

Abstract
Heat shock proteins (HSPs), produced in response to stress are suppressive in disease models. We previously showed that Mycobacterium leprae HSP65 prevented development of airway hyperresponsiveness and inflammation in mice. Our goal here was to define the mechanism responsible for the suppressive effects of HSP. In one in vivo approach, BALB/c mice were sensitized to ovalbumin (OVA) followed by primary OVA challenges. Several weeks later, HSP65 was administered prior to a single, provocative secondary challenge. In a second in vivo approach, the secondary challenge was replaced by intratracheal instillation of allergen-pulsed bone marrow-derived dendritic cells (BMDCs). The in vitro effects of HSP65 on BMDCs were examined in co-culture experiments with CD4+ T cells. In vivo, HSP65 prevented development of airway hyperresponsiveness and inflammation. As well, Th1 cytokine levels in bronchoalveolar lavage (BAL) fluid were increased. In vitro, HSP65 induced notch receptor ligand Delta1 expression on BMDCs and HSP65-treated BMDCs skewed CD4+ T cells to Th1 cytokine production. Thus, HSP65-induced effects on allergen-induced airway hyperresponsiveness and inflammation were associated with increased Delta 1 expression on DCs, modulation of DC function, and CD4+ Th1 cytokine production.
Keywords: HSP65, asthma, dendritic cells, T cells
INTRODUCTION
Asthma is the most common chronic airway disease in industrial countries and despite advances in disease management, the socio-economic burden caused by disease exacerbations or steroid refractory asthma remains high (1). The introduction of new therapeutic modalities in asthma has been limited by an absence of benefits in major proportions of asthmatics, highlighting the need for better targeting of relevant pathophysiologic pathways.
Asthma is a chronic airway disease characterized by persistent airway hyperresponsiveness and airway inflammation as a result of cellular and molecular responses (2). As Th2-type CD4+ T cells have been shown to be a dominant cell type in the airways of asthmatics, allergic asthma has been viewed as an imbalance between Th1 and Th2 cells (3), although this notion is not universally accepted (4). Antigen presenting cells (APCs), dendritic cells (DCs) in the lung, govern the direction of T lymphocyte differentiation and cytokine responses through Notch signaling pathways (5, 6). In mammals, four Notch receptors (Notch1-4) have been found on T lymphocytes, and two ligand families, Delta-like family (Delta1, Delta 3, and Delta 4) and the Jagged family (Jagged1 and Jagged2) have been identified on the surface of APCs (5, 6). In experimental models of asthma, the ligation of Notch receptor on CD4+ T cells by Jagged1 expressed on DCs augmented IL-4 production and resulted in the development of airway hyperresponsiveness and airway inflammation. In contrast, Delta-like ligands have been associated with suppression of lung allergic responses (7–9).
Heat shock proteins (HSPs) are a highly conserved group of functional proteins produced by prokaryotic and eukaryotic cells in response to a variety of stressors including inflammation. Their primary function is folding and unfolding of protein substrates (10–13). HSPs also recognize cellular abnormalities and deliver them to APCs to generate peptide-specific T lymphocyte responses by binding pathogen-associated molecular pattern (PAMP) receptors or by modulating PAMP-induced stimulation (14). HSPs are classified on the basis of their monomeric molecular weights (15). HSP60 and HSP70 have been shown to be involved in binding and presenting antigens to the immune system (16, 17). HSPs have been shown to modulate the function of APCs, including DCs (18, 19).
In a mouse model of experimental asthma, we previously showed that HSP65 derived from Mycobacterium leprae prevented the development of allergic inflammation and airway hyperresponsiveness (20). Thus, in the context of asthma, HSPs may modulate DC function and skew the lymphocyte response from a Th2-type to one where Th1 cytokines predominate.
Based on these descriptions, we hypothesized that HSP65 may prevent allergen-induced airway inflammation and airway hyperresponsiveness, even in established disease, by modulating DC regulation of Th differentiation through Notch ligand expression. In the present study, we defined the efficacy of HSP65 derived from Mycobacterium leprae in a secondary allergen challenge model, linking attenuation of lung allergic responses to alterations in Notch ligand expression on DCs.
MATERIALS AND METHODS
Animals
Female BALB/c mice, 6–8 wks of age and free of pathogens were purchased from Harlan Laboratory (Indianapolis, ID). All mice were housed under specific pathogen and ovalbumin (OVA)-free conditions and maintained on a 12 hour light-dark cycle with food and water ad libitum. All experimental animals used in this study were under a protocol approved by the Institutional Animal Care and Use Committee of National Jewish Health.
Sensitization and challenge to allergen
The experimental protocol for sensitization and challenge to allergen was modified from previously described procedures (21). Briefly, BALB/c mice were sensitized intraperitoneally with 10 µg of OVA (BP2535-5, Fisher Scientific, Pittsburgh, PA) emulsified in 1 mg of alum (Imject Alum; Pierce, Rockford, IL) or sham-sensitized with saline on days 0 and 14. Mice received airway primary allergen challenges by exposure to OVA aerosols (0.2% in saline) for 20 minutes on days 28, 29, and 30 with an ultrasonic nebulizer (NE-U07, OMRON, Kyoto, Japan). This was followed by a single, secondary, or provocative allergen challenge (1% OVA in saline for 20 minutes) 2 weeks after the last primary allergen challenge. Forty-eight hours after the last allergen challenge, airway responsiveness was measured followed by collection of samples.
Preparation and treatment with HSP65
The recombinant Mycobacterium leprae HSP65 (LIONEX Diagnostics and Therapeutics, Braunschweig, Germany) was purified from E. coli to more than 98% purity as confirmed by SDS electrophoresis, immunoblotting, and N-terminal sequencing and was without flagellin contamination. Endotoxin was removed by endotoxin removing gel (Thermo Scientific, Rockford, IL); after removal, the endotoxin levels were <0.2 EU/mg of HSP protein as detected using the ToxinSensorTM Chromogenic LAL Endotoxin Assay Kit (GenScript, Piscataway, NJ).
In in vivo experiments, purified HSP65 (100 µg) was administered by intraperitoneal injection 2 hours prior to secondary OVA challenge. For in vitro experiments, 1 or 10 µg/ml of HSP65 was added to the cultures of BMDCs for 24 hours.
