

Abstract
The repair of abasic sites that arise in DNA from hydrolytic depurination/depyrimidination of the nitrogenous bases from the sugar-phosphate backbone and the action of DNA glycosylases on deaminated, oxidized and alkylated bases is critical to cell survival. Apurinic/Apyrimidinic Endonuclease-1/Redox Effector Factor-1 (APE-1; aka, APE1/Ref-1) is responsible for the initial removal of abasic lesions as part of the base excision repair pathway. Deletion of APE-1 activity is embryonic lethal in animals and is lethal in cells. Potential inhibitors of the repair function of APE-1 were identified based upon molecular modeling of the crystal structure of the APE-1 protein. We describe the characterization of several unique nM inhibitors using two complementary biochemical screens. The most active molecules all contain a 2-methyl-4-amino-6,7-dioxolo-quinoline structure that is predicted from the modeling to anchor the compounds in the endonuclease site of the protein. The mechanism of action of the selected compounds was probed by fluorescence and competition studies, which indicate, in a specific case, direct interaction between the inhibitor and the active site of the protein. It is demonstrated that the inhibitors induce time-dependent increases in the accumulation of abasic sites in cells at levels that correlate with their potency to inhibit APE-1 endonuclease excision. The inhibitor molecules also potentiate by 5-fold the toxicity of a DNA methylating agent that creates abasic sites. The molecules represent a new class of APE-1 inhibitors that can be used to probe the biology of this critical enzyme and to sensitize resistant tumor cells to the cytotoxicity of clinically used DNA damaging anticancer drugs.
Abasic sites created by hydrolytic depurination/depyrimidination and excision of lesions by base excision repair (BER*) DNA glycosylases are both cytotoxic and mutagenic.1,2 It is estimated that more than 104 abasic sites are formed per mammalian cell per day.3,4 The repair of abasic lesions in mammalian cells is predominantly mediated by the initial action of Apurinic/Apyrimidinic Endonuclease-1/Redox Effector Factor-1 (APE-1),5 which cleaves the phosphodiester linkage that is 5′ to the abasic site, leaving a single strand break (SSB) with 3′-hydroxyl and 5′-deoxyribose phosphate (dRP) termini.6 This repair intermediate is then processed by Polβ, which removes the 5′-DRP structure to afford a 5′-phosphate and then adds the appropriate complementary base to the 3′-terminus.7 In the final step, DNA ligase seals the nick. While cells and animals can survive without the different DNA glycosylases, albeit with increased sensitivity to DNA damaging agents,8–11 the genetic deletion of APE-1, which is expressed ubiquitously, is lethal in cells.12 In mice, embryos terminate at post-implantation following blastocyst formation, and without developmental defects.13,14 Heterozygous mice are viable but become sensitized to DNA damaging agents that induce the formation of abasic sites.15–17 Deletion of Polβ, which is also critical in BER,18 causes neonatal lethality due to defective neurogenesis characterized by apoptotic cell death in the developing central and peripheral nervous systems,19 indicating the critical need for cells to maintain functional BER during embryogenesis. Zebrafish knockdown of AP endonuclease (Apex) using siRNA, also terminate during development.20 Of interest is the observation that Pol β appears to be translationally coupled to Apex since the mRNA for the polymerase is present in the null fish but the protein is absent.21 Whether this is also the case in mammalian cells is not known.
The endonuclease function of APE-1 is located toward the C-terminus of the protein. The N-terminal domain is associated with the redox center (a.k.a., Ref-1) that regulates the activity of specific transcriptional factors by maintaining them in a reduced state.22–26 In addition, APE-1 has been linked to several other functions, including RNA processing27 and in Ca2+-dependent gene expression and regulation.28 The lethality of APE-1 knockouts has been attributed to loss of the repair activity, and the mechanism of cell death involves apoptosis.29 Over-expression of APE-1 makes cells resistant to alkylating agents.12 There is also evidence that APE-1 expression can be induced by genotoxic agents, including cancer drugs.30 These data raise the question of whether APE-1 expression is associated with tumor resistance to DNA damaging agents. In this regard, the lethality of clinically used anticancer treatments can be enhanced by a temporal decrease in APE-1 using antisense technology.31–34 Therefore, molecules that modulate APE-1 activity could be important adjuvants to clinically used DNA damaging antineoplastic agents. Recently, it has been reported that inhibitors of APE-1 endonuclease activity can create a synthetic lethality in cells defective in double-strand break repair, i.e., BRCA1, BRCA2 and ATM.35 This result is not unexpected since homologous recombination (HR) mutants are particularly sensitive to methylation damage repaired by BER.36,37 In fact, yeast cells that lack HR tolerate DNA alkylation damage better if there is no BER, indicating the biological consequences of BER in the absence of HR.38 This result with APE-1 induced synthetic lethality is similar to the interaction between BRCA defective cells and PARP inhibitors.39,40
A number of small molecule inhibitors of APE-1 that have been identified and characterized.41–47 In many cases the inhibitors identified in screens are dicarboxylic acids or related analogues (Figure 1). These molecules potentially mimic the phosphate linkages flanking the abasic lesion on the DNA (Figure 2), which participate in salt bridges with the cationic face of the enzyme. Included in these inhibitors are a series of arylstibonic acids, though extremely potent in biochemical experiments, lacked activity in cells.44 Lucanthone inhibits APE-1 activity41 and binds to the protein,48 but also interacts with a number of other cellular targets, including DNA, so the mechanism of action remains uncertain.49 The dimethoxyquinone, E3330,50 (Figure 1), which also has a number of cellular activities,51 has been shown to inhibit both the endonuclease and redox activity of APE-1.52–54 Of note is that NMR data indicate that the quinone molecule binds near the active site associated with the endonuclease activity. 54 How the redox and endonuclease activities, which are in physically distinct regions of the protein, are inhibited by a single molecule remains unclear. Another approach to block APE-1 activity is to chemically modify the abasic lesion using aldehyde reactive compounds, such as methoxylamine. Methoxylamine reacts to form an imine with the aldehyde group in the ring-open form of the abasic lesion (Figure 2), which prevents the protein mediated cleavage of the abasic site.55,56 Though methoxylamine will react with any cellular aldehyde or ketone, it is being clinically evaluated in a phase-1 trial as an adjunct therapy in combination with DNA alkylating agents.57
Figure 1.

Structures of some previously reported inhibitors of APE-1 (and their calculated IC50 values): A (ref 46), B (ref 46), C (ref 45), D (ref 44), E (ref 44), F (ref 85), G (ref 85), H (ref 79), I (ref 48), J (ref 86).
Figure 2.
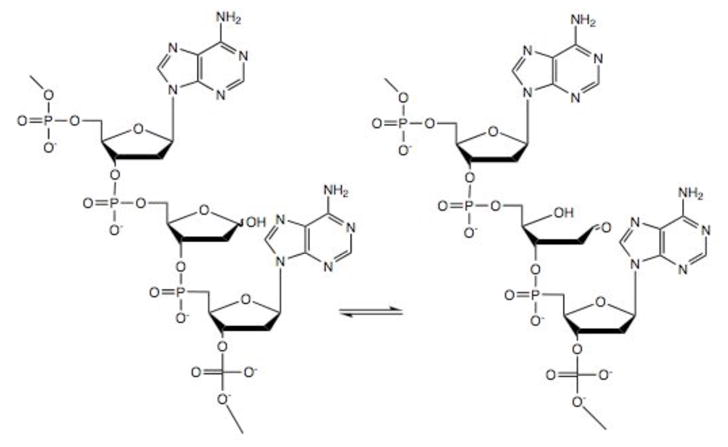
Structure of closed and ring-opened abasic site in DNA.
