

Abstract
We examined the function of interleukin-10 (IL-10) in regulating changes in macrophage phenotype during muscle growth and regeneration following injury. Our findings showed that the Th1 cytokine response in inflamed muscle is characterized by high levels of expression of CD68, CCL-2, TNF-α and IL-6 at 1-day post-injury. During transition to the Th2 cytokine response, expression of those transcripts declined while CD163, IL-10, IL-10 receptor-1 and arginase-1 increased. Ablation of IL-10 amplified the Th1 response at 1-day post-injury, causing increases in IL-6 and CCL2, while preventing a subsequent increase in CD163 and arginase-1. Reductions in muscle damage that normally occurred between 1 and 4-days post-injury did not occur in IL-10 mutants. In addition, muscle regeneration and growth were greatly slowed by loss of IL-10. Furthermore, myogenin expression increased in IL-10 mutant muscle at 1-day post-injury, suggesting that the mutation amplified the transition from the proliferative to the early differentiation stages of myogenesis. In vitro assays showed that stimulation of muscle cells with IL-10 had no effect on cell proliferation or expression of MyoD or myogenin. However, co-culturing muscle cells with macrophages activated with IL-10 to the M2 phenotype increased myoblast proliferation affecting MyoD or myogenin expression, showing that M2 macrophages promote the early, proliferative stage of myogenesis. Collectively, these data show that IL-10 plays a central role in regulating the switch of muscle macrophages from a M1 to M2 phenotype in injured muscle in vivo and this transition is necessary for normal growth and regeneration of muscle.
Introduction
Changes in muscle use or acute damage produce a rapid and highly-structured inflammatory response that is dominated by successive waves of invasion of myeloid cells that can strongly influence both the magnitude of muscle damage and the rate at which repair occurs (1). Regardless of the perturbation that leads to inflammation, the muscle experiences an initial, rapid invasion by neutrophils and CD68 high macrophages. In an apparently non-adaptive response, these inflammatory cells amplify damage to the muscle, producing lesions to muscle cell membranes that can lead to necrosis of muscle fibers. These lesions are primarily attributable to the actions of free radicals generated by myeloperoxidase and inducible nitric oxide synthase (iNOS) (2 - 6). However, within days of increased muscle use or acute injury, the numbers of CD68high macrophages and neutrophils decline while the numbers of macrophages that express CD163 and CD206 increase and remain elevated as muscle repair, regeneration and growth proceed (7 - 10).
In addition to their capacity to exacerbate muscle damage, macrophages have been implicated in promoting muscle regeneration following acute muscle injuries or during increased muscle loading. For example, genetic ablation of chemokine receptor-2 (CCR2) or its ligand (CCL2) greatly reduced the numbers of macrophages in muscles that were injured by freezing (11, 12), ischemia (13, 14) or cardiotoxin injection (15), and those reductions in macrophages were associated with slower muscle regeneration and growth of the injured tissue. Furthermore, transplantation of wild-type bone marrow into irradiated, CCR2-mutant mice prior to muscle injury was sufficient to restore macrophage invasion and normalize muscle growth to wild-type rates (16). Although other myeloid cells can express CCR2, ablation of CCR2-mediated signaling does not affect chemoattraction of neutrophils or lymphoid cells to injured muscle (17), providing strong support that the reported treatment effects of disrupted CCR2 signaling reflect a disruption of macrophage functions in promoting muscle regeneration. Similarly, selective depletion of circulating CD11b-expressing cells prior to muscle injury by toxin injection reduced muscle regeneration (18), lending support to the view that CD11b+ leukocytes, most likely macrophages, promote muscle regeneration.
In vitro observations indicate that macrophages may positively affect muscle growth and regeneration by modulating both the proliferation and differentiation of a population of muscle stem cells, called satellite cells, that normally reside in muscle in a quiescent state. Upon increased muscle loading or injury, satellite cells are activated to proliferate, after which some of the activated cells withdraw from the cell cycle to enter a stage of early differentiation followed by terminal differentiation when they fuse to form muscle fibers (19). Each of these stages of myogenesis is characterized by shifts in the expression of developmentally-regulated genes. For example, during the proliferative stage of myogenesis, muscle cells show elevated expression of the transcription factor MyoD (20, 21). During early differentiation, muscle cells withdraw from the cell cycle and the expression of myogenin, also a transcription factor, increases (20, 21). This transition in the myogenic program may be influenced by macrophages. For example, when satellite cells are placed in co-cultures with macrophages, satellite cell proliferation increases (22 - 25) while the proportion of muscle cells that express myogenin is reduced (26).
Although some in vitro observations of macrophage/satellite cell co-cultures show that macrophages can promote proliferation and inhibit differentiation of muscle cells, other findings indicate that some macrophage populations can promote muscle differentiation. For example, if muscle cells are co-cultured with CD163+ macrophages, muscle cell fusion to form myotubes is increased (26), which is a morphological indicator of increased differentiation. CD163, a hemoglobin/haptaglobin receptor, is selectively expressed on macrophages that are activated to an anti-inflammatory, M2 phenotype (27 - 30). In injured tissues, CD163+ macrophages are in a state of alternative activation in which they secrete anti-inflammatory cytokines that can deactivate neutrophils and CD68high macrophages and thereby reduce tissue damage. Thus, in vitro findings suggest that M2 macrophages could improve repair and regeneration of muscle by deactivation of cytotoxic neutrophils and CD68high macrophages and also by directly influencing muscle cell differentiation. Similarly, in vivo observations support a role for CD163+ macrophages in promoting muscle growth following increased muscle loading or injury. Depletion of macrophages from injured muscles at the stage when CD68high macrophages decline and CD163+ macrophages increase produced reductions of muscle repair and greatly slowed muscle growth and regeneration (31).
Collectively, these observations indicate that the transition of macrophage populations in injured muscle from a CD68high/CD163- population to a CD68 low/CD163+ population reflects transition to a macrophage population that can promote muscle growth and regeneration following injury. If that were true, then modulation of the expression of signaling molecules that promote the CD163+ M2 phenotype would affect the course of muscle differentiation, regeneration and growth. Although several cytokines can drive macrophages to the M2 phenotype, interleukin-10 (IL-10) provides an especially good candidate molecule for regulating changes in macrophage phenotype that influence muscle growth and regeneration. IL-10 is a strong activator of CD163 expression in vitro (29) and ablation of IL-10 expression in dystrophic muscle reduces the numbers of CD163+ macrophages in the muscle, indicating that IL-10 is also capable of modulating muscle macrophage phenotype in vivo (32). In addition, IL-10 promotes phagocytosis by macrophages (32) and induction of phagocytosis can produce a phenotype switch from pro-inflammatory M1 macrophages to the anti-inflammatory M2 phenotype, at least in vitro (18).
In the present investigation, we test whether ablation of IL-10 expression affects muscle injury, regeneration or growth that occurs following muscle damage caused by increased muscle loading. We anticipate that null mutation of IL-10 in mice experiencing muscle reloading after disuse atrophy will attenuate the shift of macrophages to the M2 phenotype that is caused by increased muscle loading and help us identify the functional importance of that shift in the context of muscle growth and regeneration. Finally, we test whether IL-10 has direct effects on muscle cell growth or differentiation in vitro, to assess whether IL-10-mediated influences on muscle can reflect direct actions on muscle cells in addition to effects mediated through the myeloid compartment.
Materials and methods
Animals
All experimental protocols involving the use of animals were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of California, Los Angeles Institutional Animal Care and Use Committee. Adult C57BL/6J mice and IL-10 null mice (B10.129P2(B6)-Il10tm1Cgn/J) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mutant mice were back-crossed onto the C57BL/6J background for at least 8 generations by the supplier.
Hindlimb muscle unloading and reloading
In the present investigation, we use the rodent hindlimb unloading followed by reloading model to cause muscle damage, regeneration and growth that occur over a well-delineated time-course (33). In this manipulation, rodent hindlimbs are elevated for a period of time so that they are no longer weight-bearing which causes a rapid loss of mass in some muscles as they adapt to the unloaded condition. The soleus muscle is most rapidly affected and can lose 30 to 40% of its mass in 10 days through mechanisms that resemble those that occur in the muscles of patients subjected to prolonged bedrest (34). Reloading the hindlimbs by returning the animals to normal weight-bearing and ambulation provides a reproducible model for studying muscle adaptation to increased loading. During reloading, muscle experiences growth and regeneration, that are characterized by the appearance of central-nucleated muscle fibers that show renewed expression of developmental genes, and a rapid increase in fiber cross-sectional area, so that the fibers return to their original, pre-suspension size within 4 to 7 days of reloading. This response of soleus muscle to reloading permits the in vivo analysis of the contributions of specific leukocyte populations to injury, regeneration and growth that result from increased muscle use.
Four-month-old, female mice were subjected to 10 days of hindlimb unloading using a previously described device (33), followed by no reloading or by 1-day or 4-days of reloading by normal weight-bearing. At the end of the treatment period, mice were euthanized with inhalation of isoflurane and soleus muscles were rapidly dissected and collected. Ambulatory, control mice were subjected to normal cage activity until euthanized for tissue collection.
Immunohistochemistry
One soleus muscle from each mouse was dissected and then rapidly frozen in isopentane cooled in liquid nitrogen. Frozen, cross-sections were cut from the midbelly of each muscle at a thickness of 10 μm. The frozen sections were air-dried and fixed in cold acetone for 10 minutes and endogenous peroxidase activity was quenched with 0.03% hydrogen peroxide. Sections were blocked in 3% bovine serum albumin (BSA) and 2% gelatin in 50 mM Tris buffer pH 7.2 for 1 hour and then immunolabeled with rat anti-CD68 (Serotec) or rat anti-CD163 (Serotec) for 3 hours or with mouse anti-MyoD (BD Bioscience), mouse anti-myogenin (BD Bioscience) overnight. Negative control sections were prepared for each primary antibody using isotype control IgG in place of the specific primary antibody. Sections were washed with 50 mM sodium phosphate pH 7.2 containing 150 mM sodium chloride (phosphate-buffered saline; PBS) and then incubated with biotin-conjugated secondary antibody (Vector) for 1 hour and avidin D-conjugated horseradish peroxidase (Vector) for 30 minutes. Immunoreactive cells were visualized by using the enzyme substrate, 3-amino-9-ethylcarbazole, which yields red reaction products in the presence of peroxidase (AEC kit, Vector). The number of immunolabeled cells per volume of muscle tissue was then determined by first measuring the total volume of each section using a stereological, point-counting technique to determine section area (31) and then multiplying that value by the section thickness (10 μm). The numbers of immunolabelled cells in each section were then counted and expressed as the number of cells per unit volume of each section.
