

Abstract
Background
One primary goal of transcriptomic studies is identifying gene expression patterns correlating with disease progression. This is usually achieved by considering transcripts that independently pass an arbitrary threshold (e.g. p<0.05). In diseases involving severe perturbations of multiple molecular systems, such as Alzheimer’s disease (AD), this univariate approach often results in a large list of seemingly unrelated transcripts. We utilised a powerful multivariate clustering approach to identify clusters of RNA biomarkers strongly associated with markers of AD progression. We discuss the value of considering pairs of transcripts which, in contrast to individual transcripts, helps avoid natural human transcriptome variation that can overshadow disease-related changes.
Methodology/Principal Findings
We re-analysed a dataset of hippocampal transcript levels in nine controls and 22 patients with varying degrees of AD. A large-scale clustering approach determined groups of transcript probe sets that correlate strongly with measures of AD progression, including both clinical and neuropathological measures and quantifiers of the characteristic transcriptome shift from control to severe AD. This enabled identification of restricted groups of highly correlated probe sets from an initial list of 1,372 previously published by our group. We repeated this analysis on an expanded dataset that included all pair-wise combinations of the 1,372 probe sets. As clustering of this massive dataset is unfeasible using standard computational tools, we adapted and re-implemented a clustering algorithm that uses external memory algorithmic approach. This identified various pairs that strongly correlated with markers of AD progression and highlighted important biological pathways potentially involved in AD pathogenesis.
Conclusions/Significance
Our analyses demonstrate that, although there exists a relatively large molecular signature of AD progression, only a small number of transcripts recurrently cluster with different markers of AD progression. Furthermore, considering the relationship between two transcripts can highlight important biological relationships that are missed when considering either transcript in isolation.
Introduction
Alzheimer’s disease (AD) is an irreversible brain disease that begins with mild memory impairment but eventually progresses to severe brain dysfunction and dementia. The prevalence of AD is rising dramatically due to an increasingly ageing population [1]. Early diagnosis is challenging as it can be difficult to discriminate the initial manifestations of the disease from cognitive decline that occurs as a function of normal aging [2]. Intensive efforts are being made to better understand AD and identify appropriate treatments, however the molecular mechanisms underlying the disease are still far from being understood.
In 2004, Blalock and colleagues [3] made an important contribution towards finding a set of molecular biomarkers that correlate with the progression of AD in one region of the brain. Using microarray technology, they assessed RNA transcript levels in post-mortem hippocampal tissue from 9 controls and 22 patients with varying degrees of AD severity. Participants were categorized, based primarily on Mini-Mental State Examination (MMSE) score [4], into one of four clinical groups: Control, Incipient AD, Moderate AD or Severe AD (see Materials and Methods for details of classification).
A total of 22,286 probe sets for protein coding RNA (i.e. mRNA) and non-coding RNA (ncRNA) were used to interrogate the transcriptome. After excluding probe sets with signals below detection thresholds and probe sets targeting unidentified transcripts (e.g. expressed sequence tags), Blalock and colleagues assessed the Pearson’s correlation of each of the remaining 9,921 probe set values with MMSE score and neurofibrillary tangle (NFT) count. This analysis revealed 3,413 genes that are significantly correlated (at p<0.05) with MMSE score, NFT count or both [3].
In 2010, Gomez Ravetti and colleagues (including two authors of this contribution) re-analyzed the publicly available dataset of Blalock using a different method, uncovering a 1,372-probe set signature that presents a remarkably high consensus with established phenotypic markers of AD progression [5]. Instead of just assessing correlations between gene expression and clinical measures of AD (such as MMSE score or NFT count), they employed an integrated approach based on combinatorial optimization techniques [6], [7] and Information Theory. Briefly, divergence of the gene expression profile of each individual sample from the average characteristic profile of the “Control” group was computed using the Jensen-Shannon divergence [8]. Similarly, this approach was used to compute the convergence of the gene expression profile of each individual sample to the average characteristic profile of the “Severe AD” group. This allowed the authors to identify genes with expression levels that correlate with the characteristic molecular progression from normal cognition to severe AD. In the current report, we use these quantifiers (which we will simply term as JSDcontrol and JSDsevere, respectively) as measures of AD progression, in addition to three common phenotypic markers (MMSE score, NFT count and Braak staging; obtained from [3]). We will refer collectively to these measures of AD progression as ‘progression markers’.
The main objective of this work is to identify a reduced set of pairs of RNA transcripts that strongly cluster with markers of AD progression. The analysis of these clusters will also help to identify individual transcripts that recurrently appear in many pairs, and may thus guide the selection of candidate molecules for further research. Towards this end, we apply a large-scale graph-based clustering approach to datasets derived from the 1,372-probe set signature identified by Gomez Ravetti et al. [5] to identify molecular features that correlate with the different ‘progression markers’. These datasets include the original 1,372-probe set signature, all pair-wise ratios that can be computed from the 1,372-probe set signature and an expanded dataset containing all pair-wise differences, summations, ratios and products that can be computed from the original signature (giving a total of 3,762,024 probe set combinations). We will refer to these probe set pairs as ‘metafeatures’ [9].
To cluster such a large number of metafeatures, we re-implemented and enhanced the MSTkNN algorithm in [10] by using the external memory (EM) approaches in [11], [12], [13]. External memory algorithms are known for their efficiency in handling large-scale data sets [14], [15], [16] and the MSTkNN algorithm is a graph based data clustering algorithm that has successfully been applied in several applications including the analysis of stock market time series [17], a gene expression dataset [18], a prostate cancer trial dataset [19] and has been integrated with a combinatorial optimization based graph visualization layout [20]. Our previous work indicates that the MSTkNN algorithm produces meaningful clusters (see [17], [18], [20]) and our proposed modification to this algorithm is still capable of producing reasonable clustering structure in terms of homogeneity and separation (Table 1).
Table 1. Performance comparison of the EM MSTkNN with k-Means, SOM, CLICK and the original MSTkNN algorithms, in terms of homogeneity and separation.
| Data | Methods/Algorithm | Parameter | Havg | Savg | #Clusters | Time |
| AD Signature data set(n = 1,372) | k-Means | k = 5 | 0.179 | 0.121 | 5 | <0.5 min |
| k = 120 | 0.394 | 0.172 | 120 | <1 min | ||
| SOM | 2X5 grid | 0.185 | 0.183 | 6 | <0.5min | |
| 5X5 grid | 0.217 | 0.142 | 14 | <1 min | ||
| CLICK | – | 0.606 | 0.245 | 5 | <1 min | |
| MSTkNN | – | 0.780 | 0.369 | 226 | <0.5 min | |
| EM MSTkNN(this paper) | – | 0.789 | 0.370 | 228 | <0.2 min | |
| AD ratios data set(n = 941,885) | k-Means, SOM,CLICK, MSTkNN | – | Not Available | Not Available | Not Available | Not Available |
| EM MSTkNN(this paper) | – | 0.812 | 0.420 | 40,139 | 30 min | |
| AD ratios- sums-diffs-prods dataset(n = 3,763,403) | EM MSTkNN(this paper) | – | 0.879 | 0.521 | 121,611 | 120 min |
The implementations of the k-Means, SOM, CLICK algorithms are obtained from the Expander microarray data cluster tool in [124]. The homogeneity and separation are computed using the definition in [124]. The AD ratio metafeatures data set is generated by taking pair-wise ratios between the features in 1,372-probe AD signatures [5] and including MMSE score, NFT count, Braak staging, JSDcontrol and JSDsevere as five progression markers. The other data set contains four different types of metafeatures (ratios, summations, differences and products) and the aforementioned progression markers.
After clustering all the metafeatures together with the different progression markers, we attempt to uncover pairs of probe sets that jointly cluster with each progression marker. Additionally, we identify some probe sets in these pairs that not only cluster with different progression markers but also relate to genes that share common biological pathways. We annotate these pairs of markers with the most recent information available. We also look at the expression of some of these markers in a different transcriptomic study that involves several regions of the brain in search for consensus among different studies.
Results
Results for each of the datasets are presented in the following order: 1. composition of the clusters containing the aforementioned progression markers, 2. results of functional analyses carried out using publicly available tools (iHop, Gather, GIM) and 3. a bibliographic (Pubmed) characterization of certain highlighted probe sets. Probe sets are highlighted for several reasons, including having a strong correlation with one or more progression markers or appearing in a metafeature with a probe set for another gene which has a potential role in AD or in other brain disease. The clustering outcomes are given in File S1, File S2 and File S3. Where a particular probe set has been highlighted and discussed in our previous publication on this dataset [5], we refer the reader to this paper rather than re-iterating discussion points in the present report.
1,372-probe set signature (File S1)
First, we analysed the 1,372-probe set signature from [5] using the proposed clustering algorithm and found a total of 228 clusters. We then identified the clusters that contain the proposed progression markers (Figure 1 and Table 2).
Figure 1. Visualization of the clustering outcome of the 1372-probe set signature.

The figure shows only the clusters that contain the progression markers (hexagonal nodes). We note that the probe set for PTEN, whose product has been recently observed to localize with intracellular NFTs [36], has values that correlate strongly with the Jensen-Shannon divergence of the severe profile (JSDsevere).
Table 2. Clustering outcomes for the 1372-probe set signature.
| Progression Marker | Gene Symbol. Probe Set ID | Correlation Coefficient | KEGG Pathway |
| MMSE | ATP5C1. 213366_x_at | 0.764201 | ATP synthesis |
| COX4I1. 202698_x_at | 0.590462 | Oxidative phosphorylation | |
| SPDEF. 214403_x_at | −0.48268 | ||
| MKS1. 218630_at | −0.69599 | ||
| PTMS. 218044_x_at | −0.72683 | ||
| TTN. 208195_at | −0.73718 | Calcium signaling pathway, Focal adhesion | |
| C19orf50. 200076_s_at | −0.80389 | ||
| KLHL20. 204177_s_at | −0.81714 | ||
| C10orf76. 55662_at | −0.82583 | ||
| ITGB8. 205816_at | −0.86993 | ECM-receptor interaction, Focal adhesion | |
| NM_024849. 220531_at | −0.88067 | ||
| NFT | COX6B1. 201441_at | 0.564201 | Oxidative phosphorylation |
| MMP11. 203876_s_at | −0.290462 | ||
| Braak | CR1. 206244_at | 0.363241 | Complement and coagulation cascades |
| CASP9. 210775_x_at | −0.240432 | Apoptosis, MAPK signaling pathway | |
| TNKS. 216695_s_at | −0.287898 | ||
| JSDcontrol | MLLT4. 208512_s_at | 0.723242 | Adherens junction, Tight junction |
| ALDOB. 217238_s_at | 0.480468 | Carbon fixation, Glycolysis | |
| JSDsevere | BTG3. 215425_at | 0.464201 | ATP synthesis |
| PTEN. 211711_s_at | 0.250462 | Phosphatidylinositol signaling system, Tight junction | |
| TREX1. 34689_at | 0.236458 |
For each progression marker, probe sets have been ordered according to their Spearman’s rank correlation with the progression marker. Gene symbols in boldface indicate that they were previously discussed in [5] and gene symbols with underlined boldface represent the cases for which a putative relationship exists in the published literature between the gene and AD.
Cluster containing MMSE score
A total of 11 probe sets clustered with MMSE score. Among the gene transcripts targeted by these probe sets, TTN (Titin/Connectin) may have a role in AD progression through its ability to form amyloid aggregates [21]. The genes ATP5C1, COX4I1, KLHL20, ITGB8 and C10orf76 were highlighted by the analysis in Gomez Ravetti et al. [5] and we refer the reader to this paper for extensive discussion of these genes in the context of AD. We identified several probe sets that uniquely cluster with MMSE score and target transcripts for genes not previously associated with AD. These included PTMS (parathymosin), a chromatin remodelling protein essential for cell cycle progression and proliferation of normal and malignant cells, MKS1 (Meckel syndrome, type 1), mutations in which are associated with a malformation of central nervous system known as Meckel syndrome, and SPDEF and a probe set for C19orf50.
Cluster containing NFT count
Two probe sets clustered with NFT count. These probe sets targeted transcripts for COX6B1 and MMP11, both of which were highlighted in Gomez Ravetti et al. [5] and will not be discussed in detail here.
Cluster containing braak staging
Three probe sets clustered with the Braak staging values. These probe sets targeted transcripts for CASP9 (caspase 9), which was previously highlighted by the analysis in Gomez Ravetti et al. [5], TNKS (tankyrase) and CR1 (complement receptor 1), which is genetically associated with the risk of AD [22], [23], [24], [25], [26], [27], [28], [29], [30] and entorhinal cortex volume in young healthy adults [31] (see also [32]).
Clusters containing JSDcontrol and JSDsevere
Two probe sets clustered with JSDcontrol - these related to the genes ALDOB (aldolase B, fructose-bisphosphate) and MLLT4 (myeloid/lymphoid or mixed-lineage leukemia, translocated to, 4), which is reportedly involved in the formation and remodelling of synapses in hippocampus [33]. Three probe sets clustered with JSDsevere. These related to the genes BTG3 (BTG family, member 3), which is related to prognosis of neuropathic symptoms in paraneoplastic neuropathy [34], TREX1 (three prime repair exonuclease 1), mutations in which can cause a neurovascular disorder involving progressive cognitive decline due to brain degeneration [35], and PTEN (phosphatase and tensin homolog). Altered distribution of PTEN has been reported in degenerating neurons in AD; specifically, a delocalization from the nucleus to the cytoplasm and accumulation in intracellular neurofibrillary tangles [36].
Comparing clustering outcomes with statistics-based outcomes
An alternative method for selecting probe sets highly correlated with progression markers is to simply perform regression analysis and identify the probe sets with the highest correlation coefficient or lowest p-value. In essence this is a univariate approach, where the selection of a particular probe set is independent of which other probe sets are selected. In contrast, the MSTkNN-based clustering method used here is a multivariate approach which considers the interrelationships of different probe sets in its selection. As such, we would expect the MSTkNN-based method to identify features of potential biological relevance that are missed by univariate approaches.
