

Abstract
The integrity of the p53 tumor suppressor pathway is compromised in the majority of cancers. In 7% of cancers, p53 is inactivated by abnormally high levels of MDM2—an E3 ubiquitin ligase that polyubiquitinates p53, marking it for degradation. MDM2 engages p53 through its hydrophobic cleft and blockage of that cleft by small molecules can re-establish p53 activity. Small molecule MDM2 inhibitors have been developed, but there is likely to be a high cost and long time period before effective drugs reach the market. An alternative is to repurpose FDA-approved drugs. This report describes a new approach, called Computational Conformer Selection, to screen for compounds that potentially inhibit MDM2. This screen was used to computationally generate up to 600 conformers of 3,244 FDA-approved drugs. Drug conformer similarities to 41 computationally-generated conformers of MDM2 inhibitor nutlin 3a were ranked by shape and charge distribution. Quantification of similarities by Tanimoto combo scoring resulted in scores that ranged from 0.142 to 0.802. In silico docking of drugs to MDM2 was used to calculate binding energies and to visualize contacts between the top-ranking drugs and the MDM2 hydrophobic cleft. We present 15 FDA-approved drugs predicted to inhibit p53/MDM2 interaction.
Keywords: MDM2, cancer, nutlin, FDA, repurposing, repositioning, p53, drugs
Introduction
The p53 tumor suppressor is a transcription factor that enhances transcription of genes required for DNA repair, cell cycle arrest, apoptosis and angiogenesis in response to cellular stress [1]. The p53 gene is mutated in a wide range of human cancers at an overall frequency of approximately 50% [2]. In cancers with wild-type p53 genes, defects in the p53 pathway have been observed [3]. One defect is the overexpression of MDM2, a negative regulator of p53. MDM2 overexpression is due to gene amplification at an overall frequency of 7% of human cancers [4, 5]. MDM2 overexpression can occur in cancers without gene amplification due to increased transcription and increased expression from MDM2 promoter elements responsive to Smad3/4 and SP1 [6, 7]. MDM2 is an E3 ligase that binds to and ubiquitinates p53, targeting the tumor suppressor for degradation by the 26S proteasome [8–10]. As part of its feedback pathway, p53 mediates transcription of the MDM2 gene, the product of which maintains p53 at relatively low levels in normal proliferating cells [11]. If cells are stressed (by, for example, DNA damage or oncogene activation), p53 and MDM2 become phosphorylated and interact with factors that inhibit p53-MDM2 complex formation. In the absence of bound MDM2, the p53 level rises in the cell nucleus and activates transcription of appropriate genes that ultimately prevent cancer. When the p53 level is suppressed, cells undergo DNA replication in spite of the fact that the genome is damaged resulting in mutations [12]. If the mutations deactivate tumor suppressors or activate proto-oncogenes, cells can become incapable of responding to growth-restraint signals and cancer ensues.
In some cancers with a wild-type p53 genotype, abnormally high MDM2 levels accumulate and inhibit the p53 pathway. This scenario occurs in up to 20% of soft tissue tumors [4] and with an overall frequency of 7% in 19 different tumor types [5]. One approach to combating these cancers is to relieve the suppression of wild-type p53 by inhibiting MDM2. Early studies using antisense oligonucleotides showed that MDM2 may be a viable druggable target [13].
One well-studied small molecule MDM2 inhibitor is nutlin 3a, a cis-imidazoline compound with an IC50 of 90 nM for inhibition of p53 binding to MDM2 [14, 15]. Nutlin 3a treatment of wild type p53-expressing cultured cancer cells increases p53 levels and causes p53-mediated transactivation, cell cycle arrest, and apoptosis [16]. However, the magnitude and selectivity of these p53 downstream effects are variable [15]. Nutlin 3a and other small molecules that target the N-terminal p53 binding pocket of MDM2 decrease the rate of tumorigenesis of wild-type p53-expressing cancer cells in nude mouse-bearing human cancer xenografts [14, 16, 17].
Nutlin 3a was isolated by screening a library of cis-imidazoline compounds for inhibition of p53 binding to MDM2 by surface plasmon resonance [14]. Nutlin 3a belongs to a class of compounds composed of two halogen-derivatized phenyl rings in cis configuration attached to positions 4 and 5 of an imidazoline scaffold. Roche, the manufacturer of these compounds, has a nutlin 3a-like compound called RG7112 in Phase I clinical trials for the treatment of advanced solid tumors and hematologic cancers [18]. Although nutlins are a promising start for drug development, there may be many hurdles to overcome prior to approval for clinical use. An alternate approach to development of novel MDM2 inhibitors is to determine if FDA-approved compounds can be repurposed as MDM2 inhibitors [19].
In this report, we present an in silico approach to drug repurposing, called Computational Conformer Selection (CCS), to screen and identify compounds with similar shape and charge distribution as nutlin 3a across different nutlin 3a conformers. Conformers of FDA-approved drugs are computationally generated and compared with the shape and charge distribution of nutlin 3a. Highly ranked drugs are docked to the N-terminal p53-binding domain of MDM2 using the Autodock Vina docking program [20] yielding binding energies. Here we report the ranked order of the 3,244 drugs from the ZINC database of FDA-approved drugs [21].