Assay for muscle fiber injury
Muscle fibers experiencing membrane lesions were identified by IgG staining as previously described (35). Presence of IgG in the cytosol indicates the presence of muscle membrane lesions large enough to allow the unregulated transit of large molecules through lesions in the cell membrane. Frozen sections were air-dried and blocked in 1% gelatin diluted in PBS and then labeled with FITC-conjugated mouse anti-IgG (Vector) for 1 hour. After washing with PBS, the staining was visualized by epifluorescence microscopy and the number of injured fibers showing cytosolic fluorescence and total number of fibers were counted. The extent of muscle injury was expressed as the number of injured fibers relative to the total number of fibers in the muscle cross-section.
Assay for muscle fiber regeneration
Centrally-located nuclei are morphological markers of muscle fibers undergoing regeneration. The number of fibers containing central nuclei in complete cross-sections of soleus muscles was counted in unfixed sections that were air-dried and stained with hematoxylin. Central-nucleated, regenerative fibers are expressed as the percentage of the total number of fibers in each section.
Measurement of muscle fiber cross-sectional area
Muscle fiber atrophy or growth was analyzed by measuring the muscle fiber cross-sectional area using a digital imaging system (Bioquant). In each soleus muscle cross-section, 250 muscle fibers were randomly chosen and measured for each of 6 mice in each treatment group. The average of the group of 250 fibers was calculated and that value was used as a single datum that was then used to calculate the mean and standard error of the mean (sem) for fiber cross-sectional area for each treatment group.
RNA isolation and quantitative real-time PCR
During tissue collection, one soleus muscle from each animal was rapidly frozen in liquid nitrogen and used for RNA isolation. Total RNA was isolated with Trizol according to the manufacturer’s protocol (Invitrogen). 500 ng of total RNA from each sample was reverse transcribed to cDNA using Super Script Reverse Transcriptase II (Invitrogen). Quantitative real-time PCR (QPCR) reactions with cDNA were performed using SYBR Green master mix (Biorad). The real-time amplification of genes was measured with an iCycler thermocycler system and iQ5 optical system software (Biorad). Data were normalized to β-actin transcript levels. The PCR primers used are listed in Table 1.
Table 1.
Primers used to assay expression levels of immune-cell and muscle-cell specific genes.
| Gene | Accession Number | 5’→3’ | Amplicon size (bp) | |
|---|---|---|---|---|
| Arginase-1 | NM_007482 | Fwd | CAATGAAGAGCTGGCTGGTGT | 153 |
| Rev | GTGTGAGCATCCACCCAAATG | |||
| CCL-2 | NM_011333 | Fwd | GCTCAGCCAGATGCAGTTAAC | 153 |
| Rev | CTCTCTCTTGAGCTTGGTGAC | |||
| CCR-2 | NM_009915 | Fwd | CCTGTAAATGCCATGCAAGTTC | 165 |
| Rev | GTATGCCGTGGATGAACTGAG | |||
| CD68 | NM_009853 | Fwd | CAAAGCTTCTGCTGTGGAAAT | 140 |
| Rev | GACTGGTCACGGTTGCAAG | |||
| CD163 | CD163 | Fwd | GCAAAAACTGGCAGTGGG | 164 |
| Rev | GTCAAAATCACAGACGGAGC | |||
| HMOX-1 | NM_010442 | Fwd | CAGCATGCCCCAGGATTTG | 76 |
| Rev | CTCAGCATTCTCGGCTTGG | |||
| IL-4 | NM_021283 | Fwd | GGATGTGCCAAACGTCCTC | 126 |
| Rev | GAGTTCTTCTTCAAGCATGGAG | |||
| IL-6 | NM_031168 | Fwd | GAACAACGATGATGCACTTGC | 154 |
| Rev | CTTCATGTACTCCAGGTAGCTATGGT | |||
| IL-10 | NM_010548 | Fwd | CAAGGAGCATTTGAATTCCC | 157 |
| Rev | GGCCTTGTAGACACCTTGGTC | |||
| IL-10R1 | NM_008348 | Fwd | CAAGCCCTTCCTATGTGTGG | 166 |
| Rev | AGAAAGGCTCAGGCATTGTC | |||
| IL-10R2 | NM_008349 | Fwd | GGACAACTTACTGCATTC | 157 |
| Rev | CACCAGGACGGAGACTATG | |||
| IL-12 | NM_008351 | Fwd | CGCAGCACTTCAGAATCACA | 129 |
| Rev | TCTCCCACAGGAGGTTTCTG | |||
| IRF-5 | NM_012057 | Fwd | GACAACACCATCTTCAAGGC | 120 |
| Rev | GTCACGGCTTTTGTTAAGGGC | |||
| MyoD | NM_010866 | Fwd | GAGCGCATCTCCACAGACAG | 178 |
| Rev | AAATCGCATTGGGGTTTGAG | |||
| myogenin | NM_031189 | Fwd | CCAGTACATTGAGCGCCTAC | 163 |
| Rev | ACCGAACTCCAGTGCATTGC | |||
| TNFα | NM_013693 | Fwd | CTTCTGTCTACTGAACTTCGGG | 163 |
| Rev | CACTTGGTGGTTTGCTACGAC | |||
| β-actin | NM_007393 | Fwd | CAACCGTGAAAAGATGACCC | 157 |
| Rev | GTAGATGGGCACAGTGTGGG |
Macrophage isolation
Peritoneal macrophages were isolated from 3 to 6 month old C57BL/6J mice. Mice received an intraperitoneal injection of 12% sodium casein in 0.9% sodium chloride and were euthanized with isoflurane 3 days later. Immediately after euthanization, the peritoneal cavity of the mouse was opened and rinsed with PBS. The peritoneal exudates were then collected and filtered through a 70 μm cell strainer (BD Bioscience). Filtered peritoneal cells were centrifuged and resuspended in proliferation medium (Dulbecco’s modified eagle’s medium [DMEM, Sigma-Aldrich] supplemented with 10% fetal bovine serum [FBS] and 1% penicillin and streptomycin [P/S; GIBCO]). Cell suspensions were overlaid on an equal volume of histopaque-1077 (Sigma-Aldrich) and centrifuged at 400 g for 30 minutes. Macrophages were collected from the interface of histopaque and DMEM and then were centrifuged and resuspended in DMEM media for later use. Some isolated cells were adhered to microscope slides by centrifugation at 14 g for 3 minutes using a Cytospin (Shandon) and stained with rat anti-F4/80 (0.2 μg/ml) to confirm that they were macrophages. Rat anti-F4/80 was prepared previously by ammonium sulfate precipitation of immunoglobulins from F4/80 hybridoma cultures (HB-198, American Type Culture Collection).
Assay for muscle cell proliferation or differentiation
Peritoneal macrophages were seeded at 4.0 × 105 cells/well in six-well plates and stimulated for 24 hours with IL-10 (10 ng/ml) to induce the M2 phenotype or IFNγ and TNFα (10 ng/ml of each) to induce the M1 phenotype. After stimulation, cytokines were washed away with DMEM. C2C12 cells that were grown previously in proliferation medium under sub-confluent conditions were then plated on top of stimulated macrophages at 4.0 × 105 cells/well in differentiation media in which 10% FBS was replaced by 2% horse serum. Fresh differentiation media were added to the co-cultures every 24 hours. Myoblast proliferation was assayed by harvesting cells from 2-day-old co-cultures in 0.05% trypsin-EDTA, after which the cells were pelleted, resuspended in PBS and counted using a hemocytometer. Macrophages remained adherent to the culture dishes during trypsinization. Myoblast differentiation was assayed by collecting cell lysates from 4-day co-cultures in reducing sample buffer (RSB; 80 mM Tris, pH 6.8, 0.1 M dithiothreitol and 70 mM sodium dodecyl sulfate (SDS)) and protease inhibitor cocktail (1:100, Sigma-Aldrich) for western blot analysis.
Western blot analysis
Cell lysates collected in RSB with protease inhibitor cocktail were electrophoresed in polyacrylamide gels and then transferred electrophoretically to nitrocellulose membranes. Thirty micrograms of sample were used in each lane for all gels and blots analyzed. Following transfer, the membranes were stained with Ponceau S solution (Sigma-Aldrich) to confirm the equal loading of samples. Nitrocellulose membranes were blocked with 3% non-fat milk and then incubated with mouse anti-MyoD (BD Bioscience), mouse anti-myogenin (BD Bioscience), rabbit anti-CD163 (Santa Cruz Biotech) or rabbit anti-mouse inducible nitric oxide synthase (iNOS; Upstate Biotechnology) for 3 hours at room temperature. The antibodies were diluted in 50 mM Tris, pH 7.6, containing 150 mM NaCl, 0.1% NaN3, 0.05% Tween 20, and 3% BSA. After washing with PBS buffer containing 0.1% Tween-20, the blots were incubated with a specific secondary antibody conjugated with horseradish peroxidase (Amersham) for 1 hour at room temperature. After washing, the signal was detected by enhanced chemiluminescence (Amersham) and a fluorochem imaging system (Alpha Innotech).
Statistics
Data are presented as mean ± sem. One-way analysis of variance (ANOVA) (GraphPad InStat version 2.03) was used to test whether differences between groups were significant at p < 0.05. Comparisons of two groups of values were analyzed using unpaired, two-tailed, T-test. Differences were considered significant at p < 0.05.
