison using Arabidopsis membrane protein samples, quantification results obtained using i-Tracker were found to be in agreement with results obtained using ProQuant (commercial software distributed by ABI)[56], indicating robust performance.
Other published software for analyzing iTRAQ datasets are iTRAQ reporter ion counter[57], Multi-Q[58] and Libra[59]. These programs work with spectra in dta and mgf file formats. Libra is a part of Trans Proteomic Pipeline (TPP)[59] - a collection of integrated proteomics software tools freely distributed by the Institute for Systems Biology, Seattle, WA.
2.3 BIOINFORMATICS METHODS FOR METABOLIC LABELING
2.3 A Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC)
Jurgen Cox et al.[60] describe MaxQuant, an integrated suite of algorithms for protein quantification specifically designed to take advantage of high-resolution MS data generated by experiments using SILAC labeling. SILAC incorporates stable isotopes into cellular proteins by metabolic incorporation of labeled amino acids in cell culture. Global proteome profiles are obtained by fragmentation and LC-MS/MS analysis. MaxQuant analyzes the data in the following steps
Calculation of peptide masses. Two dimensional (2D) peaks are reconstructed on MS spectra by fitting Gaussian curves, and are then assembled into 3D peak hills over the m/z – retention time plane. The mass of a peptide is calculated as the intensity-weighted average of all MS peak centroids in the 3D peaks within the isotope patterns belonging to a SILAC pair or triplet. Mass precision is calculated for each 3D peak by bootstrap replication. By integrating multiple mass measurements and correcting for linear and nonlinear mass offsets, the accuracy of peptide mass estimation is in the parts per billion (ppb) range, sixfold better than other methods.
Determination of isotopic peaks. A graph-theoretical approach is used to detect isotope clusters of 3D peaks. First, an undirected graph is constructed from the 3D peaks. An edge is inserted between two peaks if the difference in their masses is equal to the difference in isotope masses of an averigine, within a given mass accuracy. The longest subgraph that is consistent in terms of charge states of the peaks is then determined. This approach allows one to distinguish between overlapping isotope patterns caused by different charge states and very similar masses of co-eluting peptides. Isotope-labeled (SILAC) peptide pairs are identified using correlation analysis of their intensities, as well as the difference between measured and theoretical isotope patterns. This is done under assumption that there may be at most three labeled amino acids per peptide.
Peptide and protein identification. For each spectrum that has been associated with its SILAC counterpart, Mascot database search results are filtered using information about the number of arginines and lysines in the sequence, derived from isotopic patterns known beforehand. To determine a list of identified peptides for a given FDR, all peptide candidates are sorted according to their posterior error probability (PEP), which is estimated based on a search against a database with forward and reversed sequences. Peptides are accepted until the specified fraction of reverse hits / forward hits is accumulated, starting with the peptides with the smallest PEP. Peptide hits are then assembled into protein hits. Each protein is assigned a PEP by multiplying PEPs of its peptides. A list of identified proteins with a given FDR is produced similar to the list of peptides. A protein may then be quantified based on its unique peptides, or using all of its peptides.
Protein quantification. Protein ratios are calculated as the median of all SILAC peptide ratios, and are supplied with significance scores.
MaxQuant has been used to automatically quantify more than 5,000 proteins in the mouse stem cell proteome, and has also been used to quantify several other proteomes in similar depth. However, in experiments using SILAC, argentine-to-proline conversion may lead to inaccurate ratio estimations[61]. Park and colleagues have suggested a method to account for this effect[62].
Bakalarski[63] et al. have investigated signal-to-noise ratio (S/N) characteristics of MS data obtained from complex mixtures, and how they affect the accuracy of peptide quantification. Their study was conducted using test mixtures prepared from Jurkat cell lysate and labeled using SILAC. Hybrid linear ion trap / Fourier transform ion cyclotron resonance (FT-ICR) and Orbitrap spectrometers were used for MS analyses and peptide quantification and S/N calculation were performed using the automated software suite Vista, (http://vista.hms.harvard.edu/). The S/N of a peptide’s chromatogram is defined as the ratio of the maximum peak intensity to the noise level. Noise level is estimated by the median intensity observed in the local vicinity of the peak. It was shown that the vast majority of identified peptides have chromatograms with low S/N (less than 10), and that there is an association between S/N level and accuracy of quantification. For example, the correlation coefficient for the standard deviation of ratio values and their S/N level was −0.67 for a 1:1 mixture. For the same mixture, the standard deviation of ratio values is 0.22 for S/N > 10, and 0.83 for S/N < 10.
A mass precision algorithm to distinguish signals from noise and interfering peaks has been also proposed in that study. The algorithm is based on the observation that the difference between masses of true light and heavy peptide peaks (mass precision) is more consistent than the difference between species’ theoretical and spectral masses (mass accuracy). The algorithm was shown to decrease the standard deviation of ratio values especially for spectra with low S/N level. For example, in a 1:1 mixture, the standard deviation decreased from 0.83 to 0.47 for spectra with S/N < 10.
The authors of the study describe a scoring method to assess the credibility of peptide quantification results. The method is based on a random forest classifier and the heuristic score built using 47 features of MS spectra, including S/N level, number of observations across the chromatographic peak, mass accuracy statistics, and unlabeled/labeled pair co-elution. The method has been used in a study of metastatic prostate cancer[64], as and substrate profiling of DNA repair kinases[65,66].
Bakalarski et al. suggested using S/N levels to estimate abundance ratios of those peptides for which only one type of species (labeled or unlabeled) is present on the collected mass spectra. This may happen when signal intensity of a certain species does not reach the detection threshold of the instrument, due to its low abundance in the corresponding sample. While in this instance it is not possible to calculate abundance ratios based on the area or height of the light and heavy peaks, the S/N of the observed chromatographic peak can alternatively be used to estimate the abundance ratio. For example, if an upregulated protein is present in a mixture in 100-fold excess of its basal state and is detected with a S/N level of 50, the signal generated from its basal counterpart will not be distinguishable. Nevertheless, it is clear that in this case the abundance ratio between the species in this case must be at least 50-fold. S/N characteristics and quantitative performance of Orbitrap and FT-ICR mass spectrometers have been compared and found to be quite similar.
2.3 B 15N-Labeling
Metabolic labeling by 15N-enriched amino acids was the first labeling technique used in quantitative proteomics[32]. The technique has been applied to study differential protein expression in yeast[67], mouse brain[33], human tissues[31] and bacteria[68,69]. To calculate peptide ratios in 15N-labeling experiments, MacCoss et al. developed a correlation based algorithm, named RelEx[24],. Intensity dependencies of heavy and light peptide pairs are assumed to be linear and are fit to a straight line via a correlation algorithm. The line with the minimum residual error is obtained, and the slope of the fitted line is an estimate of the peptide abundance ratio. Pan et al. expanded this approach further to assign a “score” to a ratio estimation[69]. They used parallel paired covariance algorithm for peak detection. A data matrix is created from the measured ion intensities of the heavy and light peptides, and principal component analysis of the peak profile is used for ratio estimation and to score the ratio with respect to the S/N profile. It was shown that the profile S/N ratio is inversely correlated with variability and bias of peptide abundance ratios. When peptide ratios were assembled into a protein ratio using maximum likelihood point estimation, it was shown that this approach is more accurate than averaging ratios[68]. The accuracy of the point estimation and the precision and confidence level of the interval estimation were benchmarked on standard mixtures of labeled proteomes. This approach has been implemented in the algorithm, ProRata[68].
