

Abstract
Cholangiocarcinoma is a tumor that originates from the neoplastic transformation of the epithelial cells of the intrahepatic or extrahepatic bile ducts. This type of cancer is difficult to diagnose, extremely aggressive, and has very poor prognosis. It is also relatively resistant to chemotherapy and radiation therapy. Its pathogenesis is poorly understood, however it is known that the tumor microenvironment is a very important factor in the regulation of tumor angiogenesis, invasion, and metastasis. The current knowledge about the mechanisms by which these events are regulated as well as the role of the tumor microenvironment in the pathogenesis and classification of cholangiocarcinoma will be discussed.
Keywords: Cholangiocarcinoma, pathogenesis, tumor microenvironment, tumor angiogenesis
Introduction
Cholangiocarcinoma is a tumor that arises from the malignant transformation of the epithelial cells of the intrahepatic or extrahepatic bile ducts. This type of liver cancer has very poor prognosis and is extremely aggressive with symptoms unobservable until there is a blockage of the bile duct by the tumor. Treatment of cholangiocarcinoma by chemotherapy and radiation therapy is not very effective; leaving surgical resection of the tumor as the only treatment option. Further study of the factors that lead to tumor initiation, promotion, and progression is necessary to be able to design alternative treatments for this devastating illness. The tumor microenvironment is a very important factor in the regulation of tumor angiogenesis, invasion, and metastasis; however, the mechanisms by which these events are regulated remain mostly unknown. The goal of this review is to discuss the current knowledge about the role of the tumor microenvironment in the pathogenesis and classification of cholangiocarcinoma as well as recent advancements in the development of therapies directed at the tumor microenvironment.
Cholangiocarcinoma origin, epidemiology and risk factors
Cholangiocarcinoma originates from the neoplastic transformation of cholangiocytes into intrahepatic, perihilar, or distal extrahepatic tumors (1). Usually, cholangiocarcinomas are adenocarcinomas and have poor prognosis with restricted treatment alternatives. This is partly due to the late onset of symptoms and relative resistance to the therapies currently available (2).
The incidence of intrahepatic and extrahepatic cholangiocarcinoma varies by geographic region, with the highest being in Asian countries. Intrahepatic cholangiocarcinoma mortality rates have continuously increased since 1970, conversely, deaths due to extrahepatic cholangiocarcinoma have been decreasing in most countries. Men are slightly more likely to develop cholangiocarcinoma, while incidence increases with age in both sexes (3). As mentioned above, cholangiocarcinoma incidence varies by geographic region. This is partly due to the distribution of risk factors by region and ethnic groups (4). Regional risk factors share the involvement of chronic inflammation and biliary irritation (5). In Asian countries, prevalence of this disease is associated with infection by pathogens that include liver flukes, Hepatitis B, and Hepatitis C. Meanwhile, in Western countries, 90% of patients diagnosed with cholangiocarcinoma lack any of the reported risk factors (4). However, certain risk factors are associated with the remaining 10% of cases, these include chronic inflammation, primary sclerosing cholangitis, obesity, hepatolithiasis, bacterial infection, and/or bile stasisrelated chronic cholangitis (6-8).
Tumor microenvironment
The tumor microenvironment is made up of neoplastic epithelial cells, a biologically complex stroma constituted of various types of stromal cells and the extracellular matrix (9). Stromal cells, particularly inflammatory cells, vascular endothelial cells, and fibroblasts, have been shown to actively support tumor growth in murine models of tumorigenesis (10-13). Additionally, the microenvironment has been shown to play a role in neoplastic transformation, progression, metastasis and invasion of cancer cells (10,13). Also, resistance to radiotherapy and chemotherapy is influenced by the interaction between the cancer cells and the tumor microenvironment (14,15). There is evidence that shows that the interaction between the cancer cells and stromal cells of the microenvironment is bi-directional and dynamic. Neoplastic cells can secrete factors that recruit and activate stromal cells into the tumor microenvironment in a paracrine fashion. Stromal cells that have been recruited and activated can then release factors into the extracellular milieu that can stimulate or inhibit tumor growth (16-18). Vascular endothelial cell proliferation and recruitment leading to the formation of new blood vessels provides the tumor with the nutrient supply necessary for its growth and metastasis. Cancer-associated fibroblasts can stimulate angiogenesis as well as promote tumor growth and invasion. The immune cells present in the tumor microenvironment, tumor-associated macrophages in particular, can confer resistance to toxic insults in addition to promoting growth. Finally, the proliferation of lymph endothelial cells leading to the increase in lymphatic vessel density can promote tumor metastasis (Figure 1). The individual roles of the components of the tumor microenvironment in cholangiocarcinoma will be further discussed below.
Figure 1.

Effects of stromal support cells on tumor growth and metastasis. B (B-cell), EMT (epithelial-mesenchymal transition), M (monocyte), T (T-cell), TAM (tumor associated macrophage).