Experimental Section
Creating the nutlin 3a-conformer model
Nutlin 2 from PDB #1RV1 structure was used as the starting point. The nutlin 2 isopropyl group was substituted for the ethyl group at position 2 on the methoxy-phenyl ring. Chlorine atoms were substituted for the bromine atoms at position 4 on the two phenyl rings. A ketone oxygen was added to position 3 of the piperazine ring and the ethanol group was removed from position 4 of the piperazine ring.
Conformer generation and rapid overlay chemical structure software (ROCS) program
Conformers of nutlin 3a and ZINC database drugs were created by OMEGA with the maximum number of conformers for each drug set to 600 (-maxconfs 600) [22]. Rapid Overlay of Chemical Structures (ROCS) software was used to measure similarities between nutlin 3a and drug conformers [23, 24]. The number of random starting points for compound alignments was set to “-randomstarts 40”. Default parameters of ROCS were changed to “rank by ColorTanimoto” to emphasize charge distribution. Preliminary analysis of ROCS-mediated FDA drug comparisons to nutlin 3a showed that the Tanimoto combo score was strongly dominated by the shape parameter. In the default setting, the ROCS software program produced Tanimoto shape scores ranging from 0.103 to 0.644 for FDA drug conformers when the nutlin 3a conformers were queried. On the other hand, for the same comparisons the color (charge distribution) scores for FDA drug conformers ranged from 0 to 0.350. To create a better balance between Tanimoto shape and Tanimoto color, the default ROCS matching setting was changed to “rank by ColorTanimoto” to place greater emphasis on the color parameter. With this change, the Tanimoto shape scores ranged from 0.071 to 0.538 and the Tanimoto color scores ranged from 0.006 to 0.373. Unexpectedly, ROCS generated repeats of a few drug conformers (see Additional Supporting Information online for details on repeats).
Docking measurements
Drugs were prepared for docking by obtaining structure data format (sdf) files from the ZINC database [21]. The software program PyRx (version 0.7) was used to convert the sdf files to PDBQT files. PDBQT files were used for docking to MDM2 with AutoDoc Vina (version 1.1). The macromolecule docking target was amino acids 25–109 of MDM2 (Protein Data Bank #1RV1). Water molecules, heteroatoms (such as metals) and nutlin 2 were removed from 1RV1 prior to docking. The coordinates of the search space for MDM2 were maximized to allow the entire macromolecule to be considered for docking. The search space coordinates were: Center X: 13.5812 Y: 0.8458 Z: 19.5482, Dimensions (Å) X: 44.1592 Y: 34.3006 Z: 28.3009. Two-dimensional images highlighting the compounds and interacting MDM2 amino acids were created with LIGPLOT (version 4.5.3) [25]. RMSD values of compounds and solvent excluded areas within MDM2 were calculated by Chimera software (version 1.4.1) [26].
Results and Discussion
A structural model for nutlin 3a bound to MDM2
Our approach to computationally screen drugs that target MDM2 is based on the idea that such compounds would exhibit shape and charge distribution similar to nutlin 3a across its different conformers. To test if this approach was valid, a feasible structural model for nutlin 3a bound to MDM2 was required. Since no nutlin 3a structures are available, a model was developed using the structure of nutlin 2 bound to MDM2 [14]. Nutlin 2 (Figure 1A) shares, spatially, 90% of its heavy atoms with nutlin 3a (Figure 1B), suggesting that both compounds likely bind to MDM2 in a similar fashion. The nutlin 3a model was built from the crystal structure of nutlin 2 bound to MDM2 (PDB 1RV1-mean resolution = 2.18 ± 1.21 Å) (Figure 1C). The nutlin 3a model/MDM2 complex was energy minimized using WebLabViewer Pro software program (version 4.0). The energy minimized nutlin 3a model is called the nutlin 3a conformer model (nutlin 3a-CM) (Figure 3D).
Figure 1.