In the above description, we grouped the bioinformatics methods by specific quantification workflows. However, it should be noted that many of the algorithms can be used for ICAT, SILAC and metabolic labeling. They only need to extract ion chromatograms based on mass specificity of different labeling methods and they often use the same downstream data processing techniques. For example, ASAPratio, MaxQuant, RelEx and ProRata can be used for any of these labeling strategies. Our grouping is only “historical”, meaning it is based on the original workflows for which the programs were developed.
3 METHODS FOR LABEL-FREE QUANTIFICATION
3.1 Spectral Counting
Spectral counting of peptides has been shown to be informative for semi-quantitative estimations of protein concentration[26,70] in shotgun[71,72] proteomics. This strategy is based on the notion that the MS/MS scanning rate of a particular peptide signal is related to its abundance level. Peptide spectral counts are integrated into a protein abundance index[73,74] to estimate protein abundance levels by dividing the number of observed peptides by the number of all possible tryptic peptides (that fit the mass range of a mass spectrometer) from a protein.
Spectral counting has been used to estimate protein ratios in both labeling[67] and label-free[75] approaches, and for both relative[67] and absolute[28] quantification. However, this method has mostly been applied to label-free quantification datasets collected on QSTART, LCQ and LTQ ion-trap instruments.
One problem with spectral counting is that it strongly depends on the quality of database identifications. Spectral counts can be influenced by poor peptide fragmentation, wrong database identification and poor reproducibility of LC-MS-MS/MS experiments, and selection criteria used for generating peptide lists. Also, peptide sequences that are common between different proteins create ambiguity when assembling them into protein indices. Errors in peptide identification propagate into quantifications of protein abundance. Some of these problems have been addressed by incorporating database score dependence into spectral counting[76,77].
Protein length and the number of potential peptides are important factors in using spectral counts for protein quantification. To account for protein length, Washburn and colleagues have introduced a normalized spectral abundance factor (NSAF)[78]. NSAF averages spectral counts for a protein by the protein’s length. It has been shown that the variance of NSAF is dependent on its average following a power law. This allows application of a power law global error model (PLGEM)[79] to analyze statistical significance of the computed protein ratios.
Marcotte et al.[28] introduced normalization of the spectral counts with respect to the expected number of unique peptides for a protein. This normalization is intended to account for different ionization efficiency, solvent conditions, and other factors that affect peptide fragmentation in the mass spectrometer. The method has been implemented in an algorithm, APEX[27]. The method exploits proportionality between two quantities: the theoretically expected fraction of all unique peptides in a tryptic mixture, contributed by a given protein, and the observed fraction of all mass spectra, contributed by the same protein. This proportionality allows one to estimate the number of molecules of a given protein in the mixture being analyzed. A suggested protein expression index uses the probability of correct protein identification calculated by ProteinProphet[80-82] and the expected number of unique peptides to be observed for a given protein. The latter can be roughly estimated by the number of possible tryptic peptides of the protein that fall within the given mass/charge window. A more accurate estimate of this number can be obtained using a machine learning approach. In this approach, a forest of random decision trees is trained to predict whether a tryptic peptide will be observed in a proteomic experiment. Training was done on 4,023 tryptic peptides derived from 40 of the most abundant yeast proteins; 714 of those peptides have been observed in the proteomic experiment conducted by the authors. Each peptide is described by 22 features: its length, molecular weight, and frequencies of the 20 amino acids. The reported accuracy of the classifier on the training set, for two classes, was 86%. Use of the machine learning approach to estimate the expected number of unique tryptic peptides of a protein was shown to improve estimations of protein abundance by 30%[27]. Measurements produced by the APEX method have been validated in a number of experiments related to absolute and relative quantification of proteins. These results are consistent with those obtained by western blotting, flow cytometry, and 2D gel electrophoresis.
3.2 LABEL-FREE QUANTIFICATION VIA RIC
A different technique for peptide quantification uses reconstructed ion chromatograms, RICs, rather than spectral counting. Reconstructed ion chromatogram describes ion intensity at a given mass-to-charge value over time. The use of the RICs reduces dependence on reproducible, consistent peptide identification. Often, only one identification and one elution time are sufficient to reconstruct the ion chromatogram of a peptide. The area under the RIC is used as a measure of peptide abundance level. Higgs et al.[83,84] have developed a set of algorithms for processing LC-MS datasets for label free quantification. An important element of these algorithms is the time alignment of RICs between different MS runs. Peptide elution times can change between experiments, due to fluctuations in temperature and column aging[85]. Higgs et al. used curve fitting to obtain time warping functions. Other approaches have used dynamic programming to globally optimize pair-wise correlations between full survey scans of different MS runs[86,87]. Higgs et al. developed their algorithm in the R[88] environment for statistical computing. Another approach for analyzing datasets from LCQ (a three-dimensional ion trap mass spectrometer) instruments is Serac[89]. This program computes peptide ratios from both reconstructed ion chromatograms and spectral counts.
High mass accuracy and resolution mass spectrometers are the best choice platforms for label-free quantification. High mass resolution allows for better separation of co-eluting peptides, while high mass accuracy improves the confidence of peptide identifications made from databases. Label-free strategies are applied using Orbitrap[90], LTQ-FT[91] and time-of-flight mass analyzers. The high mass accuracy facilitates peak detection and extraction of ion chromatograms. Developed for LTQ FT mass spectrometers, Quoil[92] analyzes high mass accuracy data to normalize ion intensities across chromatographic runs.
High mass accuracy and resolution allow the application of workflows using pattern matching[93,94]. Smith and co-workers have shown the power of using high mass accuracy data from FTICR-MS for high throughput pattern-based processing[95,96]. The accurate mass and time tag (AMT) approach capitalizes on the fact that multiple peptide species are highly unlikely to have both the same mass and LC retention time, especially in relatively simple genomes. A database consisting of sequence, mass, and retention time is built from multiple experiments. The database is then used to assign the identity of an LC-MS peak using AMT tag.
Another pattern matching approach has been implemented in PEPPer[94], a platform of several algorithms that attempt to address the problems associated with chromatographic reproducibility and to reduce reliance on identification-based quantitative methods. The algorithms use data derived from disparate data acquisition strategies to perform timed base-independent propagation of peptide identities onto accurate mass LC-MS features. Identical species across multiple LC-MS experiments are recognized by identity-independent clustering. Successful application of PEPPer was demonstrated in a study of complex biological mixtures[94].
