

Abstract
Cleft palate is a frequent and recognizable birth defect attributed to a variety of etiologies including genetic abnormalities and environmental exposures. Bone morphogenetic proteins (BMPs) are involved in embryonic signaling important for a number of developmental processes including bone formation and palate morphogenesis. Recently, haploinsufficency of BMP2 was associated with syndromic forms of cleft palate (CP). Here, we report on a multigenerational family with a history of cleft palate as a result of a 2.3 Megabase (Mb) deletion of chromosome 20p12.3, including the BMP2 gene. In addition to a submucous cleft palate, the proband’s clinical phenotype included failure to thrive (FTT), global developmental delays (DD), and dysmorphic features. The affected father exhibited an overt cleft palate, with a facial gestalt and minor dysmorphic features similar to the proband. The father was otherwise healthy with no history of FTT or DD, suggesting high penetrance, yet variable expressivity for haploinsufficiency of BMP2. The findings presented here provide further evidence for the role of BMP2 in syndromic forms of cleft palate.
Keywords: Cleft palate, microarray analysis, BMP2, 20p12.3, microdeletion
INTRODUCTION
Orofacial clefting is a frequent and recognizable birth defect with an incidence of approximately 1 in 500 to 1 in 1000 [Croen et al., 1998; Genisca et al., 2009; Jones, 1988]. The relatively high incidence of clefting defects is a reflection of the complexity of palatal morphogenesis and its regulation, which is sensitive to gene dosage effects and environmental factors. Orofacial clefts are classified as either cleft lip with or without cleft palate (CL/P) or cleft palate only (CP), and the etiology of these two clefting defects is considered distinct. Both CL/P and CP arise as isolated defects or in conjunction with other major malformations (syndromic). Approximately 85% of CL/P cases occur as an isolated abnormality [Genisca et al., 2009], whereas isolated CP is believed to account for somewhere between 50 and 75% of all CP cases [Croen et al., 1998; Genisca et al., 2009; Jones, 1988]. The syndromic forms of both CL/P and CP have been attributed to a variety of etiologies including chromosomal abnormalities, Mendelian single gene disorders, and environmental factors. Causative genetic mutations have been identified in a number of individuals with syndromic clefting defects, however delineating clear genotype to phenotype correlations for the responsible genes has been difficult due to the genetic heterogeneity of this phenotype.
Bone morphogenetic proteins (BMPs) are important in a number of developmental processes, with their activity in bone formation first being described in 1965 [Urist, 1965]. In addition to critical roles in osteogenesis, BMPs function in heart, kidney, dentin, and eye formation, nervous system development, spermatogenesis, and oocyte development (Reviewed in [Bragdon et al., 2011]). Haploinsufficiency of bone morphogenetic protein 2 (BMP2) was recently proposed to play a crucial role in the formation of cleft palate [Sahoo et al., 2011]. In this work, two individuals with similar clefting defects were reported to have microdeletions that encompassed only the BMP2 gene. However, prior reports of deletions of 20p12.3 encompassing BMP2 did not describe cleft palate in the affected individuals, but rather described cardiac defects including Wolff-Parkinson-White syndrome and, when the deletion included the JAG1 gene in addition to BMP2, Alagille syndrome [Lalani et al., 2009; Le Gloan et al., 2008; Robert et al., 2007].
Here, we report on a child exhibiting syndromic cleft palate with a paternally-inherited deletion of chromosome 20p12.3 identified by chromosomal microarray. The father also exhibited cleft palate and shared a facial gestalt and minor dysmorphic features with the proband. This deletion encompasses four known genes including BMP2 and provides further evidence for haploinsufficiency of BMP2 in cleft palate formation.
CLINICAL REPORT
The proband presented to the genetics clinic at age 5 months for a history of dysmorphic features. He was the first child of nonconsanguineous Caucasian parents. He was born after a 37 week gestation, with birthweight of 2608 g (5th centile), length of 47 cm (10th centile) and head circumference of 32 cm (5th centile). At the time of his first visit to genetics, he was noted to have a bifid uvula and microretrognathia and was ultimately diagnosed with a submucous cleft palate. Additional findings at that time included widened palpebral fissures, a long philtrum and digital hypoplasia of second and third toes (Fig 1A, B, C). A microarray was ordered and revealed an ~2.3 Mb deletion on chromosome 20 involving band p12.3 (see Results).
Figure 1. Facial profiles and toe hypoplasia in proband and family members.