Angiogenesis
Angiogenesis is defined as the physiological process by which new blood vessels are formed from pre-existing ones. Formation of new blood vessels is necessary to supply nutrients and oxygen to support tumor growth (19). Angiogenesis is accomplished by the organized release of angiogenic factors from the tumor cells, such as vascular endothelial growth factor (VEGF), epidermal growth factor (EGF) and fibroblast growth factor (FGF). These angiogenic factors bind their receptors on the endothelial cell surface leading to augmented vascular permeability, which results in the extravasation of plasma proteins and the dissociation of pericyte coverage (20,21). Endothelial cell proliferation and migration to initiate the formation of new blood vessels follows (22). Localized degradation of the extracellular matrix is a necessary step in the formation of the new blood vessels. This degradation is executed by the matrix metalloproteinases, cathepsin B and other degradation enzymes, in addition to increased matrix protein expression including fibronectin and laminin (23-25). Tumor cells or cancer-associated fibroblast are the main source of these essential extracellular matrix proteins (17).
Tumor-associated angiogenesis in intrahepatic cholangiocarcinoma has been observed by the immunohistochemical study of microvessel density and lymphatic microvessel density. Lower 5-year survival rates, higher recurrence rates, and increased nodal spread were seen in patients that had tumors with increased microvessel density (26). The angiogenic factors, nerve growth factor-β(NGF-β) and vascular endothelial growth factor-C (VEGF-C), are overexpressed by 57.1% and 46.4%, respectively in cholangiocarcinoma samples (27). VEGF-A and VEGF receptors (VEGFRs), angiogenic factors angiopoietin-1, -2, and thrombospondin-1, EGF, EGF receptors (EGFR), and basic fibroblast growth factor are also overexpressed in human cholangiocarcinoma cell lines and samples (28-32). Secretion of these factors may individually or cooperatively increase angiogenesis, evidenced by increased microvessel density. An example of this is VEGF-A, which contributes to the neovascularization of extrahepatic cholangiocarcinoma (33).
Angiogenic factors may act via an autocrine manner to elicit diverse effects on cholangiocytes and cholangiocarcinoma growth (31,33-37). In fact, the proliferative effects that estrogen has on cholangiocarcinoma cell lines are thought to be due to the upregulation of VEGF expression, since VEGF blockage diminishes the estrogenic effects on proliferation (38). Collectively, theses data suggest that agents capable of blocking angiogenesis can potentially have a direct effect on proliferation of cholangiocarcinoma cell in addition to their anti-angiogenic effects. In support of this notion, inhibition of VEGFR and EGFR signaling with vandetanib (ZD6474, tyrosine kinase inhibitor) can be an important approach for the management of the subset of cholangiocarcinoma that lack KRAS mutations and/or have EGFR amplification (37). The orally active, specific inhibitor of EGFR tyrosine kinase, ZD1839 (IRESSA) has clinical activity against cholangiocarcinoma. This inhibitor stabilizes the cell cycle inhibitor p27Kip1 and improves radiosensitivity in cholangiocarcinoma cell lines (36). Curcumin is a natural phenol found in tumeric. In a hamster model of cholangiocarcinoma, curcumin resulted in suppression VEGF expression and a decrease in the microvessel density. Curcumin can also have antiproliferative and proapoptotic effects on cholangiocarcinoma cell lines independent from the angiogenic effects (39,40). Inhibitors of histamine synthesis, H3 histamine receptor agonists, and Endothelin-1, among others have similar effects (41-43). The interplay between angiogenesis, angiogenic factors, and cholangiocarcinoma growth and progression are illustrated in Figure 2.
Figure 2.
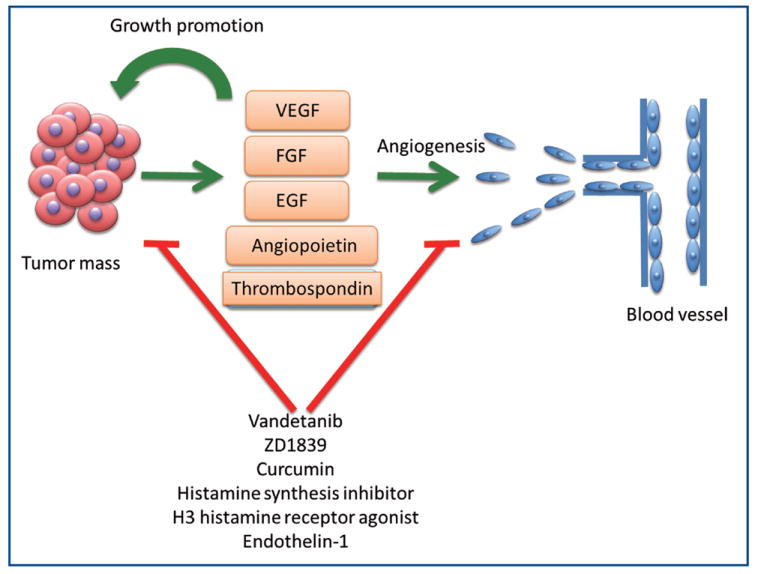
Interactions between cholangiocarcinoma and angiogenic factors that regulate proliferation and angiogenesis. VEGF (vascular endothelial growth factor), FGF (fibroblast growth factor), EGF (epidermal growth factor).