In conclusion, there are a number quantification tools that are based on spectral counting or RIC. These are valuable strategies for analyses of low to medium resolution mass spectral datasets. These quantification approaches attempt to address time[86,87,97-99] or peptide[100-103] alignments, normalizations[104-106] of ion signals between different MS runs due to fluctuations in mass spectrometer measurements, and the stochastic nature of MS/MS sampling[107]. However, as technology is still developing and consistent sampling of low abundance proteins remains a challenge, bioinformatics approaches that can accurately estimate the error rates of ratio estimations would be very useful.
4 SIGNIFICANCE OF RATIO ESTIMATION
Figure 2 outlines the structure of data obtained in typical proteomic studies. A study includes one or several experiments, each of which produces data about a protein sample obtained in an LC-MS-MS/MS run. Mass spectra produced in a single experiment are associated with peptides, and the latter are associated with proteins. Thus, a set of proteins is identified in one experiment, represented by a set of peptides. If two experiments are concerned with two samples taken from the same mixture of proteins, they may produce varying results. That is, there may be proteins and peptides identified in one experiment, that are not identified in another experiment (usually these are low-abundant species), and abundance ratios calculated for common species in separate experiments may be different.
Figure 2.
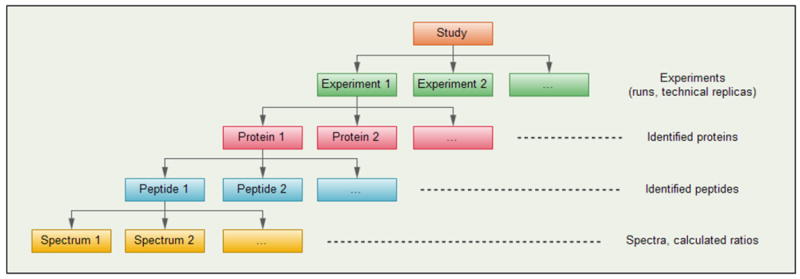
Common structures of the data obtained in quantitative proteomics. Top-down approach: a specific mixture of proteins is analyzed in each experiment (run), each of which produces a specific set of identified proteins and peptides, where each peptide is represented by a set of spectra. Bottom-up: ratios calculated at the level of spectra are grouped by peptides and proteins, and are related to the specific mixture analyzed in a given experiment.
Four types of variations have been associated with ratio measurements in quantitative proteomics experiments[7,108]: biological (variances due to fluctuations between individual biological subjects), instrumental (variations due to fluctuations in mass, abundance and time measurements), processing variations (variations due to processing steps, such as digestion, labeling and mixing) and treatment (actual differences between the treatment and control samples due to treatments). Careful experimental design should be used to minimize artifacts adversely affecting ratio estimations[109]. Statistical tests assign significance levels to ratio estimations and help to control error rates.
There are several potential strategies for dealing with data obtained from multiple experiments. One might choose to analyze data separately within each experiment, since there is a bias in the data specific to each experiment. On the other hand, it is advantageous to be able to combine data from different experiments to increase sample size and allow for more reliable analysis. Data from different experiments can be combined using data normalization as done, for example, in work by Wang et al.[110]. Another way is to use methods based on analysis of variance (ANOVA). ANOVA provides a very efficient way to combine data from different sources while taking into account specific types of biases presented in the data. In proteomic applications, ANOVA allows one to determine protein abundance (abundance ratio) related to biological phenomena, while ruling out biases caused by technical and instrumental factors[111-117].
In protein quantification, ANOVA is generally used in the following way. A variable of interest, measured on different spectra, for different peptides, proteins, and subsets of the data (see Figure 2) is modeled as a sum of predictors, associated with relevant levels of data hierarchy, and a random error term. For example, ion peak intensity y(i, j, k) can be modeled as
| (3) |
where α(i) is a portion of the intensity defined by peptide i, i =1, 2, …, I; β(j) is a portion defined by experiment j, j = 1, 2, …, J; γ(i, j) is a portion defined by peptide i in experiment j (interaction term), and ε(i, j, k) is a random error for peptide i, experiment j and spectrum k, k =1, 2, …, K(i, j). The outcome variable (left part of equation (3)) and predictors, also called explanatory variables (right part of equation (3)), are defined by experimental design and the goal of study.
An ANOVA model can be used to test whether factors accounted for in the model introduce a bias in the peak intensity measurements. For example, the model in Eq. (3) can be used to test whether β(1) = β(2) = … = β(J), that is, whether given experiments do not introduce a bias in ion peak intensity measurements. If different experiments correspond to different treatment groups, this can answer the question whether proteins are being differently expressed between these groups.
ANOVA requires that data meet several assumptions. First, samples in each group must be independent and normally distributed. Second, variances of groups must be equal. In the case of model (3), these assumptions must hold for I groups {y(i, ., .)}, J groups [118], and K(i, j) groups {y(i, j, .)}, for every i, j. It should be noted that ANOVA is known to be robust to moderate deviations from these assumptions.
ANOVA also places another requirement on the data: an observed variable should be present throughout each level of data hierarchy. For example, in model (3) ion peak intensity y for peptide i should be defined for each experiment j. In practice, however, certain peptides may not be observed in all experiments. In this case, we should either exclude peptide i from analysis, or somehow impute its missing intensities (on imputing missing values see, for example, papers by Karpievitch et al.[119] and Wang et al.[110]).
Hill et al.[120] used an ANOVA approach for the analysis of iTRAQ reporter ion peak areas. They give a rigorous description of a model that accounts for most major sources of variability in peak areas, including different proteins, peptides, conditions, experiments, and tags. The model allows investigators to obtain abundance ratio estimates and their confidence intervals. The model has been used to analyze two test mixtures of eight proteins in separate iTRAQ experiments conducted on MALDI TOF mass spectrometer. Calculated ratios were found to be consistent with the ratios obtained using ABI’s GPS software, and ANOVA-based results may seem to be more straightforward for interpretation. The authors also discus differences between their approach and that proposed by Keshamouni et al.[121], who used a model based on logarithms of peptide ratios instead of ion peak intensities, coupled with averaging of replicates to produce a single measurement.
Oberg et al.[122] extended the approach presented by Hill et al.[123] to analyze data from six iTRAQ experiments comparing serum protein profiles in patients with three histologic subtypes of acute cardiomyopathy. The authors describe strategies to overcome computational difficulties arising from large number of proteins and peptides identified in the experiments, and ways to deal with missing values.
Karpievitch et al.[124] describe a statistical model which relates the logarithm of peak intensity with effects attributable to proteins, peptides, and groups of subjects. The model was applied to experiments utilizing AMT strategy and LC-FTICR mass spectrometry, although the proposed methodology is also applicable to labeling experiments. In this model, parameters are estimated using the maximum likelihood method. The model also performs data filtering and imputes missing values. Proteins for which no collection of peptides can produce an identifiable model, with non-zero information matrix determinant, are filtered out. After that, a greedy search algorithm is used to select a subset of peptides for each remaining protein, providing optimal information content, and filtering out the rest peptides. Missing values are imputed by random numbers generated using the estimated model. The model has been applied to simulated data, human serum samples from healthy and Type 1 diabetes subjects, and salmonella mutant virulence data.