Preparation of BMDCs and protocol for transfer of OVA-pulsed BMDCs
BMDCs were generated from bone marrow of naive BALB/c mice as described previously (22). Briefly, bone marrow cells were obtained from femurs and iliac bones of mice and placed in DC culture medium (RPMI 1640 containing 10% heat-inactivated FCS, 50 µM 2-mercaptoethanol, 2 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin (GIBCO, Carlsbad, CA), 10 ng/ml recombinant mouse GM-CSF, and 10 ng/ml recombinant mouse IL-4 (R&D Systems, Minneapolis, MN). On day 8, non-adherent cells were recovered. These cells were >95% CD11c+. BMDCs were pulsed with OVA (200 µg/ml) in the presence or absence of HSP65 for 24 hours and washed three times with PBS. As controls, OVA-pulsed BMDCs were cultured with PBS instead of HSP65 or were not pulsed with OVA. BMDCs (2×106 cells) were administered intratracheally into sensitized and primary allergen challenged mice in lieu of secondary allergen challenge. To determine the direct effects of HSP65 on BMDC function, the cells were cultured with HSP65 (0, 1, or 10 µg/ml) or LPS (1 µg/ml) as a positive control for 24 hours followed by assays of cells or culture supernates.
CD4+ T cell preparation and co-culture with BMDCs and HSP65
CD4+ T cells were isolated as previously described (23). Spleen cells from sensitized and secondary allergen challenged mice were harvested by mincing the tissues and passing them through a stainless steel sieve. After washing with PBS, mononuclear cells (MNCs) were isolated by Histopaque gradient centrifugation (Sigma-Aldrich). Purification of CD4+ T cells was conducted by negative selection using a mouse CD4+ T cell recovery column kit (Cedarlane Laboratories, Burlington, NC) in accordance with the manufacturer’s instructions. Purity of CD4+ T cell populations after purification exceeded 95% as assessed by flow cytometry.
BMDCs were pretreated with HSP65 (1 or 10 µg/ml) or vehicle alone for 24 hours, and then co-cultured with isolated CD4+ cells in the presence of OVA (200 µg/ml) at a ratio of 1:10 for 24 hours. Supernates were collected and evaluated by ELISA.
Assessment of airway responsiveness, bronchoalveolar lavage (BAL) fluid, and lung histology
Airway responsiveness was assessed as previously described by measuring changes in airway resistance in response to increasing doses of inhaled methacholine (MCh, Sigma-Aldrich, St. Louis, MO) in anesthetized and ventilated mice (21). Data are expressed as the percent change from baseline lung resistance (RL) values obtained after inhalation of saline. Immediately after measurement of airway hyperresponsiveness, lungs were lavaged via the tracheal tube as described (21). Numbers of total leukocytes in BAL fluid were determined and cell differentiation was performed on cytospin slides prepared with Wright-Giemsa stain. After BAL was obtained, the lungs were fixed in 10% formalin, embedded in paraffin, and cut into 5-µm sections. The number of inflammatory and mucus-containing cells were quantitated as previously described with some modification (24). Tissue sections were evaluated using the NIH ImageJ (Version 1.45) available at http://rsbweb.nih.gov/ij/download.html. For detection of inflammatory cells, sections were stained with hematoxylin and eosin (H&E), and the number of inflammatory cells per micrometer square of perivascular and peribronchial areas was determined. In addition, mucus-containing cells stained with periodic acid-Schiff (PAS) were quantitated and expressed as PAS-positive areas per micrometer of basement membrane.
Preparation of RNA and real-time PCR
Total RNA was extracted from HSP65 or LPS-treated BMDCs using a total RNA isolation kit (Macherey-Nagel, Bethlehem, PA). One microgram of total RNA was used in each reaction primed with oligo-dT to obtain cDNA. Then, 3 µl of cDNA was used as the template for real-time PCR (9). Real-time cDNA primers for Delta1 and Jagged1 were obtained from Taqman gene expression assay (Applied Biosystems, Carlsbad, CA). The real-time PCRs were performed on an ABI 7700 sequence detection system (Applied Biosystems, Carlsbad, CA) with cycling parameters of 50°C for 2 minutes, 95°C for 10 minutes, and 40 repeats at 95°C for 15 seconds and 60°C for 1 minute. The cycle threshold method was performed for relative quantification of mRNA expression (7).
Measurement of cytokines
Cytokine levels in the BAL fluid and cell culture supernatants were measured by ELISA as previously described (24). IL-5, IL-10, IL-12, IL-13, and IFN-γ cytokine ELISAs were performed according to the manufacturers’ instructions (eBioscience, San Diego, CA). The lower limits of detection were 4 pg/ml for IL-5 and IL-13, 10 pg/ml for IL-10, IFN-γ, and TNF-±, and 15 pg/ml for IL-12.
Statistical analysis
Results were expressed as the mean±SEM. The t test was used to determine differences between two groups. For comparisons between multiple groups, the Tukey-Kramer test was used. Nonparametric analysis using the Mann-Whitney U test or Kruskal-Wallis test was also used to confirm that the statistical differences remained significant even if the underlying distribution was uncertain. The p values for significance were set to 0.05 for all tests.
RESULTS
HSP65 Treatment Inhibits the Development of Airway Hyperresponsiveness and Airway Inflammation Following Secondary Allergen Challenge
HSP65 or PBS were administered by intraperitoneal injection just prior to secondary allergen challenge of sensitized and primary allergen challenged mice. As shown in Figures 1A and 1B, PBS-treated mice developed increases in RL to inhaled MCh and eosinophil numbers in BAL fluid. Mice treated with HSP65 developed significantly lower airway responsiveness and BAL eosinophilia, accompanied by significantly increased levels of IL-10, IL-12, and IFN-γ (Fig. 1C), and decreased levels of IL-4, IL-5, and IL-13 in BAL fluid compared to vehicle-treated mice.
Figure 1. Effect of systemic HSP65 administration on airway responses in a secondary allergen challenge model.