Using the crystal structure of APE-1,58 we have computationally constructed molecules that would sterically block its endonuclease site. Based upon the modeling, potential inhibitor molecules were identified and sequentially assayed in two complementary biochemical screens. Fluorescence studies, including displacement assays, indicate that one of the inhibitors directly interacts with the APE-1 endonuclease site. The toxicity of a range of inhibitors was evaluated in a resistant T98G glioma cell line. To complement the inhibition and toxicity studies, we measured the levels of abasic lesions in the untreated T98G cells and in cells treated with the APE-1 inhibitors alone or with a methylating agent that selectively generates N3-methyladenine lesions59 that are processed via abasic intermediates.60,61
EXPERIMENTAL PROCEDURES
Materials
Potential APE-1 inhibitor compounds were obtained via the Developmental Therapeutics Program of the National Cancer Institute, National Institute of Health, Bethesda, MD. Chemicals, solvents and CelLytic NuCLEAR Extraction kit were purchased from Sigma Aldrich Chemicals (St. Louis, MO). Oligonucleotide sequences were custom-synthesized by Integrated DNA Technologies (Coralville, IA) or Sigma Aldrich (St. Louis, MO). Nickel nitrilotriacetate (Ni-NTA) sepharose resin was procured from Qiagen Inc. (Valencia, CA). pENTR, pDEST17, Escherichia coli DH5α, pre-cast polyacrylamide gels, DNAzol reagent, cell culture components and CyQUANT Cell Proliferation Assay Kit were obtained from Invitrogen (Carlsbad, CA). Human glioblastoma (T98G) cells were obtained from American Type Culture Collection (ATCC) (Manassas, VA). CellTiter 96 AQueous One Solution Reagent was obtained from Promega Corp, Madison, WI. DNA Damage Quantification kit (abasic site counting) was obtained from Dojindo Molecular Technologies (Rockville, MD). Escherichia coli C41(DE3) cells were obtained from Lucigen Corp. (Middleton, WI). His-Pur Cobalt resin, Trypan Blue stain, BCA Protein assay kit, dialysis cassettes and molecular biology grade buffers, as well as plastic- and glass-ware were obtained from Fisher Scientific (Pittsburgh, PA). The sequence-specific alkylating agent, 1-methyl-4-[1-methyl-4-(3-methoxysulfonylpropanamido)pyrrole-2-carboxamido]pyrrole-2-carboxamido}propane (MeLex) was synthesized as previously described.59
Virtual screening strategy for small-molecule inhibitors of APE-1
The 3D structural model of APE-1 was constructed based on the X-ray crystallographic data for 1DEW in the PDB58 and was further modeled by refining the low-resolution region using our previously reported protocols.62 all computations were performed using Sybyl 8.2 (Tripos Associates) on a Dell 32 CPU Linux Cluster. After the generation of Surflex-Dock protomol using the reported binding residues and default parameters, a virtual docking screen63 was carried out on the reported 3D virtual chemical compound library.64 For each compound, 20 optimal conformers were generated by docking calculations. The conformer with the best total Hammerhead score (8.21)65,66 was chosen for further biochemical and cellular studies.
Cloning, expression and purification of APE-1
The coding sequence for the human apurinic endonuclease (ape-1) was previously amplified by Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) and cloned into pENTR, a directional TOPO-cloning Gateway entry vector (R.W. Sobol et al., unpublished). The ape-1 coding sequence was sub-cloned into the destination vector pDEST17 via the LR reaction, which introduced an N-terminal polyhistidine (6x) coding sequence. The pDEST17-ape-1 construct, amplified and purified from Escherichia coli DH5α cells was used to transform ‘OverExpress’ E. coli C41(DE3) cells for expression and purification of recombinant (His)6-APE-1 protein.
Transformed E. coli C41(DE3) was grown overnight at 37 °C and constant shaking (100 rpm) in 5 mL Luria Bertani (LB) medium containing 100 μg mL−1 ampicillin. The overnight culture was used to seed 500 mL LB medium and the cells were allowed to grow at 37 °C with constant shaking (100 rpm, 2 h) until the culture reached mid-log phase (λ600nm ~0.5). Isopropyl β-D-1-thiogalactopyranoside (IPTG) at 0.8 mM was used to induce APE-1 expression, at 25 °C, under constant stirring (100 rpm, ~ 2 h). At 16 h post-induction, the cells were harvested by centrifugation (5000 rpm, 10 min), washed free of media with 1x phosphate buffered saline (PBS) and resuspended in 5.77 mM sodium phosphates (4.3 mM Na2HPO4 + 1.47 mM NaH2PO4, pH 7.4), 2.7 mM KCl, 330 mM NaCl, 10% (v/v) glycerol, 0.5 % (v/v) Tween-20, 0.1 % (w/v) lysozyme, 10 units mL−1 Benzonase (nuclease), and incubated for 30 min at 25 °C. Intact cells were lysed by ultrasonication (5 min, 30% amplitude), centrifuged at 10,000 rpm, for 30 min, and clear supernatant (cell lysates) collected. APE-1 protein in cell lysates was purified by immobilized metal affinity chromatography using Ni- or Co-chelated resins to bind the to the N-terminus His6 tag. Immobilized APE-1 protein was eluted using a 10–250 mM gradient of imidazole in 10 mM HEPES (pH 7.4), 50 mM KCl. Pooled fractions containing APE-1 protein was dialyzed against 10 mM HEPES (pH 7.4), 250 mM KCl, 10% (v/v) glycerol and protein concentration estimated colorimetrically (BCA protein assay kit) as well as by UV absorbance (λ280nm). Homogeneity of purified proteins was checked by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by staining with 0.05% Coomassie Brilliant Blue R-250, 50% (v/v) methanol, 10% (v/v) acetic acid. Purified proteins were stored at −80 °C (long-term) or kept at −20 °C during regular use.
Molecular beacon assay for screening of APE-1 inhibitors
A fluorescence-based molecular beacon assay to measure APE-1 protein activity and inhibition was performed as described previously44,67 with some modifications.68 APE-1 (2 nM) in 10 mM HEPES (pH 7.8) 0.5 mM MgCl2, 100 mM NaCl, 100 mM KCl, 0.0005% (w/v) BSA, was incubated with 20 nM 5′-(F)-CGACTXTTGAATTGACACGCCATGTCGATCAATTCAATAGTCG-(D)-3′ [X = tetrahydrofuran (THF) abasic site modification; F, 6-carboxyfluorescein (6-FAM); and D, DABCYL] that forms a hairpin with a 15 base pair stem and a 13 base loop. The THF modification is a known substrate for APE-1 excision. APE-1 mediated cleavage of the phosphodiester backbone at the THF-abasic site led to release of the 5′ 6-FAM (fluorophore) containing pentanucleotide, disrupting the Förster Resonance Energy Transfer (FRET) mediated quenching of the 6-FAM by DABCYL in the native hairpin oligomer. Since the signal was generated by continuous liberation of the fluorophore-labeled excision fragment, progress of this reaction could be monitored in real-time using a Cary Eclipse fluorescence spectrometer (Varian, Palo Alto, CA) or a Synergy H1 multi-well plate reader (BioTek, Winooski, VT). Reactions were optimized to yield ~90% substrate turnover in 10 min, and fluorescence intensities measured every 12 s at 495 nm excitation and 520 nm emission. Data from the first 1–3 min of the assays (linear phase) were used to determine rate of substrate turnover (V0). The Michaelis constant (KM) was deduced experimentally, by incubating 2 nM APE-1 protein with 2–500 nM hairpin DNA substrate and measuring V0 - the experimentally observed V0 were analyzed as a function of substrate concentration [S] via non-linear regression using Prism (v.4; GraphPad Software, Inc., La Jolla, CA) and the KM was calculated as per the algorithm in the software package. To assay inhibition, 2 nM APE-1 protein was pre-incubated with 0.05–10.00 μM of the candidate inhibitors at 25 °C for 60 min, before initiating the excision assay by addition of hairpin DNA substrate. V0 was calculated at various concentrations of inhibitors and represented as inverse function of increasing inhibitor concentration [I]. Data were plotted on Prism and analyzed by non-linear regression to obtain values for inhibitor concentration at 50% inhibition (IC50), and the inhibitory constant (Ki) was approximated by the formula, Ki = [IC50]/(1+ [S]/KM)is.69 All assays were carried out in three independent sets to obtain mean data and standard error of mean.
Gel-based assay for APE-1 inhibitors
In addition to the real-time fluorescence assay for studying inhibition of APE-1 activity, we also employed a gel-based assay to visualize and quantify repair activity of APE-1 and its inhibition. Briefly, 20 nM APE-1, in 10 mM HEPES (pH 7.8) 0.5 mM MgCl2, 100 mM NaCl, 100 mM KCl, 0.0005% (w/v) BSA, was incubated with 100 nM of a duplex DNA that was formed by annealing 5′-(F)-CGATCATCACTXTTGAGACTGACACTGACC-3′, which contained the THF abasic site modification (X), a 5′ 6-FAM fluorophore (F), and the complementary sequence, 5′-GGTCAGTGTCAGTCTCAATAGTGATGATCG-3′. Cleavage by APE-1 at the THF abasic site led to release of the 5′-fluorophore tagged 11-nucleotide fragment, which could be resolved from the uncleaved 30-nucleotide fragment (intact substrate) by gel electrophoresis under denaturing conditions. To assay inhibition of APE-1 activity, the APE-1 protein was pre-incubated with 5–100 μM of the candidate inhibitors for 60 min, before initiation of the excision assay by addition of duplex DNA substrate. Reaction products were separated on a 20% denaturing (urea) polyacrylamide gel, and the bands were visualized by fluorescence emission of the 6-FAM under ultraviolet illumination using a GelDoc EZ Imager (BioRad, Hercules, CA) and quantified using the Image Lab Software (BioRad, Hercules, CA). Inhibitory activity was calculated by co-relating fluorescence intensity of the 11-nucleotide fragment product as the (inverse) function of inhibitor concentration. All assays included a positive control (complete digestion of duplex DNA in absence of any inhibitor) as well as a negative control (uncleaved duplex DNA), which corresponded to 100% and 0% substrate turnover, respectively. Substrate turnover in presence of candidate inhibitors were calculated as percentage of positive control. Data were plotted on Prism, analyzed by non-linear regression and the IC50 values were obtained from the dose response curves. All assays were carried out in three independent measurements.