Results
Increased muscle use following periods of disuse induces an initial Th1 cytokine response followed by a Th2 cytokine response
Previous investigations demonstrated that CD68high macrophages appeared at elevated numbers in the initial inflammatory infiltrate in muscle experiencing increased loading, but their numbers then decline while a CD163+ macrophage population subsequently increased (7 - 9). Our immunohistochemical observations confirm the early invasion of CD68high macrophages into muscle that has experienced 1 day of reloading, and show that these macrophages can invade the cytoplasm of injured muscle fibers (Figure 1). We also confirmed that their decline between days 1 and 4 of reloading was accompanied by an increase in CD163+ macrophages that do not invade damaged muscle fibers (Figures 2 and 3). CD68+ and CD163+ macrophages were found co-distributed in the same inflammatory lesions at each stage of reloading that was assayed (Supplemental figure 1).
Figure 1.
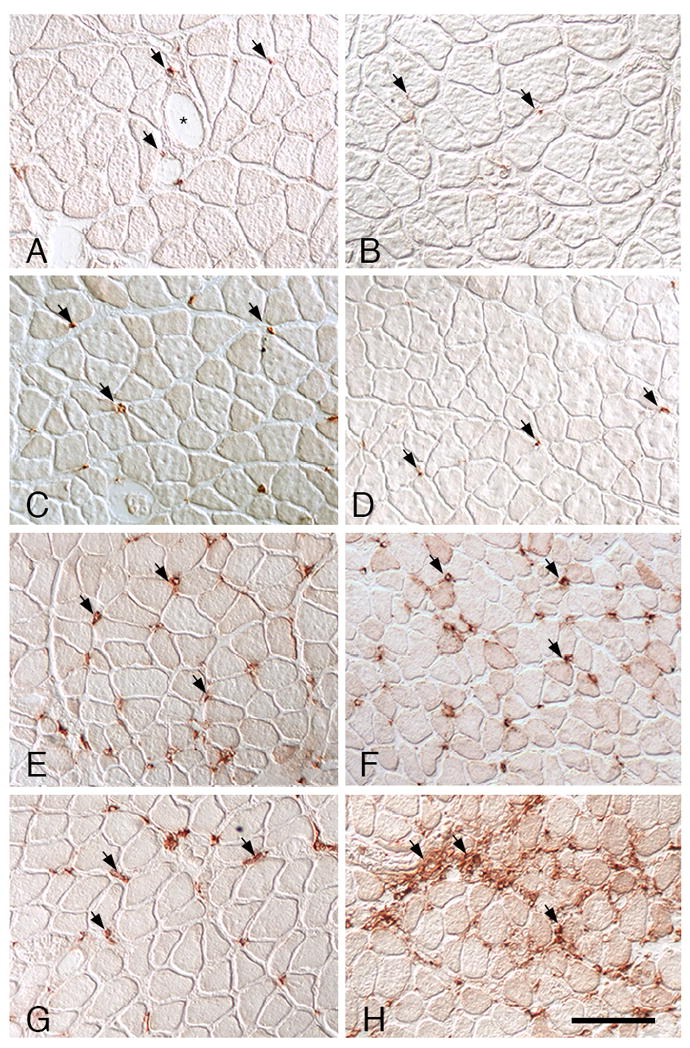
CD68+ macrophage distribution in soleus muscles. A, C, E, G: Wild-type muscles. B, D, F, H: IL-10 null mutant muscles. A, B: Ambulatory control muscles showing CD68+ macrophages (arrows) in the endomysium surrounding muscles. CD68+ macrophages in wild-type muscles were frequently near blood vessels (*). C, D: In muscles experiencing unloading without subsequent reloading, the numbers and distribution of CD68+ macrophages were similar as observed in ambulatory controls. Muscle fiber diameters are reduced because of atrophy during unloading. E, F: At 1-day of muscle reloading following unloading, both wild-type and IL-10 null muscles show identical increases in the numbers of CD68+ macrophages. G, H: At 4-days of reloading, CD68+ macrophage numbers have begun to decline in wild-type muscle but remain elevated in IL-10-null muscle. All images are at the same magnification; bar = 100 μm.
Figure 2.
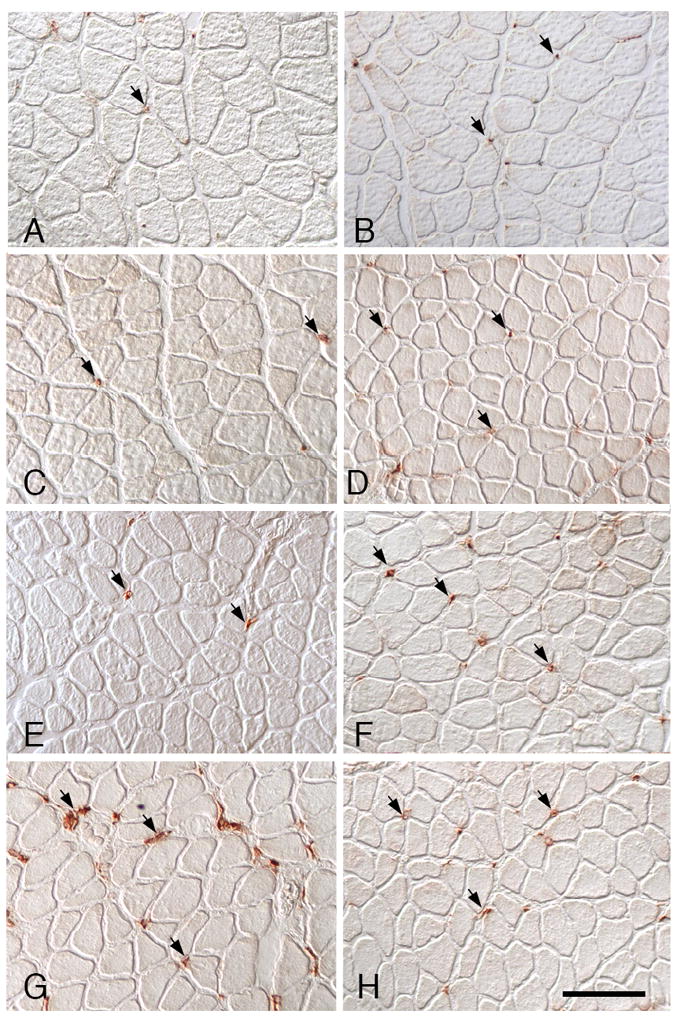
CD163+ macrophage distribution in soleus muscles. A, C, E, G: Wild-type muscles. B, D, F, H: IL-10 null mutant muscles. A, B: Ambulatory control muscles showing CD163+ macrophages (arrows) in the endomysium surrounding muscles. C, D: In muscles experiencing unloading only, the numbers and distribution of CD163+ macrophages did not differ from ambulatory controls and fibers are atrophied. E, F: At 1-day of muscle reloading, both wild-type and IL-10 null muscles show numbers of CD163+ macrophages that are similar to ambulatory controls. G, H: At 4-days of reloading, CD163+ macrophage numbers have increased substantially in wild-type muscle, but show little increase in IL-10-null muscles. All images are at the same magnification; bar = 100 μm.
Figure 3.
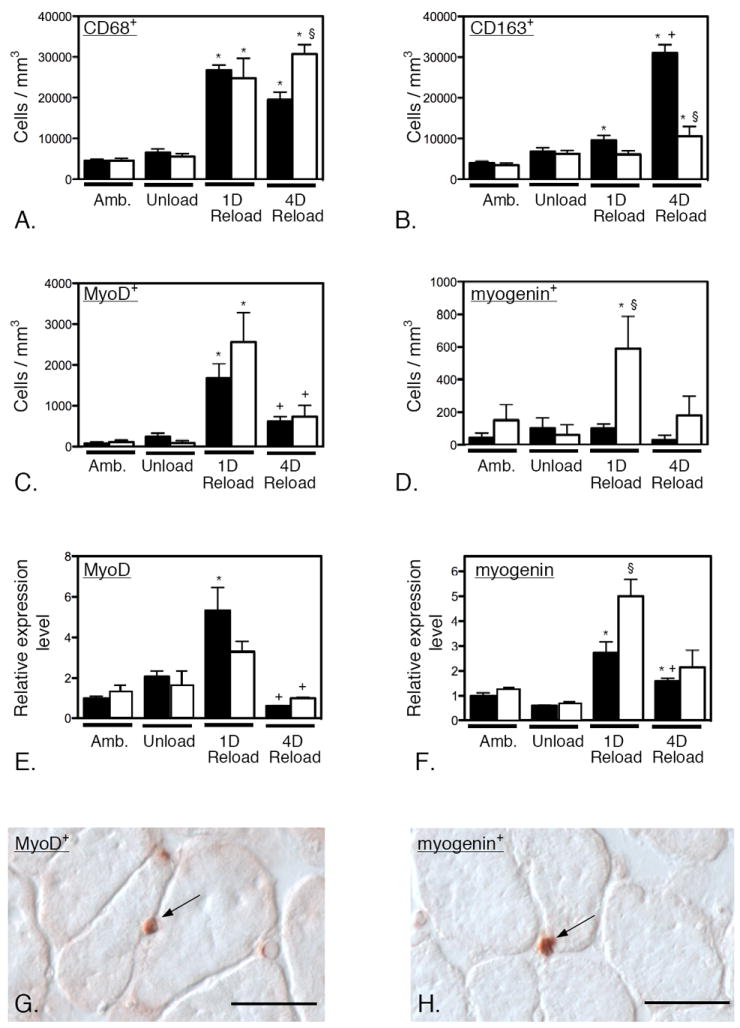
Null mutation of IL-10 amplifies CD68high macrophage numbers, reduces M2 macrophage numbers and perturbs myogenin expression in injured muscle. A – D: Cell counts of CD68+ (A), CD163+ (B), MyoD+ (C) and myogenin+ (D) cells in soleus muscles of wild-type and IL-10 null mutants over the time-course on muscle unloading and reloading. A. Numbers of CD68+ macrophages are elevated at 1-day of reloading in wild-type and mutant muscles and CD68+ cell numbers begin to decline after 1-day of muscle reloading of wild-type muscle. However, IL-10 null mutants do not experience a decline in CD68+ cells at 4-days of reloading. B. Numbers of CD163+ macrophages are elevated slightly in wild-type muscles at 1-day reloading, but not in mutant muscles at that time-point. Numbers of CD163+ cells are elevated in both wild-type and mutant muscles at 4-days reloading, but the numbers are greatly amplified in wild-type muscles. C. The numbers of MyoD-expressing satellite cells show similar increases in both wild-type and mutant muscles at 1-day of reloading and similar reductions in numbers at 4-days reloading. D. The numbers of myogenin-expressing satellite cells show large increases in mutant muscle at 1-day of reloading, although their numbers do not change significantly in wild-type muscles at that time-point. E and F. QPCR data showing changes in the levels of expression of MyoD (E) and myogenin (F) over the course of muscle unloading and reloading. The changes in gene expression for the two transcripts resemble the changes in numbers of MyoD+ and myogenin+ cells shown in Figures 3C and 3D. * indicates significantly different from ambulatory control muscle of same genotype at p < 0.05. § indicates significantly different from wild-type muscle under the same treatment conditions at p < 0.05. + indicates significantly different from 1-day reloaded muscle of same genotype at p < 0.05. Each bar represents the mean and sem for the muscles collected from 5 mice in each data set. All data in each set were normalized relative to expression levels in ambulatory, wild-type muscles which were set at 1.0. G. Image showing an anti-MyoD labeled satellite cell (arrow) at the surface of a muscle fiber in 1-day reloaded, IL-10 null muscle. H. Image showing an anti-myogenin labeled satellite cell (arrow) at the surface of a muscle fiber in 4-days reloaded, IL-10 null muscle. Bars = 40 μm.