5 PERSPECTIVE
Methods for quantitative proteomics will continue to evolve and improve the reliability of ratio estimations. This relates to both experimental techniques and their analyses. Key to this evolution will be advances in separation science. Currently, many spectra exhibit co-elutions, where two or more different species elute at the same time and m/z ratio. These co-elutions are a major challenge to estimating ratios, as they distort natural isotopic profiles. Development of improved separation techniques will result in mass profiles with better resolution and will improve the accuracy of computed ratios. Quantitative proteomics will also be positively affected as mass spectrometers advance to improve mass accuracy, resolution, sensitivity and acquisition speed. Improved mass accuracy and resolution will reduce ambiguity in determining peak positions. In combination with confident identification of peptides from database searches, higher mass accuracy and resolution will result in improved reliability of ratio estimations. Increases in the sensitivity of mass spectrometers will also help to increase the dynamic range of proteome that can be sampled in experiments. Often, only high-to-medium abundance proteins are analyzed. Many low-abundant proteins are not identified and quantified. With increased sensitivity, it will be possible to analyze peptides from even these low abundance proteins. Shorter acquisition times will translate into more interrogated peptides and more proteins being identified. In theory, shorter acquisition times should also improve the reproducibility of proteomics experiments.
Tools for bioinformatics will continue to evolve to take advantage of mass informatics features and their use in specific applications (both labeling and label-free). Sophisticated algorithms will be developed for processing high-mass accuracy data with statistical and machine learning techniques for controlling error. There are several concerns related to the application of bioinformatics tools to data from labeling experiments. Among these are: peak detection, noise filtering, background subtraction, accounting for fluctuations in measurements of spectral features and saturation of signal. In the case of label-free quantification, bioinformatics tools must also address signal normalization between different chromatographic runs. We suggest that isotope labeling techniques will provide better accuracy of ratios, especially for low-abundance species. However, there are some situations where labeling is not feasible (for example, with human samples) and label-free techniques will provide a means of estimating ratios in these situations.
The Human Proteome Organization’s Proteomics Standard Initiative[125] is improving processing, storage, visualization and dissemination of experimental results by developing standard file formats for spectra (mzML), database search results (mzIndentML) and quantitative data (mzQuantML). This standardization will also improve integration of other tools being developed for such similar purposes as peak detection and noise filtering.
6 SUMMARY
A multitude of methods have been developed to allow use of mass spectrometry-based proteomics as an essential technique for large scale protein identification and quantification. Improvements in labeling, peptide/protein separations, mass accuracy and resolution provide an optimistic perspective for this approach. In this work, we have reviewed a large number of bioinformatics tools available for processing datasets from diverse quantification workflows. Common features of these bioinformatics approaches are the use of high mass accuracy and resolution information, improved signal processing for noise reduction and application of statistical tests to control error rates in ratio estimations. It is believed that the continued improvement computational methods for accurate data processing will be essential for successful applications of quantitative proteomics platforms.
Acknowledgments
RGS acknowledges fruitful discussions with Drs. A. Brasier, A. Kurosky, J. E. Wiktorowicz and Y. Zhao.
ABBREVIATIONS
- Da
Dalton
- FDR
false discovery rate
- H
heavy
- L
light
- LC-MS
liquid chromatography - mass spectrometry
- LTQ
Thermo Fisher Scientific linear quadrupole ion trap
- MS/MS
tandem mass spectrometry
- m/z
mass-to-charge ratio
- S/N
signal-to-noise ratio
- Th
the Thomson unit of m/z
- RIC
reconstructed ion chromatogram
- NSAF
normalized spectral abundance factor
- ICAT
Isotope Coded Affinity Tag
- iTRAQ
Isobaric Tag for Relative and Absolute Quantification
- SILAC
Stable Isotope Labeling with Amino Acids in Cell Culture
- PEP
posterior error probability
- LCQ
Thermo Fisher Scientific three-dimensional quadrupole ion trap
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- 3.Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol Biol. 2007;359:37–52. doi: 10.1007/978-1-59745-255-7_3. [DOI] [PubMed] [Google Scholar]
- 4.Hwang D, Zhang N, Lee H, Yi E, Zhang H, Lee IY, Hood L, Aebersold R. MS-BID: a Java package for label-free LC-MS based comparative proteomic analysis. Bioinformatics. 2008 doi: 10.1093/bioinformatics/btn491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson NL, Anderson NG, Pearson TW, Borchers CH, Paulovich AG, Patterson SD, Gillette M, Aebersold R, Carr SA. A human proteome detection and quantitation project. Mol Cell Proteomics. 2009;8:883–86. doi: 10.1074/mcp.R800015-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhai B, Villen J, Beausoleil SA, Mintseris J, Gygi SP. Phosphoproteome analysis of Drosophila melanogaster embryos. J Proteome Res. 2008;7:1675–82. doi: 10.1021/pr700696a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007;389:1017–31. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 8.McLafferty FW, Horn DM, Breuker K, Ge Y, Lewis MA, Cerda B, Zubarev RA, Carpenter BK. Electron capture dissociation of gaseous multiply charged ions by Fourier-transform ion cyclotron resonance. J Am Soc Mass Spectrom. 2001;12:245–49. doi: 10.1016/S1044-0305(00)00223-3. [DOI] [PubMed] [Google Scholar]