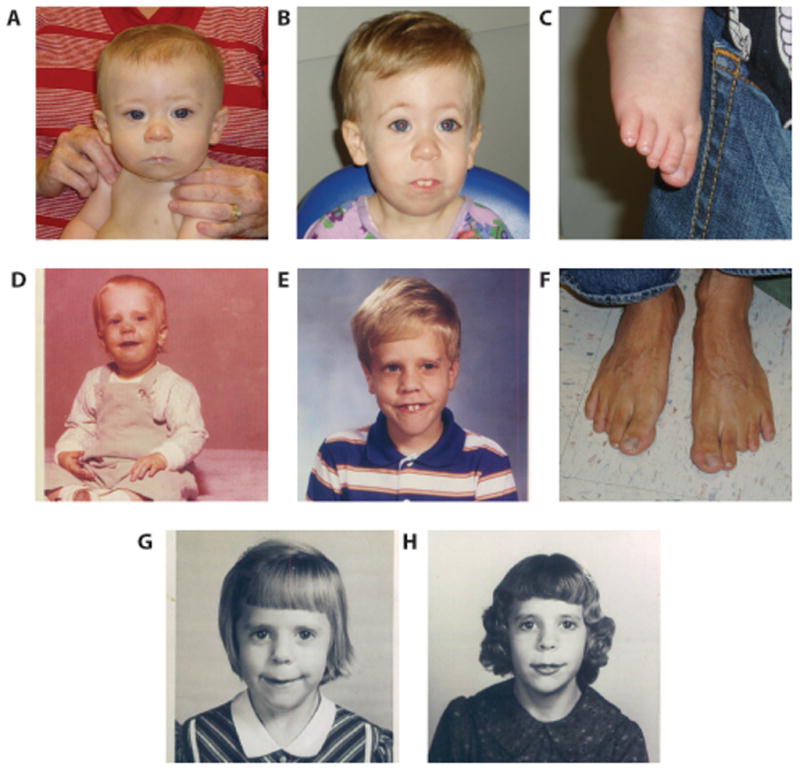
(A,B) Proband at 5 months and 3 years, 4 months of age. Note the long philtrum and pronounced central incisors. (C) Proband’s right foot showing hypoplasia of the second and third toes (D,E) Proband’s father at 17 months and 9 years of age. Note the similar facial features including long philtrum and pronounced teeth. (F) Father’s feet showing third and fourth digit hypoplasia of the left foot. (G,H) Paternal grandmother at approximately 7 years and 11 years of age.
An initial echocardiogram to rule out midline cardiac anomalies was carried out when the child was 7 months of age and was significant for a patent ductus arteriosus (PDA). A follow-up echocardiogram performed when the child was 1 year 10 months revealed normal results. Given the association of Wolf-Parkinson-White (WPW) with microdeletions at 20p12.3, an electrocardiogram (EKG) was also performed when the child was 1 year 10 months. This revealed a borderline prolonged QT interval. However, repeat EKGs at 2 years 11 months and 3 years 10 months were normal.
The child has a long-standing history of failure to thrive (FTT). On physical exam at 2 years 5 months, weight was 8.3 kg (<3rd centile, 50th centile for 7-month-old male), length was 80 cm (<3rd centile, 50th centile for 16- month-old male), and head circumference was 43.8 cm (<3rd centile, 50th centile for 7-month-old male). On physical exam at 4 years 4 months, height was 87.2 cm (<3rd centile, 50th centile for 2-year-old male), weight was 9.5 kg (<3rd centile, 50th centile for 10-month-old male), and head circumference was 44.8 cm (<3rd centile, 50th centile for 8 month-old-male).
When the child was 1 year 10 months, an MRI of brain was normal. At 2 years 2 months, a complete neurodevelopmental evaluation diagnosed him with receptive and expressive language delays and gross motor delays, consistent with global developmental delays. There is a history of self-stimulatory behaviors including tongue clicking, hand flapping, echolalia, and repetitive speech. There is no history of seizures or staring spells. Additional pertinent medical history includes a normal newborn hearing screen, normal pediatric ophthalmologic exam, and normal renal ultrasound. Thoracolumbar spine films in the first and second years of life were consistent with mild dextroscoliosis of the mid-thoracic spine. Pronounced central incisors were also noted in this child.
The proband’s father was noted to have a similar facial gestalt including a long philtrum and pronounced central incisors, a history of cleft palate, sensorineural hearing loss requiring the use of aids, and digital hypoplasia of toes 2–3 bilaterally (Fig 1D, E, F). There is no history of developmental delays or cardiac abnormalities and his EKG has been documented as normal and specifically with no evidence for WPW. The father reports his general health as good. Interestingly, the proband’s paternal grandmother has similar facial features as both the proband and his father; however, medical history was uninformative regarding cleft palate (Fig 1G, H).
