

Abstract
The use of a cationic cyclization reaction as a probe of glycosylation mechanism is developed and applied to the 4,6-O-benzylidene-protected mannopyranoside system. Cyclization results in the formation of both cis-and trans-fused tricyclic systems invoking an intermediate glycosyl oxocarbenium ion reacting through a boat conformation. Competition reactions with isopropanol and trimethyl methallylsilane are interpreted as indicating β-O-mannosylation to proceed via an associative SN2-like mechanism, whereas α-O-mannosylation and β-C-mannosylation are dissociative and SN1-like. Relative rate constants for reactions going via a common intermediate can be estimated.
Glycosylation is the substitution of a leaving group in a glycosyl donor by an acceptor alcohol, often with the aid of a promoter, for which there are two extreme mechanisms, uni- and bimolecular nucleophilic substitution bridged by a continuum of more or less tightly bound ion pairs.1 Reaction mechanisms are typically based on combinations of stereochemical and kinetic evidence, and are supported whenever possible by the characterization of any predicted intermediates and by computational work. In glycosylation stereochemical evidence in terms of the anomeric selectivity of a coupling is readily available. Kinetic evidence on the other hand is rare,1a,2 particularly when it is required for both anomers, and is difficult to obtain because of the multicomponent nature of most glycosylation reactions. Furthermore, the most widely invoked mechanism for glycosylation involves the intermediacy of a glycosyl oxocarbenium ion, a species which, despite much effort,3 has never been observed other than in silico4 or in a mass spectrometer.5 We seek to develop methods for the determination of reaction kinetics for individual glycosylations so as to facilitate their rational optimization and provide evidence for or against the involvement of glycosyl oxocarbenium ions.2c In view of the frequent difficulties faced in obtaining absolute kinetic data for glycosylations, we conceived that relative kinetics would be helpful and that such data might be obtained through the use of a competing cyclization reaction as a clock. We report on the implementation of such a competition kinetic scheme and through it on the distinction between the mechanisms of α- and β-O and β-C mannopyranosylation in the presence of 4,6-O-benzylidene acetal group.
Mayr has developed a series of reference scales for the characterization of cationic electrophiles and neutral nucleophiles and has discussed their potential use to predict changes between SN1 and SN2 reactions.6 These scales, however, make use of the intermolecular trapping of a series of chromophoric cations and, consequently, are not readily adaptable to our purposes. The diffusion-controlled azide clock reaction has been developed for the determination of the kinetics of acetal hydrolysis in aqueous solution and used to estimate the lifetimes of various glycosyl oxocarbenium ions under those conditions,7 but these methods have not been applied to actual glycosidic bond forming reactions conducted at low temperature in organic solution. Cognizant of the impact of rearrangements as clocks for the determination of relative kinetics in the field of radical chemistry,8 and drawing on experience with cyclization of activated glycosyl donors onto protecting groups9 and parallels with the intramolecular aglycone delivery method of glycosidic bond formation,10 we considered that ring closure onto appropriately designed substituents would provide a suitable clock reaction for the determination of relative kinetics in glycosylation reactions.
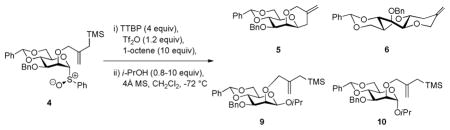
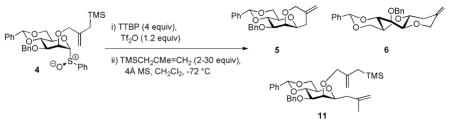
To avoid complications from the formation of stereogenic centers during the cyclization reaction we designed a system based on an intramolecular Sakurai reaction11 and, so, on the use of 2-O-(2-trimethylsilylmethyl)allyl ethers. Accordingly, regioselective benzylation of diol 1 in the standard manner12 gave the 3-O-benzyl ether 2,13 which was alkylated with 2-chloromethyl-3-trimethyl-silyl-1-propene and sodium hydride to give the desired trimethylsilylmethallyl ether 3 in moderate yield (Scheme 1). Preferring the use of the glycosyl sulfoxides14 over the thioglycosides for mechanistic work because of the simpler activation protocol and cleaner reaction mixtures, 3 was then oxidized with m-CPBA to give the sulfoxide 4 as a 16:1 diastereomeric mixture in which the major isomer is assigned the (R)S isomer consistent with the precedent.15 Activation of 4 at −72 °C in dichloromethane in the presence of 2,4,6-tri-tert-butylpyrimidine (TTBP)16 gave, after quenching at −72 °C, two cyclization products (Scheme 1). The major product 5 was identified as the anticipated cis-fused system, whereas the minor isomer was the unexpected trans-fused product 6. Both cyclization products were confirmed by X-ray crystallography (Figure 1), which reveals the pyranose ring of 5 to adopt the 4C1 chair conformation, while that of the minor trans-fused isomer 6 takes up the 1S5 twist boat conformation.