We assayed for changes in expression levels of CD68 and Th1 cytokines that are associated with classical activation of macrophages to the M1 phenotype, and found that there is a large, significant elevation of CD68 expression at 1-day reloading in wild-type muscle that is largely attenuated by 4-days of reloading (Figures 3 and 4), consistent with changes in the numbers of CD68high macrophages that occurs during this period of reloading (31). This change of expression of CD68 coincided with changes in expression of TNF-α, IL-6, and CCL2 (Figure 4), all of which are cytokines that typify a Th1 cytokine response (36). Other transcripts associated with a Th1 cytokine response were not concurrently elevated (CCR2, IRF-5 and IL-12; Figure 4). In contrast, CD163 expression was not significantly elevated at 1-day of reloading, but was elevated at 4-days in wild-type muscle. This elevation of CD163 coincided with a tremendous increase in arginase-1 (Arg1) expression, a marker for M2 activation of macrophages (37), along with substantial increases in the expression of IL-10 and its receptor, IL-10R1 (Figure 5). However, expression of heme oxygenase-1 (HMOX-1), which can be activated by CD163 ligation (38), was not elevated at 4-days (Figure 5). Collectively, these data indicate that the CD68high/CD163-, early-invading macrophages are characteristic of a Th1 cytokine response and that they are subsequently replaced by a population of CD68lo/CD163+ M2 macrophages.
Figure 4.
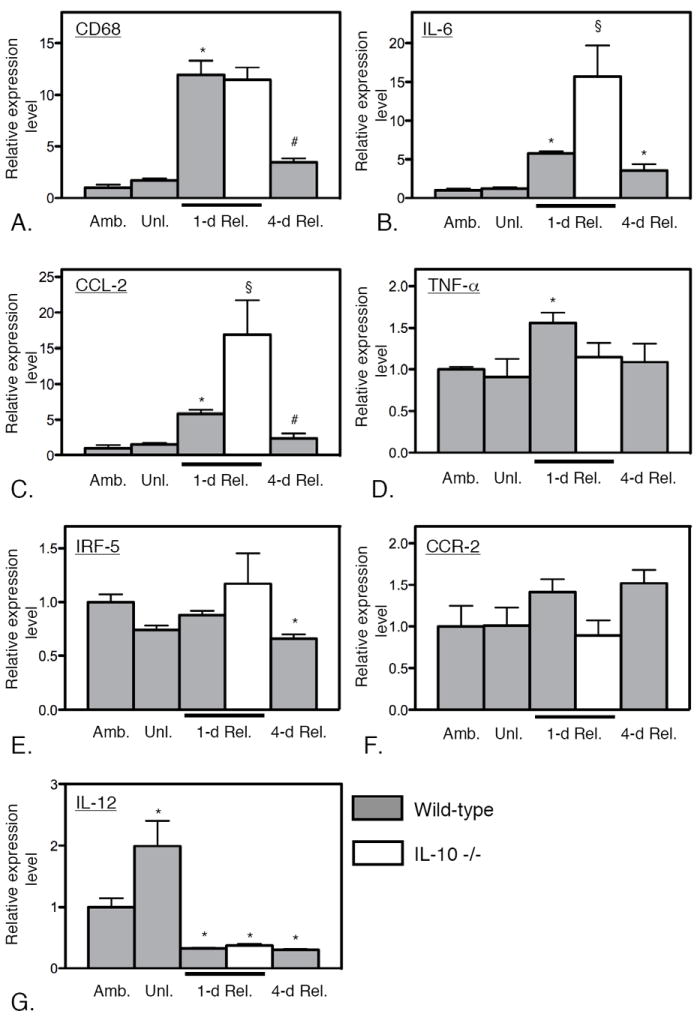
Levels of expression of transcripts related to the Th1 cytokine response during muscle unloading and reloading. Expression levels of CD68 (A), IL-6 (B), CCL-2 (C) and TNF-α (D) were all elevated at 1-day of reloading in wild-type muscles, reflecting a Th1 cytokine response, although IRF-5 (E), CCR-2 (F) and IL-12 (G) were not elevated at that stage. The transcripts that were elevated at 1-day reloading were all significantly reduced by 4-days of reloading, reflecting resolution of the Th1 cytokine response. Null mutation of IL-10 amplified the Th1 cytokine response at 1-day of reloading, reflected in significant elevations of IL-6 and CCL-2. * indicates significantly different from ambulatory control muscle of same genotype at p < 0.05. § indicates significantly different from wild-type muscle under the same treatment conditions at p < 0.05. # indicates significantly different from 1-day reloaded muscle of same genotype at p < 0.05. Each bar represents the mean and sem for the muscles collected from 6 mice in each data set. All data in each set were normalized relative to expression levels in ambulatory, wild-type muscles which were set at 1.0.
Figure 5.
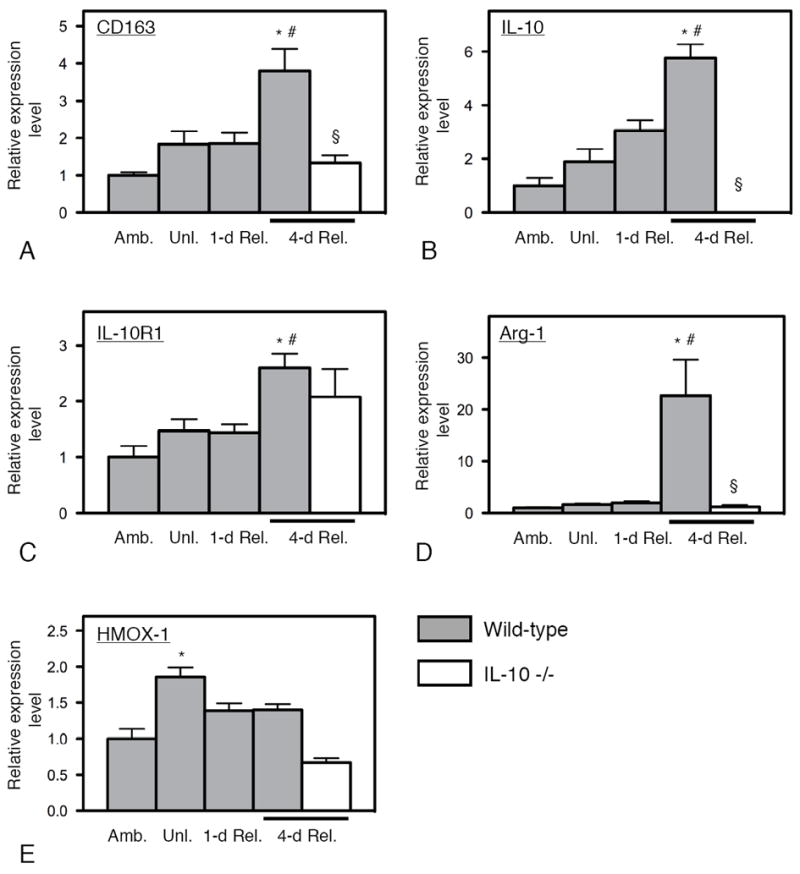
Levels of expression of transcripts related to the Th2 cytokine response during muscle unloading and reloading. Expression levels of CD163 (A), IL-10 (B), IL-10R1 (C) and Arg-1 (D) were all elevated at 4-days of reloading in wild-type muscles, reflecting a Th2 cytokine response, although HMOX-1 (E) was not elevated at that stage. Null mutation of IL-10 attenuated the Th2 cytokine response at 4-days of reloading, reflected in significant reductions of CD163 and Arg-1. * indicates significantly different from ambulatory control muscle of same genotype at p < 0.05. § indicates significantly different from wild-type muscle under the same treatment conditions at p < 0.05. # indicates significantly different from 1-day reloaded muscle of same genotype at p < 0.05. Each bar represents the mean and sem for the muscles collected from 6 mice in each data set. All data in each set were normalized relative to expression levels in ambulatory, wild-type muscles which were set at 1.0.