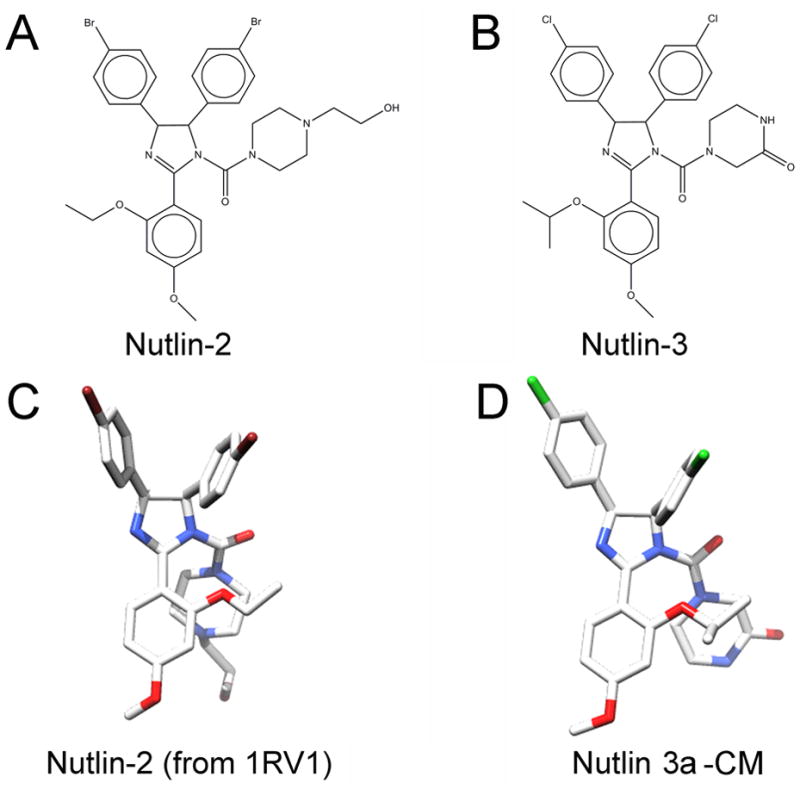
Chemical structures of nutlin 2 and nutlin 3. Nutlin 2 structure was taken from a co-crystal structure of nutlin 2 with the MDM2 N-terminal binding domain. Nutlin 3a-CM was created from nutlin 2 structure and energy minimized within 1RV1.
Figure 3.
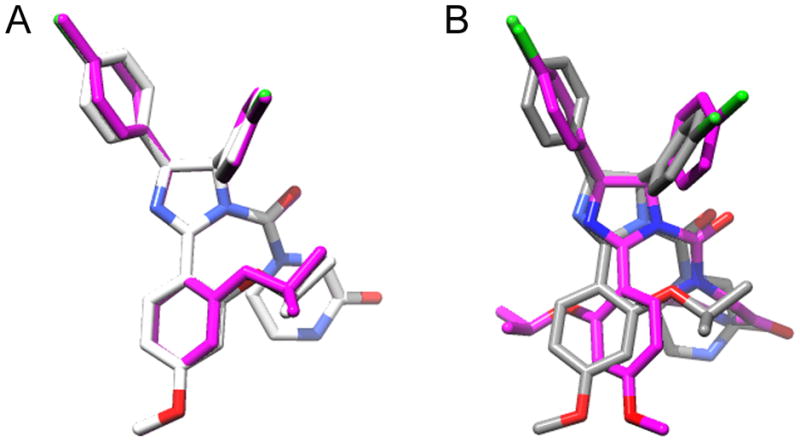
Nutlin 3A conformations. A. Comparison of nutlin 3a-CM (CPY) and docked nutlin 3a-CM (magenta) structures. B. Comparison of nutlin 3a-CM (CPY) and top scoring OMEGA generated nutlin 3a (magenta).
A comparison of the structure of nutlin 2 and nutlin 3a-CM revealed that the root mean square deviation (RMSD) of bound molecules was 0.73 Å for shared heavy atoms indicating that energy minimization did not dramatically alter the nutlin 3a-CM conformation. Given the low RMSD value, one would expect nutlin 3a-CM to bind to MDM2 with a binding energy comparable to nutlin 2. Autodock Vina was used to separately dock the two nutlins to MDM2 (PDB 1RV1). The average binding energies reported (out of 9 dockings) was −6.8 ± 0.7 kcal/mol for nutlin 2 and −7.7 ± 0.6 kcal/mol for nutlin 3a-CM. The top-scoring docked nutlin 2 (from PDB 1RV1) and nutlin 3a-CM conformers generated after docking indicate that both nutlins bind with three functional groups inserted into the N-terminal p53-binding hydrophobic pocket of MDM2 (Figure 2). The three functional groups of nutlins simulate three p53 amino acid side chains that are critical for engagement with MDM2: Phe19, Trp23, Leu26 [27]. The three functional groups are two singly halogenated-substituted phenyl rings and an alkyloxy group at position 3 of the methoxy-phenyl ring. The slightly better binding energy of nutlin 3a-CM is likely due to increased van der Waals interactions resulting from its bulkier alkoxy group. The docked nutlins closely resemble the crystal structure showing nutlin 2 bound to MDM2 [14]. The docking software allows ligand flexibility for optimal fitting in the protein pocket. Nutlin-3a-CM docked without much perturbation from the original nutlin 3a-CM conformation (RMSD = 0.23 Å) (Figure 3A).
Figure 2.

Nutlin 2 (blue) and nutlin 3a-CM (CPY) docked to the MDM2 N-terminal domain.
Generation of nutlin 3a conformers
Our approach to drug repurposing is to identify small molecule conformers from the drug database that resemble low energy conformers of nutlin 3a. Low energy conformers of nutlin 3a were generated by the software program OMEGA [22] which uses a rule-based algorithm in combination with variants of the Merck force field 94 to produce conformations [28, 29]. Using default parameters, OMEGA generated 41 conformations of nutlin 3a from the nutlin 3a sdf file obtained from PubChem (PubChem ID: 11433190).