Scheme 1.

Preparation and activation of sulfoxide 4; formation of tricyclic compounds 5 and 6.
Figure 1.

X-Ray crystallographic structures of tricycles 5 and 6
We rationalize the formation of the trans-fused product 6 by invoking a mannosyl oxocarbenium ion 8 that exists in equilibrium with the α-glycosyl triflate 717 and that accesses the B2,5 conformation previously computed.2c,18 In this conformation the 2-O-silylmethylallyl ether is able to access both the β-face of the cation, leading to the cis-fused product 5,19 and the α-face resulting in the formation of the trans-isomer 6 initially as the OS2 twist boat that then relaxes to the observed 1S5 conformer (Scheme 2). The observation of product 6 provides very strong evidence in support of the existence of a mannosyl oxocarbenium ion in equilibrium with the covalent glycosyl triflate.
Scheme 2.

Mechanism of formation of the tricycles 5 and 6
With the concept established we turned to the deployment of cyclization of the sulfoxide 4 as a clock for a glycosylation reaction. Activation of 4 at −72 °C, in the presence of 1-octene as scavenger of the various electrophilic byproducts, was followed by the rapid addition of isopropanol and led to the formation of the β- and α-mannosides 9 and 10, respectively, along with the two cyclization products 5 and 6 (Table 1). In a series of experiments the ratios of the individual glycosides 9 and 10 formed over the amount of the combined cyclization products 5 and 6 produced were determined as a function of the amount of isopropanol added resulting in the data presented in Table 1 and plotted in Figure 2.
Table 1.
O-Glycosylation in competition with cyclization
| |||
|---|---|---|---|
| Entrya | i-PrOHb | 9/(5+6)c | 10/(5+6)c |
| 1 | 0.8 (0.014) | 2.17 | 0.15 |
| 2 | 1.2 (0.020) | 3.66 | 0.28 |
| 3 | 1.5 (0.026) | 5.36 | 0.44 |
| 4 | 2.5 (0.043) | 10.99 | 0.99 |
| 5 | 3 (0.051) | 13.09 | 1.28 |
| 6 | 4 (0.068) | 15.75 | 1.14 |
| 7 | 5 (0.085) | 19.38 | 1.53 |
| 8 | 8 (0.136) | 24.34 | 1.60 |
Experimental conditions: TTBP (4 equiv), 1-octene (10 equiv), molecular sieves 4 Å, Tf2O (1.2 equiv) at − 72 °C;
equiv (conc, M);
Molar ratios were determined by UHPLC/UV/MS
Figure 2.

O- and C-Glycoside to cyclized products ratio as a function of nucleophile concentration
Comparable experiments were also conducted using trimethyl methallylsilane as external nucleophile resulting in a competition between cyclization and C-glycoside formation (Table 2 and Figure 2). Consistent with earlier results from our laboratory on the reaction of strong C-nucleophiles with 4,6-O-benzylidene protected mannopyranosyl donors,20 only a single β-anomer of the C-glycoside 11 was formed in the course of these experiments.
Table 2.
