

Abstract
Rhombencephalosynapsis (RES) is an uncommon cerebellar malformation characterized by fusion of the hemispheres without an intervening vermis. Frequently described in association with Gómez-López-Hernández syndrome, RES also occurs in conjunction with VACTERL features and with holoprosencephaly (HPE). We sought to determine the full phenotypic spectrum of RES in a large cohort of patients. Information was obtained through database review, patient questionnaire, radiographic and morphologic assessment, and statistical analysis. We assessed 53 patients. 33 had alopecia, 3 had trigeminal anesthesia, 14 had VACTERL features and 2 had HPE with aventriculy. Specific craniofacial features were seen throughout the cohort, but were more common in patients with alopecia. We noted substantial overlap between groups. We conclude that although some distinct subgroups can be delineated, the overlapping features seen in our cohort suggest an underlying spectrum of RES-associated malformations rather than a collection of discrete syndromes.
Keywords: Rhombencephalosynapsis, Gómez-López-Hernández syndrome, Congenital Triangular Alopecia, Holoprosencephaly, Aventriculy, VACTERL, Developmental Field Defect
INTRODUCTION
Rhombencephalosynapsis (RES) is an uncommon but likely under-recognized cerebellar malformation characterized by continuity of the cerebellar hemispheres across the midline without an intervening vermis. This condition usually occurs sporadically but is presumed to have genetic underpinnings (Table I), though no consistent genetic causes have been identified and no animal models exist. Disruption of dorsal-ventral patterning has been proposed as the cause of RES [Sarnat, 2000, Yachnis, 2002], but the mechanisms involved have yet to be elucidated. The prevalence of RES is unknown, but Ishak et al [2012] recently reported that 5 of 56 patients with aqueductal stenosis had unrecognized RES, suggesting that RES may be considerably more common than previously believed. Initially described as an isolated brain malformation, RES is increasingly recognized in conjunction with other anomalies.
Table I.
Genetic changes described in association with RES.
| Genetic changes described in association with RES | |
|---|---|
| Recurrence in two subsequent pregnancies | Pasquier et al., 2009 |
|
| |
| Consanguinity | Chemli et al., 2007 |
| Pasquier et al., 2009a | |
| Romanengo et al., 1997 | |
| Sandalcioglu et al., 2006 | |
| Toelle et al., 2002 | |
|
| |
| Interstitial deletion of 2q | Truwit et al., 1991 |
|
| |
| Unbalanced subtelomeric translocation: t(2p:10q) | Lespinasse et al., 2004 |
|
| |
| Tetrasomy 9p | di Vera et al., 2008 |
|
| |
| Microduplication 1p | Pasquier et al., 2009 |
|
| |
| Microduplication 7q | Pasquier et al., 2009 |
|
| |
| Complex rearrangement of 22q13.3 | Ramocki et al, 2011* |
The authors also reported partial RES in conjunction with HPE in two sisters with truncating mutations of ZIC2. However, the presence of RES was disputed by Guleria [2011] and could not be confirmed by our review.
The first syndrome to be associated with RES was described by Gómez [1979] and López-Hernández [1982], though the nature of the underlying brain malformation was not evident at the time. Key features of Gómez-López-Hernández syndrome (GLH) include RES, parietal-occipital scalp alopecia and trigeminal anesthesia, the latter often leading to recurrent corneal and facial scarring. Distinctive craniofacial features have been described in GLH, including a towering skull with short anterior-posterior and side-to-side dimensions (turricephaly)[Gómez, 1979; López-Hernández, 1982], a flat facial contour or midface retrusion, hypertelorism and low-set ears with increased posterior angulation [López-Hernández, 1982]. GLH has also been associated with variable degrees of motor developmental delay, intellectual disability, short stature, and behavioral difficulties [Brocks et al., 2000; Gomy et al., 2008].
Recently, some of the features once considered integral to a diagnosis of GLH have been called into question [Sukhudyan et al., 2010b]; the term GLH is now often applied to RES with alopecia but without trigeminal anesthesia or characteristic craniofacial features. Sporadic reports have also linked RES with holoprosencephaly (HPE) [Pasquier et al., 2009a; Ramocki et al., 2011; Siebert et al., 2005] and features of the VACTERL association [Aydingoz et al., 1997; de Jong and Kirby 2000; Pasquier et al., 2009b; Toelle et al., 2002]. Seeking to better define the full clinical spectrum of RES, we assessed a large cohort of patients with RES for associated anomalies.
METHODS
Patient acquisition
We performed a comprehensive search for patients with RES in our database of more than 6800 subjects with brain malformations or other developmental brain disorders maintained by the senior author (WBD). Most patients were referred by their families or physicians for imaging review. In some, the diagnosis of RES had been made previously, while in others it was noted upon our review of their imaging studies. Several patients in the database had previously been ascertained by reviewing MRIs of children with aqueductal stenosis in whom RES had not been recognized [Ishak et al., 2012]. Patients whose families elected to enroll in research underwent a formal consent process, supplied DNA samples and provided additional clinical information.
Assessment of RES and additional structural brain abnormalities
Imaging studies were reviewed by 4 authors (Dobyns, Doherty, Ishak, Tully) to confirm the diagnosis of RES. Scans were also assessed for major additional malformations of the forebrain and the ventricular system. Subtle abnormalities such as fusion of the colliculi and absent mammillary bodies, which are frequently seen in conjunction with RES but often not mentioned in routine radiology reports, are described in a related article by Ishak et al [2012].
Evaluation of GLH features
Alopecia was assessed directly during clinic visits, through patient pictures and by parental questionnaire. Trigeminal anesthesia was assessed directly when possible (corneal swab or saline drops to elicit blink reflex), through review of medical records, or by parental questionnaire. In questionable situations, a history of repeated corneal injuries or facial scarring was considered indicative of trigeminal anesthesia. Morphologic features were assessed by sending photographs of patients to nine authors (Adam, Allanson, Cunniff, Curry, Dobyns, Glass, Gripp, Hunter, Sanchez-Lara) for subjective assessment of head shape (turricephaly, brachycephaly), forehead shape (high, wide), facial contour (flat contour, midface retrusion), eye placement (subjectively wide-set, telecanthus), and ear placement (low set, increased posterior angulation). Respondents could answer Yes, No, or Unable to Determine for each variable and were given the opportunity to comment on other features. A patient was deemed to have the feature in question if a majority of the 9 evaluating geneticists agreed that it was present, with a minimum consensus of 3 when some respondents were unsure. When skull shape could not be reliably assessed because of hair, a neuroradiologist made the determination based on MRI or CT images. We created a composite morphology score for each patient based on these responses. Patients were given 1 point for a positive finding in each of the 5 categories (head shape, forehead shape, facial contour, eye placement and ear placement). For each patient, these points became a numerator; the denominator was the total number of categories in which evaluators reached consensus. Thus, a patient who was determined to have a high forehead but no other features would receive a morphology score of 1/5. A patient who was rated as having a flat facial contour and telecanthus but in whom forehead shape and ear placement could not be determined would receive a score of 2/3.