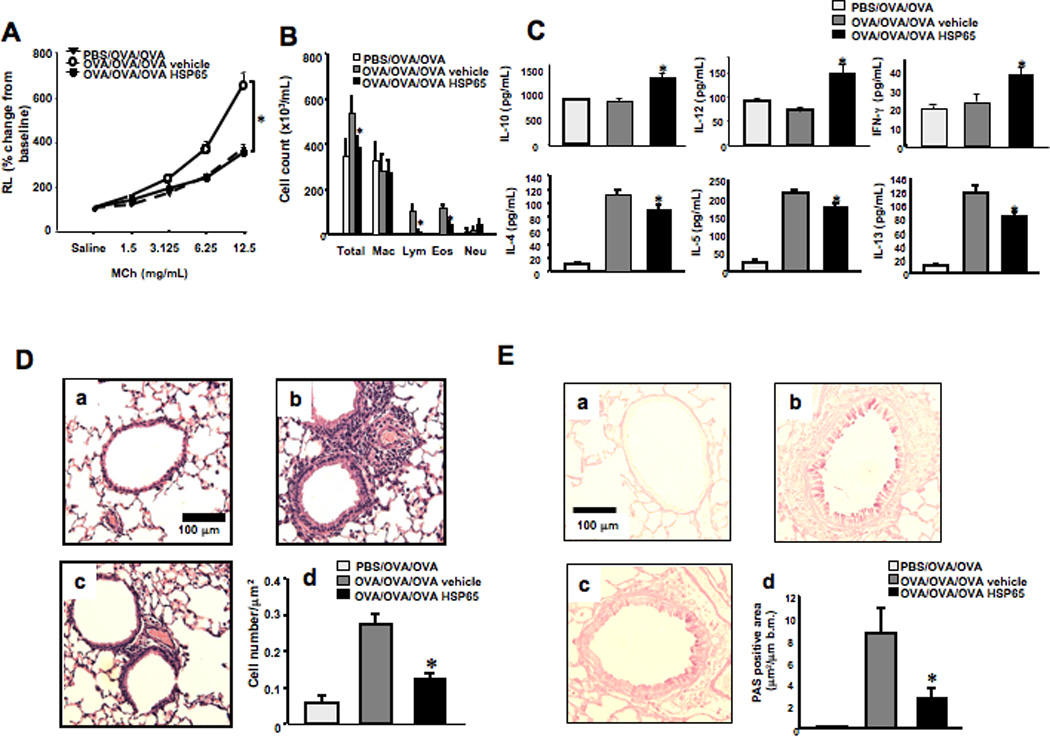
The effects of intraperitoneal injection of HSP65 were determined in a secondary allergen challenge model. Control groups received vehicle. (A) Lung resistance changes in response to increasing doses of MCh, expressed as a percent of baseline (saline) values. (B) Cell composition in BAL fluid. Macro; macrophages, Lympho; lymphocytes, Eos; eosinophils, Neu; neutrophils. (C) BAL fluid cytokine levels. (D) Lung tissue histology (H&E) and quantitation of inflammatory cells. (E) Periodic acid-Schiff (PAS) staining and quantitation of goblet cells. Panel a: Sham sensitization and primary and secondary OVA challenge, Panel b: secondary challenge following vehicle treatment, and panel c: secondary challenge following HSP65 treatment. Mice were sham sensitized followed by primary and secondary OVA challenge (PBS/OVA/OVA) or OVA sensitized followed by primary and secondary OVA challenge (OVA/OVA/OVA). (n=8). *p<0.05 compared to OVA/OVA/OVA vehicle.
Histopathological analysis of lung tissue sections stained with H/E stain revealed that the number of inflammatory cells including eosinophils in the peribronchial and perivascular areas were increased in vehicle-treated mice after secondary allergen challenge (Fig. 1D, panel b) compared to sham sensitized and challenged mice (panel a); these cell numbers were significantly decreased in HSP65-treated mice (panel c). Similar to the changes in inflammatory cell numbers, goblet cell metaplasia developed in vehicle-treated mice following secondary allergen challenge and HSP65 treatment significantly reduced the number of goblet cells (Fig. 1E).
HSP65-Treated BMDCs Alter Th Responses by Increasing Delta 1 Expression
In view of the increases in levels of IL-10, IL-12, and IFN-γ in BAL fluid following HSP65 treatment, we examined the effect of HSP65 on BMDC function. BMDCs were cultured in the presence (or absence) of HSP65 (1 or 10 µg/mL) or LPS (1 µg/mL), a potent stimulus of DC cytokine production (25), as a positive control. Addition of 1 or 10 µg/ml HSP65 triggered significant increases in DC production of TNFα, almost to the levels induced by LPS (Fig. 2A). Similarly, production of IL-12 and IL-10 was also increased following addition of HSP.
Figure 2. Cytokine release and Notch ligand expression on BMDCs after treatment with LPS or HSP65.
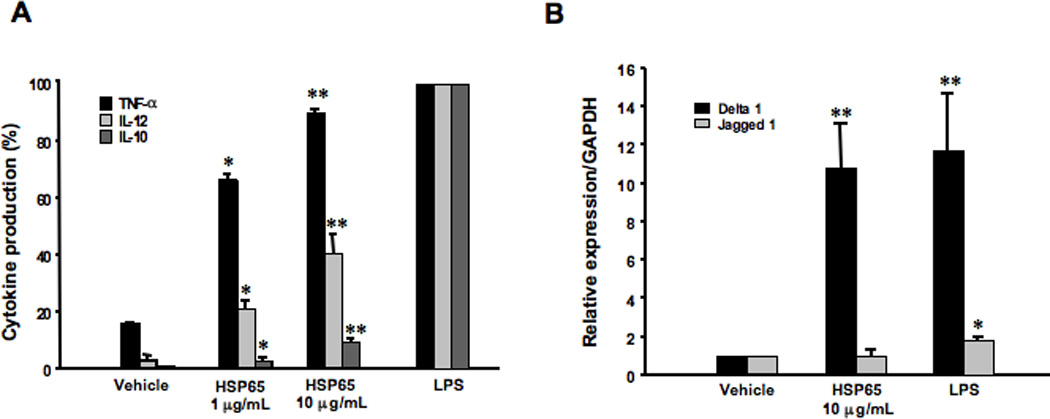
BMDCs from naive mice were incubated with LPS (1 µg/ml), HSP65 (1 or 10 µg/ml), or PBS as a vehicle for 24 hours. Culture supernates were recovered to measure cytokine levels and mRNA was isolated from cells. (A) Cytokine levels. To compensate for the variability in cytokine release by cultured BMDCs from experiment to experiment, the results of cytokine assays were expressed as a percent of the cytokine levels detected after LPS stimulation. (B) Notch ligand expression. The relative expression levels of Notch ligands (Delta1 or Jagged1) were determined by quantitative real-time PCR. cDNA contents were normalized to levels of GAPDH. Results are from three independent experiments and the results for each group are expressed as means±SEM. *p<0.05 compared to vehicle-treated BMDCs.