Null mutation of IL-10 attenuates the shift of muscle macrophages to the M2 phenotype
The finding that the expression of IL-10, its receptor and two major indicators of macrophage activation to the M2 phenotype (CD163 and Arg1) increased in muscle experiencing 4-days of reloading following a period of atrophy, led us to test whether the shift to the M2 phenotype in reloaded muscle was regulated by IL-10. Because IL-4 expression levels remained very low throughout the unloading and reloading process and did not differ significantly between any two time-points assayed (data not shown), we inferred that IL-4-mediated signaling was not an important modulator of macrophage phenotype switching in this model. Expression levels of markers of the M1 phenotype were assessed by QPCR in both wild-type (N = 6) and IL-10 -/- muscle (N = 6) after 1-day of reloading, showing that IL-10 ablation produced elevated expression of two Th1-associated transcripts, CCL2 and IL-6 (Figure 4). However, the expression levels of CD68 and TNFα were not significantly affected by the IL-10 mutation. Similarly, expression levels of markers of the M2 phenotype were assayed in muscle after 4-days of reloading, showing large, significant reductions in the expression levels of CD163 and Arg1, although the expression level of IL10R1 was not significantly affected (Figure 5). Together, these data show that IL-10 plays a significant role in regulating induction of the M2 macrophage phenotype switch in reloaded muscle, although IL-10-mediated signaling does not influence the expression of all Th1 or Th2 cytokines in injured muscles in vivo.
Null mutation of IL-10 slows muscle fiber repair and regeneration in vivo
We assayed whether the attenuated shift to a M2 macrophage phenotype in IL-10 null mutants influenced muscle fiber injury and regeneration that occur during the first 4-days of muscle reloading. Sections of soleus muscles were immunolabeled with FITC-conjugated anti-mouse IgG to assay for the presence of extracellular protein (immunoglobin) in the muscle fiber cytoplasm, indicating the presence of muscle membrane lesions that permitted unregulated transit of large molecules across the cell membrane. The number of muscle fibers containing cytosolic IgG did not differ between wild-type and IL-10 -/- mice in 1-day reloaded muscles (Figure 6). In wild-type mice, there was an 80% reduction in the percentage of IgG+ muscle fibers from 1-day to 4-days reloading, which indicates that the majority of muscle membrane repair occurred between 1-day and 4-days reloading, which is consistent with previous findings (31). However, the number of injured muscle fibers in IL-10 -/- mice was not reduced at 4-days of reloading compared to 1-day of reloading (Figure 6C), indicating that null mutation of IL-10 either impairs muscle membrane repair during reloading or that recurring membrane damage persists for longer periods in the absence of IL-10.
Figure 6.
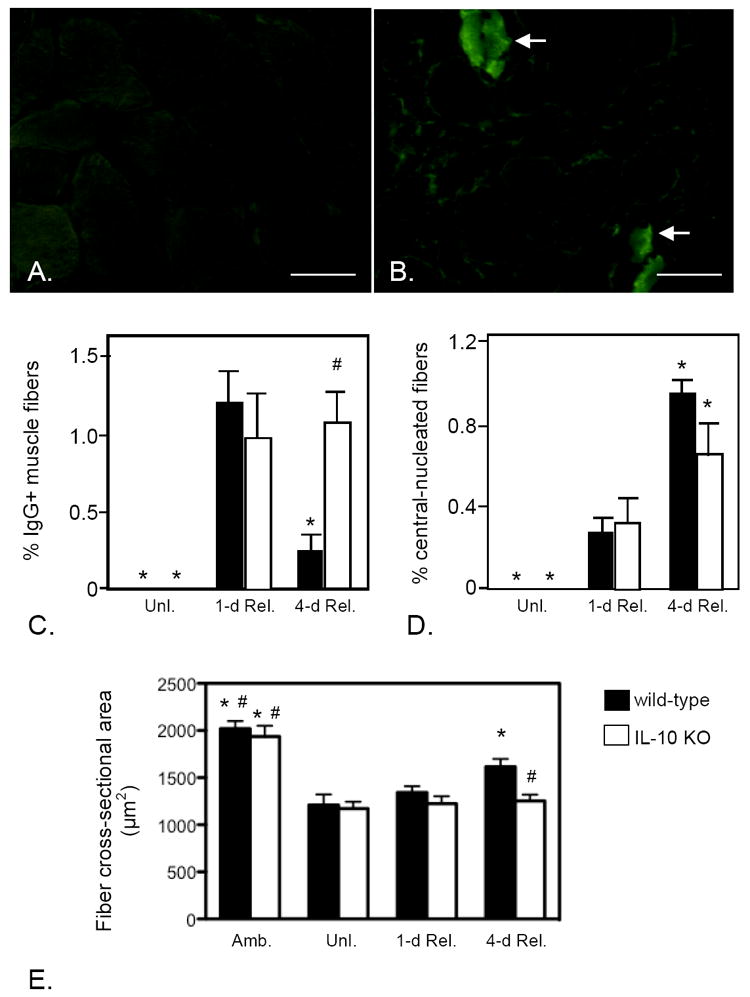
IL-10 mutants experience reductions in muscle fiber repair, regeneration and growth during reloading. A, B: Cross-section of soleus muscles from wild-type (A) or IL-10 null mutant (B) mice from 4-days reloaded mice labeled with FITC-conjugated, anti-mouse IgG. The presence of IgG in the muscle fiber cytosol (arrows) indicates the presence of membrane lesions large enough to allow influx of IgG from the extracellular space. Bars = 100 μm. C: Quantification of the number of IgG+ muscle fibers in unloaded (Unl.), 1-day and 4-day reloaded muscles indicates that membrane lesions caused by muscle reloading are repaired between days 1 and 4 of reloading in wild-type, but not IL-10 mutant, muscles. D. Quantification of the proportion of central-nucleated muscle fibers in reloaded muscles shows that the increase in regenerative muscle fibers that occurs in wild-type muscles between in 1-day and 4-days reloading is diminished in IL-10 null mutants. E. Muscle fiber growth during reloading was assayed by measuring changes in cross-sectional area of the muscle fibers in soleus muscle sections. Note that fiber size in ambulatory controls (Amb.) did not differ between wild-type and IL-10 mutant muscles and that mutation of IL-10 did not affect the atrophy of fibers that occurred during unloading (Unl.). However, fiber growth that occurred during 4-days of reloading did not occur in IL-10 mutants. Each bar represents the mean and sem for the muscles collected from 6 mice in each data set. * indicates significantly different from 1-day reloaded muscle of the same genotype at p < 0.05. # indicates significantly different from 4-days reloaded wild-type muscle at p < 0.05.
Null mutation of IL-10 also impairs muscle regeneration during muscle reloading. No significant difference in muscle fiber central-nucleation was observed between IL-10 -/- and wild-type mice at 1-day reloading (Figure 6D). However, there was a 4-fold increase in central-nucleation between 1-day and 4-day reloading in wild-type mice, suggesting that muscle experiences regeneration during this period of reloading. However, the increase in central-nucleation at 4-day reloading was less in IL-10 -/- muscles (Figure 6D), indicating that IL-10 mediates processes that promote muscle regeneration.
Loss of IL-10-mediated signaling perturbs muscle differentiation and reduces growth during regeneration
Our qualitative observations of muscle fiber histology (Figures 1 and 2) suggested that muscle fibers in IL-10 -/- mice regained their normal size during reloading more slowly than wild-type muscle fibers, suggesting possible defects in growth and differentiation. We assayed changes in muscle growth in IL-10 null mutant mice by measuring changes in muscle fiber size during muscle unloading and reloading. Consistent with previous findings in rodents subjected to hindlimb unloading (31), 10 days of unloading in wild-type mice caused about 40% atrophy in soleus muscle. No significant increase in the cross-sectional area of muscle fibers was observed at 1-day reloading compared to fiber size of mice that were assayed immediately following completion of unloading. However, there was a rapid increase in the fiber cross-sectional area from 1-day to 4-day reloading in wild-type mice but that increase did not occur in IL-10 -/- mice (Figure 6E). We assayed muscle differentiation by quantifying the expression of key, myogenic transcription factors by QPCR and by immunohistochemistry. Neither the expression level of MyoD or the number of MyoD-expressing cells was significantly affected by IL-10 mutation at any stage of muscle unloading or reloading (Figure 3). However, IL-10 null mutant mice displayed a greatly increased expression of myogenin and significantly more myogenin expressing cells in 1-day reloaded muscle (Figure 3), showing that normal patterns of muscle differentiation are disrupted by null mutation of IL-10 during muscle reloading.
M2 macrophages promote muscle cell proliferation without direct effects on differentiation in vitro
Our in vivo data show that null mutation of IL-10 increased myogenin expression in reloaded muscle, indicating that IL-10 could affect muscle differentiation either by direct actions on myogenic cells or by acting through M2 macrophages. We tested these possibilities by analyzing the effects of IL-10 or IL-10-activated M2 macrophages on muscle cell proliferation and differentiation in vitro. Although we were unable to detect IL-10R expression in myoblasts at the protein level, expression of both IL-10R1 and IL-10R2 were confirmed by RT-PCR in proliferative myoblasts, although IL-10 R1 was barely detectable (Figure 7A).
Figure 7.