Assessment of VACTERL features
Patient records and radiologic scans were assessed for vertebral anomalies, anal atresia, cardiac malformations, tracheoesophageal fistula, renal anomalies, radial dysplasia, or other limb defects. Although the VACTERL association is defined as the presence of 3 or more features, we noted the presence of any VACTERL feature in our patients.
Statistical Analysis
We performed a Fisher’s exact test to determine whether alopecia correlated with VACTERL features. We performed an analysis of variance (ANOVA) and a Student’s t-test to determine whether mean composite morphology scores varied among specific groups of patients. We used SAS, version 9.3 (SAS Institute, Inc., Cary, NC) for all statistical analyses.
RESULTS
Overview
We assessed our cohort of 53 patients for the presence of GLH features, VACTERL features, and major additional brain anomalies (Table II). These features are seen in overlapping groups of patients (Fig 2).
Table II.
Clinical characteristics of 53 patients with RES.
| Pt ID | Age | Sex | GLH Features | VACTERL Features | Other Brain Malfs | Notes | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Alo | TA | CMS | Vert SD | Cardiac/Renal/GI/GU | Limb | |||||
| RES Without Alopecia or VACTERL (13 patients) | ||||||||||
| 1 | 10m | F | - | 2/5 | - | - | - | - | ||
| 3 | 22m | F | - | - | 1/5 | - | - | - | - | |
| 10 | 26y | M | - | - | 0/5 | - | - | - | - | |
| 11 | 6y | F | - | - | 2/4 | - | - | - | - | Congenital pyriform aperture stenosis |
| 16 | 44y | F | - | - | 0/5 | - | - | - | - | |
| 22 | 7y | M | - | - | NA | Bilateral microtia, aural atresia, L corneal clouding attributed to infection | ||||
| 23 | 2y | M | - | - | 3/5 | - | - | - | - | |
| 26 | 5y | F | - | - | 1/5 | - | - | - | - | Gestational DM |
| 38 | 13m | F | - | - | NA | - | - | - | - | |
| 40 | 9y | F | - | - | NA | - | - | - | - | L corneal clouding attributed to scarring from incomplete eye closure, no TA on exam |
| 42 | 2y | F | - | - | NA | - | - | - | - | |
| 46 | 15y | M | - | - | 2/5 | - | - | - | Urachal cyst | |
| 47 | 16y | M | - | - | 0/5 | - | - | - | ||
| RES with Alopecia (23 patients) | ||||||||||
| 2 | 3y | F | L | - | 3/5 | - | - | - | - | |
| 14 | 5y | M | R | - | 0/5 | - | - | - | Poss MIF-like HPE | |
| 17 | 4y | M | L | - | 3/5 | - | - | - | - | |
| 18 | 7y | F | L>R | - | 3/5 | - | - | - | - | R squamosal CSO s/p surgery, eyebrow tuft |
| 28 | 4y | M | R>L | - | 2/5 | - | - | - | - | |
| 41 | 16m | M | L | - | 2/5 | - | - | - | - | |
| 4 | 22m | M | B | - | 3/4 | - | - | - | - | |
| 6 | 5y | M | B | - | 2/5 | - | - | - | - | Gestational DM. B SN hearing loss. Chordee. |
| 7 | 6y | F | B | - | 4/5 | - | - | - | P PMG | B SN hearing loss |
| 9 | 14y | M | B | - | 2/5 | - | - | - | - | |
| 12 | 4y | M | B | - | 0/3 | - | - | - | - | |
| 13 | 2y | M | B | - | 3/5 | - | - | - | - | |
| 24 | 5y | M | B | - | 2/5 | - | - | - | - | Idiopathic renal failure |
| 25 | 27m | M | B | - | 3/5 | - | - | - | - | Neurofibromatosis with NF1 mutation |
| 27 | 2y | M | B | - | 3/5 | - | - | - | - | |
| 31 | 6y | F | B | - | 2/5 | - | - | - | - | |
| 32 | 5y | M | B | - | 2/5 | - | - | - | - | |
| 34 | 4y | M | B | - | 3/5 | - | - | - | - | |
| 39 | 3y | M | B | - | 3/5 | - | - | - | Gestational DM | |
| 45 | 4y | F | B | - | 3/5 | - | - | - | - | CSO |
| 48 | 9y | M | B | - | NA | - | - | - | - | CSO s/p surgery |
| 51 | 4y | F | B | - | NA | - | - | - | - | |
| 52 | 3y | F | B | - | NA | - | ||||
| RES with Alopecia and Trigeminal Anesthesia (3 patients) | ||||||||||
| 8 | 3y | F | B | B | 4/5 | - | - | - | - | Multi-suture CSO s/p surgery |
| 44 | 31y | M | B | B | 3/5 | - | - | - | - | CSO s/p surgery |
| 53 | 9y | F | B | B | NA | - | ||||
| RES with VACTERL Features (+/− Alopecia) (12 patients) | ||||||||||
| 5 | 5y | F | B | - | 4/4 | - | Duplicated renal collecting system, retro-esophageal cyst | Radial aplasia B | - | TAR syndrome with 1q21.1 deletion |
| 19 | 6y | M | R | - | 1/3 | Y | ASD, VSD, bicuspid AV | Radial aplasia R | - | Posterior skull defect. Possible CSO. |
| 20 | 4y | M | - | - | 1/5 | - | - | PPD L hand | - | Temporal tuft |
| 21 | 3y | M | - | - | NA | - | Absent R kidney, fused/duplicated L kidney | - | - | |
| 29 | 17y | M | R | - | 0/5 | Y | Absent L kidney, PUV | - | - | Gestational DM |
| 30 | 3y | M | L>R | - | 4/5 | Y | Absent L kidney, Hypoplastic AA, VSD | - | - | |
| 33 | 12y | F | - | - | 5/5 | Y | - | - | - | |
| 36 | 19m | F | B | 3/5 | Y | TOF, RSAA, horseshoe kidney | - | |||
| 37 | 4y | F | - | - | NA | - | TAPVR, anal atresia | - | - | |
| 43 | 10m | F | B | - | 2/5 | Y | VSD | - | - | |
| 49 | 23y | M | - | NA | - | - | PPD L hand | - | Temporal tuft, posterior skull defect | |
| 50 | 5y | F | NA | NA | NA | NA | Absent kidney, body wall defect | PPD R foot | - | Mother with pre-existing DM |
| RES with Posterior HPE, Aventriculy and VACTERL Features (+/− Alopecia) (2 patients) | ||||||||||
| 15 | 3y | F | R | - | 1/3 | Y | - | - | Post HPE, AV | Mother with pre-existing DM. Microcephaly. Forme fruste cleft lip. |
| 35 | 2d | M | - | NA | 2/5 | Y | TOF | Fused/duplicated R hallux | Post HPE, AV | Mother with pre-existing DM. Microcephaly. Malformed ears. |
Abbreviations: Alo, Alopecia. TA, trigeminal anesthesia. CMS, composite morphology score. DM, diabetes mellitus. MIF, middle interhemispheric fusion [variant of holoprsoencephaly]. HPE, holoprosencephaly. CSO, craniosynostosis. SN [hearing loss], sensorineural. P PMG, perisylvian polymicrogyria. ASD, atrial septal defect, VSD, ventricular septal defect. [Bicuspid] AV, aortic valve. PUV, posterior urethral valves. [hypoplastic] AA, aortic arch. TOF, Tetralogy of Fallot. RSAA, right-sided aortic arch. TAPVR, total anomalous pulmonary venous return.