METHODS AND RESULTS
Clinical chromosomal microarray analysis was carried out on genomic DNA isolated from peripheral blood of the proband and his father using a custom-designed oligonucleotide plus SNP array, according to standard protocols (Oxford Gene Technology, Oxford, UK). This array allows the detection of both copy number changes and large stretches of absence of heterozygosity (AOH >10 Megabase (Mb)). The oligonucleotide probes are distributed in a whole genome plus targeted design with an average probe spacing of 25 kilobases (kb) genome-wide and a minimum of 10 probes per targeted gene or region [Baldwin et al., 2008]. Data analysis was carried out using CytoSure Interpret software (version 3.4.8, OGT, Oxford, UK).
We identified a 2.26 Mb (minimum; chr20:6,265,251–8,523,663; build hg18, 2006) to 2.32 Mb (maximum; chr20:6,236,616–8,555,628) deletion of chromosome 20p12.3 in both the proband and his father (Fig 2). No clinically significant copy neutral regions of AOH were identified. FISH using two BAC clones from the deleted region (RP11–184L8 and RP11–137P21) confirmed the loss as an interstitial deletion in both individuals (data not shown). Both the minimum and the maximum deletion include four known genes: BMP2, HAO1 (hydroxyacid oxidase 1), TMX4 (thioredoxin-related transmembrane protein 4), and PLCB1 (phospholipase C, Beta-1).
Figure 2. Microdeletions of 20p12.3.
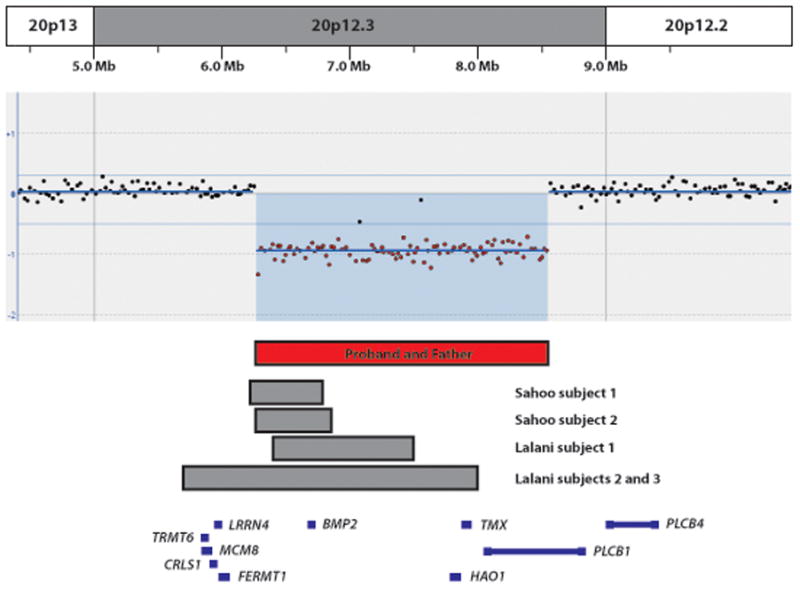
Chromosomal microarray analysis of the proband revealed a ~2.3 Mb deletion of chromosome 20p12.3 (chr20:6,265,251–8,523,663; build hg18, 2006). The relevant region of chromosome 20 with the genomic coordinates is shown above the microarray results for the proband. The black dots represent probes with normal Log2 ratios and the red dots represent probes with Log2ratios meeting our criteria for losses (Log2 ratio≤ −0.5). A comparison to previously reported microdeletions of 20p12.3 that include BMP2 and are less than 3 Mb in size [Sahoo et al., 2011 and Lalani et al., 2009] is also shown. The red and gray rectangles represent the minimum deleted region for the current case (red) and previously reported cases (gray). UCSC genes are shown as blue bars and labeled with the gene symbol. Note that scale is approximate.
DISCUSSION
We describe a boy with a paternally inherited ~2.3 Mb deletion of chromosome 20p12.3 including the BMP2 gene. Both the father and the proband presented with a spectrum of cleft palate, toe hypoplasia, and a similar facial gestalt, including retromicrognathia and a long philtrum. In addition to these overlapping clinical features, the proband presented with a more severe phenotype including global developmental delays and failure to thrive. The size of the deletion in the father and son are exactly the same size at the resolution of the array, which suggests variable expressivity of deletions of this region.