Cancer-associated fibroblasts
Under homeostatic conditions, fibroblasts have a low proliferative index and secrete factors necessary to maintain a normal physiological state. Normal fibroblasts provide biochemical signals that restrain epithelial tumor cells within their basement membrane (44,45). As a result of tissue injury, stromal cells quickly and transiently alter their phenotype and proliferation rate. In tumorigenesis, the normally reversible wound healing response lacks the regulatory mechanisms that allow it to return to normal homeostasis. Thus, stromal dynamics are altered because of this lack of downregulation. Tumor-dependent changes in signaling and plasticity of the stroma elicit alterations that result in a ‘primed’ stroma capable of supporting and inciting tumor initiation or progression (45).
The stroma of cholangiocarcinoma tumors is made up of mostly cancer-associated fibroblasts (also known as myofibroblasts) (46). An increase in α-smooth muscle actin-positive fibroblasts correlated with decreased survival times and increased tumor sizes in resected cholangiocarcinoma tissue(47,48). The origin of these cancer-associated fibroblasts is currently unknown, though hepatic stellate cells, portal fibroblasts and circulating bone marrow-derived precursor cells, among others, have been suggested as possible sources (48-50). Cancer-associated fibroblasts in cholangiocarcinoma tumors appear to be a heterogeneous population. It is likely that these fibroblasts have multiple sources of origin. Genetic screening to elucidate the differences in gene expression between cholangiocarcinoma-derived cancer-associated fibroblasts and non-malignant liver fibroblasts showed differential expression of several genes associated with angiogenesis, cell proliferation and motility. Particularly the cell adhesion molecule, periostin, was significantly upregulated and correlated with shorter survival time in patients and increased cell proliferation and invasive properties in vitro (51). The extracellular matrix protein tenascin-C is specifically expressed by cholangiocarcinoma-derived cancer-associated fibroblasts. It is expressed mainly in the stroma near the invasion front of the tumor and is associated with poor prognosis in intrahepatic cholangiocarcinoma (52,53). Additionally, thrombospondin-1 expression by cancer-associated fibroblasts correlated with increased metastasis (31,54). The stromal-derived factor 1, a chemokine, is released from stromal fibroblasts leading to the stimulation of invasion and migration of cholangiocarcinoma cells by interacting with the chemokine receptor, CXCR4 (55). Recent studies have shown that myofibroblast-derived platelet-derived growth factor protects cholangiocarcinoma cells from TRAIL-induced cell death via a hedgehog dependent mechanism in vitro and in vivo (56). The signaling molecules and their known functions secreted by cholangiocarcinoma-derived cancer-associated fibroblasts are summarized in Figure 3.
Figure 3.
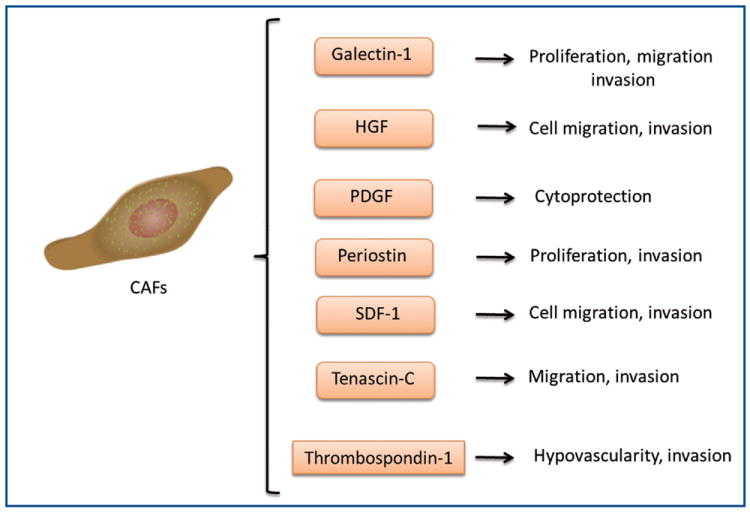
Effects of signaling molecules secreted by cholangiocarcinoma-derived cancer-associated fibroblasts on cholangiocarcinoma progression. CAF (cancer-associate fibroblasts), HGF (hepatocyte growth factor), PDGF (platelet-derived growth factor), SDF-1 (stromal derived factor-1).
Cancer-associated fibroblasts seem to play an important role in the growth and invasion of cholangiocarcinoma. Targeting the molecular signals released by these cells, in addition to strategies to suppress cholangiocarcinoma cell proliferation, could aid in cholangiocarcinoma treatment.