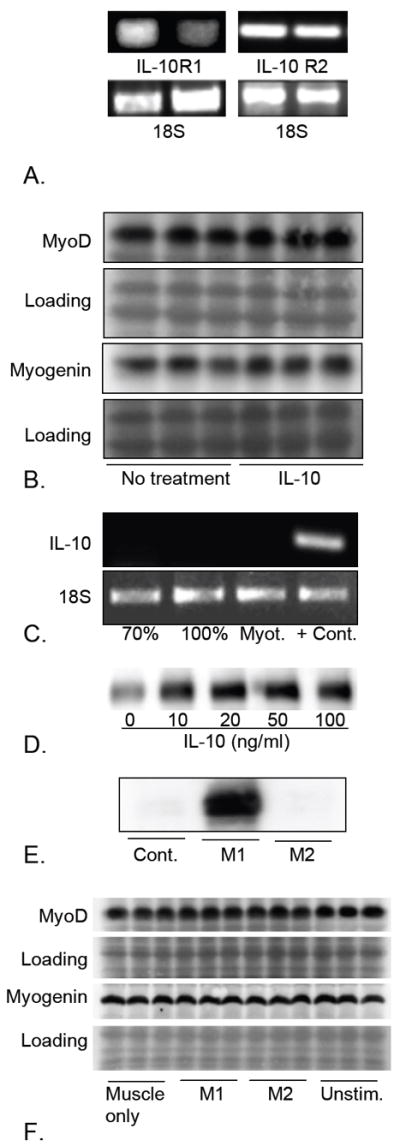
MyoD and myogenin expression are unaffected by direct stimulation with IL-10 or co-culture with IL-10 stimulated M2 macrophages. A. RT-PCR was used to confirm that C2C12 myoblasts can express both IL-10R1 and IL-10R2. The 18S ribosomal subunit was used as a loading control. B. Western blots of muscle cell extracts following treatment with 10 ng/mg IL-10 shows that direct application of IL-10 for 24 hours did not affect expression of MyoD or myogenin. Ponceau red staining of membranes that were subsequently used for antibody incubations was used to confirm uniform loading of the gels and transfer of proteins to the membrane (loading). The western blots that are shown are representative of three independent experiments. C: RT-PCR showed that muscle cells in vitro do not express IL-10, showing that lack of treatment effect with exogenous IL-10 was not attributable to saturation by endogenous IL-10. Lane 1 sample was obtained from 70% confluent cultures of proliferative muscle cells. Lane 2 was from 100% confluent cultures. Lane 3 was obtained from differentiated myotube (Myot.) cultures, 2-days after transfer to differentiation medium. Lane 4 used RNA isolated from inflamed, dystrophic muscle from mice in the mdx line, as a positive control. The 18S ribosomal subunit is used as a loading control. The results are representative of those obtained from 3 independent experiments. D. Western blot of extracts of macrophages stimulated with IL-10 for 24 hours. IL-10 induction of CD163 expression indicates increased activation to the M2 phenotype. The blot is representative of 3 independent experiments. E. Western blot of extracts of macrophages stimulated with TNF-α and IFNγ for 24 hours. Induction of iNOS indicates activation to the M1 phenotype. F. Western blot of extracts of myoblasts cultured in the absence of macrophages (Muscle only), or in the presence of M1 macrophages activated by TNF-α and IFNγ (M1), or in the presence of M2 macrophages stimulated with IL-10 (M2) or macrophages that were not treated with cytokines (Unstim.) No detectable changes in the levels of expression of MyoD or myogenin were observed in any co-culture conditions, compared to myoblasts alone. The results are representative of those obtained from 3 independent experiments. Ponceau red staining of the western blot membranes (Loading) was used to confirm uniform loading and transfer of samples.
We assayed whether IL-10 or IL-10 activated macrophages influenced myoblast proliferation in vitro. Consistent with previous findings (32), we found that the direct application of IL-10 to myoblasts in vitro did not affect myoblast proliferation, but that co-culture of myoblasts with IL-10-activated M2 macrophages caused significant increases in myoblast proliferation (myoblasts alone increased 5.5-fold in 48 hours [sem = 0.28]; myoblasts co-cultured with IL-10-stimulated macrophages increased 7.4-fold [sem = 0.20]; p < 0.01). We then tested whether direct stimulation of myoblasts with IL-10 affected early stages of muscle differentiation but found no detectable changes in the expression of MyoD or myogenin when muscle cell extracts were assayed by western blots (Figure 7B). Finally, we tested whether the lack of effect of direct stimulation of muscle with exogenous IL-10 in vitro could result from a saturation of IL-10 effects on muscle by endogenous IL-10 by assaying whether muscle cells in vitro expressed IL-10. However, no IL-10 mRNA was detectable in cultures of sub-confluent myoblasts, confluent myoblasts or differentiated myotubes (Figure 7C).
Because perturbations of IL-10 expression in vivo influenced expression of myogenin but direct application of IL-10 to muscle cells in vitro did not affect MyoD or myogenin expression, we assayed whether co-culturing muscle cells with IL-10-stimulated M2 macrophages affected the expression of MyoD or myogenin. Macrophages were stimulated with IL-10 to drive them to an M2 phenotype or stimulated with both IFNγ and TNFα to drive them to an M1 phenotype before co-culturing with C2C12 cells. Western blot analysis of CD163 confirmed that IL-10 stimulation promoted the M2 phenotype and showed that maximum induction of CD163 was achieved by 10 ng/ml (Figure 7D). Western blot analysis of iNOS expression confirmed induction of the M1 macrophage phenotype (Figure 7E). However, we found that neither M1 nor M2 macrophages affected expression levels of MyoD or myogenin (Figure 7F).
Collectively, these findings indicate that the elevation of myogenin expression that occurs in reloaded, IL-10 null mutant muscles reflects the capacity of M2 macrophages to maintain myoblasts in a proliferative state, preventing entry into the post-mitotic stage of myogenesis at which myogenin expression increases.
Discussion
The results of the present investigation show that IL-10 plays a dominant role in regulating macrophage transition to a CD163+ M2 phenotype in regenerative muscle. In the absence of IL-10, no significant elevation of CD163 expression occurred in regenerative muscle and the increase in CD163+ M2 cells during the transition from 1-day to 4-days reloaded muscle was nearly abolished. Our data also indicate that IL-10 is required in regenerative muscle for induction of Arg-1 expression, another specific indicator of activation of the M2 phenotype. We also observed that the expression of some transcripts associated with the M1 phenotype, in particular, CCL2 and IL-6, was greatly amplified by IL-10 ablation at 1-day of muscle reloading, indicating that IL-10 has a suppressive effect on the Th1 cytokine response in injured muscle. The impact on muscle of perturbing induction of the M2 macrophage phenotype was substantial; muscle fiber growth was significantly slowed, muscle membrane lesions persisted and muscle regeneration was reduced, which emphasize the important role of IL-10-activated macrophages in modulating muscle repair.
Previous observations have already established that shifts in macrophage phenotype coincide with transitions in the stage of myogenesis in regenerating muscle following acute injury. For example, CD68 high macrophages reach maximum numbers at about 2-days following muscle injury and then decline (7, 39). The number of CD163+ macrophages begins to increase in regenerative muscle at about 2-days post-injury and reaches maximum levels at about 4-days (7, 40). Suggestively, that shift in macrophage phenotype coincides with changes in expression of myogenic, regulatory transcription factors. Injection of cardiotoxin into soleus muscle increases MyoD expression at 2-days post-injury (41) and myogenin expression is elevated in muscle fibers at about 1-day following the increase in MyoD (42). Other muscle injury models show a similar time-course in the elevation of MyoD at about 2-days post-injury followed by the elevated expression of other, developmentally-regulated transcripts (43).
The transition during myogenesis to a stage at which myogenin expression is elevated is developmentally important because it represents the transition from a proliferative population of myogenic cells that are unable to undergo terminal differentiation to a population that has permanently withdrawn from the cell cycle and enters early stages of differentiation (44). We had anticipated that if shifts in macrophage phenotype and the stage of myogenesis that occur in injured muscle in vivo were linked, then disruptions in macrophage phenotype switching would prevent the transition to early differentiation of myogenic cells that would be reflected in lower myogenin expression. Although our findings show that obstructing the shift to the M2 phenotype disrupts normal patterns of muscle differentiation and slows muscle growth, we unexpectedly found that amplifying the Th1 cytokine response at 1-day reloading by ablating IL-10 expression increased the number of myogenin expressing satellite cells. In addition, our data and previous findings (32) show that IL-10-stimulated M2 macrophages increase the proliferation of myoblasts in co-cultures, also indicating a role for M2 macrophages in promoting the proliferative stage of myogenesis rather than mediating the transition to early differentiation. Furthermore, our data show that the significant reduction of myogenin expression that occurs between 1-day and 4-days of reloading in wild-type muscles coincides with the large increase in CD163+ M2 macrophages and elevations of IL-10 expression, supporting the interpretation that M2 macrophages suppress myogenin expression in vivo. These findings contrast with a previous report that showed that co-cultures of muscle cells with macrophages that had been activated with the corticosteroid dexamethasone together with IL-10 caused no change in myoblast proliferation but increased myogenin expression (18). Those findings suggest that the influence of dexamethasone on M2 macrophages overwhelmed the influence of IL-10 on macrophage mediated effects on muscle cells, at least in the muscle co-culture experiments.
The effect of IL-10 ablation on myogenin expression in injured muscle in vivo may be mediated indirectly by changes in the expression of other cytokines that can influence myogenin expression. For example, the large increase in myogenin expression and increase in numbers of myogenin-expressing satellite cells that we observed in IL-10 mutants at 1-day reloading coincided with large elevations in IL-6 expression. However, the relationships between IL-6 and myoblast proliferation and differentiation are complex; IL-6 is well-established as a muscle mitogen (25) that plays a significant role in muscle fiber growth (45) but can also induce muscle wasting (46). Nevertheless, IL-6 treatments of myoblasts in vitro have been reported to increase myogenin expression (47) and a recent investigation showed a strong, positive effect of IL-6 on myogenin expression in vivo (48). Unlike wild-type mice, myogenin expression did not increase in skeletal muscle of IL-6 null mutant mice at 1-day of muscle reloading (48), although interpretation of the finding is complicated by the higher baseline level of expression of myogenin in the muscles of IL-6 mutants. Similarly, the increase in myogenin expression in reloaded muscles of IL-10 null mice may be attributable to the loss of elevated TNF-α expression in the injured muscles of mutants. Application of TNF-α to muscle cells in vitro can greatly diminish myogenin expression (49, 50, 51) that can result from activation of c-jun N-terminal kinase (52).