To investigate the similarities and differences in outcomes of multivariate and univariate approaches, we compared the probe sets identified by our clustering method with those having the highest, most significant correlations by regression analysis (see Figure S1). For example, we compared the 11 probe sets clustered with MMSE to the 11 probe sets most significantly correlated with MMSE by regression analysis. While there were some similarities in the probe sets identified, there were also important differences. Notably, some genes of particular relevance to AD were selected by the clustering method but not by filtering based on statistical significance. For example, clustering with Braak staging identified a probe set targeting CR1 which, as mentioned above, is widely proposed as a genetic risk factor for AD, however this probe set was not identified by the statistical method.
941,885 Ratio Metafeatures Data Set (File S2)
Next, we conducted a cluster analysis using the proposed external memory clustering algorithm on the 941,885 metafeatures generated by calculating the ratio of each pair of probe sets from the 1,372-probe set signature [5]. There were a total of 40,139 clusters in the data set, from which we again identified the clusters that contain the previously mentioned progression markers.
Cluster containing MMSE score
A total of 32 ratio metafeatures (containing 35 different probe sets) clustered with MMSE score (Table 3). The cluster contained metafeatures involving probe sets for five genes previously studied in the context of AD (TTN [37], [38], PPIA [39], [40],[41], VSNL1 [42], [43], [44], [45], [46], [47], [48], NEFL [49], [50], [51] and FXYD6 [52]). We also identified, in different clustered metafeatures, probe sets for 15 other genes (FOXO1, ACACA, CASK, PTEN, PTN, ATP5C1, LDHA, PRKAR2B, CPT2, DDX1, ITGBL1, TCF7L2, C10orf76, the non-coding RNA TUG1 and SCFD1) previously highlighted in the analysis of Gomez Ravetti et al. [5]. Additionally, we found 12 metafeatures in which both of the probe sets comprising the metafeature relate to genes in a common pathway (Table 3). These gene pairs were PRKCB1/PTEN (phosphatidylinositol signaling system), FOXO1/ACACA and PRKAR2B/ACACA (insulin signaling pathway), ACACA/LDHA (propanoate metabolism), COX6A1/ATP5C1 (oxidative phosphorylation), TCF7L2/ACTN1 (adherens junction), CASK/PTEN (tight junction), C10orf76/PPIA (ECM-receptor interaction), ACTN1/VCL, ITGB8/TTN, ITGB8/PTN and TTN/PRKCB1 (focal adhesion).
Table 3. Ratio metafeatures clustered with MMSE score.
| Metafeature (Gene Symbol. Probe Set ID) | Correlation Coefficient | Common KEGG Pathways |
| PPIA.212661_x_at/C3orf60.209177_at | 0.884365 | |
| PIGO.209998_at/CASK.211208_s_at | 0.760958 | |
| PURA.213806_at/AJ225093.211835_at | 0.673877 | |
| PRKCB1.209685_s_at/PTEN.222176_at | −0.68675 | Phosphatidylinositol signaling system |
| FOXO1.202724_s_at/ACACA.212186_at | −0.78654 | Insulin signaling pathway |
| CASK.211208_s_at/PTEN.222176_at | −0.83565 | Tight junction |
| RPL23A.203012_x_at/ATP5C1.213366_x_at | −0.83758 | |
| CASK.211208_s_at/ATP5C1.213366_x_at | −0.86355 | |
| ACACA.212186_at/LDHA.200650_s_at | −0.87642 | Propanoate metabolism |
| PRKAR2B.203680_at/ACACA.212186_at | −0.88001 | Insulin signaling pathway |
| COX6A1.200925_at/ATP5C1.213366_x_at | −0.88233 | Oxidative phosphorylation |
| RPL23A.203012_x_at/PPIA.211765_x_at | −0.88328 | |
| ACTN1.208636_at/VCL.200930_s_at | −0.88344 | Focal adhesion |
| ITGB8.205816_at/TTN.208195_at | −0.88642 | Focal adhesion |
| ITGB8.205816_at/PTN.209466_x_at | −0.88642 | Focal adhesion |
| CPT2.204264_at/ATP5C1.213366_x_at | −0.89851 | |
| TTN.208195_at/PRKCB1.209685_s_at | −0.90065 | Focal adhesion |
| SDC1.201287_s_at/DDX1.201241_at | −0.90504 | |
| TTN.208195_at/NEFL.221805_at | −0.91592 | |
| ZNF34.219801_at/FXYD6.217897_at | −0.9181 | |
| AL359052.214927_at/ATP5C1.205711_x_at | −0.9188 | |
| NUCKS1.217802_s_at/PPIA.212661_x_at | −0.91944 | |
| TCF7L2.212761_at/ACTN1.208636_at | −0.92353 | Adherens junction |
| TTN.208195_at/VSNL1.203798_s_at | −0.93006 | |
| CASK.211208_s_at/PTN.209466_x_at | −0.9301 | |
| NM_024849.220531_at/MRPL16.217980_s_at | −0.93682 | |
| AJ225093.211835_at/ATP5C1.205711_x_at | −0.93986 | |
| C10orf76.55662_at/PPIA.212661_x_at | −0.94211 | |
| ITGB8.205816_at/SDC1.201287_s_at | −0.95719 | ECM-receptor interaction |
| ITGB8.205816_at/PPIA.212661_x_at | −0.95911 | |
| CASK.211208_s_at/PPIA.211765_x_at | −0.95641 | |
| TUG1.222244_s_at/SCFD1.215548_s_at | −0.95643 |
Metafeatures are ordered by Spearman’s rank correlation with MMSE score. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature.
Cluster containing NFT count
Four ratio metafeatures (containing 8 different probe sets) clustered with NFT count (Table 4). The probe sets comprising these metafeatures did not target genes in common pathways but four of them targeted genes previously highlighted in the analysis of Gomez Ravetti et al. [5]: ICA1, COX6B1, ATP5C1 and ITGB8.
Table 4. Ratio metafeatures clustered with NFT count.
| Metafeature(Gene Symbol.Probe Set ID) | Correlation Coefficient |
| ICA1.207949_s_at/RUNX2.216994_s_at | −0.78797 |
| TSPAN9.220968_s_at/NRG1.206343_s_at | −0.87831 |
| PTMS.218044_x_at/GRPEL1.212434_at | −0.87839 |
| ITGB8.205816_at/ABAT.209460_at | −0.87857 |
Metafeatures are ordered by Spearman’s rank correlation with NFT count. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature.
Cluster containing braak staging
A total of 27 ratio metafeatures clustered with Braak staging values (Table 5). Three of these were comprised of probe sets targeting genes that can be mapped to common KEGG pathways: CR1/SERPINA5 (complement and coagulation cascades), MDH2/ACO2 (TCA cycle) and ITGB5/TNXB (ECM-receptor interaction). Of the various probe sets identified in this cluster, only one targets a gene previously implicated in AD - CR1 (discussed above). Metafeatures in the cluster also contained probe sets targeting six other genes previously highlighted by the analysis of Gomez Ravetti et al. [5]: PTN, PPT1, RIMS2, ASTN1, ITGB5 and PAX6. Many of the metafeatures that showed a positive correlation with Braak staging were dominated by a probe set for LRRC48 (leucine rich repeat containing 48), a gene which is currently poorly characterized.
Table 5. Ratio metafeatures clustered with Braak staging.
| Metafeature (Gene Symbol. Probe Set ID) | Correlation Coefficient | Common KEGG Pathways |
| LRRC48.208140_s_at/SLC6A1.205152_at | 0.885391 | |
| LRRC48.208140_s_at/CHRDL1.209763_at | 0.875476 | |
| LRRC48.208140_s_at/PTN.209466_x_at | 0.868268 | |
| LRRC48.208140_s_at/PTPN4.205171_at | 0.853365 | |
| LRRC48.208140_s_at/CAST.212586_at | 0.847157 | |
| LRRC48.208140_s_at/MAOA.212741_at | 0.837757 | |
| LRRC48.208140_s_at/PPT1.200975_at | 0.837151 | |
| C16orf57.218060_s_at/RIMS2.206137_at | 0.832873 | |
| LRRC48.208140_s_at/ACO2.200793_s_at | 0.830536 | |
| LRRC48.208140_s_at/TIMM23.218118_s_at | 0.828653 | |
| LRRC48.208140_s_at/ASTN1.213197_at | 0.815831 | |
| PTTG1IP.200677_at/RANBP9.202583_s_at | 0.786333 | |
| CSF2RA.211286_x_at/BBS4.212745_s_at | 0.775829 | |
| LRRC48.208140_s_at/GABBR1.203146_s_at | 0.761019 | |
| LRRC48.208140_s_at/PRC1.218009_s_at | 0.748013 | |
| TRA@.209671_x_at/NKX3-1.209706_at | 0.673362 | |
| RHOQ.212122_at/TBCE.203715_at | 0.668935 | |
| ITGB5.201125_s_at/IFNGR2.201642_at | 0.591936 | |
| LRRC48.208140_s_at/SLC6A1.205152_at | −0.46804 | |
| CR1.206244_at/SERPINA5.209443_at | −0.66797 | Complement and coagulation cascades |
| AL520908.217833_at/LRRC48.208140_s_at | −0.68605 | |
| MDH2.213333_at/ACO2.200793_s_at | −0.76804 | TCA cycle |
| PAX6.205646_s_at/LRRC48.208140_s_at | −0.77464 | |
| SFN.33322_i_at/LRRC48.208140_s_at | −0.78278 | |
| BMX.206464_at/RGS3.220300_at | −0.82257 | |
| ITGB5.201125_s_at/TNXB.216333_x_at | −0.86804 | ECM-receptor interaction |
| C15orf39.204494_s_at/LRRC48.208140_s_at | −0.89186 |
Metafeatures are ordered by Spearman’s rank correlation with Braak staging. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature. Most of the positively correlated metafeatures in this cluster are dominated by LRRC48 (See File S2 for details).
Cluster containing JSDcontrol
A total of 93 ratio metafeatures (containing a total of 94 different probe sets) clustered with JSDcontrol (Table 6), see also File S2 for complete list). Three of these metafeatures comprised probe sets targeting genes in common KEGG metabolic pathways: LDHA/GOT2 (cysteine metabolism), NDUFA10/ATP5C1 (oxidative phosphorylation) and ATP5C1/ATP5H (ATP synthesis). The probe sets contained in the clustered metafeatures targeted six genes that have previously been studied in the context of AD: PPP2CA [53] , [54], [55], [56] , SERPINI1 [57], [58], [59], [60], [61], OPA1 [62], [63], [64], [65], [66], PPIA and CSF1 [67], [68], [69], [70], [71], [72]. Additionally, we identified probe sets targeting 27 other genes previously highlighted by the analysis of Gomez Ravetti et al. [5]: NUFIP1, ATP6V1D, UQCRQ, DDX1, WASF1, ATP5C1, COX4I1, SNRK, PPP3CA, LDB2, COX7AP2, LAMTOR2, LDHA, PBX1, CAPRIN2, SLC25A6, SCFD1, DOPEY1, CSPG5, TUBG2, NRXN1, CADPS2, CRYM, FZD5, MAPK1, CASP9, PTN and ICA1. Most of the metafeatures in this cluster that showed a positive correlation with the divergence from control to severe AD were dominated by KLK3 (kallikrein 3), also known as prostate specific antigen, a well-known blood biomarker of prostate cancer [73]. To determine whether correlations involving KLK3 levels were influenced by gender, we stratified our dataset by gender. We performed the clustering again in both gender-specific datasets and found that KLK3 was completely absent in the same cluster of the dataset comprising only females. We therefore suggest that further study of KLK3 in relation to AD should be done in male patients only.
Table 6. Ratio metafeatures clustered with JSDcontrol.
| Metafeature (Gene Symbol. Probe Set ID) | Correlation Coefficient | Common KEGG Pathways |
| KLK3.204582_s_at/MAST3.213045_at | 0.852532 | |
| KLK3.204582_s_at/NUFIP1.205136_s_at | 0.792759 | |
| KLK3.204582_s_at/ATP5H.210149_s_at | 0.79046 | |
| KLK3.204582_s_at/AW242701.213411_at | 0.781264 | |
| KLK3.204582_s_at/GOT2.200708_at | 0.774367 | |
| KLK3.204582_s_at/KIAA1467.213234_at | 0.769769 | |
| KLK3.204582_s_at/FMO5.205776_at | 0.758274 | |
| KLK3.204582_s_at/MDH2.213333_at | 0.758067 | |
| KLK3.204582_s_at/PPP2CA.208652_at | 0.757663 | |
| KLK3.204582_s_at/ATP6V1D.208898_at | 0.744079 | |
| MAPK1.208351_s_at/KLK3.204582_s_at | −0.71785 | |
| LDHA.200650_s_at/GOT2.200708_at | −0.72409 | Cysteine metabolism |
| CASP9.210775_x_at/KLK3.204582_s_at | −0.72609 | |
| DNAJA4.220395_at/KLK3.204582_s_at | −0.74448 | |
| ICA1.207949_s_at/CSF1.211839_s_at | −0.75766 | |
| MDH2.213333_at/GOT2.200708_at | −0.77348 | |
| TRIM26.202702_at/KLK3.204582_s_at | −0.84302 | |
| AKR1B1.201272_at/KLK3.204582_s_at | −0.86002 | |
| NDUFA10.217860_at/ATP5C1.205711_x_at | −0.86735 | Oxidative phosphorylation |
| ATP5C1.205711_x_at/ATP5H.210149_s_at | −0.87348 | ATP synthesis |
We have selected 20 metafeatures (10 most positively correlated and 10 most negatively correlated) clustered with JSDcontrol and ordered them by Spearman’s rank correlation with JSDcontrol. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature. Most of the positively correlated metafeatures in this cluster are dominated by KLK3 (kallikrein 3) (See File S2 for details).
Cluster containing JSDsevere
Nine ratio metafeatures clustered with JSDsevere (Table 7). For four of these metafeatures, both probe sets comprising the metafeature targeted genes that can be mapped to a common KEGG pathway. These metafeatures were MLLT4/PTEN (tight junction), PRKCB1/ATP2B2 (calcium signaling pathway), TGFB2/PPP2CA (TGF-β signaling pathway), CYP3A4/CPT2 (fatty acid metabolism). Metafeatures in this cluster contained probe sets for three genes previously investigated in the context of AD (CYP3A4 [74], [75], [76], ATP2B2 [77], [78] and PPP2CA) and four other genes previously highlighted by the analysis of Gomez Ravetti et al. [5] (PTEN, PRKCB1, FCAR and CPT2).