Tumor-associated macrophages
Tumor initiation and progression are intimately related to inflammation and the immune system. A major risk factor for the development of various tumor types is chronic inflammation of the target organ. Within a tumor, tumor-associated macrophages (TAMs) are the primary immune cell found. Macrophages have the ability to secrete pro- or anti-inflammatory mediators depending on the stimuli (57). Macrophages activated with TNF-α have anti-tumor activity and signal tissue destruction, this is known as an M1 phenotype. The M2 phenotype characterized by the initiation of tissue repair, remodeling, and tumor promotion can be induced by interleukin-4 activation (58). Most TAMs are the M2 phenotype, as a result of multiple signals expressed within the tumor microenvironment which include interleukin-10, transforming growth factor-β, and colony stimulating factor-1. These immunomodulatory signals have been reported to be secreted by myeloid-derived suppressor cells, IL-10+ B lymphocytes, Th2 helper T cells, and the tumors themselves (57). Alternatively activated TAMs have reduced anti-tumor activities, and increase the production of angiogenic mediators that include VEGF and IL-10, in addition to M2-specific genes known to be involved in the promotion of cell proliferation. These events are summarized in Figure 4. Strategies that inhibit the M2 phenotype and induce M1 signals can reestablish the anti-tumor functions of TAMs and aid in the removal of protective signals that originate from the M2-TAMs, perhaps activating the innate immune response thus leading to a reduced tumor size (57,59).
Figure 4.

Differential activation of macrophages and their effect on tumor growth. M1 (pro-inflammatory), M2 (anti-inflammatory).
Chronic inflammation and cholangiocarcinoma seem to be intimately related (60). Cholangiocarcinoma cells are known to overproduce many inflammatory cytokines, however, IL-6 is the most studied to date (61). How TAMs are involved in cholangiocarcinoma development and progression is still unclear. Recent studies have shown that the density of infiltrating macrophages (stained for MAC387, a specific macrophage marker) was high in more than half of the studied tumor samples. This high density of MAC387-positive cells correlates with poor survival rates, despite the lack of evidence that the MAC387-positive cells are of the M2 phenotype (62). Cholangiocarcinoma patients have increased circulating CD14+/CD16+ monocytes, which are thought to be precursors of resident macrophages. This increase correlated with the higher density of MAC387-positive infiltrating macrophages. Additionally, the circulating CD14+/CD16+ monocytes expressed higher levels of angiogenic factors that include VEGF and CXCL3 (63). In an a separate study, it was found that M2 macrophages infiltrate intrahepatic cholangiocarcinoma (stained for CD163, an M2 marker); their number closely correlates with neovascularization and infiltration of FOXP3+ regulatory T cells. This study also found that treating macrophages in vitro with the supernatant from CCA cells lead to macrophage polarization toward the M2 phenotype and secretion of VEGF-A, IL-10 and TGFβ (64). Collectively, these data support the hypothesis that TAMs may play a role in cholangiocarcinoma progression. However, the regulation of the crosstalk between M2 TAMs and cholangiocarcinoma cells is not understood and needs to be further studied.
Lymphangiogenesis
Tumor metastasis is a major cause of the lethality present in this disorder and the spread of tumor cells generally occurs through lymphatic vasculature. In the lymph nodes, a higher incidence of tumor foci is found which leads to an unfavorable prognostic factor in most cancers. Previously, it was thought that the spread of tumor cells via the lymphatic system was a passive process where detached tumor cells would enter pre-existing lymphatic vessels in the tumor area (65). We now know that new lymphatic vessels are formed in the tumor microenvironment and that this correlates with lymphatic metastasis (66). Lymphangiogenesis is regulated by growth factors similar to those that control angiogenesis. VEGF-C and VEGF-D are secreted from the tumors and in turn activate VEGFR-3, which is expressed on lymphatic endothelium (67). VEGFR-3 activation induces the proliferation of lymphatic endothelial cells in vitro and the formation of new lymphatic vessels in vivo (68,69). Additional lymphangiogenic factors include VEGF-A, fibroblast growth factor-2, angiopoietin-2, and platelet-derived growth factor-BB (70-73).
Since the factors mentioned above have overlapping angiogenic and lymphangiogenic activities, agents that block angiogenesis may also block lymphangiogenesis. Inhibitors that block VEGF-C/VEGF-D/VEGFR3 signaling could potentially block lymphangiogenesis in addition to angiogenesis, leading to blockage of lymphogenous metastatic spread (74-76). Supporting this, it has been shown that blocking the interaction between VEGF-D and its receptors with a monoclonal neutralizing antibody resulted in the inhibition of angiogenesis, lymphangiogenesis, and metastatic spread through the lymphatics in a murine tumor model (77).
Lymphangiogenesis in the context of cholangiocarcinoma remains poorly understood and controversial. Recent studies suggest there may be a correlation between lymphangiogenesis and lymph node metastases and prognosis. Patients with cholangiocarcinoma tumors that have low lymphatic vessel density have a longer survival rate compared to patients that have higher lymphatic vessel density (78). Furthermore, intrahepatic cholangiocarcinoma tumors that show high lymphatic vessel density correlate with higher nodal spread and recurrence rate (26). On the other hand, other studies show that in intrahepatic cholangiocarcinoma tumors, lymphangiogenesis does not correlate with lymph node metastasis but correlates with VEGF-C expression and the presence of myofibroblast that express the same markers as lymphendothelial cells, possibly explaining the divergence in conclusions (79). NGF has been associated with tumor progression and growth as well as VEGF expression in various other cell types (80-84). NGF-β expression and its correlation with lymphangiogenesis, lymph node metastasis, and VEGF-C expression in hilar cholangiocarcinoma were studied by Xu et al. This study concluded that high NGF expression indeed correlates with VEGF-C overexpression, lymphatic vessel density, and lymph node metastasis; this suggests that NGF might play a role in the stimulation of lymphangiogenesis in cholangiocarcinoma tumors.