The ROCS software program was utilized to compare nutlin 3a-CM with 41 OMEGA-generated nutlin conformers. The Tanimoto shape scores ranged from 0.325 to 0.748 and the Tanimoto color (charge distribution) scores ranged from 0.278 to 0.472 resulting in a top Tanimoto combo score of 1.189. Structural alignment of the top scoring nutlin 3a generated by OMEGA (nutlin 3a-OG) and nutlin 3a-CM gave an RMSD of 2.52 Å (Figure 3B). The high RMSD value occurs because OMEGA rotated the phenyl methoxy ring of nutlin 3a 159° compared to nutlin 3a-CM to reduce steric hindrance between the isopropyloxy group and the piperizine ring. OMEGA also bent the piperizine ring so that carbon atoms 3, 4 and 5 on the piperizine ring are far from the phenyl methoxy ring. The high RMSD value obtained reflects these structure differences. ROCS analysis of the other 40 OMEGA-generated conformers confirmed that these OMEGA-generated conformers were more dissimilar to nutlin 3a-CM than nutlin 3a-OG (data not shown).
Autodock Vina was used to dock nutlin 3a-OG into MDM2. The average binding energy of nutlin 3a-OG docked to MDM2 was −7.1 kcal/mol ± 0.4 (9 dockings) compared to nutlin 3a-CM with a −7.7 kcal/mol binding energy. The less favorable binding energy is due to the fact that the binding conformation of nutlin 3a-OG differs from the binding conformation of nutlin 3a-CM (Figure 4). The bound nutlin 3a-OG isopropyloxy group is directed away from the hydrophobic pocket instead of into the hydrophobic pocket. This result calls into question how nutlin 3a is able to produce a conformation predicted to bind into the three sub-pockets of the hydrophobic cleft of MDM2. Because an experimental structure of nutlin 3a bound to MDM2 is unavailable, we must consider the possibility that nutlin 3a binds to MDM2 in a manner similar to nutlin 3a-OG. Another possibility is that Nutlin 3a-OG may in fact bind to MDM2 analogously to nutlin 2, but Auto Dock Vina is not capable of simulating the docking process accurately. Regardless of how nutlin 3a binds to MDM2’s hydrophobic cleft, in vitro competition experiments shows that nutlin 3a prevents p53 peptide from binding to MDM2 with an IC50 of 90 nM [14] indicating that nutlin 3a is an effective MDM2 inhibitor.
Figure 4.
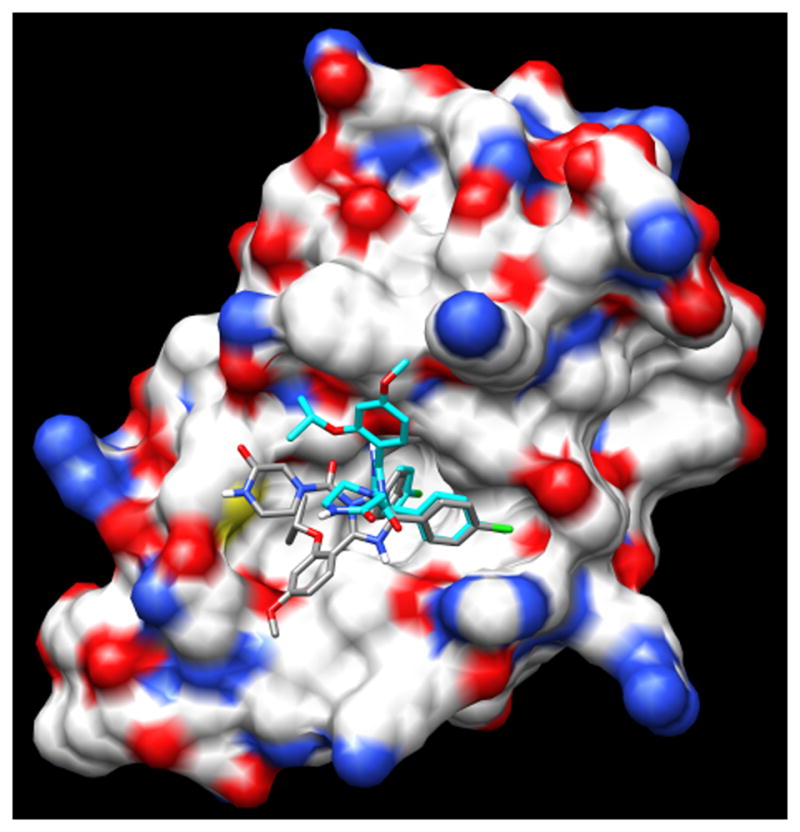
Nutlin 3A-CM (CPY) and docked OMEGA generated nutlin 3a (blue).