Table 7. Ratio metafeatures clustered with JSDsevere.
| Metafeature (Gene Symbol. Probe Set ID) | Correlation Coefficient | Common KEGG Pathways |
| MLLT4.208512_s_at/PTEN.211711_s_at | 0.842239 | Tight junction |
| CYP3A4.205998_x_at/PTEN.211711_s_at | 0.777368 | |
| MLLT4.208512_s_at/CCDC6.204716_at | 0.753547 | |
| PRKCB1.209685_s_at/ATP2B2.204685_s_at | −0.2207 | Calcium signaling pathway |
| CPNE3.202118_s_at/AL520908.217833_at | −0.46933 | |
| TGFB2.209909_s_at/PPP2CA.208652_at | −0.67022 | TGF-beta signaling pathway |
| FCAR.211307_s_at/AF043586.216394_x_at | −0.78256 | |
| CYP3A4.205998_x_a/CPT2.204264_at | −0.87018 | Fatty acid metabolism |
| N25732.204131_s_at/AF043586.216394_x_at | −0.9031 |
Metafeatures are ordered by Spearman’s rank correlation with JSDsevere. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature.
Estimation of False Discovery Rate
Investigating a very large data space, such as that occupied by the many possible metafeatures, will inevitably lead to a number of false positive findings. In order to estimate the false discovery rate at different correlation coefficient thresholds, and therefore demonstrate the validity of our approach in identifying more than just random events, we performed a simple a Monte-Carlo permutation test by randomly permuting the MMSE scores of the 17 samples and computing the correlation of each metafeatures with the permuted MMSE labels. The results after 1,000 permutations reveal that, at all thresholds tested, there is clearly a higher number of strongly correlated metafeatures among our ratio metafeatures dataset than would be expected by chance alone (see Figure S2, Figure S3, Figure S4 and Figure S5).
3,763,403 Ratio-sum-difference-product Metafeatures Data Set (File S3)
We next applied our clustering algorithm to a data set of 3,763,403 metafeatures. This dataset was produced by calculating all pair-wise differences, summations, ratios and products of the 1,372-probes identified in Gomez Ravetti et al. [5].
The algorithm created a total of 121,611 clusters for the data set. We identified one larger cluster containing all of the progression markers. Due to the large number of metafeatures in the cluster, we focused only on the metafeatures with the strongest positive and negative correlations with each of the progression markers. We refer to File S3 for the details of this cluster.
Metafeatures correlating with MMSE Score
From the list of 50 metafeatures most strongly correlated (25 positively and 25 negatively) with MMSE score, we identified five that involve probe sets that target genes in common KEGG pathways (Table 8). This list of metafeatures also involved probe sets targeting genes previously investigated in the context of AD (PPIA, TTN, FXYD6, VSNL1, SERPINI1(see above), PLCB1 [79], [80], [81], IL15 [82], [83], [84], NRG1 [85], [86], [87], SERTAD2 [88]) and genes highlighted by the analysis in Ravetti et al. [5] (ICA1, ITGB8, GABBR2, CSPG5, ATP2B2, C10orf76, PRKAR2B, ACACA, MYT1L, KLHL20, PTEN, LDHA, AFF1, TUG1, RBM19, CPT2, ZBTB20, ITGBL1). We refer the reader to the paper of Gomez Ravetti et al. [5] for discussion of these genes in the context of AD.
Table 8. Ratio-sum-difference-product metafeatures clustered with MMSE score.
| Metafeature (Gene Symbol. Probe Set ID) | Correlation Coefficient | Common KEGG Pathways |
| ICA1.207949_s_at-ITGB8.205816_at | 0.940945 | |
| GABBR2.209990_s_at+PPIA.212661_x_at | 0.9268 | |
| LTBP1.202728_s_at-ITGB8.205816_at | 0.923973 | |
| RRAD.204803_s_at-ITGB8.205816_at | 0.922066 | |
| CSPG5.39966_at*ATP5C1.213366_x_at | 0.917444 | |
| ATP5C1.213366_x_at*FXYD6.217897_at | 0.911438 | |
| MKL2.218259_at+PPIA.212661_x_at | 0.909957 | |
| PPIA.212661_x_at+ATP2B2.204685_s_at | 0.908852 | |
| ATP5C1.213366_x_at+KIAA1107.214098_at | 0.903328 | |
| ICA1.207949_s_at-C10orf76.55662_at | 0.902256 | |
| PPIA.212661_x_at+KCNQ2.205737_at | 0.900168 | |
| PRKAR2B.203680_at*ACACA.212186_at | 0.8986 | Insulin signaling pathway |
| ATP5C1.205711_x_at*FXYD6.217897_at | 0.898298 | |
| VCL.200930_s_at-C10orf76.55662_at | 0.897422 | |
| PPIA.212661_x_at+ARF3.200734_s_at | 0.895376 | |
| RIBC2.206526_at-ITGB8.205816_at | 0.894556 | |
| CSPG5.39966_at+ATP5C1.213366_x_at | 0.894256 | |
| AW851559.216056_at-ZBTB20.222357_at | 0.893367 | |
| MYT1L.210016_at+PPIA.212661_x_at | 0.893136 | |
| PPIA.212661_x_at+AW514267.214945_at | 0.892278 | |
| RRAD.204803_s_at-KLHL20.204177_s_at | 0.891174 | |
| PTEN.222176_at*TTN.208195_at | 0.891049 | Focal adhesion |
| ATP5C1.213366_x_at+FXYD6.217897_at | 0.890893 | |
| AI708767.211978_x_at*FXYD6.217897_at | 0.890212 | |
| ATP5C1.213366_x_at*ACACA.212186_at | 0.888843 | |
| U82303.216702_x_at+NM_018601.220880_at | −0.90422 | |
| U82303.216702_x_at+PTEN.211711_s_at | −0.90498 | |
| ITGB8.205816_at*SDC1.201287_s_at | −0.90504 | ECM-receptor interaction |
| ZNF34.219801_at-NRG1.206343_s_at | −0.90504 | |
| U82303.216702_x_at+SERTAD2.202656_s_at | −0.90546 | |
| JPH2.220385_at+NM_024849.220531_at | −0.90598 | |
| ZBTB20.222357_at-NRG1.206343_s_at | −0.90613 | |
| U82303.216702_x_at+RBM19.205115_s_at | −0.90613 | |
| PTMS.218044_x_at+NM_024849.220531_at | −0.90683 | |
| U82303.216702_x_at+TPP1.214195_at | −0.90775 | |
| KLHL20.204177_s_at+AL359052.214927_at | −0.90825 | |
| ITGB8.205816_at+TTN.208195_at | −0.90941 | Focal adhesion |
| KLHL20.204177_s_at+UMOD.206716_at | −0.90994 | |
| TSPAN9.220968_s_at-NRG1.206343_s_at | −0.91157 | |
| C10orf76.55662_at+NM_018601.220880_at | −0.91218 | |
| C10orf76.55662_at+OTUB2.219369_s_at | −0.91235 | |
| ITGB8.205816_at+NM_014163.220695_at | −0.91264 | |
| SDC1.201287_s_at+FLJ23172.217016_x_at | −0.91266 | |
| PRKCB1.209685_s_at+ACTN1.208636_at | −0.91327 | Focal adhesion |
| BE138647.214314_s_at+AL049242.216101_at | −0.91548 | |
| TTN.208195_at/NEFL.221805_at | −0.91592 | |
| KLHL20.204177_s_at+C19orf50.200076_s_at | −0.91666 | |
| ZNF34.219801_at/FXYD6.217897_at | −0.9181 | |
| AL359052.214927_at/ATP5C1.205711_x_at | −0.9188 | |
| KLHL20.204177_s_at+SDC1.201287_s_at | −0.9188 |
We have selected 50 metafeatures (25 most positively correlated and 25 most negatively correlated) and ordered them by Spearman’s rank correlation with MMSE score. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature (see File S3 for details).
Metafeatures correlating with NFT count
From the list of 50 metafeatures most strongly correlated with NFT count, four comprised probe sets targeting genes in common pathways (Table 9). There were also probe sets targeting genes previously investigated in the context of AD (PPIA, TTN, MCL1 [89], UPF1 [90], RGS4 [91], [92], [93], [94]) and some other genes highlighted by the analysis in Gomez Ravetti et al. [5] (GNA14, C10orf76, MMP11, TCF7L2, COX6B1, PRKCI, ICA1).
Table 9. Ratio-sum-difference-product metafeatures clustered with NFT count.
| Metafeature (Gene Symbol. Probe Set ID) | Correlation Coefficient | Common KEGG Pathways |
| CCDC121.220321_s_at+U62966.207560_at | 0.939664 | |
| CGA.204637_at+PRL.205445_at | 0.918887 | |
| AL049435.213817_at+U62966.207560_at | 0.912476 | |
| AL049242.216101_at+U62966.207560_at | 0.912476 | |
| RNF121.219021_at+GNA14.220108_at | 0.910216 | |
| U62966.207560_at+WWP1.212637_s_at | 0.90442 | |
| MED13L.212207_at+U62966.207560_at | 0.897981 | |
| F9.207218_at+U62966.207560_at | 0.897533 | |
| U62966.207560_at+C10orf76.55662_at | 0.890537 | |
| U62966.207560_at+U66059.216597_at | 0.884725 | |
| U62966.207560_at+OTUB2.219369_s_at | 0.884725 | |
| MMP11.203877_at+PRL.205445_at | 0.884093 | |
| U62966.207560_at+TEAD3.209454_s_at | 0.880455 | |
| U62966.207560_at+SPAG1.210117_at | 0.871197 | |
| TEAD1.214600_at+TOX3.216623_x_at | 0.867647 | |
| TTN.208195_at/CAST.212586_at | 0.867043 | Focal adhesion |
| TTN.208195_at*LOC100131599.213222_at | 0.866901 | |
| TTN.208195_at*AP3S1.202442_at | 0.866901 | |
| CYP3A7.211843_x_at+PRL.205445_at | 0.865975 | |
| U62966.207560_at+RNMT.202684_s_at | 0.865512 | |
| TCF7L2.212761_at*GNA14.220108_at | 0.864466 | |
| LOC286434.222196_at+U62966.207560_at | 0.86248 | |
| LOC286434.222196_at+PRL.205445_at | 0.861916 | |
| MCL1.214057_at+U62966.207560_at | 0.861243 | |
| ITGB8.205816_at+CAST.212586_at | 0.86034 | Focal adhesion |
| EDC4.202496_at+COX6B1.201441_at | −0.85128 | |
| CAST.212586_at+COX6B1.201441_at | −0.8518 | |
| TMPRSS5.221032_s_at+COX6B1.201441_at | −0.85275 | |
| SIRT3.221562_s_at+COX6B1.201441_at | −0.8535 | |
| UPF1.211168_s_at+COX6B1.201441_at | −0.85405 | |
| COX6B1.201441_at+ACCN1.206690_at | −0.85484 | |
| COX6B1.201441_at+ACTN1.211160_x_at | −0.85501 | |
| NUP98.203195_s_at+COX6B1.201441_at | −0.85527 | |
| DET1.219641_at+COX6B1.201441_at | −0.85622 | |
| ANKRD34C.216073_at+COX6B1.201441_at | −0.85845 | |
| C20orf111.209020_at+COX6B1.201441_at | −0.86017 | |
| PRKCI.213518_at-HBG2.213515_x_at | −0.86253 | |
| CYP3A7.211843_x_at+CYP26B1.219825_at | −0.86253 | Fatty acid metabolism |
| COX6B1.201441_at+DBNDD1.222234_s_at | −0.86441 | |
| UBE3B.213822_s_at+COX6B1.201441_at | −0.86685 | |
| IRF2BP1.213771_at+COX6B1.201441_at | −0.86907 | |
| COX6B1.201441_at+PCSK1.205825_at | −0.87192 | |
| IRF2BP1.213771_at+NM_005758.206809_s_at | −0.87336 | |
| B3GALT2.210121_at+COX6B1.201441_at | −0.87405 | |
| SNCG.209877_at+COX6B1.201441_at | −0.8754 | |
| COX6B1.201441_at/ATP5C1.213366_x_at | −0.87976 | Oxidative phosphorylation |
| COX6B1.201441_at+RGS4.204339_s_at | −0.88473 | |
| ALDOB.217238_s_at-PRL.205445_at | −0.89422 | |
| ICA1.207949_s_at+NPAL3.210267_at | −0.89588 | |
| COX6B1.201441_at+SORBS2.204288_s_at | −0.89721 |
We have selected 50 metafeatures (25 most positively correlated and 25 most negatively correlated) and ordered them by Spearman’s rank correlation with NFT count. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature (see File S3 for details).
Metafeatures correlating with braak staging
From the list of 50 metafeatures most strongly correlated with Braak staging, five comprised probe sets targeting genes in common pathways (See Table 10). Only one gene previously proposed to be involved in AD, CR1 (see above), was targeted by a probe set within the 50 metafeatures. Probe sets targeting some important genes highlighted by the analysis of Gomez Ravetti et al. [5] (COX4I1, CASP9, ITGB1, RHOQ, DLGAP2, GSTA3, BCL2, COX6A1 and ATP5C1) were also found in this set of metafeatures.