Because expression of the Notch ligands such as Delta1 and Jagged1 on BMDCs has been associated with the differentiation fate of CD4+ T helper cells (7–9), we analyzed their expression in BMDCs cultured with HSP65 by real-time PCR. Expression levels of Delta1 were significantly higher in BMDCs cultured with HSP65 compared to BMDCs cultured with vehicle, similar to the levels detected after culture with LPS (Fig. 2B). This contrasted with levels of expression of Jagged 1, where little induction was seen following culture with HSP65 or LPS.
HSP65 Treatment of BMDCs Modifies Cytokine Production from CD4+ T Cells
As CD4+ T cells are potent effector cells in the development of allergic inflammation, their function was examined after co-culture with BMDCs in the presence of allergen and HSP65. CD4+ T cells isolated from secondary allergen challenged mice were incubated with OVA and BMDCs in the presence (or absence) of HSP65 for 24 hours. Following co-culture with HSP65-treated BMDCs and OVA, CD4+ T cells produced significantly lower levels of IL-5 and IL-13, and higher levels of IFN-γ compared to CD4+ T cells co-cultured with vehicle-treated BMDCs and OVA (Fig. 3).
Figure 3. Cytokine profile following culture of CD4+ T cells with HSP65-treated BMDCs.
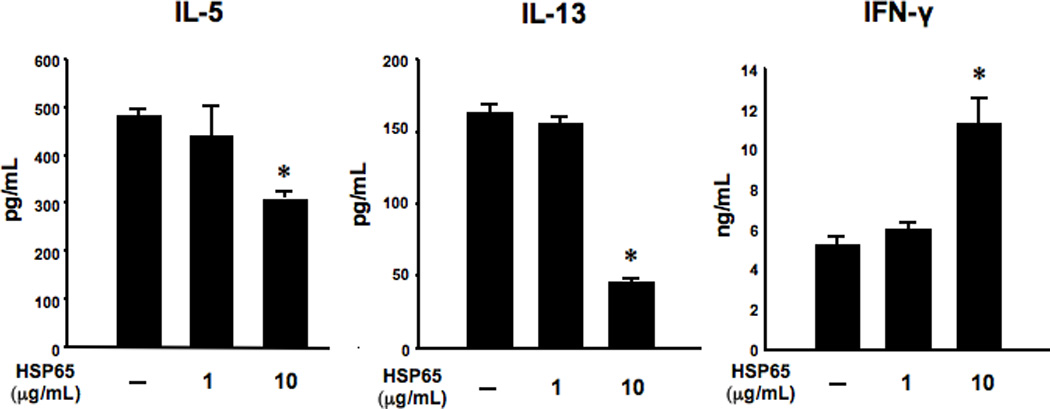
Isolated CD4+ T cells from secondary OVA challenged mice were incubated with OVA and BMDCs pretreated with HSP65 (0, 1, or 10 µg/ml) for 24 hours. Supernates were collected and evaluated by ELISA. The results are representative of three independent experiments and are expressed as means±SEM. *p<0.05 compared to vehicle-treated BMDCs.
HSP65 Treatment of BMDCs Results in Significantly Lower Airway Hyperresponsiveness and Inflammation Following Transfer into Previously Sensitized and Challenged Recipients
To determine the consequences of HSP65 treatment on BMDC function in an in vivo model, antigen-pulsed BMDCs were administered intratracheally to previously sensitized and challenged recipients as a substitute for secondary allergen challenge. As shown in Figures 4A and 4B, recipients of OVA-pulsed, vehicle-treated BMDCs developed increased airway resistance to MCh and higher eosinophil numbers in BAL fluid. In contrast, recipients of OVA-pulsed, HSP65-treated BMDCs failed to develop airway hyperresponsiveness and BAL eosinophilia. Cytokine levels in BAL fluid did not differ between groups (data not shown), similar to previously reported findings in this short-term DC transfer model (26).
Figure 4. Effect of HSP65 on OVA-pulsed BMDC function following transfer into previously sensitized and challenged mice.
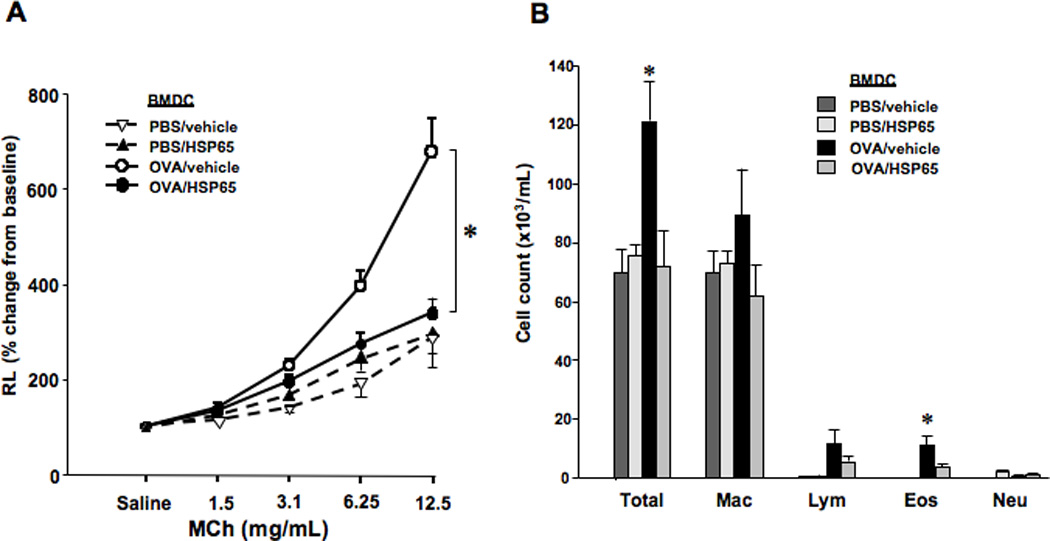
The in vivo effects of HSP65 on OVA-pulsed BMDCs were determined. All groups of mice were sensitized to OVA followed by primary OVA challenge. Two weeks after primary OVA challenge, mice received (intratracheal instillation) BMDCs pretreated with HSP65 and pulsed with OVA (OVA/HSP65) or PBS as control (PBS/HSP65), or pretreated with vehicle and pulsed with OVA (OVA/vehicle) or pulsed with PBS (PBS/vehicle). (A) Changes in airway resistance (RL). (B) Cell composition in BAL fluid. (n=8). * p<0.05 compared to OVA/vehicle.