Figure 2.
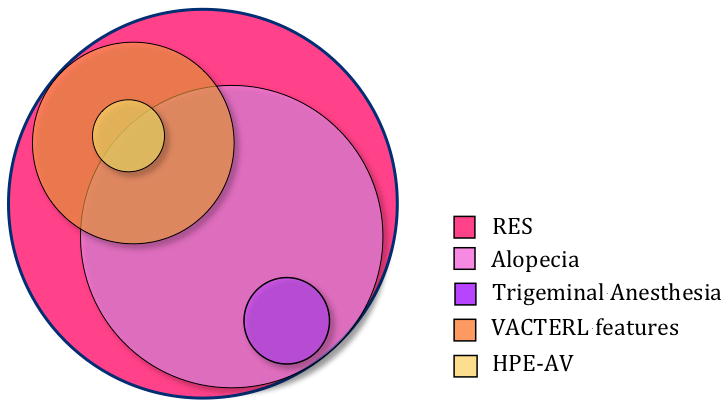
Areas of overlap among patients with RES.
GLH Features
Alopecia
In 52 patients for whom sufficient information was available, 33 (63%) had alopecia, which was bilateral in 26 patients and unilateral in seven. The degree of alopecia ranged from well-circumscribed patches of apparently hairless skin (most common) to less circumscribed areas of relatively sparse hair. We categorized patients with sparse hair as having alopecia if there was a readily appreciable difference between affected and unaffected scalp.
Trigeminal anesthesia
Trigeminal anesthesia was present in 3 of the 50 patients for whom sufficient nformation was available. In each case, there was longstanding evidence of recurrent bilateral corneal injuries and facial scarring.
Craniofacial morphology
Forty-one patients underwent morphological assessment on the basis of photographs (Fig 3). Skull shape was assessed in all 41. 3 (8%) were determined to have turricephaly. One patient had brachycephaly without turricephaly. Forehead shape was assessed in 38 patients. A high forehead was found in 22 (58%), 10 of whom were also rated as having a wide forehead (26%). A wide forehead alone was found in three patients. Facial contour was evaluated in 37 patients. Fifteen (41%) had flat facial contour or midface retrusion. Eye placement was assessed in 40 patients. Telecanthus was noted in 17 (43%); 2 also had subjectively wide-set eyes. One patient’s eyes appeared wide-set without telecanthus. Ear placement was evaluated in 38 patients. Twenty-four (63%) had low-set ears, 19 of whom also had increased posterior angulation. Two patients had ears with posterior angulation that were not low-set. Based on these features, we generated a composite morphology score for each patient as described in the Methods section (Table I). Individual morphologic features can be viewed in Supplementary eTable I (See Supporting Information online).
Figure 3. Faces of Patients with RES.

A–B: Patient 39. C–D: Patient 28. E–F: Patient 26. G–H: Patient 17. I–J, Patient 7. K–L: Patient 9. M–N: Patient 46. O–P: Patient 44.
Two patients, both with HPE, had additional dysmorphic signs: one had unilateral microtia; the other had upslanting palpebral fissures and a forme fruste cleft lip (Fig 6).
Figure 6. Additional features in patients with HPE-AV.
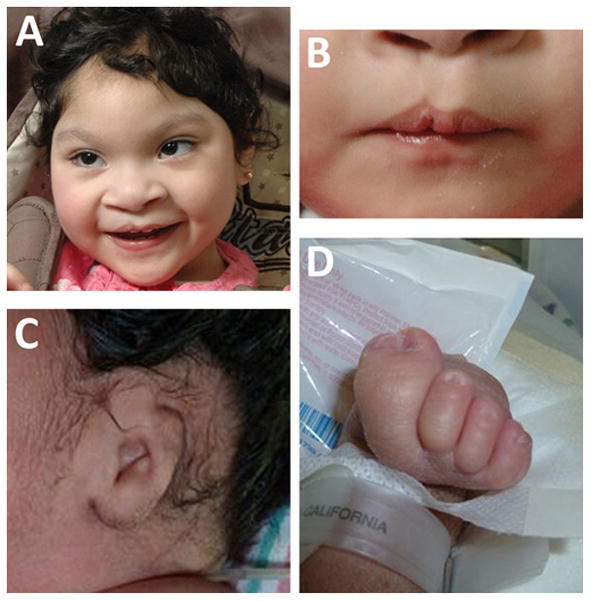
A and B: Patient 15. Broad nose, upslanting palpebral fissures and forme fruste cleft lip (A). Close-up of lip (B). C–D: Patient 35. Microtia (C). Partially duplicated, fused right hallux (D).
VACTERL Features
VACTERL features were seen in 14 (26%) of our 53 patients (Table II and Fig 4). 5 had a single VACTERL feature, four had two VACTERL features and five had three or more VACTERL features. Of the VACTERL features found in our cohort, vertebral segmentation defects were most common, seen in nine patients. Structural renal anomalies were seen in six patients, and structural cardiac defects in five. Four patients had pre-axial polydactyly or polysyndactyly; 2 patients had radial aplasia (one with TAR, one without). One patient had anal atresia and one patient had a retro-esophageal cyst. One patient also had thrombocytopenia-absent radius (TAR) syndrome with a confirmed 1q21.1 deletion. Though her bilateral absent radii are likely attributable to TAR, we included her in this category because of her two additional features (duplicated renal collecting system and retroesophageal cyst).
Figure 4. VACTERL Features.