Fluorescence and UV spectrometry binding studies
The fluorescence spectra were measured at room temperature on a Cary Eclipse fluorescence spectrometer (Varian, Palo Alto, CA). The fluorescence properties of 1 and 4 were measured in 10 mM HEPES, 0.5 mM MgCl2, 100 mM KCl, 100 mM NaCl, 2% glycerol, in absence or presence of wild type (unmodified) hairpin DNA (5′-CGAATTCGCAGGACCGAATTCG-3′) or THF-modified hairpin DNA (5′-CGAXTTCGCAGGACCGAATTCG-3′) or APE-1 protein. A solution of 0.5 μM of either DNA sequence was titrated against 1–10 μM ethidium bromide (EtBr) to determine saturation of EtBr by fluorescence (530 nm excitation with 610 nm emission) and the subsequent displacement of EtBr (4μM) by 1–20 μM 1 or 4 was also assayed. The APE protein – inhibitor binding fluorescence was assayed by titrating 0.1–20 μM of 4 against 1 μM of APE-1 protein. Displacement of 4 (6 μM) from 1 μM APE-1 protein by increasing amounts (0.75 – 30 μM) of E3330 was also measured.
The effect of the small molecules on DNA stabilization was determined by measuring UV absorption of DNA at 260 nm as a function of temperature in a Cary 300 UV-visible spectrophotometer (Varian, Palo Alto, CA) under similar conditions as previously described.70 The transition temperatures (TM) was calculated from the first derivative analysis.
Isolation and assay of APE-1 protein from nuclear extracts of glioma cells
Human glioblastoma cells (T98G) were maintained in growth medium, i.e., Eagle’s Minimum Essential Medium (EMEM) with 10% fetal bovine serum (FBS), 50 μg μL−1 gentamycin, 1x MEM non-essential amino acids and 1 mM sodium pyruvate. Cells were harvested by Trypsin-EDTA treatment, washed with 1x PBS and nuclear proteins isolated using the CelLytic NuCLEAR Extraction kit (Sigma-Aldrich) as per manufacturer’s instructions. Isolated nuclear proteins were estimated colorimetrically (BCA protein assay) and stored at −80 °C (long term) or at −20 °C during regular use. To assay APE-1 activity in T98G cells, nuclear extracts were serially diluted in 10 mM HEPES, 50 mM KCl, 1 mM DTT, 20% (v/v) glycerol and the activities measured via the molecular beacon assay (as described above). The final dilution used for all assays (EC2nM, effective concentration of 2 nM) gave ~90% turnover of 25 nM hairpin substrate DNA in 10 min, which was equivalent to the activity of 2 nM recombinant APE-1 protein in the molecular beacon assay. EC2nM of nuclear extracts was incubated with 2 to 500 nM hairpin DNA substrate and the V0 measured for each value of [S]. Finally, the apparent KM* was deduced using the software algorithm in Prism (as described previously), and the KM* was used to determine the apparent Ki*. Inhibition of EC2nM APE-1 activity by the inhibitors was assayed via the molecular beacon assay (described above).
Cytotoxic effects of APE-1 inhibitors on cultured human glioblastoma (T98G) cells
Human glioblastoma cells (T98G) were maintained in growth medium as described above. To assay short term cytotoxicity, T98G cells maintained in growth medium, were harvested by Trypsin-EDTA treatment, resuspended in fresh growth medium at a density of 104 cells mL−1, and 200 μL (2000 cells) of this suspension was seeded into the wells of a sterile 96-well tissue culture plate. The cells were allowed to attach and grow for 24 h, and then treated with serial dilutions of the drug for 72 h. After treatment, viable cells were measured by an MTS assay using the CellTiter 96 AQueous kit (Promega) as per manufacturer’s instructions. To assay long term cytotoxicity, T98G cells maintained in growth medium were harvested by Trypsin-EDTA treatment, resuspended in fresh growth medium at a density of 500 cells mL−1, and 200 μL (100 cells) of this suspension seeded into the wells of a sterile 96-well tissue culture plate. The cells were allowed to attach and grow for 24 h and treated with serial dilutions of the drug for 240 h. After treatment, viable cells were measured using the CyQUANT cell proliferation assay kit (Invitrogen) as per manufacturer’s instructions.
Cytotoxic potentiation of various DNA damaging agents
T98G cells maintained in growth medium were resuspended in fresh growth medium and seeded into 96-well plates at 2000 cells/well (short term cytotoxicity) or 100 cells/well (long term cytotoxicity) as described in the previous section. After 24 h, the cells were treated with serial dilutions of various DNA damaging drugs in absence or presence of APE-1 inhibitors. At the end of the treatment period, cell survival was assayed using CellTiter 96 AQueous or CyQUANT kits for the short- and long-term studies respectively.
Quantification of abasic sites
T98G cells maintained in growth medium, were harvested by Trypsin-EDTA treatment, resuspended in fresh growth medium at a density of 2.5 × 105 cells mL−1, and 2 mL (5 × 105 cells) of this suspension was seeded into the wells of a sterile 6-well tissue culture plate. Cells were allowed to grow for 24 h, treated with the various compounds for different time points and/or at different concentrations. At the end of each treatment, adherent cells were harvested by trypsin-EDTA treatment and pooled with live and dead cells in culture supernatant. Pooled cells in suspension were pelleted by centrifugation, washed with 1 x PBS and genomic DNA was isolated using the DNAzol reagent (Invitrogen). DNA in solution was quantified by UV absorbance (λ260 nm) using a Synergy H1 multiwell plate reader (BioTek, Winooski, VT). Abasic sites in purified DNA were then reacted with the biotin-conjugated aldehyde reactive probe (ARP), immobilized onto 96-well ELISA plates, and detected using horseradish peroxidase (HRP)-conjugated streptavidin using a colorimetric substrate as per manufacturer’s instructions (Dojindo Molecular Technologies, Rockville, Maryland).
RESULTS AND DISCUSSION
The development of small molecule inhibitors of APE-1 has been challenging because of the structure around the protein’s active site that is rich in basic amino acid residues, which make salt bridge contacts with the DNA phosphate backbone. An Arg residue on a flexible loop region moves into the DNA stack and occupies the vacancy created when the abasic lesion is extruded into the protein’s active site.58 The hydrophobic face of the synthetic THF abasic site analogue in the crystal structure of APE-1 interacts with amino acid residues that create a hydrophobic pocket; however, the physiologically relevant 2-hydroxy-THF abasic site could form an H-bond via the 1′-α-hydroxyl group that is missing in the THF lesion.
Virtual Screening for Small-molecule Inhibitors of APE-1
In silico screening was performed using the Surflex-dock program65,66 and established docking protocols63 based on the X-ray crystallographic data of APE-1 (1DEW.PDB).58 The predicted binding pocket was validated through 3D database docking searches and confirmed through experiments using the in silico screened hits (discussed below). Figure 3 shows a docking pose of inhibitor 4 (Table 1) in the predicted binding pocket with the methylenedioxy ring mimicking the furan ring of the abasic site substrate in the X-ray co-crystallographic structure of APE-1.58 Analysis of the docking results reveal H-bonding interactions of compound 4 with Asn174 (2.33 Å), Thr268 (2.67 Å) and hydrophobic interactions (2.66 ~3.0 Å) of the dioxoloquinoline moiety with Phe266 and Trp280 as well as the carbazole ring with a hydrophobic domain (Trp280, Met271 and Val278). In the X-ray structure, the hydrophobic face of the extra-helical abasic lesion packs within a complementary APE-1 pocket formed by the side chains of Phe266, Trp280, and Leu282. In addition, Asn174, Asn212 and His309 also interact through H-bonds with the phosphate that is 5′ of the abasic site target. Our docking also predicted that the basic Arg177 side chain could interact with the aromatic quinoline ring of compound 4 via a Π-cation interaction (3.42 Å).
Figure 3.

Docking results of 4 with APE-1: (a) View of compound 4 docked into the x-ray crystallographic structure of APE-1 (1DEW.pdb);58 (b) Enlarged docking pose of 4 showing methylenedioxy ring inserted inside the binding pocket and carbazole ring pointing outward, forming strong H-bonding and hydrophobic interactions; (c) Relaxed stereoview of N-ethylcarbazole ring of compound 4 pointing out with benzodiazole ring system buried into active site of APE-1.
Table 1.