DISCUSSION
In recent decades there has been an increase in the prevalence of allergic diseases in developed countries (27, 28). One factor proposed to explain this rise in allergic diseases is the decline in the incidence of many infectious diseases in developed countries as the result of improved living standards and vaccination (29). Although it is possible that increased microbial exposure could lead to a decline in allergic diseases such as asthma (30), the mechanisms are not defined. To address this issue, we hypothesized that treatment with Mycobacterium-derived HSP may inhibit the development of allergen-induced airway inflammation and airway hyperresponsiveness in a secondary allergen challenge model through modulation of DC function. Administration of HSP65 inhibited the development of airway hyperresponsiveness and inflammation when administered prior to secondary challenge of previously sensitized and challenged mice. In addition, transfer of HSP65-treated BMDCs into previously sensitized and challenged mice as a substitute for secondary allergen challenge failed to trigger airway inflammation and airway hyperresponsiveness. Upregulation of the notch ligand Delta1 was seen following HSP65 incubation with BMDCs and in co-culture experiments with CD4+ T cells, levels of IFN-γ were increased while levels of IL-5 and IL-13 were decreased relative to cultures of CD4+ T cells with untreated BMDCs. Together, these findings demonstrated that DCs are an important target of the HSP65 suppressive effects mediated, at least in part, through increased Delta1 expression on DCs and Th1 skewing of CD4+ T cell responses.
HSPs are molecular chaperones whose predominant function is the folding and unfolding of protein substrates in response to stressors such as in an infectious disease (12, 14). Elevated HSP levels are associated with many infectious diseases (31–33). Recently, upregulation of HSP was demonstrated in patients with bronchial asthma (34, 35). In an earlier study designed to link asthma suppression and HSPs (20), we used HSP65 derived from Mycobacterium leprae to define a mechanism for the observed reductions in allergen-induced airway hyperresponsiveness and airway inflammation. In the present study, we utilized a secondary allergen challenge model in order to demonstrate the efficacy of HSP65 on a background of pre-existing disease. As demonstrated here, HSP65 administration prior to a single provocative (secondary) allergen challenge to previously sensitized and challenged mice significantly reduced airway hyperresponsiveness and airway inflammation. These outcomes of HSP65 treatment were associated with significant increases in BAL cytokine levels of IL-10, IL-12, and IFN-γ as well as decreased levels of IL-4, IL-5, or IL-13 levels. It has been reported that Mycobacterium tuberculosis chaperones stimulate IL-10 and IL-12 production (36), HSP70L1 induced the production of IL-12p70 (37), and mycobacterial HSP70 treatment increased IL-10 levels in an arthritis model (38). Although these particular HSPs have differences in structure and cellular localization compared to HSP65, they share the common feature of stimulating APCs. As examples, HSP60 strongly stimulated DC maturation while HSP70L1 activated DCs (25, 37). The results in each case were increases in production of IL-10 and IL-12. IL-12 is thought to be a major APC-derived factor promoting Th1 differentiation (39) while IL-10 activity, although complex, is associated with anti-inflammatory activities (40).
Notch ligand-Notch receptor interactions also play a major role in directing APC function and in turn, the differentiation profile of Th cells. The Notch signaling pathway is a highly conserved program for cell fate decisions such as apoptosis, cell cycle arrest, and cellular polarization in all organisms (6, 41–43). Mammals express five genes that encode ligands for Notch receptors from two conserved families, Jagged (Jagged1 and Jagged2) and Delta-like (Delta1, Delta3, and Delta4) (6), and both types of ligands appear to transduce similar signaling pathways through Notch receptors (44). These ligands promote differentiation of naive CD4+ T cells into distinct effector cells; Delta promotes Th1 responses (45), and Jagged induces naive CD4+ T cells to differentiate into Th2 lineage (46). In experimental models of asthma, we showed that the Delta1-like ligand inhibited development of allergen-induced changes in lung function and inflammation while the Jagged 1 ligand enhanced responsiveness (7, 8). In view of these findings and the associated cytokine changes, we examined Notch ligand expression on BMDCs, and found that Delta1 expression on HSP65-treated BMDCs was significantly elevated with little change in Jagged1 expression. These data supported a functional link between HSP65, Notch ligand expression and production of the allergy-suppressive cytokines IL-10, IL-12, and IFN-γ, with some reduction in levels of the allergy-promoting cytokines IL-4, IL-5, and IL-13.