Similar clinical features were recently reported in two individuals with focal deletions including only BMP2 [Sahoo et al., 2011]. These individuals presented with cleft palate, digital anomalies, and facial features including a long philtrum, with the facial gestalt of Patient 1 bearing a striking similarity to the father and son described in this report. Individuals with larger deletions of 20p including BMP2 have also been described with related features including high arched palate, bifid uvula, FTT, global DD, digital anomalies, and a facies including long philtrum [Anad et al., 1990; Shohat et al., 1991; Robert et al., 2007; Kamath et al., 2009].
An additional phenotype reported in some individuals with 20p deletions is Wolff-Parkinson-White (WPW) syndrome, a pre-excitation syndrome of the heart [Anad et al., 1990; Le Gloan et al., 2008; Shohat et al., 1991; Lalani et al., 2009]. Lalani et al., report a de novo deletion encompassing only BMP2 in one individual with WPW, as well as a maternally inherited deletion encompassing BMP2 and one other gene in a male infant with WPW and dysmorphisms, including macrocephaly and a long philtrum. His mother did not have features consistent with WPW by EKG. There was no indication of WPW in the two individuals reported in the current study, nor was there any indication of WPW in the three patients in Sahoo et al., [2011]. Furthermore, neither mutation analysis in 20 individuals with WPW nor animal models, have uncovered a role for haploinsufficiency of BMP2 in WPW [Lalani et al., 2009]. Therefore, the WPW phenotype may be incompletely penetrant in cases of BMP2 haploinsufficiency, or haploinsufficiency of other dosage-sensitive genes on 20p or elsewhere may account for the observed WPW.
BMP2 is well known to play a critical role in bone formation, as well as being implicated in a wide range of functions in morphogenesis, including palate morphogenesis. Animal studies have demonstrated the expression of Bmp2 and Bmp4 in palatal shelves [Lyons et al., 1990] and the in vitro application of BMP2 increases cellular proliferation in the mandibular primordial [Barlow and Francis-West 1997]. BMP2 and its signaling pathways also play important roles in neurogenesis, as evidenced by the wide-spread expression of BMP2 and its receptor proteins, in the developing brain [Furuta et al., 1997; Sato et al., 2010; Zhang et al., 1998]. Abundant expression of BMP2 is also seen in the adult CNS, particularly in the olfactory sensory neurons [Sato et al., 2010]. A normal sense of smell was noted in both the proband and his father indicating that haploinsufficiency of BMP2 does not affect olfactory reception. Further studies are required to elucidate the precise role of BMP2 signaling in neurodevelopment. BMP2 signaling results in diverse outcomes depending on factors including the developmental stage, the cell population and the expression of other BMPs [Lim et al., 2000; Mabie et al., 1999; Nakashima et al., 2001]. This varied outcome may explain the variable expressivity of the phenotype seen in individuals with loss-of-function mutations of BMP2. Moreover, the widespread expression of BMP2 in cells of skeletal, neurological, cardiac, as well as other tissues supports the pleiotrophic effects of BMP2 haploinsufficiency.
Three additional genes are included in the deleted region in our patient: HAO1 (hydroxyacid oxidase 1), TMX4 (thioredoxin-related transmembrane protein 4), and PLCB1 (phosphoinositide-specific phospholipase C beta 1). HAO1 (also HAOX1 or GOX) is expressed primarily in liver and pancreas and encodes a protein capable of oxidizing gylcolates and other 2-hydroxyacids [Jones et al., 2000]. TMX4 is a newly identified thioredoxin that is hypothesized to assist in protein folding in the endoplasmic reticulum [Sugiura et al., 2010]. A homozygous deletion of PLCB1 was reported in one individual with early-onset epileptic encephalopathy, with both unaffected parents carrying a heterozygous deletion [Kurian et al., 2010]. The lack of a clinical significance of haploinsufficiency of these three genes, along with the absence of a clear mechanistic link between gene function and craniofacial development, implicate BMP2 as the most likely critical gene in the deleted region.
The high penetrance of BMP2 deletions is demonstrated by the presence of some physical manifestation in a four generation family [Sahoo et al., 2011], as well as the presence of related clinical manifestations in two, possibly three, generations in this report. Although the penetrance and expressivity of haploinsufficiency of BMP2 cannot be determined until more cases are collected, the data presented here in conjunction with previously published reports strongly suggest a role for haploinsufficiency of BMP2 in cleft palate, as well as digital anomalies and consistent dysmorphisms, including long philtrum and retrognathia. Therefore, studies of individuals with cleft palate plus these additional phenotypes may warrant BMP2 gene testing.