- 9.Coon JJ, Shabanowitz J, Hunt DF, Syka JE. Electron transfer dissociation of peptide anions. J Am Soc Mass Spectrom. 2005;16:880–82. doi: 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Macek B, Waanders LF, Olsen JV, Mann M. Top-down protein sequencing and MS3 on a hybrid linear quadrupole ion trap-orbitrap mass spectrometer. Mol Cell Proteomics. 2006;5:949–58. doi: 10.1074/mcp.T500042-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Eng JK, McCormack AL, Yates JR., III An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J Am Soc Mass Spectrom. 1994;5:976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 12.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 13.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–67. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 14.Sadygov RG, Good DM, Swaney DL, Coon JJ. A new probabilistic database search algorithm for ETD spectra. J Proteome Res. 2009;8:3198–205. doi: 10.1021/pr900153b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanner S, Shu H, Frank A, Wang LC, Zandi E, Mumby M, Pevzner PA, Bafna V. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem. 2005;77:4626–39. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 16.Meng F, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat Biotechnol. 2001;19:952–57. doi: 10.1038/nbt1001-952. [DOI] [PubMed] [Google Scholar]
- 17.Parks BA, Jiang L, Thomas PM, Wenger CD, Roth MJ, Boyne MT, Burke PV, Kwast KE, Kelleher NL. Top-down proteomics on a chromatographic time scale using linear ion trap fourier transform hybrid mass spectrometers. Anal Chem. 2007;79:7984–91. doi: 10.1021/ac070553t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zamdborg L, Leduc RD, Glowacz KJ, Kim YB, Viswanathan V, Spaulding IT, Early BP, Bluhm EJ, Babai S, Kelleher NL. ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic Acids Res. 2007;35:W701–W706. doi: 10.1093/nar/gkm371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kellie JF, Tran JC, Lee JE, Ahlf DR, Thomas HM, Ntai I, Catherman AD, Durbin KR, Zamdborg L, Vellaichamy A, Thomas PM, Kelleher NL. The emerging process of Top Down mass spectrometry for protein analysis: biomarkers, protein-therapeutics, and achieving high throughput. Mol Biosyst. 2010;6:1532–39. doi: 10.1039/c000896f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alban A, David SO, Bjorkesten L, Andersson C, Sloge E, Lewis S, Currie I. A novel experimental design for comparative two-dimensional gel analysis: two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics. 2003;3:36–44. doi: 10.1002/pmic.200390006. [DOI] [PubMed] [Google Scholar]
- 21.Cham MJ, Bianco L, Bessant C. Free computational resources for designing selected reaction monitoring transitions. Proteomics. 2010;10:1106–26. doi: 10.1002/pmic.200900396. [DOI] [PubMed] [Google Scholar]
- 22.Lu B, Xu T, Park SK, McClatchy DB, Liao L, Yates JR., III Shotgun protein identification and quantification by mass spectrometry in neuroproteomics. Methods Mol Biol. 2009;566:229–59. doi: 10.1007/978-1-59745-562-6_16. [DOI] [PubMed] [Google Scholar]
- 23.Li XJ, Zhang H, Ranish JA, Aebersold R. Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal Chem. 2003;75:6648–57. doi: 10.1021/ac034633i. [DOI] [PubMed] [Google Scholar]
- 24.MacCoss MJ, Wu CC, Liu H, Sadygov R, Yates JR., III A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal Chem. 2003;75:6912–21. doi: 10.1021/ac034790h. [DOI] [PubMed] [Google Scholar]
- 25.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, Sadygov RG, Yates JR., III A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 27.Braisted JC, Kuntumalla S, Vogel C, Marcotte EM, Rodrigues AR, Wang R, Huang ST, Ferlanti ES, Saeed AI, Fleischmann RD, Peterson SN, Pieper R. The APEX Quantitative Proteomics Tool: generating protein quantitation estimates from LC-MS/MS proteomics results. BMC Bioinformatics. 2008;9:529. doi: 10.1186/1471-2105-9-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vogel C, Marcotte EM. Calculating absolute and relative protein abundance from mass spectrometry-based protein expression data. Nat Protoc. 2008;3:1444–51. doi: 10.1038/nport.2008.132. [DOI] [PubMed] [Google Scholar]
- 29.Gevaert K, Impens F, Ghesquiere B, Van DP, Lambrechts A, Vandekerckhove J. Stable isotopic labeling in proteomics. Proteomics. 2008;8:4873–85. doi: 10.1002/pmic.200800421. [DOI] [PubMed] [Google Scholar]
- 30.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 31.McClatchy DB, Dong MQ, Wu CC, Venable JD, Yates JR., III 15N metabolic labeling of mammalian tissue with slow protein turnover. J Proteome Res. 2007;6:2005–10. doi: 10.1021/pr060599n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci U S A. 1999;96:6591–96. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu CC, MacCoss MJ, Howell KE, Matthews DE, Yates JR., III Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal Chem. 2004;76:4951–59. doi: 10.1021/ac049208j. [DOI] [PubMed] [Google Scholar]
- 34.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–99. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 35.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836–42. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 37.Lill J. Proteomic tools for quantitation by mass spectrometry. Mass Spectrom Rev. 2003;22:182–94. doi: 10.1002/mas.10048. [DOI] [PubMed] [Google Scholar]
- 38.Bakalarski CE, Elias JE, Villen J, Haas W, Gerber SA, Everley PA, Gygi SP. The Impact of Peptide Abundance and Dynamic Range on Stable-Isotope-Based Quantitative Proteomic Analyses. Journal of Proteome Research. 2008;7:4756–65. doi: 10.1021/pr800333e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zang L, Palmer TD, Hancock WS, Sgroi DC, Karger BL. Proteomic analysis of ductal carcinoma of the breast using laser capture microdissection, LC-MS, and 16O/18O isotopic labeling. J Proteome Res. 2004;3:604–12. doi: 10.1021/pr034131l. [DOI] [PubMed] [Google Scholar]
- 40.Yao X, Afonso C, Fenselau C. Dissection of proteolytic 18O labeling: endoprotease-catalyzed 16O-to-18O exchange of truncated peptide substrates. J Proteome Res. 2003;2:147–52. doi: 10.1021/pr025572s. [DOI] [PubMed] [Google Scholar]
- 41.Mueller LN, Brusniak MY, Mani DR, Aebersold R. An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J Proteome Res. 2008;7:51–61. doi: 10.1021/pr700758r. [DOI] [PubMed] [Google Scholar]
- 42.Miyagi M, Rao KC. Proteolytic 18O-labeling strategies for quantitative proteomics. Mass Spectrom Rev. 2007;26:121–36. doi: 10.1002/mas.20116. [DOI] [PubMed] [Google Scholar]
- 43.Halligan BD, Slyper RY, Twigger SN, Hicks W, Olivier M, Greene AS. ZoomQuant: an application for the quantitation of stable isotope labeled peptides. J Am Soc Mass Spectrom. 2005;16:302–06. doi: 10.1016/j.jasms.2004.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hicks WA, Halligan BD, Slyper RY, Twigger SN, Greene AS, Olivier M. Simultaneous quantification and identification using 18O labeling with an ion trap mass spectrometer and the analysis software application “ZoomQuant”. J Am Soc Mass Spectrom. 2005;16:916–25. doi: 10.1016/j.jasms.2005.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reynolds KJ, Yao X, Fenselau C. Proteolytic 18O labeling for comparative proteomics: evaluation of endoprotease Glu-C as the catalytic agent. J Proteome Res. 2002;1:27–33. doi: 10.1021/pr0100016. [DOI] [PubMed] [Google Scholar]
