

Abstract
Enzymatic reactions typically involve complex dynamics during substrate binding, conformational rearrangement, chemistry and product release. The non-covalent steps provide kinetic checkpoints that contribute to the overall specificity of enzymatic reactions. DNA polymerases perform DNA replication with outstanding fidelity by actively rejecting non-cognate nucleotide substrates early in the reaction pathway. Substrates are delivered to the active site by a flexible fingers subdomain of the enzyme, as it converts from an open to a closed conformation. The conformational dynamics of the fingers subdomain might also play a role in nucleotide selection, although the precise role is currently unknown. Using single-molecule Förster resonance energy transfer, we observed individual Escherichia coli DNA polymerase I (Klenow fragment) molecules performing substrate selection. We discovered that the fingers subdomain actually samples through three distinct conformations - open, closed and a previously unrecognized intermediate conformation. We measured the overall dissociation rate of the polymerase-DNA complex and the distribution among the various conformational states in the absence and presence of nucleotide substrates, which were either correct or incorrect. Correct substrates promote rapid progression of the polymerase to the catalytically competent closed conformation, whereas incorrect nucleotides block the enzyme in the intermediate conformation and induce rapid dissociation from DNA. Remarkably, incorrect nucleotide substrates also promote partitioning of DNA to the spatially separated 3′-5′ exonuclease domain, providing an additional mechanism to prevent misincorporation at the polymerase active site. These results reveal the existence of an early innate fidelity checkpoint, rejecting incorrect nucleotide substrates before the enzyme encloses the nascent base pair.
Keywords: DNA polymerase, nucleotide selection, fidelity checkpoints, single-molecule fluorescence spectroscopy, Förster resonance energy transfer
INTRODUCTION
DNA replication is a finely tuned and precisely regulated process. High-fidelity DNA polymerases replicate DNA with an extremely low error rate by selecting the correct nucleotide substrate during each cycle of nucleotide incorporation. While the selection is based on complementarity of the incoming nucleotide and templating base, the high degree of accuracy that is achieved (1 error in 105 to 108 correct incorporations) significantly exceeds the fidelity threshold based solely on the free energy difference between correct and incorrect base pairing 1. Hence DNA polymerases must actively contribute to the rejection of incorrect nucleotide substrates.
Rapid chemical kinetic studies have provided important mechanistic insights into the elementary steps in the nucleotide incorporation cycle and the origin of polymerase fidelity 2–4. These studies have revealed the existence of one or more non-covalent steps, occurring after binding of deoxynucleoside triphosphate (dNTP) substrates and preceding the covalent step of phosphoryl transfer. The non-covalent steps appear to have a functional role in nucleotide selection, serving as kinetic checkpoints before the chemical step of nucleotide incorporation 5.
DNA polymerases have an overall architecture that resembles a half-open human right hand, with fingers, palm and thumb subdomains 6. The fingers provide a binding site for dNTP substrates, while the palm and thumb grip the duplex DNA product. Crystal structures of DNA polymerases show that the fingers subdomain adopts an open conformation in binary complexes with DNA 7,8. However, in structures of ternary complexes containing complementary nucleotide substrates, the fingers are observed in a closed conformation enveloping the nascent base pair 7,8.
The open and closed conformations of DNA polymerases correspond to distinct functional states on the nucleotide insertion pathway. The open conformation allows binding of an incoming dNTP substrate, which is then delivered to the polymerase active site as the fingers switch to the closed conformation 9–11. While in the closed conformation, the triphosphate group of the incoming nucleotide is aligned for in-line attack from the 3′-hydroxyl of the extending DNA strand, resulting in covalent incorporation of the nucleotide.
Cocrystal structures of polymerase-substrate complexes, while informative, do not explain how correct nucleotide substrates are selected by the polymerase while incorrect nucleotides are rejected. In the open conformation, the templating base is sequestered in a pre-insertion site, distant from the nucleobase moiety of the incoming nucleotide 7,8. Hence, it is unlikely that the enzyme discriminates between correct (complementary to the template base) and incorrect (non-complementary) nucleotide substrates while in the open conformation. One possible mechanism of nucleotide substrate selection relies on the continual exchange of the fingers subdomain between open and closed conformations 12, such that incoming nucleotides (either correct or incorrect) are delivered to the polymerase active site to pair or mispair with the template base. In this model, nucleotide recognition occurs after the enzyme encloses the nascent base pair and only the correct nucleotide can stabilize the fingers in a long lived closed conformation 9. Alternatively, a third conformation (uncharacterized) of the polymerase-DNA complex may exist, allowing the incoming nucleotide to be “previewed” by the template base before the enzyme is committed to the closure step 5. However, the actual mechanism of nucleotide selection in DNA polymerases remains to be established.
We have devised a single-molecule Förster resonance energy transfer (smFRET) system to examine the conformational states and kinetic behavior of individual polymerase – DNA complexes during the nucleotide selection process. Our results reveal the existence of three distinct conformational states of the fingers subdomain of Escherchia coli DNA polymerase I (Klenow fragment) (Pol I KF). Two of these are the open and closed conformations, while the third emerges as a previously undocumented intermediate conformation of Pol I KF. We measured the overall dissociation rate of the polymerase–DNA complex and the distribution among the various conformational states in the absence and presence of nucleotide substrates, which were either correct or incorrect. We have also identified separate subpopulations of DNA bound at the spatially distinct polymerase and 3′-5′ exonuclease sites of KF, and determined how the partitioning between sites is influenced by the identity of the incoming nucleotide substrate. Taken together, our results imply that the newly discovered intermediate conformation of Pol I KF serves as an early fidelity checkpoint during nucleotide selection, rejecting incorrect nucleotides before the fingers subdomain fully encloses the nascent base pair.
RESULTS
A DNA-polymerase smFRET signal reports on enzyme dynamics during pre-chemical steps
The smFRET system utilizes a DNA primer/template tethered to a quartz slide (Fig. 1a). The FRET donor Alexa-Fluor 488 is attached via a 6-carbon linker to the C5 position of a modified thymine base at position 8 in the primer strand (denoted Y in Fig. 1a). The primer strand is terminated with a 2′, 3′-dideoxy modification to block covalent incorporation of dNTP substrates, creating a system to report on pre-chemical steps 9–11.