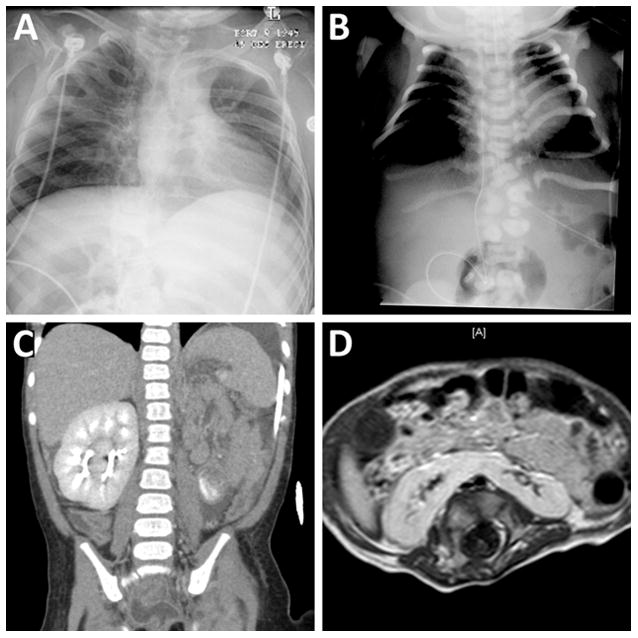
A: Patient 33. Radiograph demonstrating multiple fused ribs associated with vertebral segmentation defects. B: Patient 35. Radiograph demonstrating multiple thoracic and lumbar vertebral segmentation defects and deformed ribs. C: Patient 21. Coronal CT showing absent kidney on the left with partially duplicated kidney on the right (cross-fused renal ectopia). D: Patient 36. Axial T1-weighted image of the abdomen showing horseshoe kidney.
Several patients with VACTERL features also had alopecia, but there was no statistically significant association between these features (p = 0.51, Fisher’s exact test).
Additional Brain Malformations
Two patients had a striking form of HPE characterized by posterior-predominant fusion of the cerebral hemispheres in conjunction with absent lateral and third ventricles (Figure 5). Another patient had what appeared to be a smaller area of hemispheric continuity across the midline resembling the middle interhemispheric fusion (MIF) variant of HPE (Fig 1, images I–L). However, distortion from severe congenital hydrocephalus made this difficult to confirm. One other patient had bilateral perisylvian polymicrogyria.
Figure 5. Holoprosencephaly-Aventriculy.

A–G: Patient 15. Mid-sagittal view demonstrating dramatically infolded brain, absent 3rd ventricle and aqueduct, and mass-like fusion of the midbrain (A). Coronal T2 demonstrating gray matter continuity across the midline (HPE) and absent lateral ventricles (B). Coronal T2 demonstrating posterior predominance of HPE and continuity of cerebellar folia across the midline (C). Axial T2 demonstrating RES(D). Axial T2s showing posterior-predominant fusion, infolding and aventriculy (E–G). H–N: Patient 35. Multiple axial, coronal and mid-sagittal sections showing striking similarity of brain malformation to that of Patient 15.
Figure 1. Radiographic Features of RES.

A–D: Patient 48. Midline sagittal T1-weighted image through the cerebellum demonstrates hemispheric rather than vermian configuration (A). Axial T2 through the cerebellum shows fusion of white matter across the midline and a keyhole-shaped 4th ventricle (B). Axial T2 through though the cerebral hemispheres shows normal anatomy (C). Coronal T2 showing continuity of cerebellar folia across the midline without an intervening vermis (D). E–H: Patient 46. Midline sagittal T1-weighted image demonstrating a towering cerebellum with hemispheric architecture. Note the absence of a visible aqueduct. This patient had severe congenital hydrocephalus. Many of the supratentorial abnormalities (arrow) are likely a consequence of distortion from hydrocephalus and subsequent decompression (E). Axial T1 through the cerebellum demonstrating similar findings as the patient above (F). Axial T1 through the cerebral hemispheres shows an area of white matter continuity suspicious for mild HPE, but likely representing post-hydrocephalus distortion(G). Coronal T2 showing a towering cerebellum with upward displacement through the tentorial notch (H). I–L: Patient 14. I, Mid-sagittal and axial T1 demonstrating similar findings to Patient 46. This patient also had severe congenital hydrocephalus (J). Axial T1 showing area suspicious for HPE (arrow), though post-hydrocephalic distortion makes this difficult to confirm (K). Coronal T2 demonstrating similar findings to Patient 46 (L). M–P: normal brain.
Craniofacial differences between groups of patients (Table III)
Table III.
Patient Groups and Composite Morphology Scores
| Category | GLH Features | VACTERL Features | Major Additional Brain Anomalies | ||
|---|---|---|---|---|---|
| Alopecia | TA | Composite Morphology Score, Mean (95% CI) | |||
| RES without alopecia, VACTERL features or HPE-AV (13 patients) | N | N | 25 (5 – 45) | N | N |
| RES with alopecia but without VACTERL features or HPE-AV (23 patients) | Y, ranging from unilateral sparse patch to well-circumscribed bilateral areas of alopecia | N | 51 (39 – 64) | N | 1 patient with possible MIF-like HPE, 1 patient with perisylvian polymicrogyria |
| RES with alopecia and trigeminal Anesthesia (3 patients) | Y | Y | N | N | |
| RES with VACTERL features +/− Alopecia (12 patients) | Variably present | N | 51 (31 – 71) | Y | N |
| RES with HPE-AV and VACTERL features +/− alopecia (2 patients) | Variably present | N | Y | Posterior HPE, aventriculy | |
| All RES without alopecia, including patients with VACTERL features and HPE-AV (19 patients) | N | N | 32 (16–50) | Y/N | Y/N |
| All RES with alopecia, including patients with VACTERL features and HPE-AV (33 patients) | Y | Y/N | 51 (40–60) | Y/N | Y/N |
Abbreviations: RES, rhombencephalosynapsis. MIF, Middle Interhemispheric Fusion [variant of holoprosencephaly]. HPE, holoprosencephaly.
We divided our cohort into five groups based on clinical features: 1) RES without alopecia or VACTERL Features, 2) RES with alopecia alone, 3) RES with alopecia and trigeminal anesthesia, 4) RES with VACTERL features, and 5) RES with HPE-AV (both patients in this group also had VACTERL features). We calculated an average composite morphology score for each group. Because of small numbers, Groups 2 and 3 were combined, as were Groups 4 and 5 Analysis of variance demonstrated that Groups 2 and 3 combined had a significantly higher mean composite morphology score than Group 1 (P<0.05.). Groups 4 and 5 combined had the same mean composite morphology score as categories 2 and 3 combined, but because of smaller numbers in this group, the comparison to Group 1 did not achieve statistical significance. We also compared patients solely on the basis of alopecia (patients with VACTERL features and HPE-AV were included in both groups). Patients with alopecia had a higher mean composite morphology score than those without alopecia (p=0.03).