Structures of compounds identified from in silico screen and APE-1 endonuclease inhibition (Ki).
| Name | NCI# | Structure | Ki (μM)a |
|---|---|---|---|
| 1 | NSC332398 |
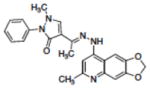
|
0.18 |
| 2 | NSC332384 |
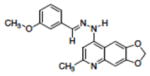
|
0.22 |
| 3 | NSC332389 |
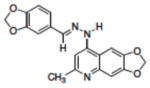
|
0.19 |
| 4 | NSC332395 |
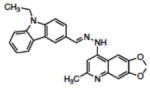
|
0.12 |
| 5 | NSC332396 |
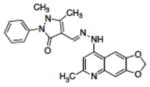
|
0.68 |
| 6 | NSC332397 |
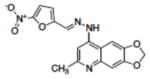
|
0.64 |
| 7 | NSC300598 |
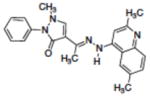
|
10.89 |
| 8 | NSC332410 |
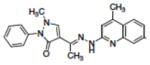
|
NDb |
| 9 | NSC89640 |
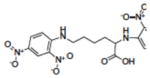
|
13.00 |
| 10 | NSC107215 |
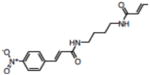
|
17.00 |
| 11 | NSC131534 |
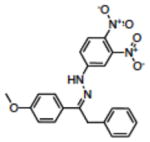
|
6.70 |
| 12 | NSC375491 |
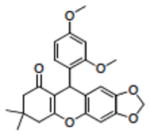
|
32.00 |
| 13 | NSC614430 |
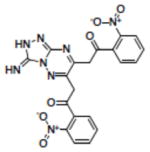
|
4.90 |
| 14 | NSC402686 |
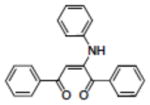
|
37.00 |
| 15 | NSC658900 |
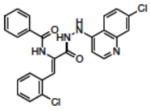
|
ND |
| 16 | NSC163444 |
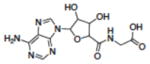
|
ND |
| 17 | NSC115605 |
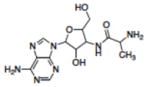
|
ND |
| 18 | NSC128335 |
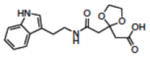
|
ND |
| 19 | NSC372329 |
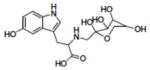
|
ND |
| 20 | NSC283787 |
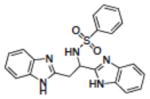
|
NDd |
| 21 | NSC11847 |
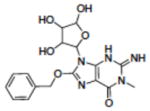
|
ND |
| 22 | NSC117589 |
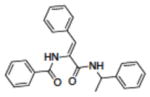
|
ND |
| 23 | NSC126939 |
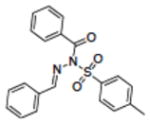
|
NDd |
| 24 | NSC296950 |
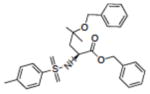
|
NDd |
| 25 | NSC343032 |
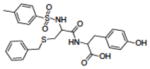
|
ND |
| 26 | NSC374123 |
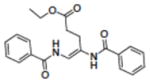
|
ND |
| 27 | NSC63636 |
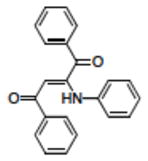
|
NDd |
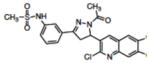
| 28 | CS-965570c |
|
ND |
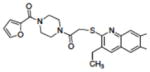
| 29 | CS-565759c |
|
ND |
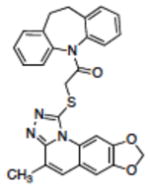
| 30 | CS-2504743c |
|
ND |
endonuclease activity.
no detectable activity (Ki > 100 μM).
Chemspider accession number
insufficient solubility in buffer containing 0.25% DMSO to determine if Ki is > 100 μM.
Screening for APE-1 Inhibitors
The initial assessment of the molecules suggested by the computational study employed a molecular beacon approach similar to that previously described.47,67,68 In this assay, the DNA hairpin substrate has a 6-FAM fluorophore on the 5′-terminus, a DABACYL fluorescence quencher on the 3′-terminus and a THF abasic site located 6 base pairs from the 5′-terminus in the stem region (Figure 4). APE-1 activity is monitored as an increase in fluorescence due to the release of the 5′-(6-FAM)-GCACT fragment as a function of time. Compounds that had good inhibitory activity (Ki ≤ 10 μM) in the molecular beacon screen were then retested using a gel-based assay to directly measure DNA excision as a function of inhibitor concentration (Figure 5). This second assay, which directly measures the amount and site of DNA nicking confirms that the endonuclease processing of the abasic lesion is inhibited and eliminates the possibility of artifacts that can arise in the fluorescence measurements or from random nuclease activity. In this assay, substrate turnover was calculated from relative intensities of cleaved vs. intact oligomers and correlated with inhibitor concentration. Using this approach, the IC50 values (Table 2) were estimated for APE-1 inhibition by 1 (4.4 μM), 2 (10.1 μM), 3 (2.9 μM), 4 (4.1 μM), 5 (34.4 μM), 8 (>100 μM) and E3330 (14.7 μM). The inhibitory activity (Ki) observed in the molecular beacon assay (Table 1, Figure 5) correlated in all cases with the IC50 activity in the gel assay (Table 2). Of the thirty compounds (Table 1) evaluated based on the modeling, six showed a Ki of < 1.0 μM in the molecular beacon assay (Table I, Figure 4). It was not possible to fully characterize some of the compounds because of their limited solubility. The relatively high hit rate in the molecular beacon assay is attributed to the success of the molecular modeling to predict structures in contrast to a random screen of a large chemical library. The importance of the benzo[d][1,3]dioxole structure and H-bond donor at position-N4 on the quinoline ring, which were predicted from the modeling, was evident from the assays. For example, compounds 1 and 8 are identical except for the methylenedioxy substitution on the quinoline ring, yet their effect on APE-1 activity differs by more than 50-fold. The dramatic difference between 1 and 7 also illustrates the important role of the dioxole ring. The appendage on the hydrazone had less impact on the in vitro inhibition. Compounds 1–5 and 8 were selected for additional evaluation based on the range of their in vitro activity.
Figure 4.

Inhibition of APE-1 endonuclease activity by 1–5, 8, 28–30, E3330 and EtBr in the molecular beacon assay (Ki values determined from this assay were calculated as described in Experimental Procedures and are shown in Table 1). (A) Sequence of THF modified hairpin DNA substrate, where, F, fluorophore and Q, quencher. .(B–D) Changes in APE-1 endonuclease activity as a function of varying concentrations of potential APE-1 inhibitor molecules: (B) 1(△) 2(□), 3 (○), 4 (◇); (C) 5 (△), 6 (□), EtBr (▼), E3330 (X); (D) 7 (○) 8 (◇); and (E) 28 (■), 29(●) and 30 (▲).
Figure 5.
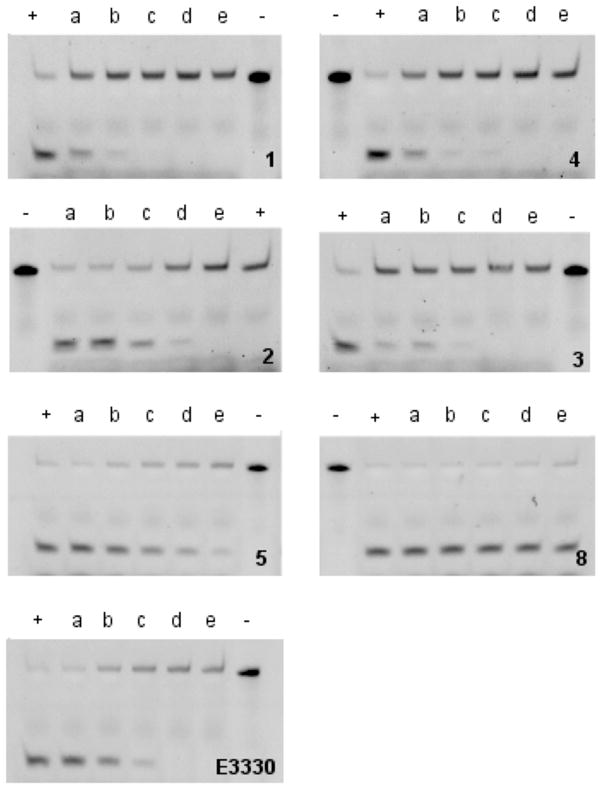
Gel based assay measuring the inhibition of APE-1 endonuclease activity by compounds 1–5, 8 and E3330 (upper band, uncleaved 30 nucleotide THF-modified DNA; lower band, endonuclease cleaved 11 nucleotide fragment): +, control (no inhibitor); −, uncleaved target; a–e, 5, 10, 25, 50 and 100 μM compound, respectively.
Table 2.