- 46.Johnson KL, Muddiman DC. A method for calculating 16O/18O peptide ion ratios for the relative quantification of proteomes. J Am Soc Mass Spectrom. 2004;15:437–45. doi: 10.1016/j.jasms.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 47.Sadygov RG, Zhao Y, Haidacher SJ, Starkey JM, Tilton RG, Denner L. Using power spectrum analysis to evaluate (18)O-water labeling data acquired from low resolution mass spectrometers. J Proteome Res. 2010;9:4306–12. doi: 10.1021/pr100642q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramos-Fernandez A, Lopez-Ferrer D, Vazquez J. Improved method for differential expression proteomics using trypsin-catalyzed 18O labeling with a correction for labeling efficiency. Mol Cell Proteomics. 2007;6:1274–86. doi: 10.1074/mcp.T600029-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Mason CJ, Therneau TM, Eckel-Passow JE, Johnson KL, Oberg AL, Olson JE, Nair KS, Muddiman DC, Bergen HR., III A method for automatically interpreting mass spectra of 18O-labeled isotopic clusters. Mol Cell Proteomics. 2007;6:305–18. doi: 10.1074/mcp.M600148-MCP200. [DOI] [PubMed] [Google Scholar]
- 50.Mirgorodskaya OA, Kozmin YP, Titov MI, Korner R, Sonksen CP, Roepstorff P. Quantitation of peptides and proteins by matrix-assisted laser desorption/ionization mass spectrometry using (18)O-labeled internal standards. Rapid Commun Mass Spectrom. 2000;14:1226–32. doi: 10.1002/1097-0231(20000730)14:14<1226::AID-RCM14>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 51.Senko MW, Beu SC, McLafferty FW. Determination of Monoisotopic Massed and Ion Populations for Large Biomolecules from Resolved Isotopic Distributions. J Am Soc Mass Spectrom. 1995;6:229–33. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 52.Eckel-Passow JE, Oberg AL, Therneau TM, Mason CJ, Mahoney DW, Johnson KL, Olson JE, Bergen HR., III Regression analysis for comparing protein samples with 16O/18O stable-isotope labeled mass spectrometry. Bioinformatics. 2006;22:2739–45. doi: 10.1093/bioinformatics/btl464. [DOI] [PubMed] [Google Scholar]
- 53.Shinkawa T, Taoka M, Yamauchi Y, Ichimura T, Kaji H, Takahashi N, Isobe T. STEM: a software tool for large-scale proteomic data analyses. J Proteome Res. 2005;4:1826–31. doi: 10.1021/pr050167x. [DOI] [PubMed] [Google Scholar]
- 54.Savitzky A, Golay MJE. Smoothing and Differentiating of Data by Simplified Least Squares Procedures. Analytical Chemistry. 1964;36:1627–39. [Google Scholar]
- 55.Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–51. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shadforth IP, Dunkley TP, Lilley KS, Bessant C. i-Tracker: for quantitative proteomics using iTRAQ. BMC Genomics. 2005;6:145. doi: 10.1186/1471-2164-6-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Griffin TJ, Xie H, Bandhakavi S, Popko J, Mohan A, Carlis JV, Higgins L. iTRAQ reagent-based quantitative proteomic analysis on a linear ion trap mass spectrometer. J Proteome Res. 2007;6:4200–09. doi: 10.1021/pr070291b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin WT, Hung WN, Yian YH, Wu KP, Han CL, Chen YR, Chen YJ, Sung TY, Hsu WL. Multi-Q: a fully automated tool for multiplexed protein quantitation. J Proteome Res. 2006;5:2328–38. doi: 10.1021/pr060132c. [DOI] [PubMed] [Google Scholar]
- 59.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N, Sun Z, Nilsson E, Pratt B, Prazen B, Eng JK, Martin DB, Nesvizhskii AI, Aebersold R. A guided tour of the Trans-Proteomic Pipeline. Proteomics. 2010;10:1150–59. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 61.Van HD, Pinkse MW, Oostwaard DW, Mummery CL, Heck AJ, Krijgsveld J. An experimental correction for arginine-to-proline conversion artifacts in SILAC-based quantitative proteomics. Nat Methods. 2007;4:677–78. doi: 10.1038/nmeth0907-677. [DOI] [PubMed] [Google Scholar]
- 62.Park SK, Liao L, Kim JY, Yates JR., III A computational approach to correct arginine-to-proline conversion in quantitative proteomics. Nat Methods. 2009;6:184–85. doi: 10.1038/nmeth0309-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bakalarski CE, Elias JE, Villen J, Haas W, Gerber SA, Everley PA, Gygi SP. The Impact of Peptide Abundance and Dynamic Range on Stable-Isotope-Based Quantitative Proteomic Analyses. Journal of Proteome Research. 2008;7:4756–65. doi: 10.1021/pr800333e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Everley PA, Bakalarski CE, Elias JE, Waghorne CG, Beausoleil SA, Gerber SA, Faherty BK, Zetter BR, Gygi SP. Enhanced analysis of metastatic prostate cancer using stable isotopes and high mass accuracy instrumentation. Journal of Proteome Research. 2006;5:1224–31. doi: 10.1021/pr0504891. [DOI] [PubMed] [Google Scholar]
- 65.Neher SB, Villen J, Oakes EC, Bakalarski CE, Sauer RT, Gygi SP, Baker TA. Proteomic profiling of CIpXP substrates after DNA damage reveals extensive instability within SOS regulon. Molecular Cell. 2006;22:193–204. doi: 10.1016/j.molcel.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 66.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao ZM, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–66. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 67.Fournier ML, Paulson A, Pavelka N, Mosley AL, Gaudenz K, Bradford WD, Glynn E, Li H, Sardiu ME, Fleharty B, Seidel C, Florens L, Washburn MP. Delayed correlation of mRNA and protein expression in rapamycin-treated cells and a role for Ggc1 in cellular sensitivity to rapamycin. Mol Cell Proteomics. 2010;9:271–84. doi: 10.1074/mcp.M900415-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pan C, Kora G, McDonald WH, Tabb DL, VerBerkmoes NC, Hurst GB, Pelletier DA, Samatova NF, Hettich RL. ProRata: A quantitative proteomics program for accurate protein abundance ratio estimation with confidence interval evaluation. Anal Chem. 2006;78:7121–31. doi: 10.1021/ac060654b. [DOI] [PubMed] [Google Scholar]
- 69.Pan C, Kora G, Tabb DL, Pelletier DA, McDonald WH, Hurst GB, Hettich RL, Samatova NF. Robust estimation of peptide abundance ratios and rigorous scoring of their variability and bias in quantitative shotgun proteomics. Anal Chem. 2006;78:7110–20. doi: 10.1021/ac0606554. [DOI] [PubMed] [Google Scholar]