C-Glycosylation in competition with cyclization
| ||
|---|---|---|
| Entrya | TMSCH2C(Me)=CH2b | 11/(5+6)c |
| 1 | 2 (0.034) | 0.06 |
| 2 | 4 (0.068) | 0.18 |
| 3 | 8 (0.136) | 0.40 |
| 4 | 12 (0.204) | 0.55 |
| 5 | 15 (0.255) | 0.69 |
| 6 | 20 (0.34) | 0.87 |
| 7 | 30 (0.51) | 1.40 |
Experimental conditions: TTBP (4 equiv), molecular sieves 4 Å, Tf2O (1.2 equiv) at − 72 °C;
equiv (conc, M);
Molar ratios were determined by UHPLC/UV/MS
From the graphical representation of the competition experiments presented in Figure 2 it is clear that the rate of formation of the β-O-mannoside 9 shows a strong and more or less linear dependence on the concentration of the nucleophile isopropanol, at least over the initial range of concentrations. The rate of formation of the α-O-mannoside 10 and of the β-C-mannoside 11, on the other hand, both exhibit a much lower dependence on concentration. These results are consistent with the formation of the β-O-mannoside 9 being first order in nucleophile, while that of the α-O-mannoside 10 and the β-C-mannoside 11 is zero order overall in nucleophile. Accordingly, a highly associative mechanism for the formation of the β-O-mannoside 9 that approximates to the SN2-like displacement of the triflate anion from the covalent intermediate 7 by isopropanol, or with the functionally indistinguishable β-face attack by isopropanol on a contact ion pair (CIP) derived by ionization of 7, is indicated in agreement conclusion derived recently from 13C primary kinetic isotope effects studies.2c The formation of the α-O-mannoside 10 and of the β-C-mannoside 11, on the other hand, is clearly the result of a dissociative SN1-like mechanism involving the formation and subsequent trapping of a mannopyranosyl oxocarbenium ion 8, in either a solvent-separated ion pair (SSIP) or as the free ion, also consistent with primary KIE measurements for the formation of α-O-mannosides. The non-zero concentration dependence of the rate of formation of the α-O-mannoside 10 and β-C-mannoside 11 arises from the product forming step when intermolecular nucleophilic attack competes with cyclization for capture of the transient oxocarbenium ion 8.
Assuming that the cyclization products 5 and 6, and the α-O-mannoside and β-C-mannosides, 10 and 11, respectively, are formed via the intermediacy of a common transient cation 8 then, employing the usual steady state approximation, the cyclization may be employed as a unimolecular clock for the determination of relative rate constants of bimolecular additions. Thus, division of the slopes for the relative rates of formation of 10 and 11 (see supporting information) leads to the conclusion that the pseudo-first order unimolecular rate constant for trapping of transient oxocarbenium ion 8 by isopropanol (k10) is approximately 6 times greater than that for trapping of the same intermediate by trimethyl methallylsilane (k11).
Transient oxocarbenium ion 8 shows different face selectivity toward isopropanol and trimethyl methallysilane being apparently α-selective, and at worst unselective,21 toward the former and β-selective toward the latter. This may reflect the fact that the transition states for attack by π-type carbon nucleophiles and σ-type alcohol nucleophiles have different steric requirements and so necessarily result in different selectivities. Indeed, the transition states for O- and C-attack on oxocarbenium ion 8 do not necessarily even involve the same conformation of the electrophile.22 Alternatively, it may be considered that this difference in selectivity arises from differing degrees of association with the counterion in the transition states for the two processes.23
In conclusion the concept of a cationic cyclization reactions as probes for mechanism in glycosylation is developed and is illustrated by application to a 4,6-O-benzylidene protected mannopyranosyl donor. Cyclization takes place via an intramolecular Sakurai reaction and results in the formation of both cis- and trans-fused tricyclic products; a fact which is best interpreted by invocation of a glycosyl oxocarbenium intermediate reacting through a B2,5 conformer. Competition experiments with external nucleophiles indicate that the β-O-mannopyranosides are formed by associative SN2-like mechanisms whereas the α-mannosides are the result of a dissociative SN1-like process. This conclusion, which is a departure from the common rationalization according to which diastereomeric ratios are analyzed in terms of two competing diastereomeric transition states, provides an obvious means of optimization for the β-isomer; it also explains why β-mannosylation of polymer-supported acceptors is relatively unselective24 while that of polymer-supported β-mannosylation donors by an excess of acceptor retains good selectivity.25 This approach to the determination of relative kinetics of glycosylation reactions, which agrees with recent results based on KIE measurements, is straightforward and is potentially applicable to a broad range of glycosyl donors.