IC50 values for compounds 1, 2, 3, 4, 5, 8 and E3330 were calculated from the in gel excision assay by quantifying the relative intensities of the APE-1 cleaved fragment vs. full length DNA substrate in the absence or presence of potential APE-1 inhibitors.
| Compound | IC50 (μM) |
|---|---|
| 1 | 4.4 |
| 2 | 10.1 |
| 3 | 2.9 |
| 4 | 4.1 |
| 5 | 34.4 |
| 8 | >100 |
| E3330 | 14.7 |
In addition, to the compounds identified from the modeling studies, E3330, a previously reported inhibitor that has been shown to interact with the endonuclease active site of APE-1,54 was tested for comparative purposes. The Ki for E3330 is our system was 2.2 μM in the molecular beacon assay (Figure 4C) and the IC50 in the gel assay was 14.7 μM (Figure 5). In order to probe the potential role of DNA intercalation as a mechanism for APE-1 inhibition (see below), ethidium bromide (EtBr) was evaluated. It showed a Ki of 1.8 μM in the molecular beacon assay (Figure 4C) but its intercalating and fluorescence properties hindered analysis in the gel assay.
Inhibition of APE-1 activity in nuclear lysates
The ability of 1–5 and 8 to block APE-1 endonuclease activity in a complex mixture of proteins from T98G cell nuclear lysates was tested using the molecular beacon assay. The APE-1 activity in the lysates was standardized to give ~90% turnover of 25 nM substrate in 10 min. This activity is similar to that of 2 nM recombinant APE-1 used in the molecular beacon assay (Figure 4). The KM and Vmax of EC2nM APE-1 activity in nuclear lysates were determined to be 24.4 nM and 8.5 fluorescence units (arbitrary) min−1, respectively. Using these values, the Ki for 1–5 was calculated to be 0.45, 0.24, 0.28, 0.21 and 1.41 μM, respectively. For comparison E3330 had a Ki of 2.93 μM. Compounds 1–4 clearly blocked excision of the target DNA with the abasic site analogue in the presence of the nuclear lysate that contain APE-1 activity (Figure 6). These results indicate that compounds 1–5 have reasonable selectivity even in the complex mixture of proteins in the nuclear lysates.
Figure 6.

Inhibition of APE-1 endonuclease activity by 1 (△), 2 (□), 3(○), 4 (◇), 5 (▲), 8 (■) and E3330 (✗) in T98G cell nuclear extracts in molecular beacon assay (see Figure 4 for details).
Interaction of inhibitors with APE-1 protein
To establish whether there is a direct interaction between an inhibitor and APE-1, the fluorescence spectra of 4 was determined in the absence and presence of increasing amounts of the protein. Compound 4 is the only potent inhibitor that is fluorescent in buffer (containing 0.5% DMSO) with an excitation at 395 nm and a broad emission maximum at 485–520 nm. In the presence of APE-1, there is a concentration-dependent enhanced fluorescence of 4 (Figure 7), which suggests a physical interaction with the protein that reduces fluorescence quenching due to interaction with solvent. The emission maximum also shifts with the 485 nm band increasing to a smaller extent than the long wavelength band, which is displaced by 10 nm to 510 nm. These data suggest direct binding between the protein and the inhibitor. E3330 is an APE-1 inhibitor for which there are NMR data showing an interaction with the protein’s active site.54 Therefore, we incubated 4 (6.0 μM) with APE-1 (1 μM) and measured the change in fluorescence upon the addition of 0.5 to 10.0 μM E3330 (Figure 7). The results show that there is an E3330 concentration-dependent displacement of 4 from the protein as indicated by the decrease in 4’s fluorescence intensity. To obtain approximately 50% decrease in the fluorescence of 6 μM 4, requires approximately 12 μM E3330. E3330 binds to APE-1 with a KD of 55 μM from the NMR study and causes a 3-fold inhibition of APE-1 endonuclease activity at 100 μM concentration in a gel based excision assay.54 The IC50 for E3330 redox inhibition is 10 μM.47 The displacement of 4 from APE-1 by E3330 indicates that 4 binds to, or near, the endonuclease site of the protein.
Figure 7.

Concentration dependent change in the fluorescence of 4 in the presence of 1 μM APE-1: (A) binding of 4 to APE-1 as measured by the increase in fluorescence (excitation at 395 nm, emission at 510 nm); KD of 6.15 μM; (B) displacement of 4 from APE-1 by E3330 measured by decrease in the fluorescence of 4.
Interaction of Inhibitors with DNA
A potential mechanism by which compounds can block APE-1 activity involves the interaction with the DNA substrate by intercalation. Such binding to DNA could sterically reduce the accessibility of the protein to the abasic site substrate. The intercalation of compounds that have planar aromatic systems is common and relevant because the abasic lesion potentially provides a high affinity site for the planar compounds to enter into the DNA base stack. The interaction of 1, which exhibits no fluorescence in solution, with an unmodified DNA oligomer and one with an abasic site causes a concentration dependent increase in fluorescence intensity (Figure 8). Compound 4 shows a similar fluorescence response. To further probe the potential role of intercalation, the effect of EtBr on the fluorescence of 1 and 4 were determined. Ethidium is a well-characterized intercalator that has a KD near 1 μM with double-stranded DNA,71 although it binds almost 10-fold stronger to DNA with a bulge.72 However, we observed similar KD’s for EtBr with the unmodified and THF modified hairpin DNA of 1.6 and 1.8 μM, respectively (data not shown). The fluorescence enhancement of 1 and 4 in the presence of DNA was significantly muted by the presence of EtBr suggesting competitive binding to the DNA. Of note, is that 1 appears to exhibit a strong preference for DNA containing the THF abasic site based on the difference in the fluorescence intensities (Figure 8). The small changes in the fluorescence of 4 with both DNAs indicate little discrimination for the THF lesion. A similar pattern was observed in the effect of 1 and 4 on the thermal stability of DNA as determined by UV melting experiments using several DNA substrates (data not shown). With the self-complementary sequence 5′-GAGAGCGCTCTC, 1 increased the TM from 36.4 to 40.8 °C while the TM for this sequence with 4 was unchanged (37.3 °C). Using the unmodified hairpin DNA has shown in Figure 8B, we observed a similar effect: ΔTM was 4 °C with 1 and 0.5 °C with 4. The stability of the hairpin with the THF abasic site increased by > 9 °C in the presence of 1, but not appreciably with 4. These data suggest weak binding between DNA and 4, while 1 clearly has a stronger interaction with both unmodified DNA and DNA with a THF abasic site.
Figure 8.

The interaction of compounds 1 and 4 with DNA was evaluated by changes in their fluorescence in the absence and presence of EtBr: (A) sequence of DNA hairpins used in the fluorescence measurements (X = THF abasic site); (B) Fluorescence of compounds 1 (395 nm excitation and 460 nm emission) and 4 (400 nm excitation and 500 nm emission) with 0.5 μM DNA was evaluated by fluorescence increases in the absence (●, 1 with unmodified DNA; ■, 1 with THF modified DNA; ◆, 4 with unmodified DNA; ▲, 4 with THF modified DNA) and presence of 4 μM EtBr (○, 1 with unmodified DNA and EtBr; □, 1 with THF modified DNA and EtBr; ◇, 4 with unmodified DNA and EtBr; △, 4 with THF modified DNA and EtBr).
In summary, both 1 and 4 can bind to DNA, presumably by intercalation, but the binding is relatively weak, as compared to EtBr, and non-specific and cannot explain their ability to inhibit APE-1 endonuclease activity at nM concentrations in the different assays. Consistent with this conclusion is the weaker inhibition of APE-1 endonuclease activity by EtBr, which has a higher affinity for DNA than 1 or 4. The overall evidence is consistent with inhibitor 4 acting by binding directly to the protein. The mechanism of action for some of the other inhibitors remains to be experimentally determined.
Toxicity
It was assumed that inhibition of APE-1 activity would be lead to toxicity since genetic deletion of the protein is lethal to cells.8 Therefore, the toxicities of compounds 1–5 and E3330 were determined in human T98G glioma cells using both short-term (3-day) MTS (Figure 9a) and long term (10-day) CyQUANT (Figure 9b) assays. The toxicities were also determined in combination with the DNA minor groove methylating agent, MeLex. MeLex efficiently and selectively generates N3-methyladenine, which is converted into an abasic site by a BER glycosylase73–75 and/or hydrolytic depurination.76 The T98G cell line is relatively resistant to DNA damaging anticancer agents due to expression of multiple drug resistance proteins and O6-alkylguanine-DNA alkyltransferase.77,78 Compound 4 was again the most active with an LD50 of 960 nM in the MTS assay and 190 nM in the clonogenic assay. The toxicities of compounds 1–3 were all approximately 500 nM in the long term assay and < 5 μM in the short term assay, while 5 was almost 20-fold less toxic than 4. Of note, E3330 was approximately 250- and 750-fold less toxic to the cells than 4 in the short- and long-term assays, respectively. The previously reported E3330 IC50 for cell growth in HEYC-2 and SKOV-3X ovarian tumor cells is ~35 μM.79 As a reference, the methylating agent, MeLex showed an LD50 of 250 and 12 μM in the short and long term cytotoxicity assays, respectively. The mechanism responsible for the toxicity of the inhibitor compounds remains to be determined, but their activities generally correlate with the Ki for in vitro APE-1 inhibition (Table I) suggesting that the decrease in APE-1 activity is involved.