- 70.Carvalho PC, Hewel J, Barbosa VC, Yates JR., III Identifying differences in protein expression levels by spectral counting and feature selection. Genet Mol Res. 2008;7:342–56. doi: 10.4238/vol7-2gmr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Washburn MP, Wolters D, Yates JR., III Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–47. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 72.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., III Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–82. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 73.Rappsilber J, Ryder U, Lamond AI, Mann M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002;12:1231–45. doi: 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ishihama Y, Schmidt T, Rappsilber J, Mann M, Hartl FU, Kerner MJ, Frishman D. Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics. 2008;9:102. doi: 10.1186/1471-2164-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Griffin NM, Yu J, Long F, Oh P, Shore S, Li Y, Koziol JA, Schnitzer JE. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat Biotechnol. 2010;28:83–89. doi: 10.1038/nbt.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Colinge J, Chiappe D, Lagache S, Moniatte M, Bougueleret L. Differential proteomics via probabilistic peptide identification scores. Anal Chem. 2005;77:596–606. doi: 10.1021/ac0488513. [DOI] [PubMed] [Google Scholar]
- 77.Allet N, Barrillat N, Baussant T, Boiteau C, Botti P, Bougueleret L, Budin N, Canet D, Carraud S, Chiappe D, Christmann N, Colinge J, Cusin I, Dafflon N, Depresle B, Fasso I, Frauchiger P, Gaertner H, Gleizes A, Gonzalez-Couto E, Jeandenans C, Karmime A, Kowall T, Lagache S, Mahe E, Masselot A, Mattou H, Moniatte M, Niknejad A, Paolini M, Perret F, Pinaud N, Ranno F, Raimondi S, Reffas S, Regamey PO, Rey PA, Rodriguez-Tome P, Rose K, Rossellat G, Saudrais C, Schmidt C, Villain M, Zwahlen C. In vitro and in silico processes to identify differentially expressed proteins. Proteomics. 2004;4:2333–51. doi: 10.1002/pmic.200300840. [DOI] [PubMed] [Google Scholar]
- 78.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res. 2006;5:2339–47. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- 79.Pavelka N, Fournier ML, Swanson SK, Pelizzola M, Ricciardi-Castagnoli P, Florens L, Washburn MP. Statistical similarities between transcriptomics and quantitative shotgun proteomics data. Mol Cell Proteomics. 2008;7:631–44. doi: 10.1074/mcp.M700240-MCP200. [DOI] [PubMed] [Google Scholar]
- 80.Nesvizhskii AI, Aebersold R. Analysis, statistical validation and dissemination of large-scale proteomics datasets generated by tandem MS. Drug Discov Today. 2004;9:173–81. doi: 10.1016/S1359-6446(03)02978-7. [DOI] [PubMed] [Google Scholar]
- 81.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 82.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 83.Higgs RE, Knierman MD, Gelfanova V, Butler JP, Hale JE. Label-free LC-MS method for the identification of biomarkers. Methods Mol Biol. 2008;428:209–30. doi: 10.1007/978-1-59745-117-8_12. [DOI] [PubMed] [Google Scholar]
- 84.Higgs RE, Knierman MD, Gelfanova V, Butler JP, Hale JE. Comprehensive label-free method for the relative quantification of proteins from biological samples. J Proteome Res. 2005;4:1442–50. doi: 10.1021/pr050109b. [DOI] [PubMed] [Google Scholar]
- 85.Nordstrom A, O’Maille G, Qin C, Siuzdak G. Nonlinear data alignment for UPLC-MS and HPLC-MS based metabolomics: quantitative analysis of endogenous and exogenous metabolites in human serum. Anal Chem. 2006;78:3289–95. doi: 10.1021/ac060245f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Prince JT, Marcotte EM. Chromatographic alignment of ESI-LC-MS proteomics data sets by ordered bijective interpolated warping. Anal Chem. 2006;78:6140–52. doi: 10.1021/ac0605344. [DOI] [PubMed] [Google Scholar]
- 87.Sadygov RG, Maroto FM, Huhmer AF. ChromAlign: A two-step algorithmic procedure for time alignment of three-dimensional LC-MS chromatographic surfaces. Anal Chem. 2006;78:8207–17. doi: 10.1021/ac060923y. [DOI] [PubMed] [Google Scholar]
- 88.R Development Core Team R. A Language and Environment for Statistical Computing. 2009 [Google Scholar]
- 89.Old WM, Meyer-Arendt K, veline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005;4:1487–502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- 90.Hu Q, Noll RJ, Li H, Makarov A, Hardman M, Graham CR. The Orbitrap: a new mass spectrometer. J Mass Spectrom. 2005;40:430–43. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 91.Syka JE, Marto JA, Bai DL, Horning S, Senko MW, Schwartz JC, Ueberheide B, Garcia B, Busby S, Muratore T, Shabanowitz J, Hunt DF. Novel linear quadrupole ion trap/FT mass spectrometer: performance characterization and use in the comparative analysis of histone H3 post-translational modifications. J Proteome Res. 2004;3:621–26. doi: 10.1021/pr0499794. [DOI] [PubMed] [Google Scholar]
- 92.Wang G, Wu WW, Zeng W, Chou CL, Shen RF. Label-free protein quantification using LC-coupled ion trap or FT mass spectrometry: Reproducibility, linearity, and application with complex proteomes. J Proteome Res. 2006;5:1214–23. doi: 10.1021/pr050406g. [DOI] [PubMed] [Google Scholar]
- 93.Zimmer JS, Monroe ME, Qian WJ, Smith RD. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom Rev. 2006;25:450–82. doi: 10.1002/mas.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jaffe JD, Mani DR, Leptos KC, Church GM, Gillette MA, Carr SA. PEPPeR, a platform for experimental proteomic pattern recognition. Mol Cell Proteomics. 2006;5:1927–41. doi: 10.1074/mcp.M600222-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Strittmatter EF, Rodriguez N, Smith RD. High mass measurement accuracy determination for proteomics using multivariate regression fitting: application to electrospray ionization time-of-flight mass spectrometry. Anal Chem. 2003;75:460–68. doi: 10.1021/ac026057g. [DOI] [PubMed] [Google Scholar]
- 96.Lipton MS, Pasa-Tolic’ L, Anderson GA, Anderson DJ, Auberry DL, Battista JR, Daly MJ, Fredrickson J, Hixson KK, Kostandarithes H, Masselon C, Markillie LM, Moore RJ, Romine MF, Shen Y, Stritmatter E, Tolic’ N, Udseth HR, Venkateswaran A, Wong KK, Zhao R, Smith RD. Global analysis of the Deinococcus radiodurans proteome by using accurate mass tags. Proc Natl Acad Sci U S A. 2002;99:11049–54. doi: 10.1073/pnas.172170199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.America AH, Cordewener JH, van Geffen MH, Lommen A, Vissers JP, Bino RJ, Hall RD. Alignment and statistical difference analysis of complex peptide data sets generated by multidimensional LC-MS. Proteomics. 2006;6:641–53. doi: 10.1002/pmic.200500034. [DOI] [PubMed] [Google Scholar]
- 98.Prakash A, Mallick P, Whiteaker J, Zhang H, Paulovich A, Flory M, Lee H, Aebersold R, Schwikowski B. Signal maps for mass spectrometry-based comparative proteomics. Mol Cell Proteomics. 2006;5:423–32. doi: 10.1074/mcp.M500133-MCP200. [DOI] [PubMed] [Google Scholar]
- 99.Listgarten J, Emili A. Statistical and computational methods for comparative proteomic profiling using liquid chromatography-tandem mass spectrometry. Mol Cell Proteomics. 2005;4:419–34. doi: 10.1074/mcp.R500005-MCP200. [DOI] [PubMed] [Google Scholar]