Conclusions
In summary, the research discussed in the present review highlights the role that the tumor microenvironment plays in the growth, progression, and metastatic invasion of cholangiocarcinoma. The interaction between stromal and cholangiocarcinoma cells via signaling mediators results in an environment that supports tumor growth and suppresses innate immunity, thus conferring resistance to cytotoxic insults (endogenous and chemotherapeutic). How the support cells in the stroma of cholangiocarcinoma tumors are recruited and activated remains unclear. Targeting the tumor microenvironment rather than the cholangiocarcinoma cells directly may lead to more effective therapeutic strategies to treat this devastating cancer.
Acknowledgments
Portions of these studies were supported by the following grants awarded to Dr. Sharon DeMorrow: American Cancer Society Research Scholar Award (RSC118760), and NIH K01 Award (DK078532) and NIH R03 Award (DK088012). This material is the result of work supported with resources and the use of facilities at the Central Texas Veterans Health Care System in Temple, TX.
Footnotes
No potential conflict of interest.
References
- 1.Alpini G, Prall R, LaRusso N. The pathobiology of biliary epithelia. In: Arias I, Boyer J, Chisari F, editors. The liver; biology and pathobiology. 4. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 421–35. [Google Scholar]
- 2.Sirica AE. Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy. Hepatology. 2005;41:5–15. doi: 10.1002/hep.20537. [DOI] [PubMed] [Google Scholar]
- 3.Patel T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer. 2002;2:10. doi: 10.1186/1471-2407-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Menachem T. Risk factors for cholangiocarcinoma. Eur J Gastroenterol Hepatol. 2007;19:615–7. doi: 10.1097/MEG.0b013e328224b935. [DOI] [PubMed] [Google Scholar]
- 5.Gores GJ. Cholangiocarcinoma: current concepts and insights. Hepatology. 2003;37:961–9. doi: 10.1053/jhep.2003.50200. [DOI] [PubMed] [Google Scholar]
- 6.Catalano OA, Sahani DV, Forcione DG, et al. Biliary infections: spectrum of imaging findings and management. Radiographics. 2009;29:2059–80. doi: 10.1148/rg.297095051. [DOI] [PubMed] [Google Scholar]
- 7.Chen MF. Peripheral cholangiocarcinoma (cholangiocellular carcinoma): clinical features, diagnosis and treatment. J Gastroenterol Hepatol. 1999;14:1144–9. doi: 10.1046/j.1440-1746.1999.01983.x. [DOI] [PubMed] [Google Scholar]
- 8.de Groen PC, Gores GJ, LaRusso NF, et al. Biliary tract cancers. N Engl J Med. 1999;341:1368–78. doi: 10.1056/NEJM199910283411807. [DOI] [PubMed] [Google Scholar]
- 9.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 10.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–7. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunha GR, Hayward SW, Wang YZ, et al. Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer. 2003;107:1–10. doi: 10.1002/ijc.11335. [DOI] [PubMed] [Google Scholar]
- 12.Olumi AF, Grossfeld GD, Hayward SW, et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–11. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tlsty TD. Stromal cells can contribute oncogenic signals. Semin Cancer Biol. 2001;11:97–104. doi: 10.1006/scbi.2000.0361. [DOI] [PubMed] [Google Scholar]
- 14.de Visser KE, Jonkers J. Towards understanding the role of cancer-associated inflammation in chemoresistance. Curr Pharm Des. 2009;15:1844–53. doi: 10.2174/138161209788453239. [DOI] [PubMed] [Google Scholar]
- 15.Shinohara ET, Maity A. Increasing sensitivity to radiotherapy and chemotherapy by usingnovel biological agents that alter the tumor microenvironment. Curr Mol Med. 2009;9:1034–45. doi: 10.2174/156652409789839107. [DOI] [PubMed] [Google Scholar]
- 16.Onimaru M, Yonemitsu Y. Angiogenic and lymphangiogenic cascades in the tumor microenvironment. Front Biosci (Schol Ed) 2011;3:216–25. doi: 10.2741/s146. [DOI] [PubMed] [Google Scholar]
- 17.Räsänen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. 2010;316:2713–22. doi: 10.1016/j.yexcr.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 18.Rojas A, Figueroa H, Morales E. Fueling inflammation at tumor microenvironment: the role of multiligand/RAGE axis. Carcinogenesis. 2010;31:334–41. doi: 10.1093/carcin/bgp322. [DOI] [PubMed] [Google Scholar]
- 19.McDougall SR, Anderson AR, Chaplain MA. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: clinical implications and therapeutic targeting strategies. J Theor Biol. 2006;241:564–89. doi: 10.1016/j.jtbi.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 20.Dvorak HF. Angiogenesis: update 2005. J Thromb Haemost. 2005;3:1835–42. doi: 10.1111/j.1538-7836.2005.01361.x. [DOI] [PubMed] [Google Scholar]