DISCUSSION
We reviewed a series of 53 individuals with RES to delineate the full range of RES-associated phenotypes and compare them to the better-known Gómez-López-Hernández syndrome. We identified one subset of patients with features of the VACTERL association and another subset with an unusual posterior form of holoprosencephaly and aventriculy; however, only 17 of 53 patients could be classified unambiguously as having a specific malformation syndrome, including GLH itself (when defined as RES with alopecia and trigeminal anesthesia).
When we analyzed the phenotypic spectrum in more depth by dividing our cohort into five categories using easily-identified objective elements (alopecia, trigeminal anesthesia, VACTERL features and HPE-AV (Table III), substantial areas of overlap became apparent (Fig 2). To determine whether the craniofacial features reported in GLH syndrome were seen among other patients with RES, we generated a composite morphology score and found a broad range of scores in all categories, but with a statistically significant association between higher scores and alopecia. We propose that the recurrent, overlapping elements associated with RES reflect disruption of a fundamental mechanism that regulates development of the involved structures. Accordingly, we contend that RES and its associated features are best viewed as a spectrum of biologically-related malformations rather than a collection of discrete syndromes, similar to the spectrum of brain and craniofacial malformations associated with classic HPE.
GLH syndrome and the RES spectrum of anomalies
In 1979, Gómez reported a girl with trigeminal anesthesia, a band of parietal-occipital alopecia and prominent cerebellar findings on examination [1979]. Two years later, López-Hernández described 2 girls with craniosynostosis, trigeminal anesthesia and bilateral parietal alopecia [1982]. MRI scans of subsequent patients defined the associated brain malformation as RES. Though a distinctive skull shape and facial features have often been considered part of GLH syndrome, Sukhudyan et al noted that the only consistent features in GLH syndrome are RES and alopecia [Sukhudyan et al., 2010], presaging our results.
Trigeminal anesthesia
Only three patients in our cohort had trigeminal anesthesia, in contrast to the patients described in a literature review by Sukhudyan et al [2010], in which 17/21 had trigeminal anesthesia. We attribute this to differences in ascertainment: previous patients in the literature were reported as examples of GLH syndrome, whereas the patients in our cohort were ascertained on the basis of their underlying brain malformation.
Alopecia
While few patients in our cohort had trigeminal anesthesia, 67% had alopecia, including several with VACTERL features or HPE-AV. The alopecia seen in conjunction with RES is distinctive, consisting of focal areas of hypotrichosis, often bilaterally symmetric, which may contain tufts of hair within them. On biopsy, these areas demonstrate preserved architecture with variably hypoplastic hair follicles, and no inflammation or scarring [Munoz et al., 1997; Pasquier et al., 2009a].
Interestingly, a clinically and histologically equivalent condition without neurologic abnormalities has been described in the dermatology literature as congenital triangular alopecia or temporal triangular alopecia (CTA, TTA) [Armstrong and Burrows, 1996; Assouly and Happle, 2010; Bargman, 1988; Elmer and George, 2002; Feuerman, 1981; Garcia-Hernández et al., 1995; Kubba and Rook, 1976; Silva et al., 2010; Tosti, 1987; Trakimas et al., 1994; Yamazaki et al., 2010]. A single report linked CTA to GLH in a child with significant gross motor delay; this patient had a brother with CTA but not RES [Purvis et al., 2007]. CTA has also been described in a mother and daughter with intellectual disability and epilepsy [Ruggieri et al., 2000]. The daughter was reported to have a Dandy-Walker malformation.
Craniofacial morphology
Most patients in our RES cohort lack a distinctive skull shape. Two of the three children with turricephaly had severe congenital hydrocephalus and macrocephaly on initial imaging. Follow-up scans demonstrated acquired turricephaly in the setting of residual ventricular enlargement. Several patients also had abnormal skull shape at birth and subsequently underwent surgical correction of craniosynostosis. Follow-up scans showed normal skull contour. Taken together, this suggests that skull shape is influenced by several factors and may change over time, rendering it less useful as a diagnostic sign. In contrast to turricephaly, differences in forehead shape, facial contour, and position of ears and eyes were common among our patients. These features were seen across the entire cohort of RES patients, though on average, patients with alopecia had higher composite morphology scores than those without.
Two additional RES associations
Two additional phenotypic motifs emerged in our cohort: features of the VACTERL association (seen in 14), and HPE-AV (seen in two). We found examples of both these RES-associated phenotypes in the literature, but with limited delineation.
RES with VACTERL features
RES has occasionally been reported in association with VACTERL features [Aydingoz et al., 1997; Toelle et al., 2002]. Hydrocephalus (without known RES) has also been described in conjunction with VACTERL features, gaining the name VACTERL-H syndrome [Beemer et al., 1990; Briard et al., 1984; Corsello and Giuffre, 1994; Evans et al., 1989; Iafolla et al., 1991]. VACTERL-H syndrome is a clinically and genetically heterogeneous condition with several reports of familial recurrence. Many of these are associated with mutations in FANCB [McCauley et al., 2011], but others occur sporadically and lack evidence of DNA fragility [Evans and Chodirker, 1993]. In 2009, Pasquier et al. published a series of 40 fetuses with RES and linked a subset to VACTERL-H, a previously unrecognized association [2009].
Our series confirms the association of VACTERL features with RES. As the majority of our patients with VACTERL features also had hydrocephalus, we affirm the link between RES and VACTERL-H and propose the term VACTERL-R for these patients.
RES with HPE-AV
Two patients in our cohort had a striking brain malformation consisting of incompletely separated cerebral hemispheres with absent lateral and third ventricles. On the basis of undivided forebrain, this malformation can be categorized as HPE, but of a very atypical form. Most HPE demonstrates an anterior-to-posterior gradient in which the frontal lobes are most severely affected. A rare type of HPE known as the middle interhemispheric fusion (MIF) variant (or syntelencephaly) involves incomplete separation of the posterior frontal and anterior parietal lobes [Barkovich and Quint, 1993; Lewis et al., 2002]. In contrast, our patients have a posterior-predominant fusion that is maximal in the occipital lobes, suggesting that the mechanism of this malformation is distinct from that of classic anterior-predominant HPE. Ventricular anomalies can be seen in HPE, but complete aventriculy is not a feature of either the common anterior-predominant or the MIF form, underscoring the unique nature of the HPE seen in conjunction with RES.
Garfinkle described an 11-year-old girl with RES, posterior-predominant HPE and aventriculy [1996]. MRI images demonstrate a brain malformation remarkably similar to that of our two HPE-AV patients. Two subsequent patients with HPE-AV have been reported, 1 with anterior-predominant HPE and a Dandy-Walker malformation [Sener, 1998], the other with posterior-predominant HPE in whom the cerebellum appears dysplastic [Kumar et al., 2006]. Sergi reported on a 23-week fetus with RES, alobar HPE and essentially absent lateral and 3rd ventricles [1997]. The fetus also had a duplicated left thumb. Kakita described RES in a 20-week fetus with aprosencephaly (which can be considered an extreme form of HPE) [2001]. This fetus also had a cleft lip, malformed ears, rib fusion anomalies and a thoracic neural tube defect.