Figure 1. Schematic illustration of labeling strategy used to probe the finger-closing conformational change in Pol I KF, based on crystal structures of homolog Bacillus stearothermophilus (Bst) Pol I.
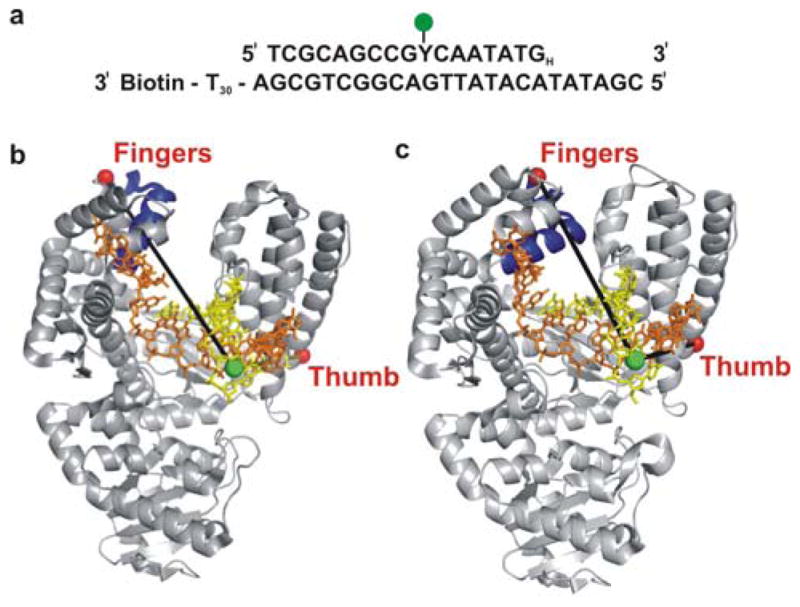
(a) The sequence of the primer-template DNA used in the experiments. The primer strand was dideoxy terminated at the 3′ end (denoted by a subscript H) to prevent nucleotide incorporation. The Y represents an amino-dT used as the labeling site for the Alexa-Fluor 488 donor dye. The template strand is 3′ biotin labeled for surface attachment. The T30 extension at the 3′ end of the template strand separates the duplex portion of the primer/template from the surface. (b) The open polymerase-DNA binary complex (PDB code 1L3S). (c) The closed polymerase-DNA-dNTP ternary complex (PDB code 1LV5). The mobile segment of the protein including the O-helix (residues 680–714 in Bst Pol I, corresponding to residues 732–766 in KF Pol I) is highlighted in dark blue. The yellow and orange oligonucleotides are the primer and template strands, respectively. The red spheres represent positions of the Alexa-Fluor 594 acceptor, attached respectively either to amino acid 744 (L744C KF) in the fingers-labeled construct or to residue 550 (K550C KF) in the thumb-labeled construct (Pol I KF residue numbers). The green sphere represents the labeling position of the donor on the DNA primer.
Two differently labeled Pol I KF proteins were prepared using the Pol I KF C907S/D424A construct. In one modification, an Alexa-Fluor 594 FRET acceptor was positioned at mutated cysteine 744 in the fingers subdomain (L744C KF, Figs. 1b & c). The FRET efficiency for the L744C KF construct reports on the proximity of the flexible fingers subdomain relative to the DNA substrate. The estimated donor-acceptor distances in the open and closed conformations, based on corresponding crystal structures (PDB codes 1L3S and 1LV5) of the KF homologue Bacillus stearothermophilus DNA polymerase I (Bst Pol I) 8, are 50.3 Å and 42.2 Å, respectively, corresponding to idealized FRET efficiencies of 0.74 and 0.89 (based on a Förster distance of 60 Å). These values overestimate the true FRET efficiencies because the extension of the donor-acceptor distances due to the dye linkers are not accounted for. Nevertheless, the FRET efficiency is expected to increase significantly as the fingers convert from an open to a closed conformation. In the other modification, the same acceptor label was placed at mutated cysteine 550 in the thumb subdomain (K550C KF, Figs. 1b & c & c). The K550C KF construct provides an internal reference, because the thumb subdomain has a fixed position in cocrystal structures of polymerase-DNA complexes in both open and closed conformations 8. Primer extension assays validated that the presence of the donor and/or acceptor dyes did not disrupt the polymerase activity, which remained similar to the activity of the non-labeled polymerase extending non-labeled DNA (Fig. S1, Supporting Information).
smFRET reveals three conformations of the fingers in binary Pol I KF – DNA complexes
TIRF microscopy was used to monitor the interaction between acceptor-labeled KF and the tethered donor-labeled DNA primer/template. We initially examined the conformational states of the finger-labeled enzyme L744C KF in the absence of dNTP substrates. In this way, it is possible to detect motions of the fingers as the polymerase binds to and positions itself at the DNA primer-template junction. The fluorescence intensity in the donor and acceptor channels was recorded for prolonged time intervals (up to 300 s). A typical set of paired fluorescence intensity time traces recorded in the donor (green) and acceptor (red) channels (Fig. 2a, upper left panel) shows perfectly anti-correlated intensity fluctuations. The corresponding smFRET efficiency trajectory (Fig. 2a, lower left panel, blue) reveals abrupt jumps as individual Pol I KF molecules bind to and dissociate from the tethered DNA. The FRET efficiency observed after each binding event is generally distinct, indicating that different conformational states are accessed in each case. Moreover, these states sometimes interconvert during a single binding period (Fig. S2, Supporting Information).
Figure 2. Fluorescence intensity time traces, smFRET efficiency trajectories and FRET efficiency histograms for finger-labeled polymerase molecules (L744C KF).