Screening for drugs that can adopt the similar conformations as nutlin 3a
Our approach was to compare conformers of compounds from a database of FDA-approved drugs to the 41 conformations of nutlin 3a generated from the nutlin 3a sdf file. Drug conformations that ranked relatively high by shape and color across 41 conformations of nutlin 3a would be considered good candidates for inhibitions of MDM2. The ZINC drug database contains molecules that are currently, or at one time were FDA-approved. The drug list shows some redundancy since it contains isomers as well as variably protonated versions of the same molecule. There are 1,125 drugs with unique molecular formulas in the ZINC database and 3,244 drugs when the isomers and alternate protonated forms are included.
The top 15 drugs were ranked by average Tanimoto combo scores, which ranged from 0.722 to 0.802 (Table 1). For all 3,244 drugs, the range was 0.142 to 0.802 (see Data S1, Supporting Information). The contribution of shape and color to the top 15 Tanimoto combo scores are also presented in Table 1. The calculated binding energies from docking of drugs to MDM2 (average of 100 dockings) are shown as well. The binding energies of the top 15 drugs ranged from −8.456 to −4.724 kcal/mol. The top ranking drug, S-bepridil, had an average binding energy of −6.6 kcal/mol, whereas the average binding energy of nutlin 3a-OG was −7.1 kcal/mol. The drug with the best binding energy is nadrolone phenyl propionate at −7.61 kcal/mol. Calculation of buried surface area of the hydrophobic revealed that nutlin 3a-CM buried 329 Å2 of MDM2. Of the top 15 drugs, the best in terms of shielding MDM2 from solvent is Nafronyl-d4, which buried 296 Å2 of solvent. In general, one expects the FDA-drugs to bury less MDM2 surface than nutlins because the former are generally smaller than the latter. To better understand the interactions of the top 15 drugs ranked by the ROCS program, we performed an analysis of the drug atom to MDM2 atom interactions that take place after docking.
Table 1.
Drugs ranked by mean Tanimoto combo score with docked scores.
| Record # | Drug name | Tanimoto Combo Score (± SD) | Tanimoto Shape Score | Tanimoto Color Score | Docked score (kcal/mol) | Calculated buried MDM2 surface (Å2) |
|---|---|---|---|---|---|---|
| nutlin 2 | −6.8 (9 docks) | 317 | ||||
| nutlin 3a-CM | −7.7 (9 docks) | 329 | ||||
| nutlin 3a-OG | −7.1 (9 docks) | 282 | ||||
| ZINC03830283 | S-bepridil/Vascor | 0.802±0.091 | 0.538 | 0.264 | −6.55 | 276 |
| ZINC03831395 | Protirelin/Thyrel TRH | 0.784±0.076 | 0.523 | 0.261 | −5.73 | 249 |
| ZINC11677473 | Azaribine/Triazure | 0.756±0.144 | 0.49 | 0.265 | −5.49 | 257 |
| ZINC01847743 | caramiphen, Ordrine AT | 0.749±0.015 | 0.43 | 0.319 | −5.82 | 218 |
| ZINC03812918 | R-bepridil | 0.748±0.057 | 0.49 | 0.258 | −6.66 | 252 |
| ZINC00001446 | Prenazone, Feprazone | 0.748±0.049 | 0.488 | 0.259 | −6.62 | 244 |
| ZINC00537935 | Lorcainide | 0.745±0.040 | 0.442 | 0.303 | −6.91 | 269 |
| ZINC00607836 | Doxapramhydrochloride/Dopram | 0.742±0.061 | 0.499 | 0.243 | −6.24 | 273 |
| ZINC01530778 | Phenylbutazone | 0.742±0.053 | 0.489 | 0.253 | −6.34 | 220 |
| ZINC03831172 | Nafronyl-d4 | 0.729±0.057 | 0.467 | 0.263 | −5.92 | 292 |
| ZINC04026871 | nandrolone phenylpropionate | 0.728±0.056 | 0.462 | 0.266 | −7.61 | 291 |
| ZINC00601229 | Azelastine | 0.726±0.064 | 0.45 | 0.276 | −7.28 | 229 |
| ZINC00000452 | Mephenoxalone | 0.724±0.041 | 0.362 | 0.363 | −4.72 | 171 |
| ZINC04534031 | Azlocillin | 0.724±0.065 | 0.504 | 0.221 | −6.55 | 281 |
| ZINC00001732 | Midazolam hydrochloride | 0.722±0.065 | 0.462 | 0.26 | −6.69 | 201 |
Data for the number of top 15 drug atoms predicted to bind to MDM2, the number of hydrogen bonds predicted to bind to MDM2, the number of MDM2 amino acids predicted to bind to drugs and the percentage of nutlin 3a-CM-binding amino acids that bind to drugs were collected (Table 2) [25]. The number of drug atoms involved in MDM2 contacts ranged from 8 to 16. The top hit, S-bepridil has 14 drug atoms that bind to MDM2. This compares favorably to nutlin 3a-CM, which has 15 drug atoms that bind to MDM2. Four of the top 15 drugs make hydrogen bond contacts to MDM2 (nutlin 3a-CM has no hydrogen bond contacts). Nutlin 3a-CM is predicted to contact nine MDM2 amino acids—all without hydrogen bonds. The number of MDM2 amino acids that bind to the top 15 drugs ranged from seven to ten. Because these drugs were selected to mimic nutlin 3a CM one might expect that the nutlin 3a-CM interacting amino acids would overlap with the drug interacting amino acids. S-bepridil interacted with five amino acids that also interacted with nutlin 3a CM. It is worth noting that the fifth-ranked drug was R-bepridil—which is predicted to have 16 atoms in contact with 10 amino acids of MDM2. Seven MDM2 amino acids overlap those that contact nutlin 3a-CM. Of the top 15 drugs, 11 are predicted to interact with the majority of the same MDM2 residues as nutlin 3a-CM. The MDM2 residues that interact with nutlin 3a-CM and the top five drugs are Leu54, Gly58, Ile61, Val93, His96, and Ile99 (Figure 5). A recent NMR study of the Nutlin 3a-MDM2 complex confirmed that at least five of these six residues interact with Nutlin 3a [30]. X-ray crystallography structure analysis of the p53-MDM2 complex shows that all six MDM2 residues bind to p53 [27].