Although the target cell or cell surface receptor for HSP has not been completely elucidated, there is accumulating evidence for interactions with cells of the innate immune system (47, 48). The stimulatory activity of HSP60, HSP70, HSP90, and gp96 on the release of proinflammatory cytokines by cells of the innate immune system has been well described (25, 49–51). HSP-like protein 1 interacts with DCs, promoting DC maturation and polarizing responses towards a Th1 pattern (37). To pursue the interactions of HSP65 on DC function, we first examined their impact on DC-T cell interactions in vitro. CD4+ Th2 cells play a major effector role in the development of allergic inflammation through the release of cytokines such as IL-4, IL-5, and IL-13 (52, 53). The transfer of Th2 cells followed by airway allergen challenge in mice was sufficient to induce airway eosinophilia and airway hyperresponsiveness (54, 55). When HSP65-treated, antigen-pulsed DCs were co-cultured with CD4+ T cells, increases in Th1 cytokines were detected in culture supernates with lower levels of Th2 cytokines, establishing a basis for the in vivo findings in secondary challenged mice. These observations were extended to in vivo studies examining DC function in an adoptive transfer model. We previously showed that antigen-pulsed BMDCs could restore the development of airway hyperresponsiveness and eosinophilic inflammation following transfer into non-sensitized mice prior to allergen challenge (26). Here, OVA-pulsed BMDCs treated with HSP65 were transferred into previously sensitized and challenged mice, as a means for exposing the mice to allergen in lieu of direct allergen challenge. Whereas recipients of untreated, OVA-pulsed BMDCs developed airway hyperresponsiveness and airway inflammation, recipients of HSP65-treated cells showed no such responses. One of the outcomes of this DC transfer model was the ability to elicit airway hyperresponsiveness after allergen challenge in the apparent absence of an accompanying robust BAL eosinophilia or elevation of Th2 cytokine levels (26). This dissociation of airway hyperresponsiveness and airway eosinophilia/Th2 cytokines has been previously reported under varying experimental conditions (56–59).
In summary, HSP65 reduced airway hyperresponsiveness and airway inflammation when administered prior to secondary allergen challenge of previously sensitized and challenged mice. The in vivo results together with the in vitro findings position DCs as a prominent target of the HSP65 suppressive effects, mediated at least in part, through increased Delta1 expression on DCs and Th1 skewing of CD4+ T cells. These results identify HSP65 as a potent modulator of allergen-induced lung allergic responses in established disease.
ACKNOWLEDGMENTS
The assistance of Ms. Diana Nabighian in the preparation of this manuscript is gratefully acknowledged.
Grant Support: This study was supported by the State of Colorado Bioscience Discovery Evaluation Grant Program and NIH grants HL-36577 and AI-77609. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH.
REFERENCES
- 1.Szefler SJ, Zeiger RS, Haselkorn T, Mink DR, Kamath TV, Fish JE, Chipps BE. Economic burden of impairment in children with severe or difficult-to-treat asthma. Ann. Allergy Asthma Immunol. 2011;107:110–119. e1. doi: 10.1016/j.anai.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Busse WW, Lemanske RF., Jr Asthma. New Engl. J. Med. 2001;344:350–362. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- 3.Ray A, Cohn L. Th2 cells and GATA-3 in asthma: new insights into the regulation of airway inflammation. J. Clin. Invest. 1999;104:985–993. doi: 10.1172/JCI8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wisniewski JA, Borish L. Novel cytokines and cytokine-producing T cells in allergic disorders. Allergy Asthma Proc. 2011;32:83–94. doi: 10.2500/aap.2011.32.3428. [DOI] [PubMed] [Google Scholar]
- 5.Amsen D, Antov A, Flavell RA. The different faces of Notch in T-helper-cell differentiation. Nat. Rev. Immunol. 2009;9:116–124. doi: 10.1038/nri2488. [DOI] [PubMed] [Google Scholar]
- 6.Yuan JS, Kousis PC, Suliman S, Visan I, Guidos CJ. Functions of notch signaling in the immune system: consensus and controversies. Annu. Rev. Immunol. 2010;28:343–365. doi: 10.1146/annurev.immunol.021908.132719. [DOI] [PubMed] [Google Scholar]
- 7.Okamoto M, Takeda K, Joetham A, Ohnishi H, Matsuda H, Swasey CH, Swanson BJ, Yasutomo K, Dakhama A, Gelfand EW. Essential role of Notch signaling in effector memory CD8+ T cell-mediated airway hyperresponsiveness and inflammation. J. Exp. Med. 2008;205:1087–1097. doi: 10.1084/jem.20072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okamoto M, Matsuda H, Joetham A, Lucas JJ, Domenico J, Yasutomo K, Takeda K, Gelfand EW. Jagged1 on dendritic cells and Notch on CD4+ T cells initiate lung allergic responsiveness by inducing IL-4 production. J. Immunol. 2009;183:2995–3003. doi: 10.4049/jimmunol.0900692. [DOI] [PubMed] [Google Scholar]
- 9.Okamoto M, Takeda K, Lucas JJ, Joetham A, Yasutomo K, Gelfand EW. Low-dose lipopolysaccharide affects lung allergic responses by regulating Jagged1 expression on antigen-pulsed dendritic cells. Intl. Arch. Allergy Immunol. 2012;157:65–72. doi: 10.1159/000324836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindquist S, Craig EA. The heat-shock proteins. Annu. Rev. Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 11.Polla BS, Perin M, Pizurki L. Regulation and functions of stress proteins in allergy and inflammation. Clin. Exp. Allergy. 1993;23:548–556. doi: 10.1111/j.1365-2222.1993.tb00893.x. [DOI] [PubMed] [Google Scholar]
- 12.Tsan MF, Gao B. Heat shock proteins and immune system. J. Leukoc. Biol. 2009;85:905–910. doi: 10.1189/jlb.0109005. [DOI] [PubMed] [Google Scholar]