This novel malformation may be linked to maternal diabetes. Our two patients with HPE-AV were both born to mothers with pre-existing diabetes mellitus, as were the individuals described by Garfinkle, Sergi and Kakita. Diabetes is a well-recognized risk factor for HPE, presumably the more common anterior-predominant form. Diabetes has also been described in conjunction with the MIF variant of HPE [Robin et al., 1996]. Its role in HPE-AV needs further investigation.
Beyond GLH Syndrome
GLH syndrome was described as a clinical entity before the nature of the underlying brain malformation was recognized. With alopecia and “GLH-like” craniofacial features seen across our cohort of RES patients (including in patients with VACTERL features and HPE-AV), choosing which patients qualify for the label GLH syndrome becomes increasingly problematic. If the term GLH is used broadly to designate all patients with RES and alopecia, it lacks clinical specificity. If the term GLH is restricted to patients with RES, alopecia and trigeminal anesthesia, it becomes more clinically precise. However, the trigeminal nuclei and nerves originate from the dorsal hindbrain, so trigeminal anesthesia may simply comprise a related developmental malformation of this region. We therefore propose that alopecia, “GLH-like” craniofacial features and trigeminal anesthesia all be considered RES-associated anomalies rather than components of a named syndrome. This approach is comparable to current practice with the common form of HPE: the brain malformation itself and its associated craniofacial features are considered part of a continuous spectrum of related malformations rather than a syndrome.
VACTERL features and HPE-AV are sufficiently clinically distinctive to justify special mention, but patients in these subgroups have much in common with the rest of the cohort: several have alopecia, and their craniofacial features overlap with those seen in patients with alopecia alone. Moreover, the HPE seen in HPE-AV is posterior-predominant, suggesting that it may be a more extreme disruption of the same (unknown) mechanism that causes RES. Although both subgroups of patients have unique clinical features, whether they differ on a genetic basis has yet to be determined.
The constellation of malformations in RES suggests that the embryologic cells of origin respond as a unit that can be disrupted by various genetic or environmental causes, a concept sometimes referred to as a developmental field defect [Martinez-Frias et al., 1998]. Since these malformations are not spatially related to each other, they presumably result from disruption of a set of interacting genes that is deployed in multiple developing regions at different times during development [Jan and Jan, 1993; Opitz et al., 2002]. The overlapping phenotypic elements seen in our RES cohort implicate a biological pathway involved in development of the cerebellum, trigeminal ganglia, skull, face and focal areas of scalp. HPE-AV suggests that this, or a related pathway, also operates in the developing cerebral hemispheres and ventricular system. VACTERL features point to a function in somitogenesis, radial ray development, and the formation of multiple organ systems. More specifically, this shared biological mechanism may play a role in pattern formation, a term used in developmental biology to describe the process by which groups of embryonic cells develop complex, spatially-oriented forms in response to genetically programmed signaling centers. Though no genes have yet been linked conclusively to RES, a number of observations support a genetic basis (Table I), although diagnostic workup in our patients did not reveal any recurrent abnormalities (Supplementary eTable II – See Supporting Information online). Our data also suggest a role for environmental influences such as diabetes.
In many ways, RES resembles classic HPE, an archetypical developmental field defect. In HPE, various genetic and environmental influences can perturb the ventral midline, resulting in a variable spectrum of characteristic forebrain and craniofacial malformations. HPE has also been seen in conjunction with VACTERL malformations [Orioli and Castilla, 2010; Siebert et al., 2005]. In contrast to HPE, however, RES involves primarily dorsal rather than ventral structures. And unlike HPE, RES is not known to occur in animals, either spontaneously or as a result of genetic or environmental manipulation. We hypothesize that RES and classic HPE are clinically analogous conditions but involve different genetic and mechanistic pathways.
CONCLUSIONS
The phenotypic profile of RES is most notable for its areas of overlap. Accordingly, the majority of patients with RES should be viewed as having RES-associated anomalies rather than a particular syndrome. Two additional phenotypic themes were seen in subgroups of patients: RES with VACTERL features (14 patients) and HPE-AV (two patients). Yet even these patients have alopecia and craniofacial features that link them to the rest of the cohort. VACTERL features and HPE-AV in the context of RES are clinically distinctive, but whether they are genetically distinct remains to be seen.
The recurring phenotypic themes observed in our RES cohort imply a fundamental biological relationship between the structures involved, most likely based upon a shared developmental pathway. Ultimately, further delineation of the RES spectrum and its key phenotypic elements will be informed by a deeper understanding of the genetic and molecular mechanisms of RES itself.
Supplementary Material
Acknowledgments
The authors would like express their gratitude to patients and their families for participating in this study, and to the many clinicians who referred them. We would also like to thank Tessa Rue for her help with statistical analysis. This work was supported by the National Institute of Neurological Disorders and Stroke via 5T32NS051171-05 (to HMT), 2R01-NS050375 (to WBD, KJM) and KL2-RR025015 (to DD). PAS is supported by the Harold Amos Faculty Development Program through the Robert Wood Johnson Foundation and the CHLA-USC Child Heath Research Career Development Program (NIH K12-HD05954).