- 21.Roberts WG, Palade GE. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997;57:765–72. [PubMed] [Google Scholar]
- 22.Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14:53–65. doi: 10.1016/0026-2862(77)90141-8. [DOI] [PubMed] [Google Scholar]
- 23.Gladson CL. The extracellular matrix of gliomas: modulation of cell function. J Neuropathol Exp Neurol. 1999;58:1029–40. doi: 10.1097/00005072-199910000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Ljubimova JY, Fujita M, Khazenzon NM, et al. Changes in laminin isoforms associated with brain tumor invasion and angiogenesis. Front Biosci. 2006;11:81–8. doi: 10.2741/1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mikkelsen T, Yan PS, Ho KL, et al. Immunolocalization of cathepsin B in human glioma: implications for tumor invasion and angiogenesis. J Neurosurg. 1995;83:285–90. doi: 10.3171/jns.1995.83.2.0285. [DOI] [PubMed] [Google Scholar]
- 26.Thelen A, Scholz A, Weichert W, et al. Tumor-associated angiogenesis and lymphangiogenesis correlate with progression of intrahepatic cholangiocarcinoma. Am J Gastroenterol. 2010;105:1123–32. doi: 10.1038/ajg.2009.674. [DOI] [PubMed] [Google Scholar]
- 27.Xu LB, Liu C, Gao GQ, et al. Nerve growth factor-beta expression is associated with lymph node metastasis and nerve infiltration in human hilar cholangiocarcinoma. World J Surg. 2010;34:1039–45. doi: 10.1007/s00268-010-0417-4. [DOI] [PubMed] [Google Scholar]
- 28.Alvaro D, Barbaro B, Franchitto A, et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am J Pathol. 2006;169:877–88. doi: 10.2353/ajpath.2006.050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harder J, Waiz O, Otto F, et al. EGFR and HER2 expression in advanced biliary tract cancer. World J Gastroenterol. 2009;15:4511–7. doi: 10.3748/wjg.15.4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogasawara S, Yano H, Higaki K, et al. Expression of angiogenic factors, basic fibroblast growth factor and vascular endothelial growth factor, in human biliary tract carcinoma cell lines. Hepatol Res. 2001;20:97–113. doi: 10.1016/s1386-6346(00)00117-0. [DOI] [PubMed] [Google Scholar]
- 31.Tang D, Nagano H, Yamamoto H, et al. Angiogenesis in cholangiocellular carcinoma: expression of vascular endothelial growth factor, angiopoietin-1/2, thrombospondin-1 and clinicopathological significance. Oncol Rep. 2006;15:525–32. [PubMed] [Google Scholar]
- 32.Yoshikawa D, Ojima H, Iwasaki M, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer. 2008;98:418–25. doi: 10.1038/sj.bjc.6604129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Möbius C, Demuth C, Aigner T, et al. Evaluation of VEGF A expression and microvascular density as prognostic factors in extrahepatic cholangiocarcinoma. Eur J Surg Oncol. 2007;33:1025–9. doi: 10.1016/j.ejso.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 34.Gaudio E, Barbaro B, Alvaro D, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270–82. doi: 10.1053/j.gastro.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 35.Glaser SS, Gaudio E, Alpini G. Vascular factors, angiogenesis and biliary tract disease. Curr Opin Gastroenterol. 2010;26:246–50. doi: 10.1097/MOG.0b013e3283369d19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yabuuchi S, Katayose Y, Oda A, et al. ZD1839 (IRESSA) stabilizes p27Kip1 and enhances radiosensitivity in cholangiocarcinoma cell lines. Anticancer Res. 2009;29:1169–80. [PubMed] [Google Scholar]
- 37.Yoshikawa D, Ojima H, Kokubu A, et al. Vandetanib (ZD6474), an inhibitor of VEGFR and EGFR signalling, as a novel molecular-targeted therapy against cholangiocarcinoma. Br J Cancer. 2009;100:1257–66. doi: 10.1038/sj.bjc.6604988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mancino A, Mancino MG, Glaser SS, et al. Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor. Dig Liver Dis. 2009;41:156–63. doi: 10.1016/j.dld.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prakobwong S, Gupta SC, Kim JH, et al. Curcumin suppresses proliferation and induces apoptosis in human biliary cancer cells through modulation of multiple cell signaling pathways. Carcinogenesis. 2011;32:1372–80. doi: 10.1093/carcin/bgr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prakobwong S, Khoontawad J, Yongvanit P, et al. Curcumin decreases cholangiocarcinogenesis in hamsters by suppressing inflammation-mediated molecular events related to multistep carcinogenesis. Int J Cancer. 2011;129:88–100. doi: 10.1002/ijc.25656. [DOI] [PubMed] [Google Scholar]