Table 10. Ratio-sum-difference-product metafeatures clustered with Braak staging.
| Metafeature (Gene Symbol. Probe Set ID) | Correlation Coefficient | Common KEGG Pathways |
| ST3GAL4.203759_at+LRRC48.208140_s_at | 0.913812 | |
| LRRC48.208140_s_at*ATP5E.217801_at | 0.891149 | |
| LRRC48.208140_s_at*COX4I1.200086_s_at | 0.891149 | |
| LRRC48.208140_s_at*DOCK4.205003_at | 0.891149 | |
| LRRC48.208140_s_at/SLC6A1.205152_at | 0.885391 | |
| PRDM2.216445_at-CASP9.210775_x_at | 0.88091 | |
| IL12B.207901_at/TNFRSF9.207536_s_at | 0.878755 | Cytokine-cytokine receptor interaction |
| C1orf89.220963_s_at+LRRC48.208140_s_at | 0.876133 | |
| COPZ2.219561_at-RANBP9.202583_s_at | 0.87554 | |
| LRRC48.208140_s_at/CHRDL1.209763_at | 0.875476 | |
| LRRC48.208140_s_at-ITGB1.216190_x_at | 0.874692 | |
| AL110206.216465_at+LRRC48.208140_s_at | 0.873511 | |
| AJ251844.216362_at+LRRC48.208140_s_at | 0.873511 | |
| SLC47A1.219525_at+PTBP1.211270_x_at | 0.871548 | |
| ZNF506.221626_at+LRRC48.208140_s_at | 0.871417 | |
| ITGB1.216178_x_at+LRRC48.208140_s_at | 0.870888 | |
| WWTR1.202133_at*TBC1D5.201813_s_at | 0.870138 | |
| MRPS11.215919_s_at+LRRC48.208140_s_at | 0.869579 | |
| GJC2.214302_x_at-CASP9.210775_x_at | 0.868789 | |
| LRRC48.208140_s_at/PTN.209466_x_at | 0.868268 | |
| LRRC48.208140_s_at-BCAP29.205084_at | 0.867814 | |
| RHOQ.212122_at+COPZ2.219561_at | 0.866957 | |
| LRRC48.208140_s_at-SLC1A4.209611_s_at | 0.865434 | |
| DNAI2.220636_at+ZNF506.221626_at | 0.864854 | |
| GJA5.214466_at+LRRC48.208140_s_at | 0.864296 | |
| CR1.206244_at/SERPINA5.209443_at | −0.66797 | Complement and coagulation cascades |
| ITGB5.201125_s_at/TNXB.216333_x_at | −0.86804 | ECM-receptor interaction |
| CASP9.210775_x_at-PRDM12.220894_x_at | −0.87283 | |
| CASP9.210775_x_at-NTRK3.217033_x_at | −0.87297 | |
| CGGBP1.214050_at-LRRC48.208140_s_at | −0.87301 | |
| CASP9.210775_x_at-RENBP.206617_s_at | −0.87347 | |
| KIAA1659.215674_at-LRRC48.208140_s_at | −0.87353 | |
| CASP9.210775_x_at-MEOX2.206202_at | −0.87395 | |
| CASP9.210775_x_at-DLGAP2.216916_s_at | −0.87605 | |
| CASP9.210775_x_at-AW408767.217608_at | −0.87684 | |
| CASP9.210775_x_at-TRBV6-4.216578_at | −0.87744 | |
| R71245.217654_at-LRRC48.208140_s_at | −0.87744 | |
| LCE2B.207710_at-LRRC48.208140_s_at | −0.8788 | |
| CASP9.210775_x_at-FETUB.214417_s_at | −0.87924 | |
| CASP9.210775_x_at+GSTA3.222102_at | −0.87933 | |
| CASP9.210775_x_at*BCL2.203685_at | −0.8806 | Apoptosis |
| CASP9.210775_x_at+C6orf106.217924_at | −0.88245 | |
| ATP5C1.205711_x_at*COX6A1.200925_at | −0.88297 | Oxidative phosphorylation |
| FLJ22222.219254_at-LRRC48.208140_s_at | −0.88461 | |
| CASP9.210775_x_at-X97875.217158_at | −0.88671 | |
| CASP9.210775_x_at-ELF3.201510_at | −0.88842 | |
| C15orf39.204494_s_at/LRRC48.208140_s_at | −0.89186 | |
| CASP9.210775_x_at-LRRC48.208140_s_at | −0.89462 | |
| CASP9.210775_x_at+DENND4A.214787_at | −0.89462 | |
| CASP9.210775_x_at-RGS3.220300_at | −0.90202 |
We have selected 50 metafeatures (25 most positively correlated and 25 most negatively correlated) and ordered them by Spearman’s rank correlation with Braak staging. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature (see File S3 for details).
Metafeatures correlating with JSDcontrol
From the list of 50 metafeatures most strongly correlated with JSDcontrol, four comprised probe sets targeting genes in common pathways (Table 11). We also identified metafeatures comprising probe sets targeting various genes highlighted in the analysis in Gomez Ravetti et al. [5] (RBM19, KCNK5, AGTR1, TUBD1, GABRQ, MMP11, ZNF669, TBXA2R, NUFIP1, LDHA, ICA1).
Table 11. Ratio-sum-difference-product metafeatures clustered with JSDcontrol.
| Metafeature (Gene Symbol. Probe Set ID) | Correlation Coefficient | Common KEGG Pathways |
| RBM19.205115_s_at-KCNK5.219615_s_at | 0.886487 | |
| KLK3.204582_s_at-LOC645961.215320_at | 0.875436 | |
| N25732.204131_s_at-MAST3.213045_at | 0.85816 | |
| KLK3.204582_s_at-KYNU.210662_at | 0.856764 | |
| RBM19.205115_s_at-AGTR1.208016_s_at | 0.856409 | |
| ZNF669.220215_at-GEMIN6.219539_at | 0.853995 | |
| KLK3.204582_s_at-BG482805.214777_at | 0.853527 | |
| KLK3.204582_s_at/MAST3.213045_at | 0.852532 | |
| TUBD1.210389_x_at-ESR1.205225_at | 0.852098 | |
| CASP4.213596_at-LOC90379.221851_at | 0.848467 | |
| KLK3.204582_s_at-MLLT4.208512_s_at | 0.846819 | |
| GABRQ.220886_at+RBMS1.203748_x_at | 0.844336 | |
| MMP11.203876_s_at-SCGB1D2.206799_at | 0.838775 | |
| CASP4.213596_at-AF043586.216394_x_at | 0.835322 | |
| RBM19.205115_s_at-AF043586.216394_x_at | 0.834155 | |
| TMBIM1.217730_at*RBM4.200997_at | 0.833134 | |
| AL080106.216121_at-BTN2A2.205298_s_at | 0.833121 | |
| ZNF669.220215_at-TBXA2R.207554_x_at | 0.832963 | |
| ZNF669.220215_at-LOC90379.221851_at | 0.832263 | |
| SLC11A1.217507_at-SCGB1D2.206799_at | 0.83103 | |
| KLK3.204582_s_at-CSH2.208342_x_at | 0.824365 | |
| CENPE.205046_at-KYNU.210662_at | 0.823506 | |
| ZNF669.220215_at-SCGB1D2.206799_at | 0.823429 | |
| TUBD1.210389_x_at-IGSF6.206420_at | 0.819997 | |
| U62966.207560_at-NUFIP1.205136_s_at | 0.817348 | |
| MTHFD1.202309_at/MDH2.213333_at | −0.67409 | Glyoxylate and dicarboxylate metabolism |
| LDHA.200650_s_at/GOT2.200708_at | −0.72409 | Cysteine metabolism |
| IRF2BP1.213771_at-AU155105.214782_at | −0.81881 | |
| LUZP4.220665_at-ZNF669.220215_at | −0.81961 | |
| TXNDC9.203008_x_at+MAST3.213045_at | −0.82019 | |
| SMAD3.205398_s_at-MLLT4.208512_s_at | −0.82047 | Adherens junction |
| ALDOB.217238_s_at+PDE4D.211840_s_at | −0.82091 | |
| BF691447.221484_at*UBP1.218082_s_at | −0.82124 | |
| BF691447.221484_at*RNMT.202683_s_at | −0.82124 | |
| SLC9A3R2.215735_s_at-PSME3.209853_s_at | −0.82473 | |
| LOC90379.221851_at-U66059.216597_at | −0.82552 | |
| TREX1.34689_at-MARCH3.213256_at | −0.82615 | |
| AW408767.217608_at-MMP11.203876_s_at | −0.82763 | |
| PURA.213806_at-CENPE.205046_at | −0.8307 | |
| C20orf111.209020_at-PSME3.209853_s_at | −0.83454 | |
| FLJ39739.217136_at-CASP4.213596_at | −0.83709 | |
| ALDOB.217238_s_at-TNFSF14.207907_at | −0.84013 | |
| DIABLO.219350_s_at+MAST3.213045_at | −0.84039 | |
| ALDOB.217238_s_at-CASP4.213596_at | −0.84169 | |
| ICA1.207949_s_at+GCNT3.219508_at | −0.84188 | |
| S80491.216974_at-MMP11.203876_s_at | −0.84634 | |
| ALDOB.217238_s_at+SMAD3.205398_s_at | −0.8545 | |
| AKR1B1.201272_at/KLK3.204582_s_at | −0.86002 | |
| KCNJ5.208397_x_at-CASP4.213596_at | −0.86111 | |
| NDUFA10.217860_at/ATP5C1.205711_x_at | −0.86735 | Oxidative phosphorylation |
We have selected 50 metafeatures (25 most positively correlated and 25 most negatively correlated) and ordered them by Spearman’s rank correlation with JSDcontrol. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature (see File S3 for details).
Metafeatures correlating with JSDsevere
From the list of 50 metafeatures most strongly correlated with JSDsevere, six comprised probe sets targeting genes in common pathways (Table 12). We also identified metafeatures comprising probe sets targeting genes previously studied in the context of AD (VSNL1, PPP2CA, CYP3A4 (see above)) and genes highlighted by the analysis in Gomez Ravetti et al. [5] (PTEN, MAPK1, COX6A1, GABRQ, FCAR, FZD5, PIP5K1C, SHANK2, CPT2).
Table 12. Ratio-sum-difference-product metafeatures clustered with JSDsevere.
| Metafeature (Gene Symbol. Probe Set ID) | Correlation Coefficient | Common KEGG Pathways |
| TREX1.34689_at+NM_005758.206809_s_at | 0.933423 | |
| ESR1.205225_at-AL050026.216626_at | 0.910083 | |
| VSNL1.203798_s_at-AKAP12.210517_s_at | 0.877939 | |
| PURA.213806_at-APOH.205216_s_at | 0.872983 | |
| PTEN.222176_at-AL050026.216626_at | 0.870794 | |
| MAPK1.208351_s_at+MLLT4.208512_s_at | 0.867901 | Adherens junction |
| TREX1.34689_at*COX6A1.200925_at | 0.864481 | |
| CNOT1.200861_at-GABRQ.220886_at | 0.856692 | |
| UBE3B.213822_s_at+ADK.204119_s_at | 0.847332 | |
| MTSS1.210360_s_at-PTEN.211711_s_at | 0.843329 | |
| S80491.216974_at-PTEN.211711_s_at | 0.842751 | |
| MLLT4.208512_s_at/PTEN.211711_s_at | 0.842239 | Tight junction |
| PPP2CA.208652_at/PTEN.211711_s_at | 0.842212 | Tight junction |
| P2RY10.214615_at-AL050026.216626_at | 0.842191 | |
| TTK.204822_at-PTEN.211711_s_at | 0.840396 | Inositol phosphate metabolism |
| FLJ39739.217136_at-PTEN.211711_s_at | 0.837966 | |
| UBE3B.213822_s_at+ATRN.212517_at | 0.836922 | |
| TREX1.34689_at+RPS24P2.217188_s_at | 0.83673 | |
| UBE3B.213822_s_at-FOLH1.217483_at | 0.835834 | |
| SCGB2A1.205979_at-APOH.205216_s_at | 0.83274 | |
| AJ302559.216818_s_at-KLK3.204582_s_at | 0.830253 | |
| TREX1.34689_at-PRKD2.38269_at | 0.82812 | |
| AJ302559.216818_s_at-PTEN.211711_s_at | 0.827864 | |
| TPD52.201691_s_at-FOLH1.217483_at | 0.827066 | |
| TREX1.34689_at+DDX18.208897_s_at | 0.8255 | |
| SI.206664_at+PTEN.211711_s_at | −0.80904 | |
| TNRC4.215045_at*AL536319.212606_at | −0.80962 | |
| FCAR.211307_s_at+PTEN.211711_s_at | −0.81072 | |
| AL050026.216626_at+OTUB2.219369_s_at | −0.81137 | |
| CMKLR1.210659_at+PTEN.211711_s_at | −0.81349 | |
| KLK3.204582_s_at+PTEN.211711_s_at | −0.81565 | |
| PAX3.216059_at+NM_018601.220880_at | −0.82244 | |
| AL050026.216626_at+CTAGE5.204055_s_at | −0.82288 | |
| FZD5.206136_at+PTEN.211711_s_at | −0.82362 | |
| CAMP.210244_at-SCGB1D2.206799_at | −0.82501 | |
| PTEN.211711_s_at*PIP5K1C.212518_at | −0.82503 | Phosphatidylinositol signaling system |
| PLA2G2F.215870_s_at-RARRES2.209496_at | −0.82717 | |
| BPI.205557_at+PTEN.211711_s_at | −0.8328 | |
| AI478300.217526_at-TREX1.34689_at | −0.83316 | |
| AL050026.216626_at+PTEN.211711_s_at | −0.83401 | |
| PTEN.211711_s_at-PTPRN2.203030_s_at | −0.83583 | |
| FOLH1.217483_at-SCGB1D2.206799_at | −0.83998 | |
| RAB14.200928_s_at+PTEN.211711_s_at | −0.84169 | |
| GALNT10.212256_at*SHANK2.213307_at | −0.84811 | |
| NM_004908.208254_at+PTEN.211711_s_at | −0.84856 | |
| SPRED2.212458_at*DDN.214788_x_at | −0.85116 | |
| FADS1.217462_at+PTEN.211711_s_at | −0.86774 | |
| CYP3A4.205998_x_a/CPT2.204264_at | −0.87018 | Fatty acid metabolism |
| N25732.204131_s_at/AF043586.216394_x_at | −0.9031 | |
| C3orf63.209285_s_at*DDN.214788_x_at | −0.90361 |
We have selected 50 metafeatures (25 most positively correlated and 25 most negatively correlated) and ordered them by Spearman’s rank correlation with JSDsevere. Genes in boldface indicate that they were previously discussed in [5] and genes with underlined boldface represent the cases for which the gene has been discussed in the context of AD in the published literature (see File S3 for details).
Comparison of Metafeature Correlations and Single Probe Set Correlations
The observation of particular probe sets recurring in multiple clustered metafeatures raises the question of whether the clustering of certain metafeatures is driven by a strong correlation between a progression marker and only one of the two individual probe sets comprising a metafeature. To investigate this possibility, we separately assessed the correlation of the two probe sets comprising a metafeature with the progression marker in question and compared this to the correlation between the metafeature and the progression marker.