References
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394–396. doi: 10.1111/j.1525-1470.1996.tb00708.x. [DOI] [PubMed] [Google Scholar]
- Assouly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107–109. doi: 10.1159/000314691. [DOI] [PubMed] [Google Scholar]
- Aydingoz U, Cila A, Aktan G. Rhombencephalosynapsis associated with hand anomalies. Br J Radiol. 1997;70:764–766. doi: 10.1259/bjr.70.835.9245891. [DOI] [PubMed] [Google Scholar]
- Bargman H. Congenital triangular alopecia. J Am Acad Dermatol. 1988;18:390. doi: 10.1016/s0190-9622(88)80163-4. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Quint DJ. Middle interhemispheric fusion: an unusual variant of holoprosencephaly. Am J Neuroradiol. 1993;14:431–440. [PMC free article] [PubMed] [Google Scholar]
- Beemer FA, Wanders RJ, Schutgens RB. VACTERL and hydrocephalus. Am J Med Genet. 1990;37:425–426. doi: 10.1002/ajmg.1320370325. [DOI] [PubMed] [Google Scholar]
- Briard ML, le Merrer M, Plauchu H, Dodinval P, Lambotte C, Moraine C, Serville F. Association of VACTERL and hydrocephalus: a new familial entity. Annales de genetique. 1984;27:220–223. [PubMed] [Google Scholar]
- Brocks D, Irons M, Sadeghi-Najad A, McCauley R, Wheeler P. Gómez-López-Hernández syndrome: expansion of the phenotype. Am J Med Genet. 2000;94:405–408. doi: 10.1002/1096-8628(20001023)94:5<405::aid-ajmg12>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Chemli J, Abroug M, Tlili K, Harbi A. Rhombencephalosynapsis diagnosed in childhood: clinical and MRI findings. Eur J Paediatr Neurol. 2007;11:35–8. doi: 10.1016/j.ejpn.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Corsello G, Giuffre L. VACTERL with hydrocephalus: a further case with probable autosomal recessive inheritance. Am J Med Genet. 1994;49:137–138. doi: 10.1002/ajmg.1320490133. [DOI] [PubMed] [Google Scholar]
- de Jong G, Kirby PA. Defects of blastogenesis: counseling dilemmas in two families. Am J Med Genet. 2000;91:175–179. [PubMed] [Google Scholar]
- di Vera E, Liberati M, Celentano C, Calabrese G, Guanciali-Franchi PE, Morizio E, Rotmensch S. Rhombencephalosynapsis in a severely polymalformed fetus with non-mosaic tetrasomy 9p, in intracytoplasmic-sperm-injection pregnancy. J Assist Reprod Genet. 2008;25:577–580. doi: 10.1007/s10815-008-9257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer KB, George RM. Congenital triangular alopecia: a case report and review. Cutis. 2002;69:255–256. [PubMed] [Google Scholar]
- Evans JA, Chodirker BN. Absence of excess chromosome breakage in a patient with VACTERL-hydrocephalus. Am J Med Genet. 1993;47:112–113. doi: 10.1002/ajmg.1320470123. [DOI] [PubMed] [Google Scholar]
- Evans JA, Stranc LC, Kaplan P, Hunter AG. VACTERL with hydrocephalus: further delineation of the syndrome(s) Am J Med Genet. 1989;34:177–182. doi: 10.1002/ajmg.1320340209. [DOI] [PubMed] [Google Scholar]
- Feuerman EJ. Congenital temporal triangular alopecia. Pediatr Dermatol. 1981;12:301–303. [PubMed] [Google Scholar]
- Garcia-Hernández MJ, Rodriguez-Pichardo A, Camacho F. Congenital triangular alopecia (Brauer nevus) Pediatric dermatology. 1995;12(4):301–303. doi: 10.1111/j.1525-1470.1995.tb00187.x. [DOI] [PubMed] [Google Scholar]
- Garfinkle WB. Aventriculy: a new entity? Am J Neuroradiol. 1996;17:1649–1650. [PMC free article] [PubMed] [Google Scholar]
- Gómez MR. Cerebellotrigeminal and focal dermal dysplasia: a newly recognized neurocutaneous syndrome. Brain Dev. 1979;1:253–256. doi: 10.1016/s0387-7604(79)80039-x. [DOI] [PubMed] [Google Scholar]
- Gomy I, Heck B, Santos AC, Figueiredo MS, Martinelli CE, Jr, Nogueira MP, Pina-Neto JM. Two new Brazilian patients with Gómez-López-Hernández syndrome: reviewing the expanded phenotype with molecular insights. Am J Med Genet Part A. 2008;146A:649–657. doi: 10.1002/ajmg.a.32173. [DOI] [PubMed] [Google Scholar]
- Guleria S. ZIC2 mutations are seen in holoprosencephaly and not partial rhombencephalosynapsis. Am J Med Genet Part A. 2011;155A:2901. doi: 10.1002/ajmg.a.34282. [DOI] [PubMed] [Google Scholar]
- Iafolla AK, McConkie-Rosell A, Chen YT. VATER and hydrocephalus: distinct syndrome? Am J Med Genet. 1991;38:46–51. doi: 10.1002/ajmg.1320380112. [DOI] [PubMed] [Google Scholar]
- Ishak GE, Dempsey JC, Shaw DW, Tully H, Adam MP, Sanchez-Lara PA, Glass I, Rue TC, Millen KJ, Dobyns WB, Doherty D. Rhombencephalosynapsis: a hindbrain malformation associated with incomplete separation of midbrain and forebrain, hydrocephalus and a broad spectrum of severity. Brain. 2012;2012 doi: 10.1093/brain/aws065. [epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Functional gene cassettes in development. Proc Natl Acad Sci USA. 1993;90:8305–7. doi: 10.1073/pnas.90.18.8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakita A, Hayashi S, Arakawa M, Takahashi H. Aprosencephaly: histopathological features of the rudimentary forebrain and retina. Acta Neuropathol. 2001;102:110–6. doi: 10.1007/s004010000352. [DOI] [PubMed] [Google Scholar]
- Kubba R, Rook A. Congenital triangular alopecia. Br J Dermatol. 1976;95:657–9. doi: 10.1111/j.1365-2133.1976.tb07042.x. [DOI] [PubMed] [Google Scholar]
- Kumar S, Jaiswal AK, Rastogi M. Aventriculi associated with holoprosencephaly. J Clin Neurosci. 2006;13:378–80. doi: 10.1016/j.jocn.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Lespinasse J, Testard H, Nugues F, Till M, Cordier MP, Althuser M, Amblard F, Fert-Ferrer S, Durand C, Dalmon F, Pourcel C, Jouk PS. A submicroscopic unbalanced subtelomeric translocation t(2p;10q) identified by fluorescence in situ hybridization: fetus with increased nuchal translucency and normal standard karyotype with later growth and developmental delay, rhombencephalosynapsis (RES) Ann Genet. 2004;47:405–17. doi: 10.1016/j.anngen.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Lewis AJ, Simon EM, Barkovich AJ, Clegg NJ, Delgado MR, Levey E, Hahn JS. Middle interhemispheric variant of holoprosencephaly: a distinct cliniconeuroradiologic subtype. Neurology. 2002;59:1860–5. doi: 10.1212/01.wnl.0000037483.31989.b9. [DOI] [PubMed] [Google Scholar]