- 13.Javid B, MacAry PA, Lehner PJ. Structure and function: heat shock proteins and adaptive immunity. J. Immunol. 2007;179:2035–2040. doi: 10.4049/jimmunol.179.4.2035. [DOI] [PubMed] [Google Scholar]
- 14.Osterloh A, Breloer M. Heat shock proteins: linking danger and pathogen recognition. Med. Microbiol. Immunol. 2008;197:1–8. doi: 10.1007/s00430-007-0055-0. [DOI] [PubMed] [Google Scholar]
- 15.van Eden W, van der Zee R, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat. Rev. Immunol. 2005;5:318–330. doi: 10.1038/nri1593. [DOI] [PubMed] [Google Scholar]
- 16.Vatner RE, Srivastava PK. The tailless complex polypeptide- ring complex of the heat shock protein 60 family facilitates cross-priming of CD8 responses specific for chaperoned peptides. J. Immunol. 2010;185:6765–6773. doi: 10.4049/jimmunol.1001720. [DOI] [PubMed] [Google Scholar]
- 17.Chen T, Cao X. Stress for maintaining memory: HSP70 as a mobile messenger for innate and adaptive immunity. Eur. J. Immunol. 2010;40:1541–1544. doi: 10.1002/eji.201040616. [DOI] [PubMed] [Google Scholar]
- 18.Jurewicz M, Takakura A, Augello A, Naini SM, Ichimura T, Zandi-Nejad K, Abdi R. Ischemic injury enhances dendritic cell immunogenicity via TLR4 and NF-kappa B activation. J. Immunol. 2010;184:2939–2948. doi: 10.4049/jimmunol.0901889. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, Su X, Zhang P, Liang J, Wei H, Wan M, Wu X, Yu Y, Wang L. Recombinant heat shock protein 65 carrying PADRE and HBV epitopes activates dendritic cells and elicits HBV-specific CTL responses. Vaccine. 2011;29:2328–2335. doi: 10.1016/j.vaccine.2010.12.124. [DOI] [PubMed] [Google Scholar]
- 20.Rha YH, Taube C, Haczku A, Joetham A, Takeda K, Duez C, Siegel M, Aydintug MK, Born WK, Dakhama A, Gelfand EW. Effect of microbial heat shock proteins on airway inflammation and hyperresponsiveness. J. Immunol. 2002;169:5300–5307. doi: 10.4049/jimmunol.169.9.5300. [DOI] [PubMed] [Google Scholar]
- 21.Takeda K, Hamelmann E, Joetham A, Shultz LD, Larsen GL, Irvin CG, Gelfand EW. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J. Exp. Med. 1997;186:449–454. doi: 10.1084/jem.186.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koya T, Kodama T, Takeda K, Miyahara N, Yang ES, Taube C, Joetham A, Park JW, Dakhama A, Gelfand EW. Importance of myeloid dendritic cells in persistent airway disease after repeated allergen exposure. Am. J. Respir. Crit. Care Med. 2006;173:42–55. doi: 10.1164/rccm.200505-783OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koya T, Miyahara N, Takeda K, Matsubara S, Matsuda H, Swasey CH, Balhorn A, Dakhama A, Gelfand EW. CD8+ T cell-mediated airway hyperresponsiveness and inflammation is dependent on CD4+IL-4+ T cells. J. Immunol. 2007;179:2787–2796. doi: 10.4049/jimmunol.179.5.2787. [DOI] [PubMed] [Google Scholar]
- 24.Tomkinson A, Cieslewicz G, Duez C, Larson KA, Lee JJ, Gelfand EW. Temporal association between airway hyperresponsiveness and airway eosinophilia in ovalbumin-sensitized mice. Am. J. Respir. Crit. Care Med. 2001;163:721–730. doi: 10.1164/ajrccm.163.3.2005010. [DOI] [PubMed] [Google Scholar]
- 25.Flohe SB, Bruggemann J, Lendemans S, Nikulina M, Meierhoff G, Flohe S, Kolb H. Human heat shock protein 60 induces maturation of dendritic cells versus a Th1-promoting phenotype. J. Immunol. 2003;170:2340–2348. doi: 10.4049/jimmunol.170.5.2340. [DOI] [PubMed] [Google Scholar]
- 26.Koya T, Matsuda H, Matsubara S, Miyahara N, Dakhama A, Takeda K, Gelfand EW. Differential effects of dendritic cell transfer on airway hyperresponsiveness and inflammation. Am. J. Respir. Cell Mol. Biol. 2009;41:271–280. doi: 10.1165/rcmb.2008-0256OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magnus P, Jaakkola JJ. Secular trend in the occurrence of asthma among children and young adults: critical appraisal of repeated cross sectional surveys. BMJ. 1997;314:1795–1799. doi: 10.1136/bmj.314.7097.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The international study of asthma and allergies in childhood (ISAAC) steering committee. Lancet. 1998;351:1225–1232. [PubMed] [Google Scholar]
- 29.Springett VH, Darbyshire JH, Nunn AJ, Sutherland I. Changes in tuberculosis notification rates in the white ethnic group in England and Wales between 1953 and 1983. J. Epidemiol. Community Health. 1988;42:370–376. doi: 10.1136/jech.42.4.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rook GA, Martinelli R, Brunet LR. Innate immune responses to mycobacteria and the downregulation of atopic responses. Curr. Opin. Allergy Clin. Immunol. 2003;3:337–342. doi: 10.1097/00130832-200310000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Waterer GW, ElBahlawan L, Quasney MW, Zhang Q, Kessler LA, Wunderink RG. Heat shock protein 70-2+1267 AA homozygotes have an increased risk of septic shock in adults with community-acquired pneumonia. Crit. Care Med. 2003;31:1367–1372. doi: 10.1097/01.CCM.0000063088.86079.03. [DOI] [PubMed] [Google Scholar]
- 32.Stebbing J, Gazzard B, Kim L, Portsmouth S, Wildfire A, Teo I, Nelson M, Bower M, Gotch F, Shaunak S, Srivastava P, Patterson S. The heat-shock protein receptor CD91 is up-regulated in monocytes of HIV-1-infected "true" long-term nonprogressors. Blood. 2003;101:4000–4004. doi: 10.1182/blood-2002-11-3353. [DOI] [PubMed] [Google Scholar]
- 33.Hashiguchi N, Ogura H, Tanaka H, Koh T, Nakamori Y, Noborio M, Shiozaki T, Nishino M, Kuwagata Y, Shimazu T, Sugimoto H. Enhanced expression of heat shock proteins in activated polymorphonuclear leukocytes in patients with sepsis. J. Trauma. 2001;51:1104–1109. doi: 10.1097/00005373-200112000-00015. [DOI] [PubMed] [Google Scholar]
- 34.Bertorelli G, Bocchino V, Zhuo X, Chetta A, Del Donno M, Foresi A, Testi R, Olivieri D. Heat shock protein 70 upregulation is related to HLA-DR expression in bronchial asthma. Effects of inhaled glucocorticoids. Clin. Exp. Allergy. 1998;28:551–560. doi: 10.1046/j.1365-2222.1998.00251.x. [DOI] [PubMed] [Google Scholar]