(a) Binary Pol+Atempl complexes, formed by binding of acceptor-labeled L744C KF molecules to donor-labeled DNA molecules with A in the template extension position, show fast sampling of three bound states, with mean FRET efficiencies of 0.41, 0.50 and 0.63. A small fraction of complexes populate a separated 0.90 FRET state. The assignment of the individual peaks to open, ajar and closed conformations of the fingers subdomain, and to a population of DNA bound at the 3′-5′ exonuclease (exo) site, are discussed in the text. (b) Correct ternary Pol+Atempl+dTTP complexes, formed in the presence of 1 mM dTTP in the solution, show stable binding in the 0.63 FRET state (left). Two separate peaks in the lower FRET region are not resolved and appear as a shoulder at 0.45 FRET, while a higher 0.9 FRET state is no longer apparent (right). (c) In the presence of 1 mM incorrect dATP nucleotides, Pol I KF frequently samples short-lived 0.4 and 0.5 FRET states, with decreased number of molecules in the 0.63 FRET state. The number of complexes populating the 0.9 FRET state increases. In all cases, the green and red traces show background corrected fluorescence intensities in the donor and acceptor channels, respectively. The blue lines show corresponding smFRET trajectories. The dashed lines positioned at 0, 0.4, 0.5 and 0.6 FRET efficiencies are for ease of visual inspection. Histograms of smFRET efficiencies compiled from multiple trajectories (right) were fitted using multiple Gaussian functions. Dashed black lines show individual Gaussian fits, with the red line corresponding to a composite sum of Gaussians. The percentage numbers on each graph indicate the fraction of complexes in different populations, obtained from the peak areas of the Gaussian fits. The numbers of individual smFRET time trajectories used to construct each histogram were 320 (a), 244 (b) and 299 (c).
A histogram of smFRET efficiencies compiled from 320 individual trajectories shows three densely populated but well resolved peaks centered at 0.41, 0.50 and 0.63 FRET efficiencies, and a smaller peak separated from this group at 0.90 FRET efficiency (Fig. 2a, right panel). Each of the observed peaks represents a distinct conformation of the L744C KF - DNA complex. The presence of three closely positioned distinct bound states within a 0.2 interval in FRET efficiency (0.41 – 0.63 FRET) is intriguing, because only two states - open and closed conformations - have been observed within a similar FRET interval previously 12. Based on available crystal structures of homologous polymerase-DNA complexes, the shortest distance between the fingers subdomain and the DNA is expected for the closed conformation 7,8. Therefore, the 0.63 FRET state probably corresponds to a closed L744C KF - DNA complex. There are two possibilities for the appearance of the 0.41 and 0.50 FRET states: (1) the 0.41 FRET state represents the open conformation, while the 0.50 FRET state represents a previously unrecognized intermediate conformation of the fingers subdomain, or (2) the 0.50 FRET state represents the open fingers conformation, whereas the 0.41 FRET state arises from a second binding mode in which KF is positioned further from the primer 3′ terminus.
To distinguish these possibilities we repeated the smFRET measurements using a K550C KF construct. This residue within the thumb subdomain maintains a fixed position in the crystal structures of both open and closed polymerase-DNA complexes (Figs. 1b & c) 7,8. If the three FRET states seen in the L744C KF construct are due to three different conformations of the fingers subdomain, then only a single species should be observed for the bound K550C KF construct. Alternatively, if the 0.41 FRET state arises from a different binding position of the enzyme, then at least two distinct FRET states must be resolved in the K550C KF - DNA complex. A typical smFRET trajectory reveals short lived binding events, as for L744C KF, but in each binding event the same FRET state is accessed (Fig. 3a, left panel). Moreover, the FRET efficiency histogram compiled from 221 individual trajectories shows just a single peak, centered at a FRET efficiency of 0.78 (Fig. 3a, right panel). This is in contrast with the three-peak behavior observed for L744C KF - DNA complexes. The higher FRET efficiency observed for K550C KF is consistent with a shorter distance from the label on the thumb to the label on DNA (32.1 Å, excluding the linker contributions), as expected from crystal structures of Bst Pol I 8. Therefore, we conclude that the 0.41, 0.50 and 0.63 FRET states in the L744C KF – DNA complex represent three conformations of the fingers subdomain: open, intermediate and closed, respectively. We denote the intermediate conformation as “ajar”, in reference to a recently reported crystal structure of Bst Pol I13 (see Discussion). The fractional occupancies of the different FRET states (Fig. 2a) show that the open conformation is the most populated state (44%), while the ajar (~19%) and closed (~24%) conformations are about evenly populated.
Figure 3. Fluorescence time traces, smFRET efficiency trajectories and FRET efficiency histograms for thumb-labeled polymerase molecules (K550C KF).

(a) Binding events of acceptor-labeled K550C KF polymerases to immobilized donor-labeled DNA molecules produced short-lived FRET bursts. A composite FRET efficiency histogram compiled from 228 trajectories shows that binary Pol+Atempl complexes populate a single 0.78 FRET state, in contrast with the three-state behavior of L744C KF. (b) Correct Pol+Atempl+dTTP complexes formed in the presence of 1 mM dTTP in the solution demonstrate that K550C molecules exhibit a prolonged period (exceeding 5 s) in a 0.80 FRET state. The composite histogram is compiled from 208 trajectories. (c) In the presence of 1 mM incorrect dATP nucleotides, K550C KF demonstrates frequent sampling of a 0.81 FRET state, reflecting formation of unstable incorrect ternary Pol+Atempl+dATP complexes. The FRET efficiency histogram (compiled from 228 trajectories) also reveals a sub-population (24%) of complexes captured in a 0.7 FRET state. In all cases, the green, red and blue traces correspond to the donor intensity, acceptor intensity and corresponding FRET efficiency trajectories, respectively. The dashed lines positioned at 0 and 0.8 FRET efficiencies are for ease of visual inspection. Gaussian fits to the histogram peaks are shown as black lines. The red line in panel c represents a composite sum of the two fitted Gaussian peaks.