The mechanism through which IL-10 promotes the shift in muscle macrophages to the M2 phenotype is not known, although several observations indicate that activation of phagocytosis may be a component of the signaling pathway. Recent findings have shown that IL-10 stimulation of macrophages isolated from skeletal muscles increases the phagocytic activity of macrophages (32), as has been shown previously for circulating macrophages (53). Furthermore, phagocytosis of cellular debris by M1 macrophages can promote their transition to the M2 phenotype in vitro (18). In addition, depletions of populations of phagocytic, muscle macrophages in vivo by the administration of clodronate-containing liposomes specifically reduced the numbers of M2 macrophages (32), possibly reflecting a decrease in the M1 to M2 phenotype switch caused by the depletion of phagocytes. However, based on in vitro observations by previous investigators, the induction of M1 to M2 phenotype transition by phagocytosis may be greatly influenced by the identity of the phagocytosed material, so that phagocytosis is not sufficient to drive the change in macrophage phenotype. For example, if the debris to be phagocytosed were generated by neutrophil lysis, then phagocytosis by macrophages caused great increases in IL-10 production, reflecting a switch to the M2 phenotype (54). However, elevation of IL-10 production does not occur if the debris resulted from lymphocyte death (54). These findings indicate that phagocytosis may promote the M2 macrophage phenotype during innate immune responses, which occurs in injured muscle, but perhaps not in an acquired immune response. Furthermore, phagocytosis of apoptotic neutrophils by M1 macrophages increased production of the Th2 cytokine TGFβ by the macrophages, while reducing expression of the Th1 cytokines IL-1β and TNF-α, reflecting a shift toward an M2 phenotype (55). However, if the neutrophils were opsonized with anti-CD45, their phagocytosis did not cause the shift in phenotype (55). Those observations indicate that phagocytosis of apoptotic neutrophils during an innate immune response can induce the M1 to M2 phenotype switch in macrophages. Interestingly, these cues for macrophage phenotype switch are in place in reloaded skeletal muscle at the time of M1 to M2 transition in vivo. The peak of apoptosis and phagocytosis of inflammatory cells occurs 2 days after the onset of muscle reloading (56) which coincides with elevation of M2 macrophage populations.
The IL-10-mediated shift in macrophage phenotype in injured muscle may be amplified by activation of mitogen-activated protein kinase (MAPK), especially p38 MAPK, through signaling that is antagonized by MAPK phosphatase-1 (MKP-1). Although IL-10 can function as either a potent activator of p38 in macrophages (57) or inhibitor of p38 (58, 59), activation of p38 by IL-10 or IL-10-independent pathways can cause further elevations in IL-10 expression and contribute to macrophage phenotype switch from M1 to M2 phenotype. For example, null mutation of MKP-1 caused supraphysiological levels of p38 activity in macrophages that induced elevated expression of IL-10 and TNF-α (60, 61) and the elevated levels of IL-10 and TNF-α are reduced by p38 inhibition (60). Furthermore, null mutation of MKP-1 increased the number of M2 macrophages in injured muscle but that increase was reduced by MAPK inhibitor (62), indicating that p38 activation contributes to the macrophage phenotype switch in injured muscle, which could elevate production of IL-10 (63), creating positive feedback for the phenotype switch. However, if IL-10 activation of p38 promotes the switch to the M2 phenotype during the regeneration of wild-type muscle, the effect appears to be independent of HMOX-1 induction. Although IL-10 can suppress the M1 macrophage phenotype by p38 activation of HMOX-1 (64), no changes in HMOX-1 expression were observed in reloaded muscle in the present investigation, even at 4-days reloading when IL-10 expression was greatly elevated.
Although this investigation illustrates that interleukin-10 is required for normal growth and regeneration of muscle following injury, the findings are not sufficient to show that further increases in IL-10 in injured muscle will improve repair. As shown recently, direct injection of IL-10 into injured, wild-type muscle slowed the growth of regenerative fibers (62). Thus, either the loss of IL-10 or the application of superphysiological levels of IL-10 can impede muscle growth and repair following injury. However, the negative effect of supraphysiological levels of IL-10 may reflect the dose-dependency of IL-10 effects on leukocyte activation. For example, IL-10 generally functions as an anti-inflammatory cytokine but supraphysiological levels of IL-10 administered during sepsis exacerbate the inflammatory response (65) and administration of exogenous IL-10 at low doses is protective against graft-versus-host disease while higher dosages promote lethality of the disease (66). Thus, developing IL-10 treatments that are useful for promoting muscle repair and regeneration following injury will require identification of therapeutically appropriate dosing with exogenous IL-10 or precise manipulations of the levels of IL-10 expression by leukocytes in vivo (67).
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 AR47721, RO1 AR47855, R01 AR/AG054451) and the Muscular Dystrophy Association, USA (157881) to J.G.T.
References
- 1.Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Amer J Physiol. 2010;298:R1173–1187. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith JK, Grisham MB, Granger DN, Korthuis RJ. Free radical defense mechanisms and neutrophil infiltration in postischemic skeletal muscle. Am J Physiol. 1989;256:H789–793. doi: 10.1152/ajpheart.1989.256.3.H789. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen HX, Lusis AJ, Tidball JG. Null mutation of myeloperoxidase in mice prevents mechanical activation of neutrophil lysis of muscle cell membranes in vitro and in vivo. J Physiol. 2005;565:403–413. doi: 10.1113/jphysiol.2005.085506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen HX, Tidball JG. Null mutation of gp91phox reduces muscle membrane lysis during muscle inflammation in mice. J Physiol. 2003;553:833–841. doi: 10.1113/jphysiol.2003.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen HX, Tidball JG. Interactions between neutrophils and macrophages promote macrophage killing of rat muscle cells in vitro. J Physiol. 2003;547:125–132. doi: 10.1113/jphysiol.2002.031450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Human Mol Genet. 2009;18:482–496. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pierre BA, St, Tidball JG. Differential response of macrophage subpopulations to soleus muscle reloading following rat hindlimb suspension. J Appl Physiol. 1994;77:290–297. doi: 10.1152/jappl.1994.77.1.290. [DOI] [PubMed] [Google Scholar]
- 8.Kasper CE. Sarcolemmal disruption in reloaded atrophic skeletal muscle. J Appl Physiol. 1995;79:607–614. doi: 10.1152/jappl.1995.79.2.607. [DOI] [PubMed] [Google Scholar]
- 9.Krippendorf BB, Riley DA. Distinguishing unloading- versus reloading-induced changes in rat soleus muscle. Muscle Nerve. 1993;16:99–108. doi: 10.1002/mus.880160116. [DOI] [PubMed] [Google Scholar]
- 10.McLennan IS. Resident macrophages (ED2- and ED3-positive) do not phagocytose degenerating rat skeletal muscle fibres. Cell Tissue Res. 1993;272:193–196. doi: 10.1007/BF00323586. [DOI] [PubMed] [Google Scholar]
- 11.Warren GL, O’Farrell L, Summan M, Hulderman T, Mishra D, Luster MI, Kuziel WA, Simeonova PP. Role of CC chemokines in skeletal muscle functional restoration after injury. Am J Physiol. 2004;286:C1031–1036. doi: 10.1152/ajpcell.00467.2003. [DOI] [PubMed] [Google Scholar]
- 12.Warren GL, Hulderman T, Mishra D, Gao X, Millecchia L, O’Farrell L, Kuziel WA, Simeonova PP. Chemokine receptor CCR2 involvement in skeletal muscle regeneration. FASEB J. 2005;19:413–415. doi: 10.1096/fj.04-2421fje. [DOI] [PubMed] [Google Scholar]
- 13.Shireman PK, Contreras-Shannon V, Ochoa O, Karia BP, Michalek JE, McManus LM. MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J Leukocyte Biol. 2007;81:775–785. doi: 10.1189/jlb.0506356. [DOI] [PubMed] [Google Scholar]
- 14.Contreras-Shannon V, Ochoa O, Reyes-Reyna SM, Sun D, Michalek JE, Kuziel WA, McManus LM, Shireman PK. Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2-/- mice following ischemic injury. Am J Physiol. 2007;292:C953–967. doi: 10.1152/ajpcell.00154.2006. [DOI] [PubMed] [Google Scholar]
- 15.Ochoa O, Sun D, Reyes-Reyna SM, Waite LL, Michalek JE, McManus LM, Shireman PK. Delayed angiogenesis and VEGF production in CCR2-/- mice during impaired skeletal muscle regeneration. Am J Physiol. 2007;293:R651–661. doi: 10.1152/ajpregu.00069.2007. [DOI] [PubMed] [Google Scholar]
- 16.Sun D, Martinez CO, Ochoa O, Ruiz-Willhite L, Bonilla JR, Centonze VE, Waite LL, Michalek JE, McManus LM, Shireman PK. Bone marrow-derived cell regulation of skeletal muscle regeneration. FASEB J. 2009;23:382–395. doi: 10.1096/fj.07-095901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu H, Huang D, Ransohoff RM, Zhou L. Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J. 2011 doi: 10.1096/fj.10-178939. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exper Med. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins CA, Partridge TA. Self-renewal of the adult skeletal muscle satellite cell. Cell Cycle. 2005;4:1338–1341. doi: 10.4161/cc.4.10.2114. [DOI] [PubMed] [Google Scholar]
- 20.Cornelison DD, Wold BJ. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Develop Biol. 1997;191:270–283. doi: 10.1006/dbio.1997.8721. [DOI] [PubMed] [Google Scholar]