Figure 9.

The toxicity of compounds 1 (■), 2 (□), 3 (●), 4 (○), 5 (▲), MeLex (△) and E3330 (✗) were determined in T98G cells : (A) short term (72 h) MTS assay; (B) 10-day Cyquant assay.
A goal of the development of APE-1 inhibitors is to determine if they can be used to enhance the activity of DNA alkylating agents. Therefore, we tested how the LD10 concentrations of compounds 1 (2.20 μM), 4 (0.25 μM) and 5 (6.50 μM) affected the toxicity of MeLex (Figure 10) in the 72 h MTS assay. The LD50 of MeLex dropped from 267 μM by itself to 54, 49 and 80 μM in the presence of 1, 4 and 5, respectively (Figure 10A). When an LD10 concentration of E3330 (6.9 μM) was combined with MeLex, the cells were only 2-fold more sensitive to MeLex (128 μM). In a long term assay the corresponding LD10 concentrations of 1 (0.17 μM), 4 (0.08 μM), and 5 (0.90 μM) caused a decrease in the LD50 of MeLex (9.9 μM) to 2.0, 1.6 and 3.6 μM respectively. E3330 (14.80 μM) had little or no contribution towards potentiating the toxicity of MeLex (Figure 10B). Overall the maximum potentiation (i.e., with 4) of MeLex was approximately 5-fold for both the short and long term assays.
Figure 10.

The potentiation of MeLex cytotoxicity was determined in T98G cells treated with different concentrations of MeLex alone (◆) or in combination with LD10 concentrations of 1 (■), 4 (○), 5 (▲), or E3330 (✗). (A) short-term (72 h) MTS assay; (B) long term (10 d) CyQUANT assay.
Some of the compounds that we tested are closely related to a series of molecules evaluated for antimalarial80 and antitubercular activity.81 The IC50 for the toxicity of compound 3 in a VERA kidney cell line was determined to be 630 nM, which compares to the 540 nM that we observed in the glioma T98G line (Table 1). The origin of the toxicity in the VERA cells was not determined but the inhibition of APE-1 would be predicted to be toxic in all cells.
Formation of Abasic Sites
To confirm that the inhibitors were actually affecting the cellular processing of abasic sites, a biotinylated aldehyde reactive probe (Biotin-ARP)82 was used to measure the level of aldehyde reactive sites (ARS) in genomic DNA isolated from T98G cells that were incubated with the different inhibitors. The biotin tagged ARS are quantified using an avidin-biotin assay, followed by colorimetric detection with horseradish peroxidase conjugated to avidin. While the method does not specifically measure abasic DNA sites, it is assumed that most of the increase in signal results from an increase in the ring-opened form of abasic sites (Figure 2). The background level of ARS in untreated or 0.25% DMSO treated T98G cells was 1/105 nucleotides, which is similar to previous reports (Figure 11a).82,83 In the presence of 0.25 μM 4 (Figure 11), which corresponds to less than the LD10 concentration due to the 25-fold increase in the number of cells used in the ARS vs. the toxicity assay (Figure 9), there was a continuous increase over the initial 6 h. The level of approximately 17 ARS/105 nucleotides was maintained until the final 18 h time point. Because the concentration of the inhibitors is below the LD10, it is unlikely that a significant percentage of the ARS measured are in dead cells. However, it cannot be ruled out that ARS levels may rise in cells entering or in apoptosis. Therefore, it is not possible to unequivocally say that the accumulation of ARS leads to cell death or vice versa. A more detailed time course for markers of cell death and the build up of ARS may resolve this issue.
Figure 11.

Formation of aldehyde reactive sites (ARS, abasic sites) was quantified in: (A) cells treated with 0.25 μM 1 (■), 4 (○), 5 (▲), 8 (◇), E3330 (✗), EtBr (▼) or 80 μM MeLex (△); (B) cells treated with combinations of 80 μM MeLex (LD10) and 0.25 μM of 1 (■), 4 (○), 5 (▲), E3330 (✗), EtBr (▼). Controls employed in both sets were untreated cells (●) or cells treated with 0.2% DMSO (◆).
At 0.25 μM, 1 showed a similar effect, but 5, which is a weaker inhibitor of APE-1 endonuclease activity (Table I), had a less dramatic effect with an increase in ARS of approximately 4-fold above background. The number of ARS with 0.25 μM 8 was the same as background. For comparison, 0.25 μM E3330 caused an increase in ARS similar to 1 and 4 at 2 h but then the number of lesions began to drop and returned to control levels by 18 h. EtBr was also evaluated at similar concentrations to determine if a strong intercalator could affect ARS levels. It had no affect on the formation of ARS. As a positive control for the formation of ARS, the cells were treated with 80 μM of MeLex. This MeLex treatment afforded a rapid induction of ARS (36/105 nucleotides) through the first 4 h that then decreased through 18 h to a value similar to that observed with 1 and 4. The rapid increase in ARS from MeLex through the first 4 h is consistent with the DNA methylation time course,81 while the gradual decrease of ARS levels is attributed to their removal by functional BER.
The effect of APE-1 inhibitors in combination with 80 μM MeLex was also determined (Figure 11b). At the time points employed in our assays, the level of ARS reflects the kinetics of 3-mA lesion formation due to MeLex, and BER processing of the lesion by MPG excision, APE-1 cleavage at the abasic site and dRP lyase or flap endonuclease removal of the aldehyde functionality from the DNA. The level of ARS induced by MeLex by itself or with the different inhibitor compounds was not significantly different at the 2 or 6 h time points. However, the number of lesions dropped off with MeLex alone or with either 0.25 μM E3330 or EtBr. In contrast, the levels of ARS continued to increase through at least 12 h when MeLex was combined with 0.25 μM of the more potent inhibitors 1 (50 sites/105 nucleotides) or 4 (63 sites/105 nucleotides). Therefore, the combination of DNA methylation and APE-1 inhibition has the effect of sustaining an elevated level of ARS, which correlates with the enhancement of MeLex toxicity.
APE-1 Redox Activity
APE-1 has a redox activity that is associated with the regulation of specific transcriptional factors. To probe whether any of the molecules identified as APE-1 endonuclease inhibitors affected the redox activity, the ability of APE-1 to affect Jun/Fos binding to an SP-1 cognate DNA sequence was determined using a gel shift assay as described previously.79 E3330 was used as a positive control. We observed no effect with 100 μM concentration of 1–5 and 8 (data not shown), which is far above the concentration required to inhibit endonuclease activity. In summary, the compounds that are potent inhibitors of APE-1 endonuclease activity increase the persistence of ARS and synergistically affect the toxicity of a DNA methylating agent that generates lesions that are repaired by BER. Structural studies are underway to establish the binding mode of the different APE-1 inhibitors that will be used to synthesize more potent and selective APE-1 inhibitors.
Acknowledgments
We thank Prema Iyer for synthesis of MeLex and Manjori Ganguly for TM measurements.
Funding: This work was supported by National Institutes of Health Grant RO1 CA29088 (BG) and GM087798 (RWS).
Footnotes
ABBREVIATIONS: APE-1, human apurinic endonuclease-1/redox effector factor-1; ARS, aldehyde reactive sites; BER, base excision repair; DMSO dimethyl sulfoxide; E3330, (2E)-2-[(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cyclohexadien-1-yl)methylene]undecanoic acid; EtBr, ethidium bromide; HEPES, 4-(2-hydroxy-ethyl)-1-piperazineethanesulfonic acid; HR, homologous recombination; MeLex, methyl 3-(1-methyl-5-(1-methyl-5-(propylcarbamoyl)-1H-pyrrol-3-ylcarbamoyl)-1H-pyrrol-3-ylamino)-3-oxopropane-1-sulfonate; MMS, methyl methane-sulfonate; PBS, phosphate buffered saline; THF, tetrahydrofuran.