The smFRET efficiency time trajectories obtained with either KF construct (L744C or K550C) show only brief periods in bound FRET states (Figs. 2a and 3a), indicating that binary KF - DNA complexes dissociate rapidly. Generally, it appears that L744C KF can dissociate from the DNA regardless of which conformational state it occupies prior to dissociation (open, ajar or closed) (Fig. 2a). Dwell-time analysis of the smFRET trajectories reveals a dissociation rate of 1.30 ± 0.14 s−1 for the L744C KF – DNA complex (an overall value for all three bound states). During the brief periods that L744C KF is bound to DNA, the polymerase mostly stays in a single conformation, although some interconversion events were captured during longer binding periods. Notably, transitions were observed between the open, ajar and closed conformations (Fig. S2, Supporting Information), indicating that these states are interconnected in a kinetic pathway. However, because relatively few interconversion events were detected prior to complex dissociation, the rate constants for conformational exchange are not well defined.
The FRET efficiency histogram for the L744C binary complex also shows a smaller separate peak at 0.90 FRET efficiency (Fig. 2a). Because the difference in FRET efficiency is large in comparison to the 0.41 FRET state associated with the open conformation of the fingers (ΔFRET ~ 0.5), it is highly unlikely that this state arises from another conformation of the fingers. Instead, the 0.90 FRET state may correspond to a subpopulation of primer/templates bound at the 3′-5′ exonuclease (exo) site, which is separated from the polymerase (pol) active site by 35 Å 14. To test this hypothesis, we introduced a mutation at the exo site (L361A) that is known to disrupt DNA binding 15. The FRET efficiency histogram for the L744C/L361A double mutant KF construct shows no species with 0.9 FRET efficiency, but is otherwise similar to the histogram of the L744C single mutant KF (Fig. S3, Supporting Information). Hence, the 0.90 FRET peak observed for L744C KF represents DNA bound at the exo site. The high FRET efficiency indicates that the donor label on the DNA has moved closer to the acceptor label on the fingers subdomain when the DNA primer terminus is bound at the exo site. The fractional population of this species (13%) is consistent with the results of previous time-resolved fluorescence anisotropy measurements of pol-exo partitioning in Pol I KF, which reveal that 7–16% of fully base paired substrates bind at the exo site, depending on the specific sequence of the DNA 16.
Ternary complexes with the correct nucleotide substrate favor a closed fingers state and stabilize the polymerase on DNA
Previous ensemble kinetic studies of Klentaq 1 (Klenow fragment analog of Thermus aquaticus DNA polymerase) and Pol I KF polymerases have shown that the presence of a correct nucleotide substrate induces fingers closure 9–11. We repeated the smFRET experiments in the presence of the correct nucleotide substrate (dTTP, complementary to the dA template base, Fig. 1a). The smFRET time trajectories for L744C KF in the presence of a saturating 1 mM concentration of dTTP show prolonged periods (exceding 5 s) in the closed conformation (0.63 FRET, Fig. 2b). The FRET efficiency histogram for the correct dTTP:dA complexes (244 individual trajectories) shows that the majority of complexes (89%) occupy the closed fingers conformation (Fig. 2b). The peaks for the open and ajar states are not individually resolved in this case but instead appear as a shoulder around 0.45 FRET efficiency (11% combined population) (Fig. 2b). We suppose that the inability to capture more ternary complexes with open and ajar conformations of the fingers subdomain in our smFRET experiments is due to the short lived duration of these states when the correct nucleotide substrate is present. Previous ensemble kinetic studies of KF have reported a rate of 140 s−1 for closure of the fingers in the presence of a correct nucleotide substrate 11. Based on this rate, the open and ajar states would not be resolved in our experiments with a 100 msec integration time.
Notably, there is no species with 0.9 FRET efficiency (exo site) in the FRET histogram (Fig. 2b), indicating that the DNA binds predominantly at the pol site in the presence of the correct nucleotide substrate. Dwell-time analysis of the 244 individual trajectories shows that the dissociation rate of the ternary complex is 0.42 ± 0.08 s−1, slower than the corresponding value for the binary complex (1.30 ± 0.14 s−1). Taken together, these results indicate that the correct dTTP nucleotide promotes binding of DNA at the pol site, favors the closed conformation of the fingers subdomain and prolongs the residence time of Pol I KF on DNA. The polymerase is presumably poised for the phosphoryl transfer reaction under these conditions, but this reaction is blocked in our experiments.
Prolonged binding periods were also observed for the thumb-labeled K550C KF construct in the presence of dTTP (Fig. 3b). The FRET efficiency histogram compiled from 208 trajectories shows a single peak centered at 0.80 FRET efficiency, similar to the binary K550C complex, as expected (Fig. 3b).
Ternary complexes with mismatched nucleotide substrates preferentially populate the ajar fingers conformation
We hypothesize that the ajar conformation of the fingers subdomain embraces a functional role in dNTP selection and may serve as a fidelity checkpoint for incoming substrates. To test this hypothesis, we next performed a series of smFRET experiments in the presence of either dATP, dCTP or dGTP (1 mM), each of which is a mismatched nucleotide for the dA templating base (Fig. 1a). A typical smFRET trajectory for the dATP:dA mismatched pair (Fig. 2c) demonstrates that the incorrect ternary complex mostly populates open and ajar states, coupled with rapid dissociation and rebinding. A FRET efficiency histogram for incorrect dATP:dA complexes (299 individual trajectories) shows a major redistribution among the open, ajar and closed conformations (Fig. 2c, right panel). The fraction of complexes occupying the ajar conformation is markedly increased (38%) compared with the binary complex (19%), while the fraction occupying the open conformation is reduced (19%) compared to the binary complex (44%). The fraction of complexes occupying the closed conformation is also reduced (18%) compared with the binary (24%) and, especially, correct ternary (89%) complexes.
In addition to the redistribution among open, ajar and closed conformations, the peak at 0.90 FRET efficiency (due to DNA bound at the exo site) shows an increased population (25%) in the incorrect dATP:dA complexes compared to the corresponding peak in the binary complex (13%). Hence, the incorrect dATP substrate also leads to increased occupancy of the exo site. In the K550C KF data, the presence of the second binding mode appears as a shoulder at ~ 0.7 FRET efficiency (24%), in addition to the previously identified 0.80 FRET state (76%) (Fig. 3c).