- 41.Fava G, DeMorrow S, Gaudio E, et al. Endothelin inhibits cholangiocarcinoma growth by a decrease in the vascular endothelial growth factor expression. Liver Int. 2009;29:1031–42. doi: 10.1111/j.1478-3231.2009.01997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Francis H, DeMorrow S, Venter J, et al. Inhibition of histidine decarboxylase ablates the autocrine tumorigenic effects of histamine in human cholangiocarcinoma. Gut. 2011 doi: 10.1136/gutjnl-2011-300007. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Francis H, Onori P, Gaudio E, et al. H3 histamine receptor-mediated activation of protein kinase Calpha inhibits the growth of cholangiocarcinoma in vitro and in vivo. Mol Cancer Res. 2009;7:1704–13. doi: 10.1158/1541-7786.MCR-09-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beacham DA, Cukierman E. Stromagenesis: the changing face of fibroblastic microenvironments during tumor progression. Semin Cancer Biol. 2005;15:329–41. doi: 10.1016/j.semcancer.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 45.Tuxhorn JA, Ayala GE, Rowley DR. Reactive stroma in prostate cancer progression. J Urol. 2001;166:2472–83. [PubMed] [Google Scholar]
- 46.Sirica AE, Dumur CI, Campbell DJ, et al. Intrahepatic cholangiocarcinoma progression: prognostic factors and basic mechanisms. Clin Gastroenterol Hepatol. 2009;7:S68–78. doi: 10.1016/j.cgh.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chuaysri C, Thuwajit P, Paupairoj A, et al. Alpha-smooth muscle actin-positive fibroblasts promote biliary cell proliferation and correlate with poor survival in cholangiocarcinoma. Oncol Rep. 2009;21:957–69. doi: 10.3892/or_00000309. [DOI] [PubMed] [Google Scholar]
- 48.Okabe H, Beppu T, Hayashi H, et al. Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann Surg Oncol. 2009;16:2555–64. doi: 10.1245/s10434-009-0568-4. [DOI] [PubMed] [Google Scholar]
- 49.Dranoff JA, Wells RG. Portal fibroblasts: Underappreciated mediators of biliary fibrosis. Hepatology. 2010;51:1438–44. doi: 10.1002/hep.23405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimoda M, Mellody KT, Orimo A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin Cell Dev Biol. 2010;21:19–25. doi: 10.1016/j.semcdb.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Utispan K, Thuwajit P, Abiko Y, et al. Gene expression profiling of cholangiocarcinoma-derived fibroblast reveals alterations related to tumor progression and indicates periostin as a poor prognostic marker. Mol Cancer. 2010;9:13. doi: 10.1186/1476-4598-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aishima S, Taguchi K, Terashi T, et al. Tenascin expression at the invasive front is associated with poor prognosis in intrahepatic cholangiocarcinoma. Mod Pathol. 2003;16:1019–27. doi: 10.1097/01.MP.0000086860.65672.73. [DOI] [PubMed] [Google Scholar]
- 53.Iguchi T, Yamashita N, Aishima S, et al. A comprehensive analysis of immunohistochemical studies in intrahepatic cholangiocarcinoma using the survival tree model. Oncology. 2009;76:293–300. doi: 10.1159/000207506. [DOI] [PubMed] [Google Scholar]
- 54.Kawahara N, Ono M, Taguchi K, et al. Enhanced expression of thrombospondin-1 and hypovascularity in human cholangiocarcinoma. Hepatology. 1998;28:1512–7. doi: 10.1002/hep.510280610. [DOI] [PubMed] [Google Scholar]
- 55.Ohira S, Sasaki M, Harada K, et al. Possible regulation of migration of intrahepatic cholangiocarcinoma cells by interaction of CXCR4 expressed in carcinoma cells with tumor necrosis factor-alpha and stromal-derived factor-1 released in stroma. Am J Pathol. 2006;168:1155–68. doi: 10.2353/ajpath.2006.050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fingas CD, Bronk SF, Werneburg NW, et al. Myofibroblast-derived PDGF-BB promotes hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011 doi: 10.1002/hep.24588. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sica A. Role of tumour-associated macrophages in cancer-related inflammation. Exp Oncol. 2010;32:153–8. [PubMed] [Google Scholar]
- 58.Mantovani A, Sozzani S, Locati M, et al. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 59.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–66. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–7. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 61.Isomoto H, Mo JL, Kobayashi S, et al. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology. 2007;132:384–96. doi: 10.1053/j.gastro.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subimerb C, Pinlaor S, Khuntikeo N, et al. Tissue invasive macrophage density is correlated with prognosis in cholangiocarcinoma. Mol Med Report. 2010;3:597–605. doi: 10.3892/mmr_00000303. [DOI] [PubMed] [Google Scholar]