Table 2.
Structure analysis of computationally docked nutlin-like inhibitors
| Record number | Drug Name | Number of drug atoms involved in MDM2 contacts | Number of hydrogen bond contacts to MDM2 | Number of MDM2 amino acids bound to drug | Percentage of nutlin 3a-CM binding amino acids |
|---|---|---|---|---|---|
| nutlin 3a-CM | 15 | 0 | 9 | 100 | |
| ZINC03830283 | S-bepridil/Vascor | 14 | 0 | 10 | 67 |
| ZINC03831395 | Protirelin/Thyrel TRH | 9 | 2 | 7 | 55 |
| ZINC11677473 | Azaribine/Triazure | 10 | 1 | 8 | 44 |
| ZINC01847743 | caramiphen, Ordrine AT | 13 | 0 | 9 | 67 |
| ZINC03812918 | R-bepridil/Vascor | 16 | 0 | 10 | 78 |
| ZINC00001446 | Prenazone, Feprazone | 10 | 0 | 9 | 67 |
| ZINC00537935 | Lorcainide hydrochloride | 12 | 0 | 7 | 67 |
| ZINC00607836 | Doxapramhydrochloride/Dopram | 14 | 0 | 8 | 55 |
| ZINC01530778 | Phenylbutazone | 11 | 0 | 9 | 55 |
| ZINC03831172 | Nafronyl-d4 | 17 | 1 | 7 | 55 |
| ZINC04026871 | nandrolone phenylpropionate | 15 | 0 | 8 | 55 |
| ZINC00601229 | Azelastine | 13 | 0 | 7 | 44 |
| ZINC00000452 | Mephenoxalone | 8 | 0 | 5 | 44 |
| ZINC04534031 | Azlocillin | 12 | 0 | 7 | 44 |
| ZINC00001732 | Midazolam hydrochloride | 10 | 0 | 7 | 55 |
Figure 5.
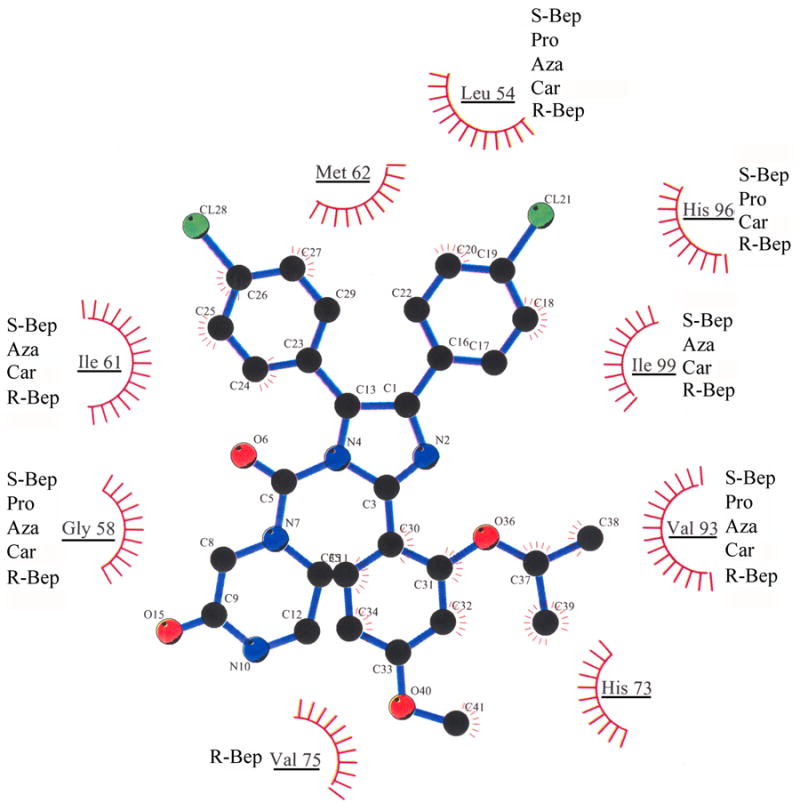
Predicted Van der Waals interactions between nutlin 3A-CM and MDM2 amino acids. MDM2 amino acids that interact with nutlin 3a-CM are underlined. Out of the top five scoring drugs, those predicted to interact with nutlin-associated MDM2 amino acids are listed next to each underlined amino acid: S-Bep, S-bepridil; Pro, Protirelin; Aza, Azaribine; Car, Caramiphen; R-Bep, R-bepridil.