Deeper analysis of the “ratio metafeatures” that clustered with MMSE scores reveals a number of metafeatures where individual probe sets (one or both) are not significantly correlated (p>0.05) with MMSE (see Table S1). This suggests that the high correlation of certain metafeatures does not simply reflect an additive effect of its two component probe sets (see Figure S6) but instead is driven by the dynamics of the interrelationship (possibly a biological interaction) between the two transcripts that are targeted.
For specific examples we refer to Figure 2 where we demonstrate an example scenario with the three probe sets targeting TTN, CASK and TUG1 and the metafeatures TTN/PKRCB1, CASK/PTEN and TUG1/SCFD1. In this example, the metafeatures show a better correlation with MMSE score than the relevant individual probe sets (Figure 2). In general, we found that if both the probe sets in a metafeature target genes in a common pathway, then the metafeature shows better correlation with the progression marker than either of the two individual probe sets. For example, TTN and PKRCB1 both appear in the ‘focal adhesion’ KEGG pathway and CASK and PTEN in both appear in ‘tight junction’ pathway.
Figure 2. Comparison of single probe set correlations and metafeature correlations.

Figure shows plots of the correlation with MMSE score of three probe sets targeting TTN, CASK and TUG1 and three metafeatures involving these probe sets (TTN/PKRCB1, CASK/PTEN and TUG1/SCFD1). In this example, the correlations between MMSE score and the metafeatures are much better than the correlation between MMSE score and the individual probe sets.
There were various instances in which the individual components of highly correlated metafeatures did not map to a common KEGG pathway. For example, a metafeature containing probe sets for TUG1 and SCFD1 shows a better correlation with MMSE score than either probe set individually. However it is not surprising that these transcripts do not map to a common pathway, as long ncRNAs such as TUG1 are relative newcomers to functional annotation.
It should be noted, however, that a high proportion of the metafeatures (>90%) showed comparatively better correlations with the progression markers than the individual probe sets comprising these metafeatures. While this is not a universal phenomenon (see Figure S6 and Table S1 for examples, where two probe sets that are highly correlated with a progression marker combine to create a poorly correlated metafeature), in view of the larger data space occupied by the metafeatures, it is logical that the metafeature analysis may yield an increased proportion of spurious, false positive results. This is supported by the observed difference in the estimated false discovery rate of two datasets of different sizes (see Table S2 and Table S3). In an attempt to avoid such results, we subsequently focus on ‘robust’ findings – probe sets that recurrently cluster with different markers of AD progression.
Robust Markers of AD Progression
We next attempted to identify probe sets that appeared recurrently in the metafeatures and also clustered with different markers of AD progression. We depict this group of probe sets in a 5-way Venn diagram (Figure 3 and Figure 4). In these figures, a null (φ) symbol means that even if an overlap is shown in the figure, there is no common transcript. From the 941,885 ratio metafeatures data set, we identified 11 probe sets that, as part of metafeatures, clustered with more than one progression marker. The genes targeted by these probe sets were PPIA, ATP5C1, LDHA, DDX1, SCFD1, ITGB8, PTEN, PRKCB1, CPT2, ICA1 and PTN. From the 3,763,403 ratios-sum-difference-product metafeatures data set, there were 13 probe sets that, as part of metafeatures, clustered with more than one progression marker. The genes targeted by these probe sets were PPIA, TTN, C10orf76, ICA1, MMP11, RBM19, LDHA, COX6A1, GABRQ, CPT2, PTEN, VSNL1 and ATP5C1. Notably, six genes were identified as clustering with more than one progression marker in both metafeature datasets: PPIA, ATP5C1, LDHA, PTEN, CPT2 and ICA1. We refer the readers to the Table S4 and Table S5, for further details of correlation of these markers to the phenotypes.
Figure 3. Venn diagram of the different transcripts clustered with progression markers in the 941,885 metafeatures data set.

This figure highlights the ‘robust correlating’ transcripts that are shared by different progression marker clusters. A null (φ) symbol here means that even if an overlap is shown in the figure, there is no common transcript. We refer the readers to Supporting Information Table S4., for further details of correlation of these markers to the phenotypes.
Figure 4. Venn diagram of the different transcripts clustered with progression markers in the 3,763,403 metafeatures data set.

This figure highlights the ‘robust correlating’ transcripts that are shared by different progression marker clusters. A null (φ) symbol here means that even if an overlap is shown in the figure, there is no common transcript. We refer the readers to Supporting Information Table S5., for further details of correlation of these markers to the phenotypes.
Validation of Robust Markers in an Alternative Dataset
In order to gain insights into whether the robust markers highlighted above are restricted to the hippocampus or show changes in other AD-affected brain regions, we utilized an independent dataset contributed by Liang and colleagues [95], [96]. This dataset contains microarray data on gene expression in neurons isolated from four different regions of control and AD brain: entorhinal cortex (EC), hippocampus (HIP), middle temporal gyrus (MTG) and posterior cingulate cortex (PC). Molecular signatures of each different region were generated as described in Materials and Methods. Several of the ‘robust’ probe set markers highlighted by our current analysis of hippocampal tissue were also selected in the molecular signatures of two or more regional neuronal populations. For example, PPIA and ATP5C1 showed expression changes in neurons isolated from the MTG and PC of AD brain relative to control brain, PTEN showed expression changes in the HIP and PC and ICA1 showed expression changes in the HIP, MTG and PC (Figure 5). In addition, Visinin-like 1 (VSNL1), highlighted in the analysis in Gomez Ravetti et al. [5] as one of the best markers of AD progression and recently proposed as one of the four best CSF biomarkers of early AD [97], showed expression changes in neurons isolated from EC, MTG and PC (Figure 5) and has been shown in an additional dataset to have altered expression in various brain regions in AD [98].
Figure 5. Validation of robust markers of AD progression in an alternative dataset.

Transcript levels for selected genes of interest were investigated in the microarray dataset of Liang and colleagues [95], [96], which assessed gene expression in healthy neurons isolated from four different regions of control and AD brain: entorhinal cortex (EC), hippocampus (HIP), middle temporal gyrus (MTG) and posterior cingulate cortex (PC). Data presented in this figure were normalized using Robust Multichip Average (RMA). In the box and whisker plots, the bottom and top of the box represent the lower and upper quartiles, respectively, and the band within the boxes represents the median, while the ends of the whiskers represent the minimum and maximum values.
Discussion
The present study has extended on our previous analysis in [5] by (i) considering variables that represent the interrelationship between two RNA transcripts (i.e. metafeatures) and (ii) applying a novel and powerful graph-based clustering approach to identify a reduced set of transcripts or transcript pairs that correlate strongly with markers of AD progression. This clustering approach is facilitated by the implementation of an external memory algorithm on a graphical processing unit, which allows clustering of massive datasets (in this case involving four million elements) that is not feasible using standard computational methods.
In addition to identifying transcripts and metafeatures that correlate with established phenotypic markers of AD severity (i.e. MMSE score, NFT count, Braak staging), we utilized two addition quantifiers of AD progression, based on Jensen-Shannon divergence, to identify transcripts and metafeatures that correlate with a putative molecular trend from control to severe AD. It remains to be seen whether these quantifiers provide a more accurate assessment of AD severity, however there are several promising attributes that suggest this is the case. Firstly, the Jensen-Shannon divergence values are based on a set of 1,372 different transcript markers, in contrast to the univariate markers of neuropathology or cognitive function. Secondly, some of the changes observed at the transcriptional level may underlie AD pathogenesis, whereas phenotypic consequences are more likely to simply reflect the molecular perturbations that drive disease pathogenesis. The results presented here indicate that a number of probe sets that are highly correlated with Jensen-Shannon divergence are also highly correlated with more traditional phenotypic markers of AD progression. Our previous studies using these quantifiers in the context of AD [5] and cancer [99] have also yielded high consensus with established markers of disease progression. Together, these findings give us confidence that metrics based on multivariate transcriptional changes can act as reliable markers of disease stage.
The analyses reported here demonstrate that, although there exists a relatively large molecular signature associated with AD progression, a relatively small number of transcripts appear recurrently in metafeatures clustered with the progression markers. This allowed a focussed investigation of a reduced set of biomarkers that have been previously studied in the context of cognitive decline and AD. Furthermore, our approach also put emphasis on a few novel markers that have not been discussed previously in relation to AD progression and warrant further investigation.
While it is outside the scope of this paper to discuss in detail all of the genes identified in the analyses, focussed discussion of some of the most robust findings is warranted. As mentioned at the end of the Results, as set of six genes (PPIA, ATP5C1, LDHA, PTEN, ICA1, CPT2) recurrently appeared in metafeatures and clustered with more than one progression marker in both metafeature datasets. Furthermore, changes in expression of several of these genes were validated in an alternative microarray dataset of neurons from different AD-affected brain regions, lending further support to the proposal that altered expression of these genes may be involved in AD pathogenesis.
Peptidylprolyl isomerase A (PPIA), also known as cyclophilin A, is believed to accelerate protein folding. Evidence from neural cell lines suggests PPIA can protect against Aβ-induced oxidative stress, possibly by acting as a ROS scavenger [100]. Proteomics studies have revealed decreased expression of PPIA in brains of patients with non-Alzheimer’s disease tauopathies [41], suggesting alterations in PPIA may be associated with the general process of neurodegeneration rather than AD specifically. Curiously, PPIA has been proposed as a suitable reference gene for PCR studies of AD brain due to its stable expression [101], [102]. The results of our analysis strongly argue against this and instead indicate that PPIA expression, particularly when considered as part of a metafeature, is strongly correlated with AD progression.
ATP synthase subunit gamma (ATP5C1) encodes a subunit of mitochondrial ATP synthase, important for catalyzing ATP synthesis in oxidative phosphorylation. While this gene has not previously been implicated in AD, its transcriptional correlation with AD progression may reflect disturbances in energy production as a result of cellular loss.
Lactate dehydrogenase A (LDHA), another metabolic gene, is responsible for catalysing the conversion of lactate to pyruvate, the final step in anaerobic glycolysis. A recent study has demonstrated that increased LDHA activity is a feature of nerve cell lines that are resistant to Aβ-induced cell death and that the phenomenon of aerobic glycolysis might contribute to the mechanisms by which certain neurons in the AD brain survive apoptosis [103].
Phosphatase and tensin homolog (PTEN) has generally been studied in the context of cancer, as it is a tumor suppressor with phosphatase activity that negatively regulates the AKT/PKB signalling pathway. However PTEN has also been shown to be necessary for proper migration of neurons and glia [104]. There is decreased expression and altered distribution of PTEN in AD brain [105], [106], where it localizes with neuritic pathology such as neurofibrillary tangles in damaged neurons [36]. PTEN affects phosphorylation and aggregation of tau [106], [107] and appears to be regulated by presenilin, as presenilin deficient neurons show a substantial reduction in PTEN [108]. Furthermore, mutations in the PTEN induced putative kinase 1 (PINK1) gene have been linked to early-onset familial Parkinson’s disease [109], [110], while ablation of PTEN in dopaminergic neurons is neuroprotective in mouse models of Parkinson’s disease [111].
Islet cell autoantigen 1 (ICA1) is believed to be an autoantigen in insulin-dependent diabetes mellitus. ICA1 is the major binding partner of PICK1 and together these proteins regulate trafficking of AMPA glutamate receptors to the synapse [112]. It has also been proposed that ICA1 participates in the process of neuroendocrine secretion through association with certain secretory vesicles [113].
Carnitine palmitoyltransferase 2 (CPT2) is involved in the oxidation of long-chain fatty acids in the mitochondria. This gene has not previously been associated with AD.
In addition, we highlight some relevant genes that correlated with more than one progression marker in one of the metafeature datasets. Protein kinase C beta, encoded by PRKCB1, is involved in a wide range of signalling pathways. Increased expression of protein kinase C beta has been observed in membrane fractions of aged Tg2576 mice, a model of AD [114]. Furthermore, one of the best biomarkers in [5], visinin-like 1 (VSNL1), is only expressed in neurons and shows decreasing expression as AD progresses. Levels of VSNL1 in the CSF have recently been proposed as an effective biomarker of early AD [97].
It is interesting to remark that the individual markers identified in this study are bringing new insights to the pathological mechanisms involved in AD but that an integrative approach is required to understand them. For example, increased VSNL1 in the CSF observed in [97] may be a consequence of increasing neuronal death rather than an marker of early AD. On the other hand, as LDHA expression is currently being considered as a possible marker of aerobic glycolysis in Aβ-resistant neurons [103], the correlation between LDHA expression and AD progression makes sense if we think that Aβ-resistant neurons will be proportionally more abundant in samples with greater disease severity.
One limitation of the present study is that the low number of samples (17) available for these analyses may have resulted in a large number of highly correlated probe sets or metafeatures that are false positives (see Figure S2, Figure S3, Figure S4, Figure S5, for a validation of our correlations). The selection of just a few features out of this large data space has been a critical task that we attempted to solve by focusing on those which appear most recurrently. Unfortunately, there are strong possibilities that even with a set of random data and a very large search space, a set of false positive markers may recurrently appear. However, since a relatively higher number of published AD studies can already be found that implicate these markers, we feel comfortable in making the claim that they warrant further investigation in future AD research.
The results presented here support the hypothesis that systematically considering relationships between two or more features (“metafeatures”) can improve biomarker discovery, particularly when explored within a multivariate framework. While univariate approaches may still provide important and complementary insights to those obtained using multivariate methods, we believe that utilizing both approaches in conjunction is likely to produce the most robust and relevant findings. Computational advances such as the external memory implementation of our clustering algorithm will hopefully make investigations of this type more commonplace, and we are currently working towards more sophisticated parallel applications that would enable the study even of larger datasets across a range of diseases.
Materials and Methods
Datasets
This analysis draws on the data set contributed by Blalock et al. [3] which can be accessed from the NCBI Gene Expression Omnibus under the accession number GSE1297. The Blalock study used Affymetrix HG-U133A microarrays to generate data on 22,286 probe sets. From this dataset, we focused our analysis on the 1,372-probe set signature identified by Gomez Ravetti et al. [5].
The Blalock study assessed gene expression in hippocampal tissue samples from 31 participants. Participants were categorized into one of four clinical groups using the MMSE criterion: “Control” (MMSE >25, n = 9),“Incipient AD” (MMSE 20–26, n = 7), “Moderate AD” (MMSE 14–19, n = 8) or “Severe AD” (MMSE <14, n = 7). Borderline cases (e.g. MMSE = 26) were resolved using NFT count and Braak staging data [115].