Acknowledgments
The authors are grateful to the family for their cooperation throughout this process. We would like to thank the members of the cytogenetics laboratory at Emory Genetics Laboratory. This work was funded in part by grant MH-074090 from the National Institutes of Health (to CLM).
References
- Anad F, Burn J, Matthews D, Cross I, Davison BC, Mueller R, Sands M, Lillington DM, Eastham E. Alagille syndrome and deletion of 20p. J Med Genet. 1990;27:729–737. doi: 10.1136/jmg.27.12.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin EL, Lee JY, Blake DM, Bunke BP, Alexander CR, Kogan AL, Ledbetter DH, Martin CL. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet Med. 2008;10:415–429. doi: 10.1097/GIM.0b013e318177015c. [DOI] [PubMed] [Google Scholar]
- Barlow AJ, Francis-West PH. Ectopic application of recombinant BMP-2 and BMP-4 can change patterning of developing chick facial primordia. Development. 1997;124:391–398. doi: 10.1242/dev.124.2.391. [DOI] [PubMed] [Google Scholar]
- Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A. Bone morphogenetic proteins: a critical review. Cell Signal. 2011;23(4):609–20. doi: 10.1016/j.cellsig.2010.10.003. [DOI] [PubMed] [Google Scholar]
- Croen LA, Shaw GM, Wasserman CR, Tolarova MM. Racial and ethnic variations in the prevalence of orofacial clefts in California, 1983–1992. Am J Med Genet. 1998;79:42–47. doi: 10.1002/(sici)1096-8628(19980827)79:1<42::aid-ajmg11>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Piston DW, Hogan BL. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development. 1997;124:2203–2212. doi: 10.1242/dev.124.11.2203. [DOI] [PubMed] [Google Scholar]
- Genisca AE, Frias JL, Broussard CS, Honein MA, Lammer EJ, Moore CA, Shaw GM, Murray JC, Yang W, Rasmussen SA. Orofacial clefts in the National Birth Defects Prevention Study, 1997–2004. Am J Med Genet A. 2009;149A:1149–1158. doi: 10.1002/ajmg.a.32854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman DC, Donley N, Christian JL. Genetic interaction between Bmp2 and Bmp4 reveals shared functions during multiple aspects of mouse organogenesis. Mech Dev. 2009;126:117–127. doi: 10.1016/j.mod.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JM, Morrell JC, Gould SJ. Identification and characterization of HAOX1, HAOX2, and HAOX3, three human peroxisomal 2-hydroxy acid oxidases. J Biol Chem. 2000;275:12590–12597. doi: 10.1074/jbc.275.17.12590. [DOI] [PubMed] [Google Scholar]
- Jones MC. Etiology of facial clefts: prospective evaluation of 428 patients. Cleft Palate J. 1988;25:16–20. [PubMed] [Google Scholar]
- Kamath BM, Thiel BD, Gai X, Conlin LK, Munoz PS, Glessner J, Clark D, Warthen DM, Shaikh TH, Mihci E, Piccoli DA, Grant SF, Hakonarson H, Krantz ID, Spinner NB. SNP array mapping of chromosome 20p deletions: genotypes, phenotypes, and copy number variation. Hum Mutat. 2009;30:371–378. doi: 10.1002/humu.20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugimiya F, Kawaguchi H, Kamekura S, Chikuda H, Ohba S, Yano F, Ogata N, Katagiri T, Harada Y, Azuma Y, Nakamura K, Chung UI. Involvement of endogenous bone morphogenetic protein (BMP) 2 and BMP6 in bone formation. J Biol Chem. 2005;280:35704–35712. doi: 10.1074/jbc.M505166200. [DOI] [PubMed] [Google Scholar]
- Kurian MA, Meyer E, Vassallo G, Morgan NV, Prakash N, Pasha S, Hai NA, Shuib S, Rahman F, Wassmer E, Cross JH, O’Callaghan FJ, Osborne JP, Scheffer IE, Gissen P, Maher ER. Phospholipase C beta 1 deficiency is associated with early-onset epileptic encephalopathy. Brain. 2010;133:2964–2970. doi: 10.1093/brain/awq238. [DOI] [PubMed] [Google Scholar]