To further investigate the five highest scoring drugs, we show them computationally docked to MDM2 (Figure 6). We note that in each case, a minimum of two functional groups of the drug bind to two of the three hydrophobic subpockets of MDM2. The three MDM2 subpockets individually bind p53 amino acids Phe19, Trp23 and Leu26 [27]. S-bepridil, Caramiphen, and R-bepridil have functional groups that bind to all three subpockets of MDM2. The combination of the potential to bind to three subpockets and their relatively small sizes suggests they are good candidates for further exploration as MDM2 inhibitors.
Figure 6.

Top five scoring drugs docked to MDM2 with AutoDock Vina. A. MDM2 with subpockets labeled where p53 residues engage: Phe19, Trp23, and Leu26. Bordered area magnified in panels B–F. B. S-bepridil; C. Protirelin; D. Azaribine; E. Caramiphen; F. R-bepridil. Insets for panels B–F show the drug functional groups predicted to bind to the MDM2 subpockets that normally interact with Phe19 (green), Trp23 (red), and Leu26 (blue) of p53.
Bepridil
The top hit, S-bepridil, marketed under the brand name Vascor, increases the contractile force of cardiac myofilament [31] by enhancing the responsiveness of the cardiac troponin C [32]. NMR and X-ray crystal structure analysis of bepridil-troponin C complexes show that S-bepridil binds to a hydrophobic depression within the N-terminal domain of troponin C [32, 33]. S-bepridil interacts with 13 hydrophobic residues in troponin C. In our computational models, S-bepridil interacts with 14 MDM2 residues and R-bepridil interacts with 16 MDM2 residues. This may make bepridil attractive as a potential inhibitor of MDM2.
Conclusion
Drug repurposing has had some success. Minoxidil, originally developed to treat hypertension, is used to combat hair loss. Viagra, designed to treat hypertension, is now indicated for erectile dysfunction and pulmonary hypertension. Rituxin was initially indicated for non-Hodgkin’s lymphoma and it is now approved for treatment of chronic lymphocytic leukemia and rheumatoid arthritis [34]. Raloxifene, originally approved for osteoporosis, has been repurposed for some breast cancers. It modulates binding of estrogen to its receptor and leads to a decrease in estrogen-mediated transcription [35]. Bisphosphonates were originally approved to reduce skeletal-related effects of chemotherapy such as pathologic fractures, spinal cord compression, hypercalcemia, and the need for bone surgery. There is now mounting evidence that they create a micro-environment in the bone that is refractory to cancer growth [36]. None of these drug repurposing examples were discovered with a bioinformatics approach. Rather, clinical side effects were noted and measured. With the development of more extensive drug structure and protein structure databases, a computational approach to repurposing drugs should be feasible. Indeed, a database of 3,665 FDA-approved and investigational drugs and their known protein targets were used to predict potential new drug-target associations (Keiser et al., 2009 Nature). Importantly, of the 184 new predicted associations, 30 were experimentally tested and 23 of these were confirmed. This progress encouraged us to use an approach that combines bioinformatics and structure analysis to repurpose drugs to target MDM2.
Our Conformation Computational Selection method introduced here is a new approach to drug repurposing. This approach is based on knowledge of the high resolution drug-target structure. OMEGA is used to create putative several drug conformations. Those conformations are then compared to potential drug conformations from a database (in our case, we used a FDA-approved drug database). ROCS is used to quantify the drug similarities in terms of shape and atom type across the different conformations. In our studies, the top 15 scoring drugs all docked to the p53 binding domain in MDM2.
Supplementary Material
Acknowledgments
We thank Sivanesan Dakshanamurthy (Lombardi Comprehensive Cancer Center, Georgetown University) for contributing the initial drug database. This project was funded by NIH grants 1 P20 CA118783 to J.M. and GM61331 to W.A.W.. R.S. was supported by the NSF grant GK-12 Program (Award No. 072426).
Footnotes
Author Contributions
J.M. developed the project, analyzed the data, and wrote the paper. W.A.W. researched the software programs, identified the drug database, helped guide the project and assisted with writing and editing. R.S. used software programs to compare nutlin conformations with FDA-approved drug conformations. A.D. conducted docking experiments. E.L. assisted A.D. in the docking experiments.
The authors have no conflict of interest to declare.