A dwell-time analysis showed that L744C KF dissociates rapidly from the DNA when the incorrect dATP is present, with an overall off-rate of 1.26 ± 0.14 s−1, significantly faster than the correct ternary complex (0.42 ± 0.08 s−1). The incorrect nucleotides dCTP and dGTP produced similar behavior to dATP (Fig. S4, Supporting Information).
To verify the selection behavior of the enzyme under more natural conditions, we also performed smFRET experiments for L744C KF in a reaction mixture with all four substrate nucleotides present, which were added in equal volume fractions (total concentration of dNTPs was kept at 1 mM). The FRET efficiency histogram obtained for this mixture demonstrated the same four peaks described above (Fig. S5, Supporting Information), but with a redistribution of species populating open (23%), ajar (21%) and closed (49%) conformations, and a significant decrease of the 0.90 FRET (7%) species (exo site).
To test whether the specific identity of the templating base has any effect on the nucleotide selection process, we performed similar experiments for L744C KF using a DNA construct with a dC templating base (otherwise identical). The results for the binary complex were similar to those obtained with the original dA template (Fig. S6, Supporting Information). Notably, addition of dGTP to the dC template produced prolonged periods (exceding 5 s) in the closed conformation (Fig. S6), similar to the behavior of the correct dTTP:dA complex (Fig. 2b). Hence, the specific identity of the template base and incoming nucleotide are unimportant, so long as these moities are complementary. Likwise, incorrect ternary complexes formed with the dC template (dATP:dC) exhibited similar behavior (Fig. S6) to incorrect complexes formed with the dA template (Fig. 2c). Hence, the behaviors reported here for correct and incorrect ternary complexes are likely to be general and independent of the specific identity of the template base.
DISCUSSION
DNA polymerases perform highly accurate DNA replication by selecting the correct nucleotide substrate and rejecting incorrect substrates during each cycle of nucleotide incorporation. Despite the critical role of correct nucleotide selection in polymerase fidelity, the underlying molecular mechanisms are still not fully understood. One current hypothesis is that all incoming nucleotides, whether correct or incorrect, bind to the fingers subdomain in the open conformation and are subsequently delivered to the polymerase active site, to pair or mispair with the template base while in the closed conformation. In this model, only the correct nucleotide is able to stabilize the fingers subdomain in the closed conformation 9,12. An alternative hypothesis is that a third (unknown) conformation of the polymerase-DNA complex allows the incoming nucleotide substrate to be “previewed” by the template base before the enzyme is committed to the closure step 5. The goal of the present study was to elucidate the mechanism of nucleotide substrate selection, and the role of the fingers subdomain, in DNA Pol I KF.
By placing an acceptor label on the fingers subdomain of Pol I KF and enabling its interaction with surface-tethered donor-labeled DNA, we created a smFRET system to monitor the conformational dynamics of the fingers as individual polymerase molecules perform nucleotide substrate selection. Using this system, we could readily resolve open and closed conformations of Pol I KF bound to DNA, by virtue of their markedly different FRET efficiencies (0.41 and 0.63, respectively). Remarkably, in between these two states, we have also resolved a third conformation of the fingers subdomain (0.50 FRET efficiency). This intermediate state is highly populated in the presence of an incorrect nucleotide substrate (dATP:dA, Fig. 2c), but practically disappears when the correct dNTP is available in the solution (dTTP:dA, Fig. 2b). As we were completing analysis of our experimental data, an X-ray crystal structure of the Pol I KF homolog Bst Pol I bound to DNA and a mismatched deoxynucleoside triphosphate was reported 13. Importantly, the structure of this polymerase caught in the act of binding a mismatched nucleotide (dTTP:dG) revealed that the enzyme adopts an “ajar” conformation in between the previously established open and closed states of the fingers (Fig. 4b). Importantly, the donor-acceptor distance predicted from this new crystal structure (PDB code 3HP6) is 47.0 Å, intermediate between the corresponding values estimated from open, 50.3 Å, and closed, 42.2 Å, structures of Bst Pol I 8. Accordingly, the FRET efficiency expected for an ajar conformation will be intermediate between that of the open and closed conformations. Hence, it is likely that the intermediate FRET state resolved in our smFRET experiments corresponds to an ajar conformation of the fingers subdomain, which also exists in Pol I KF.
Figure 4. Proposed reaction pathway steps at Pol I KF active site preceding phosphodiester bond formation.

The model is based on the results of the present smFRET study and crystal structures of the close KF homologue Bst Pol I8,13. (a) In the initial step, the complex of polymerase, DNA and incoming dNTP adopts an open conformation of the fingers (left). The structure shown is based on the binary complex of Bst Pol I (PDB file 1L3U), with a dNTP positioned adjacent to the O-helix by model building. Only the templating base, incoming dNTP, O-helix and conserved tyrosine residue (Tyr766 in Pol I KF) are shown. The open conformation exists in equilibrium with ajar and closed conformations. If the nucleotide substrate is correct, the ternary complex rapidly progresses to the closed conformation. The resulting structure (upper right) is based on the closed complex of Bst Pol I with dCTP paired with template dG (PDB code 1LV5). The intermediate ajar conformation of the fingers is only fleetingly populated in our experiments when the correct dNTP is present. In contrast, the polymerase is largely blocked in the ajar conformation when an incorrect nucleotide is bound (lower center). The structure shown is based on a ternary complex of Bst Pol I with dTTP mispaired with template dG (PDB code 3HP6). In this structure, the template base has rotated from the pre-insertion to insertion site, allowing mispairing with the incoming dNTP. (b) An overlay of three crystal structures of Bst Pol I provides a comparison of the fingers subdomain positions in open, ajar and closed conformations 8,13. The O helix of the fingers subdomain (encompassing amino residues 732–766 in KF) is shown in ribbons, except for the conserved tyrosine and two post-insertion site DNA base pairs, which are shown as sticks. Incoming nucleotides have been omitted from the ajar and closed conformations in panel (b) for clarity. All models were generated using PyMol.