References
- 1.Yu SL, Lee SK, Johnson RE, Prakash L, Prakash S. The stalling of transcription at abasic sites is highly mutagenic. Mol Cell Biol. 2003;23:382–388. doi: 10.1128/MCB.23.1.382-388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 4.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet. 1998;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 5.Wilson DM, III, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 6.Strauss PR, Beard WA, Patterson TA, Wilson SH. Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J Biol Chem. 1997;272:1302–1307. doi: 10.1074/jbc.272.2.1302. [DOI] [PubMed] [Google Scholar]
- 7.Goellner EM, Svilar D, Almeida KH, Sobol RW. Targeting DNA polymerase β for therapeutic intervention. Curr Mol Pharmacol. 2012;5:68–87. [PMC free article] [PubMed] [Google Scholar]
- 8.Glassner BJ, Weeda G, Allan JM, Broekhof JL, Carls NH, Donker I, Engelward BP, Hampson RJ, Hersmus R, Hickman MJ, Roth RB, Warren HB, Wu MM, Hoeijmakers JH, Samson LD. DNA repair methyltransferase (Mgmt) knockout mice are sensitive to the lethal effects of chemotherapeutic alkylating agents. Mutagenesis. 1999;14:339–347. doi: 10.1093/mutage/14.3.339. [DOI] [PubMed] [Google Scholar]
- 9.Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parsons JL, Elder RH. DNA N-glycosylase deficient mice: a tale of redundancy. Mutat Res. 2003;531:165–175. doi: 10.1016/j.mrfmmm.2003.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell. 2000;5:1059–1065. doi: 10.1016/s1097-2765(00)80271-3. [DOI] [PubMed] [Google Scholar]
- 12.Demple B, Fung H. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Molec Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 13.Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci USA. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, Meneses J, Pedersen RA, Chen DJ. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat Res. 1998;409:17–29. doi: 10.1016/s0921-8777(98)00039-1. [DOI] [PubMed] [Google Scholar]
- 15.Bobola MS, Finn LS, Ellenbogen RG, Geyer JR, Berger MS, Braga JM, Meade EH, Gross ME, Silber JR. Apurinic/apyrimidinic endonuclease activity is associated with response to radiation and chemotherapy in medulloblastoma and primitive neuroectodermal tumors. Clin Cancer Res. 2005;11:7405–7414. doi: 10.1158/1078-0432.CCR-05-1068. [DOI] [PubMed] [Google Scholar]
- 16.Ono Y, Furuta T, Ohmoto T, Akiyama K, Seki S. Stable expression in rat glioma cells of sense and antisense nucleic acids to a human multifunctional DNA repair enzyme, APEX nuclease. Mutat Res. 1994;315:55–63. doi: 10.1016/0921-8777(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 17.Walker LJ, Craig RB, Harris AL, Hickson ID. A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res. 1994;22:4884–4889. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobol RW, Horton JK, Kühn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase β in base excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 19.Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000;19:1397–1404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Shupenko CC, Melo LF, Strauss PR. DNA repair protein involved in heart and blood development. Mol Cell Biol. 2006;26:9083–9093. doi: 10.1128/MCB.01216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pei DS, Yang XJ, Liu W, Guikema JE, Schrader CE, Strauss PR. A novel regulatory circuit in base excision repair involving AP endonuclease 1, Creb1 and DNA polymerase beta. Nucleic Acids Res. 2011;39:3156–3165. doi: 10.1093/nar/gkq1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xanthoudakis S, Miao GG, Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc Natl Acad Sci USA. 1994;91:23–27. doi: 10.1073/pnas.91.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 24.Fritz G. Human APE/Ref-1 protein. Int J Biochem Cell Biol. 2000;32:925–929. doi: 10.1016/s1357-2725(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 25.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zou GM, Karikari C, Kabe Y, Handa H, Anders RA, Maitra A. The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: therapeutic implications in tumor angiogenesis. J Cell Physiol. 2009;219:209–218. doi: 10.1002/jcp.21666. [DOI] [PubMed] [Google Scholar]
- 27.Vascotto C, Fantini D, Romanello M, Cesaratto L, Deganuto M, Leonardi A, Radicella JP, Kelley MR, D’Ambrosio C, Scaloni A, Quadrifoglio F, Tell G. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol. 2009;29:1834–1854. doi: 10.1128/MCB.01337-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshida A, Urasaki Y, Waltham M, Bergman AC, Pourquier P, Rothwell DG, Inuzuka M, Weinstein JN, Ueda T, Appella E, Hickson ID, Pommier Y. Human apurinic/apyrimidinic endonuclease (Ape1) and its N-terminal truncated form (AN34) are involved in DNA fragmentation during apoptosis. J Biol Chem. 2003;278:37768–37776. doi: 10.1074/jbc.M304914200. [DOI] [PubMed] [Google Scholar]
- 30.Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci USA. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McNeill DR, Lam W, DeWeese TL, Cheng YC, Wilson DM., III Impairment of APE1 function enhances cellular sensitivity to clinically relevant alkylators and antimetabolites. Mol Cancer Res. 2009;7:897–906. doi: 10.1158/1541-7786.MCR-08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008;68:6425–6434. doi: 10.1158/0008-5472.CAN-08-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Wang J, Xiang D, Wang D, Xin X. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE1/Ref-1) in human ovarian cancer and indentification of the therapeutic potential of APE1/Ref-1 inhibitor. Int J Oncol. 2009;35:1069–1079. [PubMed] [Google Scholar]
- 34.Naidu MD, Mason JM, Fung H, Pena L. Radiation resistance in human glioma cell lines is determined by the DNA damage repair activity of apurinic endonuclease 1 (APE-1) J Radiat Res. 2010;51:393–404. doi: 10.1269/jrr.09077. [DOI] [PubMed] [Google Scholar]
- 35.Sultana R, McNeill DR, Abbotts R, Mohammed MZ, Zdzienicka MZ, Qutob H, Seedhouse C, Laughton CA, Fischer PM, Patel PM, Wilson DM, 3rd, Madhusudan S. Synthetic lethal targeting of DNA double strand break repair deficient cells by human apurinic/apyrimidinic endonuclease (APE1) inhibitors. Int J Cancer. 2012 doi: 10.1002/ijc.27512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowosielska A, Smith SA, Engelward BP, Marinus MG. Homologous recombination prevents methylation-induced toxicity in Escherichia coli. Nucleic Acids Res. 2006;34:2258–2268. doi: 10.1093/nar/gkl222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendricks CA, Razlog M, Matsuguchi T, Goyal A, Brock AL, Engelward BP. The S. cerevisiae Mag1 3-methyladenine DNA glycosylase modulates susceptibility to homologous recombination. DNA Repair. 2002;1:645–659. doi: 10.1016/s1568-7864(02)00072-1. [DOI] [PubMed] [Google Scholar]
- 38.Memisoglu A, Samson L. Contribution of base excision repair, nucleotide excision repair, and DNA recombination to alkylation resistance of the fission yeast Schizosaccharomyces pombe. J Bacteriol. 2000;182:2104–2112. doi: 10.1128/jb.182.8.2104-2112.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farmer H, McCabe N, Lord CJ, Turner N, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 40.Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr Op Pharmacol. 2008;8:363–369. doi: 10.1016/j.coph.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 41.Luo M, Kelley MR. Inhibition of the human apurinic/apyrimidinic endonuclease (APE1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer Res. 2004;24:2127–2134. [PubMed] [Google Scholar]
- 42.Madhusudan S, Smart F, Shrimpton P, Parsons JL, Gardiner L, Houlbrook S, Talbot DC, Hammonds T, Freemont PA, Sternberg MJ, Dianov GL, Hickson ID. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 2005;33:4711–4724. doi: 10.1093/nar/gki781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou GM, Maitra A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Ther. 2008;7:2012–2021. doi: 10.1158/1535-7163.MCT-08-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seiple LA, Cardellina JH, II, Akee R, Stivers JT. Potent inhibition of human apurinic/apyrimidinic endonuclease 1 by arylstibonic acids. Mol Pharmacol. 2008;73:669–677. doi: 10.1124/mol.107.042622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simeonov A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, McNeill DR, Austin CP, Wilson DM., III Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS One. 2009;4:e5740. doi: 10.1371/journal.pone.0005740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zawahir Z, Dayam R, Deng J, Pereira C, Neamati N. Pharmacophore guided discovery of small-molecule human apurinic/apyrimidinic endonuclease 1 inhibitors. J Med Chem. 2009;52:20–32. doi: 10.1021/jm800739m. [DOI] [PubMed] [Google Scholar]