- 35.Kariyawasam HH, Aizen M, Barkans J, Robinson DS, Kay AB. Remodeling and airway hyperresponsiveness but not cellular inflammation persist after allergen challenge in asthma. Am. J. Respir. Crit. Care Med. 2007;175:896–904. doi: 10.1164/rccm.200609-1260OC. [DOI] [PubMed] [Google Scholar]
- 36.Riffo-Vasquez Y, Spina D, Page C, Tormay P, Singh M, Henderson B, Coates A. Effect of Mycobacterium tuberculosis chaperonins on bronchial eosinophilia and hyper-responsiveness in a murine model of allergic inflammation. Clin. Exp. Allergy. 2004;34:712–719. doi: 10.1111/j.1365-2222.2004.1931.x. [DOI] [PubMed] [Google Scholar]
- 37.Wan T, Zhou X, Chen G, An H, Chen T, Zhang W, Liu S, Jiang Y, Yang F, Wu Y, Cao X. Novel heat shock protein Hsp70L1 activates dendritic cells and acts as a Th1 polarizing adjuvant. Blood. 2004;103:1747–1754. doi: 10.1182/blood-2003-08-2828. [DOI] [PubMed] [Google Scholar]
- 38.Wieten L, Berlo SE, Ten Brink CB, van Kooten PJ, Singh M, van der Zee R, Glant TT, Broere F, van Eden W. IL-10 is critically involved in mycobacterial HSP70 induced suppression of proteoglycan-induced arthritis. PLoS One. 2009;4:e4186. doi: 10.1371/journal.pone.0004186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moser M, Murphy KM. Dendritic cell regulation of TH1-TH2 development. Nat. Immunol. 2000;1:199–205. doi: 10.1038/79734. [DOI] [PubMed] [Google Scholar]
- 40.Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, Geginat J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010;21:331–344. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Morimura T, Goitsuka R, Zhang Y, Saito I, Reth M, Kitamura D. Cell cycle arrest and apoptosis induced by Notch1 in B cells. J. Biol. Chem. 2000;275:36523–36531. doi: 10.1074/jbc.M006415200. [DOI] [PubMed] [Google Scholar]
- 42.Maekawa Y, Tsukumo S, Chiba S, Hirai H, Hayashi Y, Okada H, Kishihara K, Yasutomo K. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity. 2003;19:549–559. doi: 10.1016/s1074-7613(03)00270-x. [DOI] [PubMed] [Google Scholar]
- 43.Deftos ML, He YW, Ojala EW, Bevan MJ. Correlating notch signaling with thymocyte maturation. Immunity. 1998;9:777–786. doi: 10.1016/s1074-7613(00)80643-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development. 2011;138:3593–3612. doi: 10.1242/dev.063610. [DOI] [PubMed] [Google Scholar]
- 45.Sun J, Krawczyk CJ, Pearce EJ. Suppression of Th2 cell development by Notch ligands Delta1 and Delta4. J. Immunol. 2008;180:1655–1661. doi: 10.4049/jimmunol.180.3.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liotta F, Frosali F, Querci V, Mantei A, Filì L, Maggi L, Mazzinghi B, Angeli R, Ronconi E, Santarlasci V, Biagioli T, Lasagni L, Ballerini C, Parronchi P, Scheffold A, Cosmi L, Maggi E, Romagnani S, Annunziato F. Human immature myeloid dendritic cells trigger a TH2-polarizing program via Jagged-1/Notch interaction. J. Allergy Clin. Immunol. 2008;121:1000–1005. e8. doi: 10.1016/j.jaci.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 47.Kaufmann SH. Immunity to intracellular bacteria. Annu. Rev. Immunol. 1993;11:129–163. doi: 10.1146/annurev.iy.11.040193.001021. [DOI] [PubMed] [Google Scholar]
- 48.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu. Rev. Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 49.Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000;6:435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 50.Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 2000;12:1539–1546. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 51.Singh-Jasuja H, Scherer HU, Hilf N, Arnold-Schild D, Rammensee HG, Toes RE, Schild H. The heat shock protein gp96 induces maturation of dendritic cells and down-regulation of its receptor. Eur. J. Immunol. 2000;30:2211–2215. doi: 10.1002/1521-4141(2000)30:8<2211::AID-IMMU2211>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 52.Busse WW, Coffman RL, Gelfand EW, Kay AB, Rosenwasser LJ. Mechanisms of persistent airway inflammation in asthma. A role for T cells and T-cell products. Am. J. Respir. Crit. Care Med. 1995;152:388–393. doi: 10.1164/ajrccm.152.1.7599853. [DOI] [PubMed] [Google Scholar]
- 53.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. New Engl. J. Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 54.Cohn L, Homer RJ, Marinov A, Rankin J, Bottomly K. Induction of airway mucus production By T helper 2 (Th2) cells: a critical role for interleukin 4 in cell recruitment but not mucus production. J. Exp. Med. 1997;186:1737–1747. doi: 10.1084/jem.186.10.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cohn L, Tepper JS, Bottomly K. IL-4-independent induction of airway hyperresponsiveness by Th2, but not Th1, cells. J. Immunol. 1998;161:3813–3816. [PubMed] [Google Scholar]
- 56.Joetham A, Takeda K, Taube C, Miyahara N, Kanehiro A, Dakhama A, Gelfand EW. Airway hyperresponsiveness in the absence of CD4+ T cells after primary but not secondary challenge. Am. J. Respir. Cell Mol. Biol. 2005;33:89–96. doi: 10.1165/rcmb.2004-0414OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, Locksley RM. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J. Exp. Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, Ghiran S, Gerard NP, Yu C, Orkin SH, Gerard C. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–1779. doi: 10.1126/science.1100283. [DOI] [PubMed] [Google Scholar]
- 59.Shen HH, Ochkur SI, McGarry MP, Crosby JR, Hines EM, Borchers MT, Wang H, Biechelle TL, O'Neill KR, Ansay TL, Colbert DC, Cormier SA, Justice JP, Lee NA, Lee JJ. A causative relationship exists between eosinophils and the development of allergic pulmonary pathologies in the mouse. J. Immunol. 2003;170:3296–3305. doi: 10.4049/jimmunol.170.6.3296. [DOI] [PubMed] [Google Scholar]