It is interesting to contrast our results with a recent study of Pol I KF reported by others using a different single-molecule FRET strategy 12. In that study, the donor and acceptor dyes were both covalently attached to the enzyme, at position 744 within the fingers and position 550 in the thumb. The doubly labeled L744C/K550C polymerase was monitored in solution by means of single-molecule confocal microscopy. With this experimental approach, the behavior of the unliganded enzyme could be probed, as well as complexes containing DNA and nucleotide substrates. Very fast time resolution (~ 3 msec) was achieved, although the overall observation period was limited because complexes could rapidly diffuse out of the confocal volume. A binary complex of KF with DNA showed two clearly resolved FRET states, with FRET efficiencies of 0.5 and 0.7, that were assigned to open and closed conformations of the fingers subdomain, respectively. Moreover, the closed state was strongly favored in the presence of the correct nucleotide substrate, consistent with our observations. However, an intermediate (ajar) conformation of the fingers subdomain was not resolved in the previous study. Intriguingly though, the low FRET peak was observed to shift slightly towards higher FRET efficiency as an incorrect nucleotide was introduced. This behavior suggested that a third species might also have been present but was not resolved from the open conformation. In our study, we have clearly discriminated the open and ajar conformations and shown that the latter is strongly favored by an incorrect nucleotide substrate. The large number of repetitive single-molecule events registered in our experiments was likely crucial to resolve the three distinct conformations of KF bound to DNA. In addition, by measuring FRET between the DNA substrate and the polymerase, we have been able to identify subpopulations in which DNA is bound to the pol site or exo site of Pol I KF. Notably, we have discovered that the preference for one binding mode or the other is also sensitive to the identity of the incoming nucleotide substrate. Finally, by tethering the DNA substrate we significantly extended the time window of our smFRET observations, providing additional information on the kinetics of enzyme dissociation from the DNA.
Our smFRET results reveal that the polymerase-DNA complex naturally exists in an equilibrium between open, ajar and closed conformational states. Accordingly, we propose a three-state mechanism for nucleotide substrate selection by Pol I KF. A plausible model for the species encountered on the pre-chemistry pathway is shown in Fig. 4, based on crystal structures of the KF homolog Bst Pol I 8,13. In the first step, any nucleotide substrate, whether correct or incorrect, can bind to the fingers and form an open ternary complex with the polymerase (Fig. 4a, left). In this conformation, the template base is sequestered in the pre-insertion site and is distant from the incoming dNTP. Moreover, the intervening space is occupied by a highly conserved tyrosine residue from the O-helix (Tyr766 in Pol I KF), which has been recognized as a key residue mediating fingers closure 13. Accordingly, it is unlikely that KF can discriminate between correct and incorrect nucleotides while in the open conformation. During the second step, the polymerase-DNA-dNTP complex spontaneously converts to the ajar conformation, allowing the template base to establish hydrogen bonding interactions with the incoming nucleotide (Fig. 4a, center). Hence, KF can likely discriminate between correct and incorrect nucleotides while in the ajar conformation. If the incoming nucleotide is correct, the ternary complex seamlessly progresses to the closed conformation, with the nascent Watson-Crick base pair enclosed by the fingers (Fig. 4a, right), creating a long lived ternary complex. This step occurs so rapidly with a correct nucleotide that the ajar intermediate is hardly detectable in our experiments. In contrast, if the incoming nucleotide is incorrect, the polymerase either reverts to the open conformation or remains trapped in the ajar conformation, in either case dissociating rapidly from the DNA. The ajar conformation, therefore, exhibits the hallmarks of a kinetic checkpoint that discriminates between correct and incorrect nucleotides before the polymerase encloses the nascent base pair.
In addition, our results suggest that the 3′-5′ exonuclease site also plays a role in substrate selection. The normal role of the exo site is to remove misincorporated nucleotides through hydrolytic cleavage of the terminal phosphodiester bond 1. DNA substrates naturally partition between the spatially separated pol and exo sites of KF, according to the base sequence of the DNA, especially the presence of mismatches at or adjacent to the primer terminus KF15. This partitioning is evident in our smFRET data for the binary complex of L744C KF and DNA, with 13% of the DNA population bound at the exo site (manifested as a separate peak in the FRET histogram at 0.90 efficiency), as expected for a fully base paired primer/template 16. Interestingly, the population of the exo site increased to 25% in the presence of the incorrect dATP substrate (Fig. 2c), a value that is characteristic of a mispaired primer terminus 16. This raises the intriguing possibility that the mismatched nascent base pair formed upon binding of an incorrect dNTP triggers movement of the primer terminus from the pol site to the exo site, even though the incoming nucleotide has not been covalently incorporated into the DNA. Shifting the primer terminus to the remote exo site may serve as an additional mechanism to prevent misincorporation of an incorrect substrate at the pol active site. In contrast, we found that a correct nucleotide substrate strongly favored binding of DNA at the pol site (Figs. 2b and S3), presumably in readiness for the nucleotide incorporation reaction. This is consistent with a recent study of Pol I KF-DNA complexes using low-energy CD spectroscopy of fluorescent base analogs, which revealed a shift in the distribution of DNA in favor of the polymerase site when a correct nucleotide substrate was supplied 17. Our observations suggest that the exo site plays a role in substrate selection before nucleotide incorporation, in addition to its known editing function after incorporation. This situation is reminiscent of proofreading by aminoacyl tRNA synthetases, where proofreading also occurs during selection of the incoming amino acid substrate as well as at the level of post-synthetic hydrolysis18.