- 21.Yablonka-Reuveni Z, Rivera AJ. Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Develop Biol. 1994;164:588–603. doi: 10.1006/dbio.1994.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cantini M, Carraro U. Macrophage-released factor stimulates selectively myogenic cells in primary muscle culture. J Neuropathol Exp Neurol. 1995;54:121–128. doi: 10.1097/00005072-199501000-00014. [DOI] [PubMed] [Google Scholar]
- 23.Merly F, Lescaudron L, Rouaud T, Crossin F, Gardahaut MF. Macrophages enhance muscle satellite cell proliferation and delay their differentiation. Muscle Nerve. 1999;22:724–732. doi: 10.1002/(sici)1097-4598(199906)22:6<724::aid-mus9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 24.Cantini M, Massimino M, Bruson A, Catani C, Libera LD, Carraro U. Macrophages regulate proliferation and differentiation of satellite cells. Biochem Biophys Res Commun. 1994;202:1688–1696. doi: 10.1006/bbrc.1994.2129. [DOI] [PubMed] [Google Scholar]
- 25.Cantini M, Massimino M, Rapizzi E, Rossini K, Catani C, Libera LD, Carraro U. Human satellite cell proliferation in vitro is regulated by autocrine secretion of IL-6 stimulated by a soluble factor(s) released by activated monocytes. Biochem Biophys Res Commun. 1995;216:49–53. doi: 10.1006/bbrc.1995.2590. [DOI] [PubMed] [Google Scholar]
- 26.Massimino M, Rapizzi E, Cantini M, Libera L, Mazzoeni F, Arsian P, Carraro U. ED2+ macrophages increase selectively myoblast proliferation in muscle cultures. Biochem Biophys Res Commun. 1997;235:754–759. doi: 10.1006/bbrc.1997.6823. [DOI] [PubMed] [Google Scholar]
- 27.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 28.Schaer DJ, Boretti FS, Hongegger A, Poehler D, Linnscheid P, Staege H, Muller C, Schoedon G, Schaffner A. Molecular cloning and characterization of the mouse CD163 homologue, a highly glucocorticoid-inducible member of the scavenger receptor cysteine-rich family. Immunogenetics. 2001;53:170–177. doi: 10.1007/s002510100304. [DOI] [PubMed] [Google Scholar]
- 29.Sulahian TH, Hogger P, Wahner AE, Wardwell K, Goulding NJ, Sorg C, Droste A, Stehling M, Wallace PK, Morganelli PM, Guyre PM. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine. 2000;12:1312–1321. doi: 10.1006/cyto.2000.0720. [DOI] [PubMed] [Google Scholar]
- 30.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 31.Tidball JG, Wehling-Henricks M. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J Physiol. 2007;578:327–336. doi: 10.1113/jphysiol.2006.118265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villalta SA, Rinaldi C, Deng B, Liu G, Fedor B, Tidball JG. Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Human Molec Genetics. 2011;20:790–805. doi: 10.1093/hmg/ddq523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morey-Holton ER, Globus RK. Hindlimb unloading rodent model: technical aspects. J Appl Physiol. 2002;92:1367–1377. doi: 10.1152/japplphysiol.00969.2001. [DOI] [PubMed] [Google Scholar]
- 34.Thomason DB, Booth FW. Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol. 1990;68:1–12. doi: 10.1152/jappl.1990.68.1.1. [DOI] [PubMed] [Google Scholar]
- 35.Hainsey TA, Senapati S, Kuhn DE, Rafael JA. Cardiomyopathic features associated with muscular dystrophy are independent of dystrophin absence in cardiovasculature. Neuromuscul Disord. 2003;13:294–302. doi: 10.1016/s0960-8966(02)00286-9. [DOI] [PubMed] [Google Scholar]
- 36.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 37.Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163:3771–3777. [PubMed] [Google Scholar]
- 38.Philippidis P, Mason JC, Evans BJ, Nadra I, Taylor KM, Haskard DO, Landis RC. Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ Res. 2004;94:119–126. doi: 10.1161/01.RES.0000109414.78907.F9. [DOI] [PubMed] [Google Scholar]
- 39.Frenette J, Cai B, Tidball JG. Complement activation promotes muscle inflammation during modified muscle use. Amer J Pathol. 2000;156:2103–2110. doi: 10.1016/S0002-9440(10)65081-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLennan IS. Resident macrophages (ED2- and ED3-positive) do not phagocytose degenerating rat skeletal muscle fibres. Cell Tissue Res. 1993;272:193–196. doi: 10.1007/BF00323586. [DOI] [PubMed] [Google Scholar]
- 41.Launay T, Armand AS, Charbonnier F, Mira JC, Donsez E, Gallien CL, Chanoine C. Expression and neural control of myogenic regulatory factor genes during regeneration of mouse soleus. J Histochem Cytochem. 2001;49:887–899. doi: 10.1177/002215540104900709. [DOI] [PubMed] [Google Scholar]
- 42.Yablonka-Reuveni Z, Rivera AJ. Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Develop Biol. 1994;164:588–603. doi: 10.1006/dbio.1994.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLoon LK, Nguyen LT, Wirtschafter J. Time course of the regenerative response in bupivacaine injured orbicularis oculi muscle. Cell Tissue Res. 1998;294:439–447. doi: 10.1007/s004410051195. [DOI] [PubMed] [Google Scholar]
- 44.Chargé SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 45.Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Muñoz-Cánoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008;7:33–44. doi: 10.1016/j.cmet.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 46.Haddad F, Zaldivar F, Cooper DM, Adams GR. IL-6-induced skeletal muscle atrophy. J Appl Physiol. 2005;98:911–917. doi: 10.1152/japplphysiol.01026.2004. [DOI] [PubMed] [Google Scholar]
- 47.Okazaki S, Kawai H, Arii Y, Yamaguchi H, Saito S. Effects of calcitonin gene-related peptide and interleukin 6 on myoblast differentiation. Cell Prolif. 1996;29:173–182. [PubMed] [Google Scholar]
- 48.Washington TA, White JP, Davis JM, Wilson LB, Lowe LL, Sato S, Carson JA. Skeletal muscle mass recovery from atrophy in IL-6 knockout mice. Acta Physiol. 2011;202:657–669. doi: 10.1111/j.1748-1716.2011.02281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szalay K, Rázga Z, Duda E. TNF inhibits myogenesis and downregulates the expression of myogenic regulatory factors myoD and myogenin. Eur J Cell Biol. 1997;74:391–398. [PubMed] [Google Scholar]
- 50.Layne MD, Farmer SR. Tumor necrosis factor-alpha and basic fibroblast growth factor differentially inhibit the insulin-like growth factor-I induced expression of myogenin in C2C12 myoblasts. Exp Cell Res. 1999;249:177–187. doi: 10.1006/excr.1999.4465. [DOI] [PubMed] [Google Scholar]
- 51.Chen SE, Jin B, Li YP. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Amer J Physiol. 2007;292:C1660–671. doi: 10.1152/ajpcell.00486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strle K, Broussard SR, McCusker RH, Shen WH, LeCleir JM, Johnson RW, Freund GG, Dantzer R, Kelley KW. C-jun N-terminal kinase mediates tumor necrosis factor-alpha suppression of differentiation in myoblasts. Endocrinology. 2006;147:4363–4373. doi: 10.1210/en.2005-1541. [DOI] [PubMed] [Google Scholar]
- 53.Montoya D, Cruz D, Teles RM, Lee DJ, Ochoa MT, Krutzik SR, Chun R, Schenk M, Zhang X, Ferguson BG, Burdick AE, Sarno EN, Rea TH, Hewison M, Adams JS, Cheng G, Modlin RL. Divergence of macrophage phagocytic and antimicrobial programs in leprosy. Cell Host Microbe. 2009;6:343–353. doi: 10.1016/j.chom.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fadok VA, Bratton DL, Guthrie L, Henson PM. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J Immunol. 2001;166:6847–6854. doi: 10.4049/jimmunol.166.11.6847. [DOI] [PubMed] [Google Scholar]
- 55.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clinic Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tidball JG, Pierre BA., St Apoptosis of macrophages during the resolution of muscle inflammation. J Leukocyte Biol. 1996;59:380–388. doi: 10.1002/jlb.59.3.380. [DOI] [PubMed] [Google Scholar]
- 57.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nature Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 58.Kontoyiannis D, Kotlyarov A, Carballo E, Alexopoulou L, Blackshear PJ, Gaestel M, Davis R, Flavell R, Kollias G. Interleukin-10 targets p38 MAPK to modulate ARE-dependent TNF mRNA translation and limit intestinal pathology. EMBO J. 2001;20:3760–3770. doi: 10.1093/emboj/20.14.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rajasingh J, Bord E, Luedemann C, Asai J, Hamada H, Thorne T, Qin G, Goukassian D, Zhu Y, Losordo DW, Kishore R. IL-10-induced TNF-alpha mRNA destabilization is mediated via IL-10 suppression of p38 MAP kinase activation and inhibition of HuR expression. FASEB J. 2006;20:2112–2114. doi: 10.1096/fj.06-6084fje. [DOI] [PubMed] [Google Scholar]
- 60.Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci U S A. 2006;103:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, Chang CH, Liu Y. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203:131–140. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perdiguero E, Sousa-Victor P, Ruiz-Bonilla V, Jardí M, Caelles C, Serrano AL, Muñoz-Cánoves P. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J Cell Biol. 2011;195:307–322. doi: 10.1083/jcb.201104053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Foey AD, Parry SL, Williams LM, Feldmann M, Foxwell BM, Brennan FM. Regulation of monocyte IL-10 synthesis by endogenous IL-1 and TNF-alpha: role of the p38 and p42/44 mitogen-activated protein kinases. J Immunol. 1998;160:920–928. [PubMed] [Google Scholar]
- 64.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nature Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 65.Lauw FN, Pajkrt D, Hack CE, Kurimoto M, van Deventer SJ, van der Poll T. Proinflammatory effects of IL-10 during human endotoxemia. J Immunol. 2000;165:2783–2789. doi: 10.4049/jimmunol.165.5.2783. [DOI] [PubMed] [Google Scholar]
- 66.Blazar BR, Taylor PA, Panoskaltsis-Mortari A, Narula SK, Smith SR, Roncarolo MG, Vallera DA. Interleukin-10 dose-dependent regulation of CD4+ and CD8+ T cell-mediated graft-versus-host disease. Transplantation. 1998;66:1220–1229. doi: 10.1097/00007890-199811150-00018. [DOI] [PubMed] [Google Scholar]
- 67.Cope A, Le Friec G, Cardone J, Kemper C. The Th1 life cycle: molecular control of IFN-γ to IL-10 switching. Trends Immunol. 2011;32:278–286. doi: 10.1016/j.it.2011.03.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.