Data S1. Complete drug conformer comparison rankings can be found at http://www.calstatela.edu/faculty/jmomand/compconfselspreadsheet.htm
References
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature; 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ; 2006;13:1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- 4.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 5.Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res; 1998;26:3453–3459. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bond GL, Hirshfield KM, Kirchhoff T, Alexe G, Bond EE, Robins H, Bartel F, et al. MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer Res; 2006;66:5104–5110. doi: 10.1158/0008-5472.CAN-06-0180. [DOI] [PubMed] [Google Scholar]
- 7.Araki S, Eitel JA, Batuello CN, Bijangi-Vishehsaraei K, Xie XJ, Danielpour D, Pollok KE, et al. TGF-beta1-induced expression of human Mdm2 correlates with late-stage metastatic breast cancer. J Clin Invest; 2010;120:290–302. doi: 10.1172/JCI39194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature; 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 9.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature; 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 10.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett; 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 11.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev; 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 12.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res; 1991;51:6304–6311. [PubMed] [Google Scholar]
- 13.Chen L, Agrawal S, Zhou W, Zhang R, Chen J. Synergistic activation of p53 by inhibition of MDM2 expression and DNA damage. Proc Natl Acad Sci U S A; 1998;95:195–200. doi: 10.1073/pnas.95.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science; 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 15.Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nat Rev Clin Oncol. 2010 doi: 10.1038/nrclinonc.2010.174. [DOI] [PubMed] [Google Scholar]
- 16.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A; 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol; 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber L. Patented inhibitors of p53-Mdm2 interaction (2006 – 2008) Expert Opin Ther Pat; 2010;20:179–191. doi: 10.1517/13543770903514129. [DOI] [PubMed] [Google Scholar]
- 19.Duenas-Gonzalez A, Garcia-Lopez P, Herrera LA, Medina-Franco JL, Gonzalez-Fierro A, Candelaria M. The prince and the pauper. A tale of anticancer targeted agents. Mol Cancer. 2008;7:82. doi: 10.1186/1476-4598-7-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem; 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model; 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model; 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rush TS, 3rd, Grant JA, Mosyak L, Nicholls A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction. J Med Chem; 2005;48:1489–1495. doi: 10.1021/jm040163o. [DOI] [PubMed] [Google Scholar]
- 24.Bostrom J, Greenwood JR, Gottfries J. Assessing the performance of OMEGA with respect to retrieving bioactive conformations. J Mol Graph Model; 2003;21:449–462. doi: 10.1016/s1093-3263(02)00204-8. [DOI] [PubMed] [Google Scholar]
- 25.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng; 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 26.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem; 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 27.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science; 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 28.Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comp Chem. 1996;17:490–519. [Google Scholar]
- 29.Kirchmair J, Wolber G, Laggner C, Langer T. Comparative performance assessment of the conformational model generators omega and catalyst: a large-scale survey on the retrieval of protein-bound ligand conformations. J Chem Inf Model; 2006;46:1848–1861. doi: 10.1021/ci060084g. [DOI] [PubMed] [Google Scholar]
- 30.Riedinger C, Noble ME, Wright DJ, Mulks F, Hardcastle IR, Endicott JA, McDonnell JM. Understanding small-molecule binding to MDM2: insights into structural effects of isoindolinone inhibitors from NMR spectroscopy. Chem Biol Drug Des; 2011;77:301–308. doi: 10.1111/j.1747-0285.2011.01091.x. [DOI] [PubMed] [Google Scholar]
- 31.Solaro RJ, Bousquet P, Johnson JD. Stimulation of cardiac myofilament force, ATPase activity and troponin C Ca++ binding by bepridil. J Pharmacol Exp Ther; 1986;238:502–507. [PubMed] [Google Scholar]
- 32.MacLachlan LK, Reid DG, Mitchell RC, Salter CJ, Smith SJ. Binding of a calcium sensitizer, bepridil, to cardiac troponin C. A fluorescence stopped-flow kinetic, circular dichroism, and proton nuclear magnetic resonance study. J Biol Chem. 1990;265:9764–9770. [PubMed] [Google Scholar]
- 33.Li Y, Love ML, Putkey JA, Cohen C. Bepridil opens the regulatory N-terminal lobe of cardiac troponin C. Proc Natl Acad Sci U S A; 2000;97:5140–5145. doi: 10.1073/pnas.090098997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dudley JT, Deshpande T, Butte AJ. Exploiting drug-disease relationships for computational drug repositioning. Brief Bioinform; 2011;12:303–311. doi: 10.1093/bib/bbr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grese TA, Sluka JP, Bryant HU, Cullinan GJ, Glasebrook AL, Jones CD, Matsumoto K, et al. Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc Natl Acad Sci U S A; 1997;94:14105–14110. doi: 10.1073/pnas.94.25.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koul HK, Koul S, Meacham RB. New role for an established drug? Bisphosphonates as potential anticancer agents. Prostate Cancer Prostatic Dis; 2012;15:111–119. doi: 10.1038/pcan.2011.41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.