- López-Hernández A. Craniosynostosis, ataxia, trigeminal anaesthesia and parietal alopecia with pons-vermis fusion anomaly (atresia of the fourth ventricle). Report of two cases. Neuropediatrics. 1982;13:99–102. doi: 10.1055/s-2008-1059606. [DOI] [PubMed] [Google Scholar]
- Martinez-Frias ML, Frias JL, Opitz JM. Errors of morphogenesis and developmental field theory. Am J Med Genet. 1998;76:291–6. [PubMed] [Google Scholar]
- McCauley J, Masand N, McGowan R, Rajagopalan S, Hunter A, Michaud JL, Gibson K, Robertson J, Vaz F, Abbs S, Holden ST. X-linked VACTERL with hydrocephalus syndrome: further delineation of the phenotype caused by FANCB mutations. Am J Med Genet Part A. 2011;155A:2370–80. doi: 10.1002/ajmg.a.33913. [DOI] [PubMed] [Google Scholar]
- Munoz RM, Santos AC, Graziadio C, Pina-Neto JM. Cerebello-trigeminal-dermal dysplasia (Gómez-López-Hernández syndrome): description of three new cases and review. Am J Med Genet. 1997;72:34–9. doi: 10.1002/(sici)1096-8628(19971003)72:1<34::aid-ajmg7>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Opitz JM, Zanni G, Reynolds JF, Jr, Gilbert-Barness E. Defects of blastogenesis. Am J Med Genet. 2002;115:269–86. doi: 10.1002/ajmg.10983. [DOI] [PubMed] [Google Scholar]
- Orioli IM, Castilla EE. Epidemiology of holoprosencephaly: Prevalence and risk factors. Am J Med Genet Part C. 2010;154C:13–21. doi: 10.1002/ajmg.c.30233. [DOI] [PubMed] [Google Scholar]
- Pasquier L, Marcorelles P, Loget P, Pelluard F, Carles D, Perez MJ, Bendavid C, de La Rochebrochard C, Ferry M, David V, Odent S, Laquerriere A. Rhombencephalosynapsis and related anomalies: a neuropathological study of 40 fetal cases. Acta Neuropathol. 2009a;117:185–200. doi: 10.1007/s00401-008-0469-9. [DOI] [PubMed] [Google Scholar]
- Purvis DJ, Ramirez A, Roberts N, Harper JI. Gómez-López-Hernández syndrome: another consideration in focal congenital alopecia. Br J Dermatol. 2007;157:196–8. doi: 10.1111/j.1365-2133.2007.07952.x. [DOI] [PubMed] [Google Scholar]
- Ramocki MB, Scaglia F, Stankiewicz P, Belmont JW, Jones JY, Clark GD. Recurrent partial rhombencephalosynapsis and holoprosencephaly in siblings with a mutation of ZIC2. Am J Med Genet Part A. 2011;155A:1574–80. doi: 10.1002/ajmg.a.34029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin NH, Ko LM, Heeger S, Muise KL, Judge N, Bangert BA. Syntelencephaly in an infant of a diabetic mother. Am J Med Genet. 1996;66:433–7. doi: 10.1002/(SICI)1096-8628(19961230)66:4<433::AID-AJMG9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Romanengo M, Tortori-Donati P, Di Rocco M. Rhombencephalosynapsis with facial anomalies and probable autosomal recessive inheritance: a case report. Clin Genet. 1997;52:184–6. doi: 10.1111/j.1399-0004.1997.tb02542.x. [DOI] [PubMed] [Google Scholar]
- Ruggieri M, Rizzo R, Pavone P, Baieli S, Sorge G, Happle R. Temporal triangular alopecia in association with mental retardation and epilepsy in a mother and daughter. Arch Dermatol. 2000;136:426–7. doi: 10.1001/archderm.136.3.426. [DOI] [PubMed] [Google Scholar]
- Sandalcioglu IE, Gasser T, van de Nes JA, Menken U, Stolke D, Wiedemayer H. Fusion of the cerebellar hemispheres ventral to the brainstem: a rare hindbrain-related malformation. Childs Nerv Syst. 2006;22:73–7. doi: 10.1007/s00381-004-1065-5. [DOI] [PubMed] [Google Scholar]
- Sarnat HB. Molecular genetic classification of central nervous system malformations. J Child Neurol. 2000;15:675–87. doi: 10.1177/088307380001501007. [DOI] [PubMed] [Google Scholar]
- Sener RN. Aventriculi associated with holoprosencephaly. Comput Med Imaging Graph. 1998;22:345–7. doi: 10.1016/s0895-6111(98)00029-9. [DOI] [PubMed] [Google Scholar]
- Sergi C, Hentze S, Sohn C, Voigtlander T, Jung C, Schmitt HP. Telencephalosynapsis (synencephaly) and rhombencephalosynapsis with posterior fossa ventriculocele (‘Dandy-Walker cyst’): an unusual aberrant syngenetic complex. Brain Dev. 1997;19:426–32. doi: 10.1016/s0387-7604(97)00050-8. [DOI] [PubMed] [Google Scholar]
- Siebert JR, Schoenecker KA, Resta RG, Kapur RP. Holoprosencephaly and limb reduction defects: a consideration of Steinfeld syndrome and related conditions. Am J Med Genet Part A. 2005;134A:381–92. doi: 10.1002/ajmg.a.30648. [DOI] [PubMed] [Google Scholar]
- Silva CY, Lenzy YM, Goldberg LJ. Temporal triangular alopecia with decreased follicular density. Journal Cutan Pathol. 2010;37:597–9. doi: 10.1111/j.1600-0560.2009.01388.x. [DOI] [PubMed] [Google Scholar]
- Sukhudyan B, Jaladyan V, Melikyan G, Schlump JU, Boltshauser E, Poretti A. Gómez-López-Hernández syndrome: reappraisal of the diagnostic criteria. Eur J Pediatr. 2010a;169:1523–8. doi: 10.1007/s00431-010-1259-7. [DOI] [PubMed] [Google Scholar]
- Toelle SP, Yalcinkaya C, Kocer N, Deonna T, Overweg-Plandsoen WC, Bast T, Kalmanchey R, Barsi P, Schneider JF, Capone Mori A, Boltshauser E. Rhombencephalosynapsis: clinical findings and neuroimaging in 9 children. Neuropediatrics. 2002;33:209–14. doi: 10.1055/s-2002-34498. [DOI] [PubMed] [Google Scholar]
- Tosti A. Congenital triangular alopecia. Report of fourteen cases. J Am Acad Dermatol. 1987;16:991–3. doi: 10.1016/s0190-9622(87)70127-3. [DOI] [PubMed] [Google Scholar]
- Trakimas C, Sperling LC, Skelton HG, 3rd, Smith KJ, Buker JL. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31(2 Pt 1):205–9. doi: 10.1016/s0190-9622(94)70147-4. [DOI] [PubMed] [Google Scholar]
- Truwit CL, Barkovich AJ, Shanahan R, Maroldo TV. MR imaging of rhombencephalosynapsis: report of three cases and review of the literature. Am J Neuroradiol. 1991;12:957–65. [PMC free article] [PubMed] [Google Scholar]
- Yachnis AT. Rhombencephalosynapsis with massive hydrocephalus: case report and pathogenic considerations. Acta Neuropathol. 2002;103:301–304. doi: 10.1007/s004010100454. [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360–362. doi: 10.1111/j.1346-8138.2010.00817.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.