- 100.Andreev VP, Li L, Cao L, Gu Y, Rejtar T, Wu SL, Karger BL. A new algorithm using cross-assignment for label-free quantitation with LC-LTQ-FT MS. J Proteome Res. 2007;6:2186–94. doi: 10.1021/pr0606880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Radulovic D, Jelveh S, Ryu S, Hamilton TG, Foss E, Mao Y, Emili A. Informatics platform for global proteomic profiling and biomarker discovery using liquid chromatography-tandem mass spectrometry. Mol Cell Proteomics. 2004;3:984–97. doi: 10.1074/mcp.M400061-MCP200. [DOI] [PubMed] [Google Scholar]
- 102.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics. 2006;5:144–56. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 103.Silva JC, Denny R, Dorschel CA, Gorenstein M, Kass IJ, Li GZ, McKenna T, Nold MJ, Richardson K, Young P, Geromanos S. Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem. 2005;77:2187–200. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- 104.Karpievitch YV, Taverner T, Adkins JN, Callister SJ, Anderson GA, Smith RD, Dabney AR. Normalization of peak intensities in bottom-up MS-based proteomics using singular value decomposition. Bioinformatics. 2009;25:2573–80. doi: 10.1093/bioinformatics/btp426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Callister SJ, Barry RC, Adkins JN, Johnson ET, Qian WJ, Webb-Robertson BJ, Smith RD, Lipton MS. Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J Proteome Res. 2006;5:277–86. doi: 10.1021/pr050300l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ting L, Cowley MJ, Hoon SL, Guilhaus M, Raftery MJ, Cavicchioli R. Normalization and statistical analysis of quantitative proteomics data generated by metabolic labeling. Mol Cell Proteomics. 2009;8:2227–42. doi: 10.1074/mcp.M800462-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang B, VerBerkmoes NC, Langston MA, Uberbacher E, Hettich RL, Samatova NF. Detecting differential and correlated protein expression in label-free shotgun proteomics. J Proteome Res. 2006;5:2909–18. doi: 10.1021/pr0600273. [DOI] [PubMed] [Google Scholar]
- 108.Oberg AL, Vitek O. Statistical design of quantitative mass spectrometry-based proteomic experiments. J Proteome Res. 2009;8:2144–56. doi: 10.1021/pr8010099. [DOI] [PubMed] [Google Scholar]
- 109.Oberg AL, Vitek O. Statistical Design of Quantitative Mass Spectrometry-Based Proteomic Experiments. Journal of Proteome Research. 2009;8:2144–56. doi: 10.1021/pr8010099. [DOI] [PubMed] [Google Scholar]
- 110.Wang P, Tang H, Zhang H, Whiteaker J, Paulovich AG, McIntosh M. Normalization regarding non-random missing values in high-throughput mass spectrometry data. Pacific Symposium on Biocomputing. 2006;11:315–26. [PubMed] [Google Scholar]
- 111.Karpievitch Y, Stanley J, Taverner T, Huang J, Adkins JN, Ansong C, Heffron F, Metz TO, Qian WJ, Yoon H, Smith RD, Dabney AR. A statistical framework for protein quantitation in bottom-up MS-based proteomics. Bioinformatics. 2009;25:2028–34. doi: 10.1093/bioinformatics/btp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bukhman YV, Dharsee M, Ewing R, Chu P, Topaloglou T, Le BT, Goh T, Duewel H, Stewart II, Wisniewski JR, Ng NF. Design and analysis of quantitative differential proteomics investigations using LC-MS technology. J Bioinform Comput Biol. 2008;6:107–23. doi: 10.1142/s0219720008003321. [DOI] [PubMed] [Google Scholar]
- 113.Daly DS, Anderson KK, Panisko EA, Purvine SO, Fang R, Monroe ME, Baker SE. Mixed-effects statistical model for comparative LC-MS proteomics studies. J Proteome Res. 2008;7:1209–17. doi: 10.1021/pr070441i. [DOI] [PubMed] [Google Scholar]
- 114.Clough T, Key M, Ott I, Ragg S, Schadow G, Vitek O. Protein quantification in label-free LC-MS experiments. J Proteome Res. 2009;8:5275–84. doi: 10.1021/pr900610q. [DOI] [PubMed] [Google Scholar]
- 115.Oberg AL, Mahoney DW, Eckel-Passow JE, Malone CJ, Wolfinger RD, Hill EG, Cooper LT, Onuma OK, Spiro C, Therneau TM, Bergen HR., III Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J Proteome Res. 2008;7:225–33. doi: 10.1021/pr700734f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Oberg AL, Vitek O. Statistical Design of Quantitative Mass Spectrometry-Based Proteomic Experiments. Journal of Proteome Research. 2009;8:2144–56. doi: 10.1021/pr8010099. [DOI] [PubMed] [Google Scholar]
- 117.Hill EG, Schwacke JH, Comte-Walters S, Slate EH, Oberg AL, Eckel-Passow JE, Therneau TM, Schey KL. A statistical model for iTRAQ data analysis. Journal of Proteome Research. 2008;7:3091–101. doi: 10.1021/pr070520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rodriguez H, Tezak Z, Mesri M, Carr SA, Liebler DC, Fisher SJ, Tempst P, Hiltke T, Kessler LG, Kinsinger CR, Philip R, Ransohoff DF, Skates SJ, Regnier FE, Anderson NL, Mansfield E. Analytical validation of protein-based multiplex assays: a workshop report by the NCI-FDA interagency oncology task force on molecular diagnostics. Clin Chem. 2010;56:237–43. doi: 10.1373/clinchem.2009.136416. [DOI] [PubMed] [Google Scholar]
- 119.Karpievitch Y, Stanley J, Taverner T, Huang J, Adkins JN, Ansong C, Heffron F, Metz TO, Qian WJ, Yoon H, Smith RD, Dabney AR. A statistical framework for protein quantitation in bottom-up MS-based proteomics. Bioinformatics. 2009;25:2028–34. doi: 10.1093/bioinformatics/btp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hill EG, Schwacke JH, Comte-Walters S, Slate EH, Oberg AL, Eckel-Passow JE, Therneau TM, Schey KL. A statistical model for iTRAQ data analysis. Journal of Proteome Research. 2008;7:3091–101. doi: 10.1021/pr070520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Keshamouni VG, Michailidis G, Grasso CS, Anthwal S, Strahler JR, Walker A, Arenberg DA, Reddy RC, Akulapalli S, Thannickal VJ, Standiford TJ, Andrews PC, Omenn GS. Differential protein expression profiling by iTRAO-2DLC-MS/MS of lung cancer cells undergoing epithelial-mesenchymal transition reveals a migratory/invasive phenotype. Journal of Proteome Research. 2006;5:1143–54. doi: 10.1021/pr050455t. [DOI] [PubMed] [Google Scholar]
- 122.Oberg AL, Mahoney DW, Eckel-Passow JE, Malone CJ, Wolfinger RD, Hill EG, Cooper LT, Onuma OK, Spiro C, Therneau TM, Bergen HR., III Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J Proteome Res. 2008;7:225–33. doi: 10.1021/pr700734f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hill EG, Schwacke JH, Comte-Walters S, Slate EH, Oberg AL, Eckel-Passow JE, Therneau TM, Schey KL. A statistical model for iTRAQ data analysis. Journal of Proteome Research. 2008;7:3091–101. doi: 10.1021/pr070520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Karpievitch Y, Stanley J, Taverner T, Huang J, Adkins JN, Ansong C, Heffron F, Metz TO, Qian WJ, Yoon H, Smith RD, Dabney AR. A statistical framework for protein quantitation in bottom-up MS-based proteomics. Bioinformatics. 2009;25:2028–34. doi: 10.1093/bioinformatics/btp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Orchard S, Hermjakob H, Apweiler R. The proteomics standards initiative. Proteomics. 2003;3:1374–76. doi: 10.1002/pmic.200300496. [DOI] [PubMed] [Google Scholar]