Supplementary Material
Acknowledgments
We are grateful to O. Thoison and the HPLC group at the ICSN for help with UPLC analysis and SFC purification. MH thanks the Ministère de l’Education Nationale de la Recherche et de la Technologie for a scholarship. This work was supported in part by the ICSN and NIH (GM62160).
Footnotes
Supporting Information. Full experimental procedures, spectral data for all unknown compounds, and CIF files for 5 and 6. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a) Bohe L, Crich D. CR Chimie. 2011;14:3–16. [Google Scholar]; b) Mydock LK, Demchenko AV. Org Biomol Chem. 2010;8:497–510. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]; c) Beaver MG, Woerpel KA. J Org Chem. 2010;75:1107–1118. doi: 10.1021/jo902222a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Wurst JM, Liu G, Tan DS. J Am Chem Soc. 2011;133:7916–7925. doi: 10.1021/ja201249c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gouliaras C, Lee D, Chan L, Taylor MS. J Am Chem Soc. 2011;133:13926–13929. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]; c) Huang M, Garrett GE, Birlirakis N, Bohe L, Pratt DA, Crich D. Nat Chem. 2012;4:663–667. doi: 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saito K, Ueoka K, Matsumoto K, Suga S, Nokami T, Yoshida J-i. Angew Chem Int Ed. 2011;50:5153–5156. doi: 10.1002/anie.201100854. [DOI] [PubMed] [Google Scholar]
- 4.a) Whitfield DM. Adv Carbohydr Chem Biochem. 2009;62:83–159. doi: 10.1016/S0065-2318(09)00004-3. [DOI] [PubMed] [Google Scholar]; b) Satoh H, Hansen HS, Manabe S, van Gunsteren WF, Hunenberger PH. J Chem Theor Comput. 2010;6:1783–1797. doi: 10.1021/ct1001347. [DOI] [PubMed] [Google Scholar]; c) Rhoad JS, Cagg BA, Carver PW. J Phys Chem A. 2010;114:5180–5186. doi: 10.1021/jp9100448. [DOI] [PubMed] [Google Scholar]
- 5.a) Denekamp C, Sandlers Y. J Mass Spectrom. 2005;40:1055–1063. doi: 10.1002/jms.880. [DOI] [PubMed] [Google Scholar]; b) Denekamp C, Sandlers Y. J Mass Spectrom. 2005;40:765–771. doi: 10.1002/jms.848. [DOI] [PubMed] [Google Scholar]
- 6.a) Mayr H, Bug T, Gotta MF, Hering N, Irrgang B, Janker B, Kempf B, Loos R, Ofial AR, Remennikov G, Schimmel H. J Am Chem Soc. 2001;123:9500–9512. doi: 10.1021/ja010890y. [DOI] [PubMed] [Google Scholar]; b) Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66–77. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]; c) Phan TB, Nolte C, Kobayashi S, Ofial AR, Mayr H. J Am Chem Soc. 2009;131:11392–11401. doi: 10.1021/ja903207b. [DOI] [PubMed] [Google Scholar]; d) Streidl N, Denegri B, Kronja O, Mayr H. Acc Chem Res. 2010;43:1537–1549. doi: 10.1021/ar100091m. [DOI] [PubMed] [Google Scholar]
- 7.a) Amyes TL, Jencks WP. J Am Chem Soc. 1989;111:7888–7900. [Google Scholar]; b) Horenstein BA, Bruner M. J Am Chem Soc. 1998;120:1357–1362. [Google Scholar]; c) Zhu J, Bennet AJ. J Org Chem. 2000;65:4423–4430. doi: 10.1021/jo0004106. [DOI] [PubMed] [Google Scholar]; d) Horenstein NA. Adv Phys Org Chem. 2006;41:275–314. [Google Scholar]; e) Richard JP, Amyes Tl, Toteva MM, Tsuji Y. Adv Phys Org Chem. 2004;39:1–26. doi: 10.1016/B978-0-12-386047-7.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Ingold KU, Griller D. Acc Chem Res. 1980;13:317–323. [Google Scholar]; b) Beckwith ALJ, Schiesser CH. Tetrahedron. 1985;41:3925–3941. [Google Scholar]; c) Newcomb M. Tetrahedron. 1993;49:1151–1176. [Google Scholar]; d) Newcomb M. In: In Radicals in Organic Synthesis. Renaud P, Sibi MP, editors. Vol. 1. Wiley-VCH; Weinheim: 2001. pp. 317–336. [Google Scholar]