- 63.Subimerb C, Pinlaor S, Lulitanond V, et al. Circulating CD14(+) CD16(+) monocyte levels predict tissue invasive character of cholangiocarcinoma. Clin Exp Immunol. 2010;161:471–9. doi: 10.1111/j.1365-2249.2010.04200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hasita H, Komohara Y, Okabe H, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010;101:1913–9. doi: 10.1111/j.1349-7006.2010.01614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Achen MG, Stacker SA. Molecular control of lymphatic metastasis. Ann N Y Acad Sci. 2008;1131:225–34. doi: 10.1196/annals.1413.020. [DOI] [PubMed] [Google Scholar]
- 66.Achen MG, McColl BK, Stacker SA. Focus on lymphangiogenesis in tumor metastasis. Cancer Cell. 2005;7:121–7. doi: 10.1016/j.ccr.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 67.Lymboussaki A, Partanen TA, Olofsson B, et al. Expression of the vascular endothelial growth factor C receptor VEGFR-3 in lymphatic endothelium of the skin and in vascular tumors. Am J Pathol. 1998;153:395–403. doi: 10.1016/S0002-9440(10)65583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mäkinen T, Veikkola T, Mustjoki S, et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 2001;20:4762–73. doi: 10.1093/emboj/20.17.4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Veikkola T, Jussila L, Makinen T, et al. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 2001;20:1223–31. doi: 10.1093/emboj/20.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cao R, Björndahl MA, Religa P, et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell. 2004;6:333–45. doi: 10.1016/j.ccr.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 71.Gale NW, Thurston G, Hackett SF, et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3:411–23. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- 72.Kubo H, Cao R, Brakenhielm E, et al. Blockade of vascular endothelial growth factor receptor-3 signaling inhibits fibroblast growth factor-2-induced lymphangiogenesis in mouse cornea. Proc Natl Acad Sci U S A. 2002;99:8868–73. doi: 10.1073/pnas.062040199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nagy JA, Vasile E, Feng D, et al. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J Exp Med. 2002;196:1497–506. doi: 10.1084/jem.20021244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baldwin ME, Stacker SA, Achen MG. Molecular control of lymphangiogenesis. Bioessays. 2002;24:1030–40. doi: 10.1002/bies.10173. [DOI] [PubMed] [Google Scholar]
- 75.Stacker SA, Achen MG, Jussila L, et al. Lymphangiogenesis and cancer metastasis. Nat Rev Cancer. 2002;2:573–83. doi: 10.1038/nrc863. [DOI] [PubMed] [Google Scholar]
- 76.Stacker SA, Baldwin ME, Achen MG. The role of tumor lymphangiogenesis in metastatic spread. FASEB J. 2002;16:922–34. doi: 10.1096/fj.01-0945rev. [DOI] [PubMed] [Google Scholar]
- 77.Stacker SA, Caesar C, Baldwin ME, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–91. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- 78.Thelen A, Scholz A, Benckert C, et al. Tumor-associated lymphangiogenesis correlates with lymph node metastases and prognosis in hilar cholangiocarcinoma. Ann Surg Oncol. 2008;15:791–9. doi: 10.1245/s10434-007-9774-0. [DOI] [PubMed] [Google Scholar]
- 79.Aishima S, Nishihara Y, Iguchi T, et al. Lymphatic spread is related to VEGF-C expression and D2-40-positive myofibroblasts in intrahepatic cholangiocarcinoma. Mod Pathol. 2008;21:256–64. doi: 10.1038/modpathol.3800985. [DOI] [PubMed] [Google Scholar]
- 80.Descamps S, Pawlowski V, Révillion F, et al. Expression of nerve growth factor receptors and their prognostic value in human breast cancer. Cancer Res. 2001;61:4337–40. [PubMed] [Google Scholar]
- 81.Descamps S, Toillon RA, Adriaenssens E, et al. Nerve growth factor stimulates proliferation and survival of human breast cancer cells through two distinct signaling pathways. J Biol Chem. 2001;276:17864–70. doi: 10.1074/jbc.M010499200. [DOI] [PubMed] [Google Scholar]
- 82.Lazarovici P, Gazit A, Staniszewska I, et al. Nerve growth factor (NGF) promotes angiogenesis in the quail chorioallantoic membrane. Endothelium. 2006;13:51–9. doi: 10.1080/10623320600669053. [DOI] [PubMed] [Google Scholar]
- 83.Lazarovici P, Marcinkiewicz C, Lelkes PI. Cross talk between the cardiovascular and nervous systems: neurotrophic effects of vascular endothelial growth factor (VEGF) and angiogenic effects of nerve growth factor (NGF)-implications in drug development. Curr Pharm Des. 2006;12:2609–22. doi: 10.2174/138161206777698738. [DOI] [PubMed] [Google Scholar]
- 84.Sortino MA, Condorelli F, Vancheri C, et al. Mitogenic effect of nerve growth factor (NGF) in LNCaP prostate adenocarcinoma cells: role of the high- and low-affinity NGF receptors. Mol Endocrinol. 2000;14:124–36. doi: 10.1210/mend.14.1.0402. [DOI] [PubMed] [Google Scholar]