In the present study, instead of using all 31 samples, we excluded 14 samples that had gene expression profiles similar to the representative profile of the “Control” group (n = 7) or similar to the representative profile of the “Severe AD” group (n = 7). The 17 remaining samples correspond to the central 17 columns of the supplementary material ‘File S2 (sheet:1372-probe)’ of [5] and the data set containing the signature of Gomez Ravetti et al. [5] for these 17 samples is termed 1,372-probe set signature throughout the paper. Our rationale for excluding these 14 samples is two-fold. Firstly, little information about disease progression is likely to be gained by considering participants at either extreme of a disease spectrum. Control participants will not have developed any molecular characteristics of early AD and participants with severe AD may have already progressed to the disease endpoint. Secondly, as the “Control” and “Severe AD” groups were used to generate the 1,372-probe set signature, inclusion of these samples would likely influence correlations in a biased way. By excluding these samples, we can assess correlation with AD progression in a truly independent ‘test’ set of samples.
The advantages of using pair-wise relational features (i.e. metafeatures) have recently been demonstrated by Rocha de Paula et al. [116] in the context of plasma protein biomarkers for the early detection of AD. We therefore expanded the 1,372-probe set signature by applying different operators between each possible pair of probe sets. This led to the creation of two “artificial” data sets:
The first data set includes all the pair-wise ratios of the gene expression values in 1,372-probe set data. It contains a total of 941,885 probe sets, metafeatures and progression markers. We refer to this data set as the 941,885 ratio metafeatures data set.
The second data set includes all the pair-wise differences, summations, ratios and products of the gene expression values in 1,372-probe set data. It contains a total of 3,763,403 probe sets, metafeatures and progression markers. We refer to this data set as the 3,763,403 ratio-sum-difference-product metafeatures data set.
In each of these two data sets, we have included the original 1,372-probes gene expression signature and five measures of AD progression: MMSE score, NFT count and Braak staging from Blalock et al. [3] and the Jensen-Shannon divergences, JSDcontrol and JSDsevere, from Ravetti et al. [5]. We assume here that correlation between the values associated with these measures and microarray probe set expression values can highlight important biomarkers of AD progression.
Data Clustering
We have adapted, enhanced and re-implemented the MSTkNN graph partitioning algorithm in [10] to cluster the features in the large data sets. The proposed method utilizes a graph partitioning approach that optimizes both the local minimum (by using the kNN graph) and global minimum (by using the MST). Therefore, the clusters presented are not necessarily a sorted a list of probes by their correlation to the phenotypes.
The algorithm first constructs an undirected and complete graph from the data set where each node is a biological feature and each edge represents a correlation between two features. Then, the algorithm starts the clustering process by computing two proximity graphs: a minimum spanning tree (GMST) and a k-nearest neighbour graph (GkNN); where the value for k is adaptively selected from the following equation:
| (1) |
Subsequently, the algorithm inspects all edges in GMST. If for a given edge (x,y) neither x is one of the k nearest neighbors of y, nor y is one of the k nearest neighbors of x, the edge is eliminated from GMST. This results in a new graph  = GMST – {(x,y)}. Since GMST is a tree, after the first edge is deleted
= GMST – {(x,y)}. Since GMST is a tree, after the first edge is deleted  is now a forest, as it is a graph that composed of two subtrees. The algorithm continues applying the same procedure to each subtree in
is now a forest, as it is a graph that composed of two subtrees. The algorithm continues applying the same procedure to each subtree in  thus generated (with a value of k re-adjusted by eq. (1) above where n is now the number of nodes in each subtree), until no further partition is possible. The final partition of the nodes of
thus generated (with a value of k re-adjusted by eq. (1) above where n is now the number of nodes in each subtree), until no further partition is possible. The final partition of the nodes of  induced by the forest is the result of the clustering algorithm.
induced by the forest is the result of the clustering algorithm.
Figure 6. Demonstration of the modified MSTkNN algorithm.

(a) An MSTp created from a data set with n=10 features/probe sets. Each edge is labeled with an integer value p, where the value of p is determined using a sorted list of nearest neighbors for each feature (see eq. (2)). The edge between F9 and F10 is a candidate for elimination, since it has a value of p > = 2 (b) Two connected components are identified and we apply the same procedure with the component that has more than three elements. (c) The final outcome of the clustering.
Figure 6. Demonstration of the modified MST
k
NN algorithm. (a) An MST
p
created from a data set with
n
= 10 features/probe sets. Each edge is labeled with an integer value p, where the value of p is determined using a sorted list of nearest neighbors for each feature (see eq. (2)). The edge between F9 and F10 is a candidate for elimination, since it has a value of p> 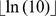 = 2 (b) Two connected components are identified and we apply the same procedure with the component that has more than three elements. (c) The final outcome of the clustering.
= 2 (b) Two connected components are identified and we apply the same procedure with the component that has more than three elements. (c) The final outcome of the clustering.
The original algorithm requires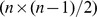 distance values (between all pairs of the n elements) as the input. For a large data set, this may be too large to fit in the computer’s in-memory and, for even larger values of n, it may not even fit in external memory. Even if we can store the distance matrix in the external memory, the computational speed will slow down dramatically because of the increased number of I/O operations. Therefore, we modified this step and instead of creating the complete graph from the distance matrix, we create a q-nearest neighbor graph (GqNN), where q =
distance values (between all pairs of the n elements) as the input. For a large data set, this may be too large to fit in the computer’s in-memory and, for even larger values of n, it may not even fit in external memory. Even if we can store the distance matrix in the external memory, the computational speed will slow down dramatically because of the increased number of I/O operations. Therefore, we modified this step and instead of creating the complete graph from the distance matrix, we create a q-nearest neighbor graph (GqNN), where q = 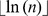 +1. This procedure reduces the input graph size, but still creates a reasonable clustering structure of the data set. The value of the q is determined from the inclusion relationship
[117] of the GMST and the family of the nested sequence of graphs (GkNN, where k > ln(n)).
+1. This procedure reduces the input graph size, but still creates a reasonable clustering structure of the data set. The value of the q is determined from the inclusion relationship
[117] of the GMST and the family of the nested sequence of graphs (GkNN, where k > ln(n)).
Next, we compute the MST of the GqNN graph. We term it as, GMSTp. We annotate each edge in GMSTp according to the following procedure: for each edge (a,b) in E(GMSTp) we assign an integer value p such that if f(a,b) is the index of b in the sorted list of nearest neighbors of a in GqNN, the value of p is given by,
| (2) |
We define the maximum value of p in the MSTp (or any of its components) as pmax and then, we partition the GMSTp with the following criteria:
C
1. If p> 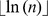 ; remove the edge,
; remove the edge,
C
2. If pmax < 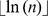 ; remove the edges with weight pmax –1, and;
; remove the edges with weight pmax –1, and;
C
3. If pmax = 1 or pmax = 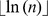 ; do not remove any edge and the result is a “cluster”.
; do not remove any edge and the result is a “cluster”.
The final output of our algorithm is a set of partitions or clusters of the input data (See Figure 6). The algorithm does not require any pre-determined value for q but it is possible to change the threshold from  to any other user-defined parameter. The complete algorithm can be found in [13]. To accelerate the data preprocessing we employed General Purpose Graphics Processing Unit (GPGPU) computing and implemented a fast and scalable approach to compute the distance metrics and the q-nearest neighbor graph (GqNN). An illustrated example of our GPU-based nearest neighbor search algorithm is given in File S4.
to any other user-defined parameter. The complete algorithm can be found in [13]. To accelerate the data preprocessing we employed General Purpose Graphics Processing Unit (GPGPU) computing and implemented a fast and scalable approach to compute the distance metrics and the q-nearest neighbor graph (GqNN). An illustrated example of our GPU-based nearest neighbor search algorithm is given in File S4.
To create the MST from large data set we adapted the EM MST algorithm in [11] and modified it to annotate the edges according eq. (4). The I/O complexity of this algorithm is O(sort(m)•log(n/M)), where n is the number of nodes of the original graph, m is number of edges and M number of nodes that fit into computer’s internal memory, respectively, and the sort(m) is the time required for sorting the m edges. After partitioning the MST, we identify the connected components using the EM connected component algorithm in [11], [12], [118]. The I/O complexity of this algorithm is O(m•log(log(n)). Unlike other clustering tools, we store the connected components/clusters in external memory and only keep the list of the components in computer’s in-memory. This eliminates the excessive use of the in-memory even when there are a large number of components or clusters. Additionally, we tuned the implementations of the adapted algorithms [11], [12],[118] for better performance with denser graphs. Since our algorithm has been implemented in external memory approach, we term our algorithm as EM MSTkNN algorithm. Please note here that our proposed method can be implemented either in-memory or external memory paradigm. To make this method further scalable, we have taken the advantages of external memory algorithms and environments.
The computational tests were performed on a Xenon Nitro T5 Supermicro server (16 CPU cores, 32 GB RAM, 4x NVIDIA Tesla C2050 “Fermi” GPU cards (1792 GPU Cores and 12 Gb RAM total) and 800GB Hard-disk) and the programs were written in C/C++ with the support of CUDA [119], STL, STXXL [120] and BOOST [121] library and compiled using the g++ and nvcc compiler on a Linux operating system with kernel version 2.6.9.
Monte-Carlo Random Permutation Test
Assessing correlations involving a large number of metafeatures and a small number of samples has the potential to lead to spurious, false positive results. To estimate the false discovery rate when assessing correlations involving metafeatures, we performed a simple a Monte-Carlo permutation test, a useful resampling test when there are many possible orderings of the samples. In this test, we randomly permuted (rearranged) the values of the progression marker in question and computed the correlation of each metafeature against it. A total of 1,000 iterations of the test were performed and the average number of metafeatures passing various correlation coefficient thresholds determined.
Functional Annotation
After performing the clustering on the expanded data sets, we identified the specific clusters that contained the progression markers (MMSE score, NFT count, Braak staging, JSDcontrol and JSDsevere) and analysed the correlation (using Spearman’s rank computation) of the clustering probe sets or metafeatures with these progression markers. If the size of the cluster was very big, we noted the top most positively and negatively correlated probe sets or metafeatures.
GATHER [122], a popular online tool for interpreting genomic signatures, was used to assess possible biological relationships between the two transcripts targeted by the probe sets comprising a metafeature. We checked each of the clustering metafeatures to determine if the relevant transcripts share any common biological pathway (KEGG pathways). Our objective here is to detect the pair of transcripts that not only appear in the same pathway but also jointly activate the progression of AD.
Validation Using an Alternative Dataset
For validation of changes in select genes, we analyzed the dataset contributed by Liang and colleagues [95], [96], which can be accessed from NCBI Gene Expression Omnibus under the accession number GSE5281. This microarray dataset was generated using Affymetrix Human Genome U133 Plus 2.0 Arrays and assessed gene expression in healthy neurons isolated by laser capture microdissection from different regions of post-mortem control and AD brain. We refer the reader to [95], [96] for full experimental details.
In the analyses presented here, we investigated gene expression in the entorhinal cortex (13 controls, 10 AD), hippocampus (13 controls, 10 AD), middle temporal gyrus (12 controls, 16 AD) and posterior cingulate cortex (13 controls, 9 AD). Microarray data were normalised with RMA in the Affymetrix Expression Console (v1.1). For each region, genetic signatures that discriminate control and AD samples were generated as described in [5]. Briefly, data were first preprocessed by discretization of the expression values using an implementation of Fayyad and Irani’s algorithm [6], an entropy-based heuristic. This was followed by a filtering step to discard probe sets that do not provide sufficient information to discriminate between the control and AD classes, based on the Minimum Description Length principle (reviewed in [123]). The matrix of discrete values returned after entropy filtering was then used to create an instance of the (α,β)-k-Feature Set problem (for details see [123]). The optimal solution to this problem was used as the genetic signature.
Supporting Information
Comparison of the clustering outcomes with stringent p -values.
(DOC)
Frequency of the highly negative correlations in the 941,885 ratio metafeatures data set.
(DOC)
Frequency of the highly positive correlations in the 941,885 ratio metafeatures data set.
(DOC)
Frequency of the highly negative correlations in the 1,372-probe AD signature data set.
(DOC)
Frequency of the highly positive correlations in the 1,372-probe AD signature data set.
(DOC)
Counter example for the correlation of probe set pairs.
(DOC)
Analysis of the 941,885 ratio metafeatures clustered with the MMSE score.
(DOC)
False discovery rate (FDR) in 1,372-probe AD signature data set.
(DOC)
False discovery rate (FDR) 941,885 ratio metafeatures data set.
(DOC)
Pair-wise comparisons of overlap between progression marker clustering outcomes in the 941,885 metafeatures data set.
(DOC)
Pair-wise comparisons of overlap between progression marker clustering outcomes in the 3,763,403 metafeatures data set.
(DOC)
Clusters containing the progression markers (from the 1,372-probe AD signature data set).
(XLSX)
Clusters containing the progression markers (from the 941,885 ratio metafeatures data set).
(XLSX)
Clusters containing the progression markers (from the 3,763,403 ratio-sum-difference-product metafeatures data set).
(XLSX)
Implementation of the proposed clustering method.
(PDF)
Funding Statement
The authors would like to thank the support of the ARC Centre of Excellence in Bioinformatics, and the University of Newcastle’s through the funding of the Priority Research Centre for Bioinformatics, Biomarker Discovery and Information-Based Medicine. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Alzheimer’s Association (2011) 2011 Alzheimer’s disease facts and figures. Alzheimer’s Dement 7: 208–244. [DOI] [PubMed] [Google Scholar]
- 2.Burns JM, Morris JC (2008) Mild cognitive impairment and early Alzheimer’s disease: detection and diagnosis: Wiley.’
- 3. Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, et al. (2004) Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A 101: 2173–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: 189–198. [DOI] [PubMed] [Google Scholar]
- 5. Gomez Ravetti M, Rosso OA, Berretta R, Moscato P (2010) Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus’ gene expression profiles in Alzheimer’s disease. PLoS One 5: e10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fayyad UM, Irani KB (1993) Multi-Interval Discretization of Continuous-Valued Attributes for Classification Learning. Morgan Kaufmann: 1022–1029.