- 9.a) Crich D, Cai W, Dai Z. J Org Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]; b) Crich D, Wu B. Org Lett. 2006;8:4879–4882. doi: 10.1021/ol061938l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Barresi F, Hindsgaul O. Synlett. 1992:759–761. [Google Scholar]; b) Stork G, Kim G. J Am Chem Soc. 1992;114:1087–1088. [Google Scholar]; c) Ito Y, Ogawa T. Angew Chem Int Ed. 1994;33:1765–1767. [Google Scholar]; d) Ishiwata A, Lee YJ, Ito Y. Org Biomol Chem. 2010;8:3596–3608. doi: 10.1039/c004281a. [DOI] [PubMed] [Google Scholar]
- 11.Schinzer D. Synthesis. 1988:263–273. [Google Scholar]
- 12.David S, Hanessian S. Tetrahedron. 1985;41:643–663. [Google Scholar]
- 13.Crich D, Li W, Li H. J Am Chem Soc. 2004;126:15081–15086. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]
- 14.a) Kahne D, Walker S, Cheng Y, Engen DV. J Am Chem Soc. 1989;111:6881–6882. [Google Scholar]; b) Crich D, Lim LBL. Org React. 2004;64:115–251. [Google Scholar]
- 15.a) Crich D, Mataka J, Sun S, Lam K-C, Rheingold AR, Wink DJ. Chem Commun. 1998:2763–2764. [Google Scholar]; b) Crich D, Mataka J, Zakharov LN, Rheingold AL, Wink DJ. J Am Chem Soc. 2002;124:6028–6036. doi: 10.1021/ja0122694. [DOI] [PubMed] [Google Scholar]
- 16.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 17.Crich D, Sun S. J Am Chem Soc. 1997;119:11217–11223. [Google Scholar]
- 18.Nukada T, Berces A, Whitfield DM. Carbohydr Res. 2002;337:765–774. doi: 10.1016/s0008-6215(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 19.The cis-fused compound 5 is not necessarily formed exclusively from the B2,5 boat conformer of oxocarbeniun 8. Other conformers unable to achieve trans-cyclization leading to 6, e.g., the 4H3 half-chair, are well suited to undergo concomitant cis-cyclization to 5.
- 20.Crich D, Sharma I. Org Lett. 2008;10:4731–4734. doi: 10.1021/ol8017038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The possibility that a minor portion of the β-O-mannoside 9 is formed by the dissociative mechanism cannot be excluded.
- 22.This difference in transition states and selectivity does not preclude the use of a clock reaction for the determination of relative rates, cf, the classical use of the 5-hexenyl radical cyclization reaction to estimate the rate of hydrogen atom abstraction from tributylstannane.8
- 23.It could also be considered that the β-O-mannoside 9 arises from reaction with the oxocarbenium ion 8, in which case the latter would exhibit a strong inherent β-selectivity toward both C- and O-nucleophiles. While we recognize this possibility we consider it unlikely on the grounds that the KIE studies2c indicate the β-O-mannosides to be formed by a highly associative pathway that is different to one for the formation of the α-O-mannosides, at least in the 4,6-O-benzylidene series.
- 24.Codee JDC, Krock L, Castagner B, Seeberger PH. Chem Eur J. 2008;14:3987–3994. doi: 10.1002/chem.200701864. [DOI] [PubMed] [Google Scholar]
- 25.Crich D, Smith M. J Am Chem Soc. 2002;124:8867–8869. doi: 10.1021/ja011406u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.