- 47.Nyland RL, Luo M, Kelley MR, Borch RF. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1) J Med Chem. 2010;53:1200–1210. doi: 10.1021/jm9014857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naidu MD, Agarwal R, Pena LA, Cunha L, Mezei M, Shen M, Wilson DM, III, Liu Y, Sanchez Z, Chaudhary P, Wilson SH, Waring MJ. Lucanthone and its derivative hycanthone inhibit apurinic endonuclease-1 (APE1) by direct protein binding. PLoS One. 2011;6:e23679. doi: 10.1371/journal.pone.0023679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Molec Aspects Med. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 50.Nagakawa J, Hishinuma I, Miyamoto K, Hirota K, Abe S, Yamanaka T, Katayama K, Yamatsu I. Protective effects of (2E)-3-[5-(2,3-dimethoxy-6-methyl-1,4-benzoquinoyl)]-2-nonyl-2-propenoic acid on endotoxin-mediated hepatitis in mice. J Pharmacol Exp Ther. 1992;262:145–150. [PubMed] [Google Scholar]
- 51.Miyamoto K, Nagakawa J, Hishinuma I, Hirota K, Yasuda M, Yamanaka T, Katayama K, Yamatsu I. Suppressive effects of E3330, a novel quinone derivative, on tumor necrosis factor-alpha generation from monocytes and macrophages. Agents Actions. 1992;37:297–304. doi: 10.1007/BF02028123. [DOI] [PubMed] [Google Scholar]
- 52.Zou GM, Luo MH, Reed A, Kelley MR, Yoder MC. Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood. 2007;109:1917–1922. doi: 10.1182/blood-2006-08-044172. [DOI] [PubMed] [Google Scholar]
- 53.Su D, Delaplane S, Luo M, Rempel DL, Vu B, Kelley MR, Gross ML, Georgiadis MM. Interactions of apurinic/apyrimidinic endonuclease with a redox inhibitor: Evidence for an alternate conformation of the enzyme. Biochemistry. 2011;50:82–92. doi: 10.1021/bi101248s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manvilla BA, Wauchope O, Seley-Radtke KL, Drohat AC. NMR studies reveal an unexpected binding site for a redox inhibitor of AP endonuclease 1. Biochemistry. 2011;50:10540–10549. doi: 10.1021/bi201071g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu L, Gerson SL. Therapeutic impact of methoxyamine: blocking repair of abasic sites in the base excision repair pathway. Curr Opin Investig Drugs. 2004;5:623–627. [PubMed] [Google Scholar]
- 56.Liu L, Yan L, Donze JR, Gerson SL. Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol Cancer Ther. 2003;2:1061–1066. [PubMed] [Google Scholar]
- 57.see http://clinicaltrials.gov/ct2/show/NCT00892385
- 58.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Chen F-X, Mehta P, Gold B. The design of groove and sequence selective alkylation of DNA by sulfonate esters tethered to lexitropsins. Biochemistry. 1993;32:7954–7965. doi: 10.1021/bi00082a017. [DOI] [PubMed] [Google Scholar]
- 60.Shah D, Kelly J, Zhang Y, Dande P, Martinez J, Ortiz G, Fronza G, Tran H, Soto AM, Marky LA, Gold B. Evidence in Escherichia coli that N3-methyladenine lesions induced by a minor groove binding methyl sulfonate ester can be processed by both base and nucleotide excision repair. Biochemistry. 2001;40:1796–1803. doi: 10.1021/bi0024658. [DOI] [PubMed] [Google Scholar]
- 61.Baldwin MR, O’Brien PJ. Human AP endonuclease 1 stimulates multiple-turnover base excision by alkyladenine DNA glycosylase. Biochemistry. 2009;48:6022–6033. doi: 10.1021/bi900517y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie XQ, Chen JZ, Billings EM. 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Protein Struct Funct Genet. 2003;53:307–319. doi: 10.1002/prot.10511. [DOI] [PubMed] [Google Scholar]
- 63.Chen JZ, Xie XQ. GPCR structure-based virtual screening approach for the CB2 antagonist search. J Comput Info Model. 2007;47:1626–1637. doi: 10.1021/ci7000814. [DOI] [PubMed] [Google Scholar]
- 64.Xie XQ, Chen J. Data-mining a small molecule drug screening representative subset from NIH PubChem database. J Comput Info Model. 2008;48:465–475. doi: 10.1021/ci700193u. [DOI] [PubMed] [Google Scholar]
- 65.Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 66.Jain AN. Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput-Aided Mol Des. 1996;10:427–440. doi: 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- 67.Maksimenko A, Ishchenko AA, Sanz G, Laval J, Elder RH, Saparbaev MK. A molecular beacon assay for measuring base excision repair activities. Biochem Biophys Res Commun. 2004;319:240–246. doi: 10.1016/j.bbrc.2004.04.179. [DOI] [PubMed] [Google Scholar]
- 68.Tang J, Svilar D, Trivedi RN, Wang XH, Goellner EM, Moore B, Hamilton RL, Banze LA, Brown AR, Sobol RW. N-methylpurine DNA glycosylase and DNA polymerase β modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro Oncol. 2011;13:471–486. doi: 10.1093/neuonc/nor011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 70.Ganguly M, Szulik MW, Donahue PS, Clancy K, Michael PStone MP, Gold B. Thermodynamic signature of DNA damage: characterization of DNA with a 5-hydroxy-2′-deoxycytidine•2′-deoxyguanosine base pair. Biochemistry. 2012;51:2018–2027. doi: 10.1021/bi3000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rentzeperis D, Alessi K, Marky LA. Thermodynamics of DNA hairpins: contribution of loop size to hairpin stability and ethidium binding. Nucleic Acids Res. 1993;21:2683–2689. doi: 10.1093/nar/21.11.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nelson JW, Tinoco I., Jr Ethidium ion binds more strongly to a DNA double helix with a bulged cytosine than to a regular double helix. Biochemistry. 1985;24:6416–6421. doi: 10.1021/bi00344a016. [DOI] [PubMed] [Google Scholar]
- 73.Sakumi K, Sekiguchi M. Structures and functions of DNA glycosylases. Mutat Res. 1990;236:161–172. doi: 10.1016/0921-8777(90)90003-n. [DOI] [PubMed] [Google Scholar]
- 74.Engelward BP, Dreslin A, Christensen J, Huszar D, Kurahara C, Samson LD. Repair-deficient 3-methyladenine DNA glycosylase homozygous mutant mouse cells have increased sensitivity to alkylation-induced chromosome damage and cell killing. EMBO J. 1996;15:945–952. [PMC free article] [PubMed] [Google Scholar]
- 75.Engelward BP, Weeda G, Wyatt MD, Broekhof JL, de Wit J, Donker I, Allan JM, Gold B, Hoeijmakers JH, Samson LD. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc Natl Acad Sci USA. 1997;94:13087–13092. doi: 10.1073/pnas.94.24.13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fujii T, Saito T, Nakasaka T. Purines. XXXIV 3 Methyladeno-sine and 3-methyl-2′-deoxyadenosine: their synthesis, glycosidic hydrolysis, and ring fission. Chem Pharmaceut Bull. 1989;37:2601–2609. [Google Scholar]
- 77.Abe1 T, Koike K, Ohga T, Kubo T, Wada M, Kohno K, Mori T, Hidaka K, Kuwano M. Chemosensitisation of spontaneous multidrug resistance by a 1,4-dihydropyridine analogue and verapamil in human glioma cell lines overexpressing MRP or MDR1. Brit J Cancer. 1995;72:418–423. doi: 10.1038/bjc.1995.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kanzawa T, Bedwell J, Kondo Y, Kondo S, Germano IM. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J Neurosurg. 2003;99:1047–1052. doi: 10.3171/jns.2003.99.6.1047. [DOI] [PubMed] [Google Scholar]
- 79.Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, II, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fattorusso C, Campiani G, Kukreja G, Persico M, Butini S, Romano MP, Altarelli M, Ros S, Brindisi M, Savini L, Novellino E, Nacci V, Fattorusso E, Parapini S, Basilico N, Taramelli D, Yardley V, Croft S, Borriello M, Gemma S. Design, synthesis, and structure-activity relationship studies of 4-quinolinyl- and 9-acrydinylhydrazones as potent antimalarial agents. J Med Chem. 2008;51:1333–1343. doi: 10.1021/jm7012375. [DOI] [PubMed] [Google Scholar]
- 81.Gemma S, Savini L, Altarelli M, Tripaldi P, Chiasserini L, Coccone SS, Kumar V, Camodeca C, Campiani G, Novellino E, Clarizio S, Delogu G, Butini S. Development of antitubercular compounds based on a 4-quinolylhydrazone scaffold. Further structure-activity relationship studies. Bioorg Med Chem. 2009;17:6063–6072. doi: 10.1016/j.bmc.2009.06.051. [DOI] [PubMed] [Google Scholar]
- 82.Atamna H, Cheung I, Ames BN. A method for detecting abasic sites in living cells: age-dependent changes in base excision repair. Proc Natl Acad Sci USA. 2000;97:686–691. doi: 10.1073/pnas.97.2.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res. 1998;58:222–225. [PubMed] [Google Scholar]
- 84.Shah S, Kelly J, Zhang Y, Dande P, Martinez J, Ortiz G, Fronza G, Tran H, Soto MA, Marky L, Gold B. Evidence in Escherichia coli that N3-methyladenine lesions induced by a minor groove binding methyl sulfonate ester can be processed by both base and nucleotide excision repair. Biochemistry. 2001;40:1796–1803. doi: 10.1021/bi0024658. [DOI] [PubMed] [Google Scholar]
- 85.Mohammed MZ, Vyjayanti VN, Laughton CA, Dekker LV, Fischer PM, Wilson DM, 3rd, Abbotts R, Shah S, Patel PM, Hickson ID, Madhusudan S. Development and evaluation of human AP endonuclease inhibitors in melanoma and glioma cell lines. Br J Cancer. 2011;104:653–663. doi: 10.1038/sj.bjc.6606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bapat A, Glass LS, Luo M, Fishel ML, Long EC, Georgiadis MM, Kelley MR. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J Pharmacol Exp Ther. 2010;334:988–998. doi: 10.1124/jpet.110.169128. [DOI] [PMC free article] [PubMed] [Google Scholar]