The results of this study provide significant new insights into the role of the fingers subdomain and the 3′-5′ exonuclease domain in nucleotide substrate selection and polymerase fidelity. Clearly, the fingers subdomain is more than a passive delivery system for incoming nucleotides, but instead plays a key role in substrate selection, discriminating between correct and incorrect substrates while in the ajar conformation. Our results also reveal that the exo domain plays a role in nucleotide substrate selection. Whereas a correct nucleotide substrate favors binding of DNA at the pol site with the fingers in a closed conformation, an incorrect nucleotide traps the fingers in the ajar conformation and sequesters a significant fraction of the DNA at the remote exo site. Together, both mechanisms help to suppress misincorporation of incorrect nucleotide substrates. The relationship between enzyme conformational dynamics and proper substrate selection as demonstrated by our current data is likely relevant to other enzymatic reactions. In conclusion, the findings of this study provide direct evidence for the existence of a fidelity checkpoint preceding the enzyme closure step and open new avenues for investigating the contribution of pre-chemical checkpoints to the overall nucleotide incorporation fidelity of DNA polymerases.
EXPERIMENTAL PROCEDURES
Expression of KF derivatives
KF mutants carrying L744C and K550C cysteine substitutions, or L744C/L361A substitutions, were generated from the D424A/C907S KF genotype construct generously provided by Dr. Catherine Joyce (Yale University) using a QuickChange kit (Stratagene). C907S and D424A mutations remove the single native cysteine and suppress 3′-5′ exonuclease activity, respectively. Expression and purification of the KF mutants was carried out as described previously 19.
Protein labeling
Labeling of KF mutants with sulfhydryl-specific dyes was based on our previous procedures 10,20. Typically, 100 nmol of KF was incubated with a 3–5 fold molar excess of Alexa-Fluor 594 maleimide (Invitrogen) in 50 mM sodium phosphate buffer, pH 7.0, for 2 hours at room temperature. The protein concentration in the reaction mixture was at 100 μM. The excess free dye was removed by use of a Sephadex-G25 gel-filtration column equilibrated with 50 mM sodium phosphate buffer, pH 7.5, 1 mM DTT and 1.7 M (NH4)2SO4. The labeled and unlabeled KF proteins were separated on a FPLC system (Akta, GE Healthcare) equipped with a Resource ISO column (GE Healthcare). The purity and specificity of labeled KF constructs was characterized by SDS-PAGE and ESI–TOF mass-spectrometry (Agilent) (Fig. S7). The degree of labeling was typically 100% after FPLC purification. Protein concentrations were calculated based on the optical absorption measured at 280 nm using extinction coefficient ε280 = 5.88 × 104 M−1 cm−1. The protein was stored at − 80° C in a buffer containing 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 mM DTT and 50 % (vol/vol) glycerol.
Oligonucleotides
The smFRET system utilizes a DNA construct consisting of a 17 nt primer annealed to a 54 nt template, producing a 7 nt single-stranded template overhang at the 5′ end with a dA templating base and a T30 linker and biotin group at the 3′ end for attachment to a streptavidin coated quartz slide surface (Fig. 1a). Primers and 5′-biotin-labeled templates were purchased from Eurofins MWG Operon. All oligonucleotides were purified by denaturing electorphoresis in 20% (weight/vol) polyacrylamide gels. Primers were dideoxy terminated by incubating 100 nmol primer, 100 units of terminal deoxynucleotidyl transferase (New England Biolabs) and 1000 nmol dideoxy nucleotide triphosphate (ddNTP) for at least 12 hours at 37° C. The reaction buffer contained 20 mM Tris-acetate, 50 mM potassium acetate, 10 mM magnesium acetate, pH 7.9 and 0.25 mM CaCl2. The reaction was halted by heating at 90° C for 10 minutes, followed by desalting using a Nap10 column (GE Healthcare) and gel purification by 20% denaturing PAGE.
smFRET measurements
smFRET data collection was performed using a custom built prism-based TIRF microscope based on an inverted Axiovert 200 microscope (Zeiss). A 488 nm laser beam from a solid state laser (Coherent, 50 mW) was shaped using a corresponding set of optics and delivered to a quartz prism positioned on a specially designed slide holder (TIRF Technologies). The laser spot size in the imaging area on the slide was ~0.07 mm2 and the excitation intensity was ~ 8 W/cm2. Primer/template duplexes were introduced into the sample chamber and allowed to bind to the streptavidin-coated surface. Binary complexes were formed by subsequently introducing 10–35 nM KF into the sample chamber, while ternary complexes additionally contained 1 mM dNTP. Fluorescence emission was collected by a water-immersion C-Apochromat 63X/1.2 W Corr UV-VIS-IR objective (Zeiss). The fluorescence signal was pre-filtered from the scattered excitation light by a dichroic mirror (z488rdc, Chroma) in the epi-detection channel, and split into donor and acceptor detection channels using a Dual-View Imager system (QImaging/Photometrics) containing a 545dcxr dichroic mirror and 525/50 (FF02-525/50, Semrock) and 620/60 (HQ620/60, Chroma) band-pass filters in the short wavelength and long wavelength channels, respectively. Fluorescence image movies were recorded by a EMCCD camera (iXonEM+, 512×512, 16 μm2 pixels, Andor Technology) for 2–5 minutes, with an integration time of 100 ms per frame, 1 pixel binning. For each slide, several complete movies (2 minute duration) were recorded in various fields of view on the slide. For each experimental sample, at least 5 independent data sets (using freshly prepared sample and a new sample chamber) were recorded and each data set was analyzed separately. Composite FRET efficiency histograms were compiled from individual traces selected from two or three independent experiments. A custom written single-molecule data acquisition package (downloaded from https://physics.illinois.edu/cplc/software/) was used in combination with IDL (ITT VIS) to record data and generate matched pairs of single-molecule fluorescence intensity traces from the imaging movies.