- 7.Cotta C, Sloper C, Moscato P (2004) Evolutionary Search of Thresholds for Robust Feature Set Selection: Application to the Analysis of Microarray Data,. Applications of Evolutionary Computing, Springer Berlin/Heidelberg: 21–30.
- 8.Grosse I, Galvan PB, Carpena P, Roldan RR, Oliver J, et al.. (2002) Analysis of symbolic sequences using the Jensen-Shannon divergence. Physical Review E 65. [DOI] [PubMed]
- 9. Johnstone D, Milward EA, Berretta R, Moscato P (2012) Multivariate Protein Signatures of Pre-Clinical Alzheimer’s Disease in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) Plasma Proteome Dataset. PLoS One 7: e34341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inostroza-Ponta M (2008) An Integrated and Scalable Approach Based on Combinatorial Optimization Techniques for the Analysis of Microarray Data [Phd Thesis]. Newcastle: The University of Newcastle, Australia.
- 11.Dementiev R, Sanders P, Schultes D, Sibeyn J (2004) Engineering an external memory minimum spanning tree algorithm. 3rd IFIP Intl Conf on Theoretical Computer Science: 195–208.
- 12. Sibeyn JF (2004) External Connected Components Algorithm Theory - SWAT 2004. 3111/2004: 468–479. [Google Scholar]
- 13.Arefin AS, Inostroza-Ponta M, Mathieson L, Berretta R, Moscato P (2011) Clustering Nodes in Large-Scale Biological Networks Using External Memory Algorithms. In: and YX, and AC, and MH, Zhou W, editors. Algorithms and Architectures for Parallel Processing - 11th International Conference, ICA3PP, Melbourne, Australia, October 24–26, 2011, Proceedings, Part II: Lecture Notes in Computer Science, Springer. 375–386.
- 14.Vitter JS (2001) External memory algorithms and data structures: Dealing with massive data. ACM Computing Surveys 33.
- 15.Chiang Y-J, Goodrich MT, Grove EF, Tamassia R, Vengroff DE, et al.. (1995) External-memory graph algorithms. SODA ’95: Proceedings of the sixth annual ACM-SIAM symposium on Discrete algorithms Society for Industrial and Applied Mathematics: 139–149.
- 16. Cormen TH, Nicol DM (1998) Performing out-of-core ffts on parallel disk systems. Parallel Comput 24: 5–20. [Google Scholar]
- 17. Inostroza-Ponta M, Berretta R, Mendes A, Moscato P (2006) An automatic graph layout procedure to visualize correlated data. Artificial Intelligence in Theory and Practice, International Federation for Information Processing 217: 179–188. [Google Scholar]
- 18.Inostroza-Ponta M, Mendes A, Berretta R, Moscato P (2007) An integrated qap-based approach to visualize patterns of gene expression similarity. Australian Conference on Artificial Life, Lectures Notes in Artificial Inteligence Springer: 482–488.
- 19. Capp A, Inostroza-Ponta M, Bill D, Moscato P, Lai C, et al. (2009) Is there more than one proctitis syndrome? A revisitation using data from the TROG 96.01 trial. Radiother Oncol 90: 400–407. [DOI] [PubMed] [Google Scholar]
- 20. Inostroza-Ponta M, Berretta R, Moscato P (2011) Qapgrid: A two level qap-based approach for large-scale data analysis and visualization. PloS One 6: e14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marsagishvili LG, Shpagina MD, Emel’ianenko VI, Podlubnaia ZA (2005) [Sarcomeric proteins of the titin family form amyloids]. Biofizika 50: 803–809. [PubMed] [Google Scholar]
- 22.Brouwers N, Van Cauwenberghe C, Engelborghs S, Lambert JC, Bettens K, et al.. (2011) Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. [DOI] [PMC free article] [PubMed]
- 23. Carrasquillo MM, Belbin O, Hunter TA, Ma L, Bisceglio GD, et al. (2010) Replication of CLU, CR1, and PICALM associations with alzheimer disease. Arch Neurol 67: 961–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chibnik LB, Shulman JM, Leurgans SE, Schneider JA, Wilson RS, et al. (2011) CR1 is associated with amyloid plaque burden and age-related cognitive decline. Ann Neurol 69: 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Corneveaux JJ, Myers AJ, Allen AN, Pruzin JJ, Ramirez M, et al. (2010) Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet 19: 3295–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu X, Pickering E, Liu YC, Hall S, Fournier H, et al. (2011) Meta-analysis for genome-wide association study identifies multiple variants at the BIN1 locus associated with late-onset Alzheimer’s disease. PLoS One 6: e16616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jun G, Naj AC, Beecham GW, Wang LS, Buros J, et al. (2010) Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol 67: 1473–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naj AC, Beecham GW, Martin ER, Gallins PJ, Powell EH, et al.. (2010) Dementia revealed: novel chromosome 6 locus for late-onset Alzheimer disease provides genetic evidence for folate-pathway abnormalities. PLoS Genet 6. [DOI] [PMC free article] [PubMed]
- 29. Schjeide B, Schnack C, Lambert J, Lill C, Kirchheiner J, et al. (2011) The role of clusterin, complement receptor 1, and phosphatidylinositol binding clathrin assembly protein in Alzheimer disease risk and cerebrospinal fluid biomarker levels. Arch Gen Psychiatry 68: 207–213. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Q, Yu JT, Zhu QX, Zhang W, Wu ZC, et al. (2010) Complement receptor 1 polymorphisms and risk of late-onset Alzheimer’s disease. Brain Res 1348: 216–221. [DOI] [PubMed] [Google Scholar]
- 31.Bralten J, Franke B, Arias-Vasquez A, Heister A, Brunner HG, et al.. (2011) CR1 genotype is associated with entorhinal cortex volume in young healthy adults. Neurobiol Aging 32: 2106 e2107–2106 e2111. [DOI] [PubMed]
- 32. Biffi A, Anderson CD, Desikan RS, Sabuncu M, Cortellini L, et al. (2010) Genetic variation and neuroimaging measures in Alzheimer disease. Arch Neurol 67: 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Majima T, Ogita H, Yamada T, Amano H, Togashi H, et al. (2009) Involvement of afadin in the formation and remodeling of synapses in the hippocampus. Biochem Biophys Res Commun 385: 539–544. [DOI] [PubMed] [Google Scholar]
- 34. Tschernatsch M, Stolz E, Strittmatter M, Kaps M, Blaes F (2005) Antinuclear antibodies define a subgroup of paraneoplastic neuropathies: clinical and immunological data. J Neurol Neurosurg Psychiatry 76: 1702–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, et al. (2007) C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet 39: 1068–1070. [DOI] [PubMed] [Google Scholar]
- 36. Sonoda Y, Mukai H, Matsuo K, Takahashi M, Ono Y, et al. (2010) Accumulation of tumor-suppressor PTEN in Alzheimer neurofibrillary tangles. Neurosci Lett 471: 20–24. [DOI] [PubMed] [Google Scholar]
- 37. Marsagishvili LG, Shpagina MD, Emel’ianenko VI, Podlubnaia ZA (2005) Sarcomeric proteins of the titin family form amyloids. Biofizika 50: 803–809. [PubMed] [Google Scholar]
- 38. Kolla V, Jeno P, Moes S, Tercanli S, Lapaire O, et al. (2010) Quantitative proteomics analysis of maternal plasma in Down syndrome pregnancies using isobaric tagging reagent (iTRAQ). J Biomed Biotechnol 2010: 952047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lian Q, Ladner CJ, Magnuson D, Lee JM (2001) Selective changes of calcineurin (protein phosphatase 2B) activity in Alzheimer’s disease cerebral cortex. Exp Neurol 167: 158–165. [DOI] [PubMed] [Google Scholar]
- 40. Shibata N, Kobayashi M (2008) The role for oxidative stress in neurodegenerative diseases. Brain Nerve 60: 157–170. [PubMed] [Google Scholar]
- 41. Yang G, Wang L, Zhu M, Xu D (2008) Identification of non-Alzheimer’s disease tauopathies-related proteins by proteomic analysis. Neurol Res 30: 613–622. [DOI] [PubMed] [Google Scholar]
- 42. Zhao C, Braunewell KH (2008) Expression of the neuronal calcium sensor visinin-like protein-1 in the rat hippocampus. Neuroscience 153: 1202–1212. [DOI] [PubMed] [Google Scholar]
- 43. Youn H, Jeoung M, Koo Y, Ji H, Markesbery WR, et al. (2007) Kalirin is under-expressed in Alzheimer’s disease hippocampus. J Alzheimers Dis 11: 385–397. [DOI] [PubMed] [Google Scholar]
- 44. Schnurra I, Bernstein HG, Riederer P, Braunewell KH (2001) The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer’s disease and promotes cell death and tau phosphorylation in vitro: a link between calcium sensors and Alzheimer’s disease? Neurobiol Dis 8: 900–909. [DOI] [PubMed] [Google Scholar]
- 45. Braunewell K, Riederer P, Spilker C, Gundelfinger ED, Bogerts B, et al. (2001) Abnormal localization of two neuronal calcium sensor proteins, visinin-like proteins (vilips)-1 and -3, in neocortical brain areas of Alzheimer disease patients. Dement Geriatr Cogn Disord 12: 110–116. [DOI] [PubMed] [Google Scholar]
- 46. Laterza OF, Modur VR, Crimmins DL, Olander JV, Landt Y, et al. (2006) Identification of novel brain biomarkers. Clin Chem 52: 1713–1721. [DOI] [PubMed] [Google Scholar]
- 47. Zhao C, Anand R, Braunewell KH (2009) Nicotine-induced Ca2+-myristoyl switch of neuronal Ca2+ sensor VILIP-1 in hippocampal neurons: a possible crosstalk mechanism for nicotinic receptors. Cell Mol Neurobiol 29: 273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao CJ, Noack C, Brackmann M, Gloveli T, Maelicke A, et al. (2009) Neuronal Ca2+ sensor VILIP-1 leads to the upregulation of functional alpha4beta2 nicotinic acetylcholine receptors in hippocampal neurons. Mol Cell Neurosci 40: 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Opii WO, Joshi G, Head E, Milgram NW, Muggenburg BA, et al. (2008) Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer’s disease. Neurobiol Aging 29: 51–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Poon HF, Farr SA, Thongboonkerd V, Lynn BC, Banks WA, et al. (2005) Proteomic analysis of specific brain proteins in aged SAMP8 mice treated with alpha-lipoic acid: implications for aging and age-related neurodegenerative disorders. Neurochem Int 46: 159–168. [DOI] [PubMed] [Google Scholar]
- 51. Somerville MJ, Percy ME, Bergeron C, Yoong LK, Grima EA, et al. (1991) Localization and quantitation of 68 kDa neurofilament and superoxide dismutase-1 mRNA in Alzheimer brains. Brain Res Mol Brain Res 9: 1–8. [DOI] [PubMed] [Google Scholar]
- 52. George A, Gordon L, Beissbarth T, Koukoulas I, Holsinger R, et al. (2010) A serial analysis of gene expression profile of the Alzheimer’s disease Tg2576 mouse model. Neurotox Res 17: 360–379. [DOI] [PubMed] [Google Scholar]
- 53. Rahman A, Grundke-Iqbal I, Iqbal K (2006) PP2B isolated from human brain preferentially dephosphorylates Ser-262 and Ser-396 of the Alzheimer disease abnormally hyperphosphorylated tau. J Neural Transm 113: 219–230. [DOI] [PubMed] [Google Scholar]
- 54. Rahman A, Grundke-Iqbal I, Iqbal K (2005) Phosphothreonine-212 of Alzheimer abnormally hyperphosphorylated tau is a preferred substrate of protein phosphatase-1. Neurochem Res 30: 277–287. [DOI] [PubMed] [Google Scholar]
- 55. Arnaud L, Chen S, Liu F, Li B, Khatoon S, et al. (2011) Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I2(PP2A)/SET. FEBS Lett 585: 2653–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu R, Zhou XW, Tanila H, Bjorkdahl C, Wang JZ, et al. (2008) Phosphorylated PP2A (tyrosine 307) is associated with Alzheimer neurofibrillary pathology. J Cell Mol Med 12: 241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fabbro S, Schaller K, Seeds NW (2011) Amyloid-beta levels are significantly reduced and spatial memory defects are rescued in a novel neuroserpin-deficient Alzheimer’s disease transgenic mouse model. J Neurochem 118: 928–938. [DOI] [PubMed] [Google Scholar]
- 58. Brorsson AC, Kumita JR, MacLeod I, Bolognesi B, Speretta E, et al. (2010) Methods and models in neurodegenerative and systemic protein aggregation diseases. Front Biosci 15: 373–396. [DOI] [PubMed] [Google Scholar]
- 59. Fabbro S, Seeds NW (2009) Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J Neurochem 109: 303–315. [DOI] [PubMed] [Google Scholar]
- 60. Nielsen HM, Minthon L, Londos E, Blennow K, Miranda E, et al. (2007) Plasma and CSF serpins in Alzheimer disease and dementia with Lewy bodies. Neurology 69: 1569–1579. [DOI] [PubMed] [Google Scholar]
- 61. Kinghorn KJ, Crowther DC, Sharp LK, Nerelius C, Davis RL, et al. (2006) Neuroserpin binds Abeta and is a neuroprotective component of amyloid plaques in Alzheimer disease. J Biol Chem 281: 29268–29277. [DOI] [PubMed] [Google Scholar]
- 62. Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 20: 2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bach-Holm D, Kessing SV, Mogensen U, Forman JL, Andersen PK, et al.. (2011) Normal tension glaucoma and Alzheimer disease: comorbidity? Acta Ophthalmol. [DOI] [PubMed]
- 64. Wang X, Su B, Lee HG, Li X, Perry G, et al. (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29: 9090–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, et al. (2008) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 105: 19318–19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chin J, Palop JJ, Puolivali J, Massaro C, Bien-Ly N, et al. (2005) Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci 25: 9694–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Akiyama H, Nishimura T, Kondo H, Ikeda K, Hayashi Y, et al. (1994) Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res 639: 171–174. [DOI] [PubMed] [Google Scholar]