- Lalani SR, Thakuria JV, Cox GF, Wang X, Bi W, Bray MS, Shaw C, Cheung SW, Chinault AC, Boggs BA, Ou Z, Brundage EK, Lupski JR, Gentile J, Waisbren S, Pursley A, Ma L, Khajavi M, Zapata G, Friedman R, Kim JJ, Towbin JA, Stankiewicz P, Schnittger S, Hansmann I, Ai T, Sood S, Wehrens XH, Martin JF, Belmont JW, Potocki L. 20p12.3 microdeletion predisposes to Wolff-Parkinson-White syndrome with variable neurocognitive deficits. J Med Genet. 2009;46:168–175. doi: 10.1136/jmg.2008.061002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gloan L, Pichon O, Isidor B, Boceno M, Rival JM, David A, Le Caignec C. A 8.26Mb deletion in 6q16 and a 4.95Mb deletion in 20p12 including JAG1 and BMP2 in a patient with Alagille syndrome and Wolff-Parkinson-White syndrome. Eur J Med Genet. 2008;51:651–657. doi: 10.1016/j.ejmg.2008.07.012. [DOI] [PubMed] [Google Scholar]
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron. 2000;28:713–726. doi: 10.1016/s0896-6273(00)00148-3. [DOI] [PubMed] [Google Scholar]
- Lyons KM, Pelton RW, Hogan BL. Organogenesis and pattern formation in the mouse: RNA distribution patterns suggest a role for bone morphogenetic protein-2A (BMP-2A) Development. 1990;109:833–844. doi: 10.1242/dev.109.4.833. [DOI] [PubMed] [Google Scholar]
- Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132(24):5601–11. doi: 10.1242/dev.02156. [DOI] [PubMed] [Google Scholar]
- Mabie PC, Mehler MF, Kessler JA. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci. 1999;19:7077–7088. doi: 10.1523/JNEUROSCI.19-16-07077.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinney DB, Krantz ID, Bason L, Piccoli DA, Emerick KM, Spinner NB, Goldmuntz E. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106:2567–2574. doi: 10.1161/01.cir.0000037221.45902.69. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Takizawa T, Ochiai W, Yanagisawa M, Hisatsune T, Nakafuku M, Miyazono K, Kishimoto T, Kageyama R, Taga T. BMP2-mediated alteration in the developmental pathway of fetal mouse brain cells from neurogenesis to astrocytogenesis. Proc Natl Acad Sci U S A. 2001;98:5868–5873. doi: 10.1073/pnas.101109698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- Robert ML, Lopez T, Crolla J, Huang S, Owen C, Burvill-Holmes L, Stumper O, Turnpenny PD. Alagille syndrome with deletion 20p12.2-p12.3 and hypoplastic left heart. Clin Dysmorphol. 2007;16:241–246. doi: 10.1097/MCD.0b013e3282358d21. [DOI] [PubMed] [Google Scholar]
- Sahoo T, Theisen A, Sanchez-Lara PA, Marble M, Schweitzer DN, Torchia BS, Lamb AN, Bejjani BA, Shaffer LG, Lacassie Y. Microdeletion 20p12.3 involving BMP2 contributes to syndromic forms of cleft palate. Am J Med Genet A. 2011;155A(7):1646–53. doi: 10.1002/ajmg.a.34063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Mikawa S, Sato K. BMP2 expression in the adult rat brain. J Comp Neurol. 2010;518:4513–4530. doi: 10.1002/cne.22469. [DOI] [PubMed] [Google Scholar]
- Shohat M, Herman V, Melmed S, Neufeld N, Schreck R, Pulst S, Graham JM, Jr, Rimoin DL, Korenberg JR. Deletion of 20p 11.23----pter with normal growth hormone-releasing hormone genes. Am J Med Genet. 1991;39:56–63. doi: 10.1002/ajmg.1320390113. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Araki K, Iemura S, Natsume T, Hoseki J, Nagata K. Novel thioredoxin-related transmembrane protein TMX4 has reductase activity. J Biol Chem. 2010;285(10):7135–42. doi: 10.1074/jbc.M109.082545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura T, Komatsu Y, Tanaka M, McCann KL, Mishina Y. Bmp2 and Bmp4 genetically interact to support multiple aspects of mouse development including functional heart development. Genesis. 2009;47:374–384. doi: 10.1002/dvg.20511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- Zhang D, Mehler MF, Song Q, Kessler JA. Development of bone morphogenetic protein receptors in the nervous system and possible roles in regulating trkC expression. J Neurosci. 1998;18:3314–3326. doi: 10.1523/JNEUROSCI.18-09-03314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]