smFRET data analysis
Individual intensity trajectories in the donor and acceptor channels were corrected for their respective background signals (an average signal recorded in the areas where no fluorescent spots were visible). In addition, the acceptor signal was corrected for 2% leakage of Alexa-Fluor 488 emission into the acceptor channel, which was determined by recording a two-channel data set with only Alexa-Fluor 488 molecules available on the slide, using the same laser power as in the polymerase experiments. The corrected intensity trajectories were used to calculate FRET efficiency trajectories, according to the formula E =IA/(γID + IA), where E is the FRET efficiency and ID and IA are the donor and acceptor intensities, respectively, and γ is a correction factor that accounts for differences in quantum yield and detection efficiency between donor and acceptor. Since γ was set to unity, the reported FRET efficiencies are apparent rather than absolute values. Each trajectory was analyzed using custom code written in MATLAB (Version R2011a, MathWorks) as described previously 21,22. Trajectories exhibiting anti-correlated fluctuations of the donor and acceptor emission and single-step photobleaching were selected for analysis. The FRET traces were binned to generate a FRET histogram for each analyzed time trace and a composite FRET histogram was then compiled from multiple trajectories, using IGOR Pro (Version 6, WaveMetrics). Individual peaks in the FRET histograms were fitted with Gaussian functions, using IGOR Pro or Origin software, and the area under each peak determined the percentage of complexes in the corresponding state. Kinetic rate constants were obtained after analyzing FRET time traces with a hidden Markov model, using the program HAMMY, as described23.
Supplementary Material
Acknowledgments
We thank Edwin Van der Schans for expert technical assistance in oligonucleotide labeling and purification, as well as mutagenesis, expression, labeling and purification of Pol I KF derivatives. This work was supported by the US National Institute of General Medical Sciences (grant GM044060 to DM).
Footnotes
Supporting Information Available: it consists of supplemental experimental procedures and seven figures combined in a single pdf file, which includes information on oligonucleotide labeling; effects of fluorophore labels on nucleotide incorporation activity; sample preparation for smFRET measurement; interconversion among different FRET states for L744C KF polymerase bound to DNA; histogram of smFRET efficiencies for binary L744C/L361A KF - DNA complexes; fluorescence intensity time traces and smFRET efficiency trajectories for L744C KF molecules binding to DNA primer/templates with dA in the template extension position in the presence of incorrect substrates dGTP and dCTP; histogram of smFRET efficiencies for L744C KF complexes in the presence of all four nucleotide substrates; fluorescence intensity time traces and smFRET efficiency trajectories for L744C KF polymerases binding to DNA molecules with dC in the template extension position; ES-TOF mass spectra of non-labeled and labeled KF derivatives.
References
- 1.Kunkel TA. J Biol Chem. 2004;279:16895. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 2.Dahlberg ME, Benkovic SJ. Biochemistry. 1991;30:4835. doi: 10.1021/bi00234a002. [DOI] [PubMed] [Google Scholar]
- 3.Eger BT, Benkovic SJ. Biochemistry. 1992;31:9227. doi: 10.1021/bi00153a016. [DOI] [PubMed] [Google Scholar]
- 4.Wong I, Patel SS, Johnson KA. Biochemistry. 1991;30:526. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 5.Joyce CM, Benkovic SJ. Biochemistry. 2004;43:14317. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 6.Ollis DL, Brick P, Hamlin R, Xuong NG, Steitz TA. Nature. 1985;313:762. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Korolev S, Waksman G. EMBO J. 1998;17:7514. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson SJ, Taylor JS, Beese LS. Proc Natl Acad Sci U S A. 2003;100:3895. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothwell PJ, Mitaksov V, Waksman G. Mol Cell. 2005;19:345. doi: 10.1016/j.molcel.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 10.Stengel G, Gill JP, Sandin P, Wilhelmsson LM, Albinsson B, Norden B, Millar D. Biochemistry. 2007;46:12289. doi: 10.1021/bi700755m. [DOI] [PubMed] [Google Scholar]
- 11.Joyce CM, Potapova O, DeLucia AM, Huang X, Basu VP, Grindley NDF. Biochemistry. 2008;47:6103. doi: 10.1021/bi7021848. [DOI] [PubMed] [Google Scholar]
- 12.Santoso Y, Joyce CM, Potapova O, Le Reste L, Hohlbein J, Torella JP, Grindley NDF, Kapanidis AN. Proc Natl Acad Sci U S A. 2010;107:715. doi: 10.1073/pnas.0910909107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu EY, Beese LS. J Biol Chem. 2011;286:19758. doi: 10.1074/jbc.M110.191130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freemont PS, Friedman JM, Beese LS, Sanderson MR, Steitz TA. Proc Natl Acad Sci U S A. 1988;85:8924. doi: 10.1073/pnas.85.23.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam WC, Van der Schans EJ, Joyce CM, Millar DP. Biochemistry. 1998;37:1513. doi: 10.1021/bi9720181. [DOI] [PubMed] [Google Scholar]
- 16.Carver TE, Jr, Hochstrasser RA, Millar DP. Proc Natl Acad Sci U S A. 1994;91:10670. doi: 10.1073/pnas.91.22.10670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Datta K, Johnson NP, von Hippel PH. Proc Natl Acad Sci U S A. 2010;107:17980. doi: 10.1073/pnas.1012277107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fersht AR. Proceedings of the Royal Society of London Series B Biological Sciences. 1981;212:351. doi: 10.1098/rspb.1981.0044. [DOI] [PubMed] [Google Scholar]
- 19.Joyce CM, Derbyshire V. Methods Enzymol. 1995;262:3. doi: 10.1016/0076-6879(95)62003-6. [DOI] [PubMed] [Google Scholar]
- 20.Thompson EH, Bailey MF, van der Schans EJ, Joyce CM, Millar DP. Biochemistry. 2002;41:713. doi: 10.1021/bi0114271. [DOI] [PubMed] [Google Scholar]
- 21.Zhuang X, Kim H, Pereira MJ, Babcock HP, Walter NG, Chu S. Science. 2002;296:1473. doi: 10.1126/science.1069013. [DOI] [PubMed] [Google Scholar]
- 22.Rueda D, Bokinsky G, Rhodes MM, Rust MJ, Zhuang X, Walter NG. Proc Natl Acad Sci U S A. 2004;101:10066. doi: 10.1073/pnas.0403575101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKinney SA, Joo C, Ha T. Biophys J. 2006;91:1941. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.