- 68. Boyd TD, Bennett SP, Mori T, Governatori N, Runfeldt M, et al. (2010) GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J Alzheimers Dis 21: 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Laske C, Stransky E, Hoffmann N, Maetzler W, Straten G, et al. (2010) Macrophage colony-stimulating factor (M-CSF) in plasma and CSF of patients with mild cognitive impairment and Alzheimer’s disease. Curr Alzheimer Res 7: 409–414. [DOI] [PubMed] [Google Scholar]
- 70. Boissonneault V, Rivest S (2009) [The hematopoietic cytokine M-CSF as a cure for Alzheimer’s disease]. Med Sci (Paris) 25: 666–668. [DOI] [PubMed] [Google Scholar]
- 71. Boissonneault V, Filali M, Lessard M, Relton J, Wong G, et al. (2009) Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer’s disease. Brain 132: 1078–1092. [DOI] [PubMed] [Google Scholar]
- 72. Wollmer MA, Nitsch RM, Hock C, Papassotiropoulos A (2006) Genetic association study on colony-stimulating factor 1 in Alzheimer’s disease. Neurodegener Dis 3: 334–337. [DOI] [PubMed] [Google Scholar]
- 73. Penney KL, Schumacher FR, Kraft P, Mucci LA, Sesso HD, et al. (2011) Association of KLK3 (PSA) genetic variants with prostate cancer risk and PSA levels. Carcinogenesis 32: 853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cacabelos R, Llovo R, Fraile C, Fernandez-Novoa L (2007) Pharmacogenetic aspects of therapy with cholinesterase inhibitors: the role of CYP2D6 in Alzheimer’s disease pharmacogenetics. Curr Alzheimer Res 4: 479–500. [DOI] [PubMed] [Google Scholar]
- 75. Magliulo L, Dahl ML, Lombardi G, Fallarini S, Villa LM, et al. (2011) Do CYP3A and ABCB1 genotypes influence the plasma concentration and clinical outcome of donepezil treatment? Eur J Clin Pharmacol 67: 47–54. [DOI] [PubMed] [Google Scholar]
- 76. Barner EL, Gray SL (1998) Donepezil use in Alzheimer disease. Ann Pharmacother 32: 70–77. [DOI] [PubMed] [Google Scholar]
- 77. Carayol J, Sacco R, Tores F, Rousseau F, Lewin P, et al. (2011) Converging Evidence for an Association of ATP2B2 Allelic Variants with Autism in Male Subjects. Biol Psychiatry 70: 880–887. [DOI] [PubMed] [Google Scholar]
- 78. Xu L, Wang Z, Xiong X, Gu X, Gao X (2011) Identification of a novel point mutation of mouse Atp2b2 induced by N-ethyl-N-nitrosourea mutagenesis. Exp Anim 60: 71–78. [DOI] [PubMed] [Google Scholar]
- 79. Sambamurti K, Sevlever D, Koothan T, Refolo LM, Pinnix I, et al. (1999) Glycosylphosphatidylinositol-anchored proteins play an important role in the biogenesis of the Alzheimer’s amyloid beta-protein. J Biol Chem 274: 26810–26814. [DOI] [PubMed] [Google Scholar]
- 80. Shimohama S, Matsushima H (1995) Signal transduction mechanisms in Alzheimer disease. Alzheimer Dis Assoc Disord 9 Suppl 215–22. [DOI] [PubMed] [Google Scholar]
- 81. Wallace MA (1994) Effects of Alzheimer’s disease-related beta amyloid protein fragments on enzymes metabolizing phosphoinositides in brain. Biochim Biophys Acta 1227: 183–187. [DOI] [PubMed] [Google Scholar]
- 82. Rentzos M, Zoga M, Paraskevas GP, Kapaki E, Rombos A, et al. (2006) IL-15 is elevated in cerebrospinal fluid of patients with Alzheimer’s disease and frontotemporal dementia. J Geriatr Psychiatry Neurol 19: 114–117. [DOI] [PubMed] [Google Scholar]
- 83. Kim SH, Lubec G (2001) Brain alpha-endosulfine is manifold decreased in brains from patients with Alzheimer’s disease: a tentative marker and drug target? Neurosci Lett 310: 77–80. [DOI] [PubMed] [Google Scholar]
- 84. Lefebvre T, Dehennaut V, Guinez C, Olivier S, Drougat L, et al. (2010) Dysregulation of the nutrient/stress sensor O-GlcNAcylation is involved in the etiology of cardiovascular disorders, type-2 diabetes and Alzheimer’s disease. Biochim Biophys Acta 1800: 67–79. [DOI] [PubMed] [Google Scholar]
- 85. Middle F, Pritchard AL, Handoko H, Haque S, Holder R, et al. (2010) No association between neuregulin 1 and psychotic symptoms in Alzheimer’s disease patients. J Alzheimers Dis 20: 561–567. [DOI] [PubMed] [Google Scholar]
- 86. Go RC, Perry RT, Wiener H, Bassett SS, Blacker D, et al. (2005) Neuregulin-1 polymorphism in late onset Alzheimer’s disease families with psychoses. Am J Med Genet B Neuropsychiatr Genet 139B: 28–32. [DOI] [PubMed] [Google Scholar]
- 87. Chaudhury AR, Gerecke KM, Wyss JM, Morgan DG, Gordon MN, et al. (2003) Neuregulin-1 and erbB4 immunoreactivity is associated with neuritic plaques in Alzheimer disease brain and in a transgenic model of Alzheimer disease. J Neuropathol Exp Neurol 62: 42–54. [DOI] [PubMed] [Google Scholar]
- 88. Kusano S, Shiimura Y, Eizuru Y (2011) I-mfa domain proteins specifically interact with SERTA domain proteins and repress their transactivating functions. Biochimie 93: 1555–1564. [DOI] [PubMed] [Google Scholar]
- 89. Desjardins P, Ledoux S (1998) Expression of ced-3 and ced-9 homologs in Alzheimer’s disease cerebral cortex. Neurosci Lett 244: 69–72. [DOI] [PubMed] [Google Scholar]
- 90. Karelson E, Mahlapuu R, Zilmer M, Soomets U, Bogdanovic N, et al. (2002) Possible signaling by glutathione and its novel analogue through potent stimulation of fontocortical G proteins in normal aging and in Alzheimer’s disease. Ann N Y Acad Sci 973: 537–540. [DOI] [PubMed] [Google Scholar]
- 91. Emilsson L, Saetre P, Jazin E (2006) Low mRNA levels of RGS4 splice variants in Alzheimer’s disease: association between a rare haplotype and decreased mRNA expression. Synapse 59: 173–176. [DOI] [PubMed] [Google Scholar]
- 92. Emilsson L, Saetre P, Jazin E (2006) Alzheimer’s disease: mRNA expression profiles of multiple patients show alterations of genes involved with calcium signaling. Neurobiol Dis 21: 618–625. [DOI] [PubMed] [Google Scholar]
- 93. Muma NA, Mariyappa R, Williams K, Lee JM (2003) Differences in regional and subcellular localization of G(q/11) and RGS4 protein levels in Alzheimer’s disease: correlation with muscarinic M1 receptor binding parameters. Synapse 47: 58–65. [DOI] [PubMed] [Google Scholar]
- 94. Saetre P, Jazin E, Emilsson L (2011) Age-related changes in gene expression are accelerated in Alzheimer’s disease. Synapse 65: 971–974. [DOI] [PubMed] [Google Scholar]
- 95. Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, et al. (2008) Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiol Genomics 33: 240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, et al. (2007) Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol Genomics 28: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tarawneh R, D’Angelo G, Macy E, Xiong C, Carter D, et al. (2011) Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol 70: 274–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Moscato P, Berretta R, Hourani M, Mendes A, Cotta C (2005) Genes related with Alzheimer’s disease: A comparison of evolutionary search, statistical and integer programming approaches. Applications of Evolutionary Computing, Proceedings 3449: 84–94. [Google Scholar]
- 99. Berretta R, Moscato P (2010) Cancer biomarker discovery: the entropic hallmark. PLoS One 5: e12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ge YS, Teng WY, Zhang CD (2009) Protective effect of cyclophilin A against Alzheimer’s amyloid beta-peptide (25–35)-induced oxidative stress in PC12 cells. Chin Med J (Engl) 122: 716–724. [PubMed] [Google Scholar]
- 101. Morimoto K, Horio J, Satoh H, Sue L, Beach T, et al. (2011) Expression profiles of cytokines in the brains of Alzheimer’s disease (AD) patients compared to the brains of non-demented patients with and without increasing AD pathology. J Alzheimers Dis 25: 59–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Leduc V, Legault V, Dea D, Poirier J (2011) Normalization of gene expression using SYBR green qPCR: A case for paraoxonase 1 and 2 in Alzheimer’s disease brains. J Neurosci Methods 200: 14–19. [DOI] [PubMed] [Google Scholar]
- 103. Newington JT, Pitts A, Chien A, Arseneault R, Schubert D, et al. (2011) Amyloid beta resistance in nerve cell lines is mediated by the Warburg effect. PLoS One 6: e19191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Marino S, Krimpenfort P, Leung C, van der Korput HA, Trapman J, et al. (2002) PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development 129: 3513–3522. [DOI] [PubMed] [Google Scholar]
- 105. Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, et al. (2005) Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J Neurochem 93: 105–117. [DOI] [PubMed] [Google Scholar]
- 106. Zhang X, Li F, Bulloj A, Zhang YW, Tong G, et al. (2006) Tumor-suppressor PTEN affects tau phosphorylation, aggregation, and binding to microtubules. FASEB J 20: 1272–1274. [DOI] [PubMed] [Google Scholar]
- 107. Kerr F, Rickle A, Nayeem N, Brandner S, Cowburn RF, et al. (2006) PTEN, a negative regulator of PI3 kinase signalling, alters tau phosphorylation in cells by mechanisms independent of GSK-3. FEBS Lett 580: 3121–3128. [DOI] [PubMed] [Google Scholar]
- 108. Zhang H, Liu R, Wang R, Hong S, Xu H, et al. (2008) Presenilins regulate the cellular level of the tumor suppressor PTEN. Neurobiol Aging 29: 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, et al. (2004) Novel PINK1 mutations in early-onset parkinsonism. Annals of neurology 56: 424–427. [DOI] [PubMed] [Google Scholar]
- 110. Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, et al. (2004) PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol 56: 336–341. [DOI] [PubMed] [Google Scholar]
- 111. Domanskyi A, Geissler C, Vinnikov IA, Alter H, Schober A, et al. (2011) Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson’s disease models. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology 25: 2898–2910. [DOI] [PubMed] [Google Scholar]
- 112. Cao M, Xu J, Shen C, Kam C, Huganir RL, et al. (2007) PICK1-ICA69 heteromeric BAR domain complex regulates synaptic targeting and surface expression of AMPA receptors. J Neurosci 27: 12945–12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Pilon M, Peng XR, Spence AM, Plasterk RH, Dosch HM (2000) The diabetes autoantigen ICA69 and its Caenorhabditis elegans homologue, ric-19, are conserved regulators of neuroendocrine secretion. Mol Biol Cell 11: 3277–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rossner S, Mehlhorn G, Schliebs R, Bigl V (2001) Increased neuronal and glial expression of protein kinase C isoforms in neocortex of transgenic Tg2576 mice with amyloid pathology. Eur J Neurosci 13: 269–278. [PubMed] [Google Scholar]
- 115. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: 239–259. [DOI] [PubMed] [Google Scholar]
- 116. Rocha de Paula M, Gomez Ravetti M, Berretta R, Moscato P (2011) Differences in abundances of cell-signalling proteins in blood reveal novel biomarkers for early detection of clinical Alzheimer’s disease. PLoS One 6: e17481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gonzlez-Barrios JMaQ, A J (2003) A clustering procedure based on the comparison between the k nearest neighbors graph and the minimal spanning tree. Statistics & Probability Letters 62: 23–34. [Google Scholar]
- 118.Schultes D (2004) External memory spanning forests and Connected Components. Available: http://algo2.iti.kit.edu/dementiev/files/cc.pdf. Accessed: 2012 Aug 27.
- 119. Nickolls J, Buck I, Garland M, Skadron K (2008) Scalable Parallel Programming with CUDA. ACM Queue 16: 40–53. [Google Scholar]
- 120. Dementiev R, Kettner L, Sanders P (2008) STXXL: Standard Template Library for XXL data sets. Softw Pract Exper 38: 589–637. [Google Scholar]
- 121.Siek JG, Lee L-Q, Lumsdaine A (2001) The Boost Graph Library: User Guide and Reference Manual: Addison-Wesley Professional.
- 122. Chang JT, Nevins JR (2006) GATHER: a systems approach to interpreting genomic signatures. Bioinformatics 22: 2926–2933. [DOI] [PubMed] [Google Scholar]
- 123. Berretta R, Costa W, Moscato P (2008) Combinatorial optimization models for finding genetic signatures from gene expression datasets. Methods Mol Biol 453: 363–377. [DOI] [PubMed] [Google Scholar]
- 124. Sharan R, Shamir R (2000) Click: A clustering algorithm with applications to gene expression analysis. AAAI Press 12: 307–316. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of the clustering outcomes with stringent p -values.
(DOC)
Frequency of the highly negative correlations in the 941,885 ratio metafeatures data set.
(DOC)
Frequency of the highly positive correlations in the 941,885 ratio metafeatures data set.
(DOC)
Frequency of the highly negative correlations in the 1,372-probe AD signature data set.
(DOC)
Frequency of the highly positive correlations in the 1,372-probe AD signature data set.
(DOC)
Counter example for the correlation of probe set pairs.
(DOC)
Analysis of the 941,885 ratio metafeatures clustered with the MMSE score.
(DOC)
False discovery rate (FDR) in 1,372-probe AD signature data set.
(DOC)
False discovery rate (FDR) 941,885 ratio metafeatures data set.
(DOC)
Pair-wise comparisons of overlap between progression marker clustering outcomes in the 941,885 metafeatures data set.
(DOC)
Pair-wise comparisons of overlap between progression marker clustering outcomes in the 3,763,403 metafeatures data set.
(DOC)
Clusters containing the progression markers (from the 1,372-probe AD signature data set).
(XLSX)
Clusters containing the progression markers (from the 941,885 ratio metafeatures data set).
(XLSX)
Clusters containing the progression markers (from the 3,763,403 ratio-sum-difference-product metafeatures data set).
(XLSX)
Implementation of the proposed clustering method.
(PDF)