

Abstract
5-thyminyl-5,6-dihydrothymine (also called spore photoproduct or SP) is the exclusive DNA photo-damage product in bacterial endospores. It is repaired by a radical SAM (S-adenosylmethionine) enzyme, the spore photoproduct lyase (SPL), at the bacterial early germination phase. Our previous studies proved that SPL utilizes the 5′-dA• generated by SAM cleavage reaction to abstract the H6proR atom to initiate the SP repair process. The resulting thymine allylic radical was suggested to take an H atom from an unknown protein source, most likely the cysteine 141. Here we show that C141 can be readily alkylated in the native SPL by iodoacetamide treatment, suggesting that it is accessible to the TpT radical. SP repair by the SPL C141A mutant yields TpTSO2− and TpT simultaneously from the very beginning of the reaction; no lag phase is observed for the TpTSO2− formation. Should any other protein residue serve as the H donor, its presence would result in TpT as the major product at least for the first enzyme turnover. These observations provide strong evidence to support C141 as the direct H atom donor. Moreover, due to the lack of this intrinsic H donor, the C141A mutant produces TpT via an unprecedented thymine cation radical reduction (proton coupled electron transfer) process, contrasting to the H atom transfer mechanism in the WT SPL reaction. The C141A mutant repairs SP at a rate which is ~3-fold slower than the WT enzyme. Formation of TpTSO2− and TpT exhibit a Vmax deuterium kinetic isotope effect (KIE) of 1.7 ± 0.2 respectively, which is smaller than the DVmax KIE of 2.8 ± 0.3 determined in the WT SPL reaction. These findings suggest that removing the intrinsic H atom donor disturbs the rate-limiting process in the enzyme catalysis. As expected, the pre-reduced C141A mutant only supports ~ 0.4 turnover, which is in sharp contrast to the > 5 turnovers exhibited by the WT SPL reaction, suggesting that the enzyme catalytic cycle (SAM regeneration) is disrupted by this single mutation.
Introduction
Due to its high energy and efficient absorption by biological macromolecules, UV light is considered to be immediately lethal to most microorganisms. However, this method fails to kill endospores due to their unique DNA photochemistry. The spore genomic DNA is bound by a group of DNA binding proteins named small acid soluble proteins (SASPs). In the resulting protein/DNA complex, the DNA adopts an A-like conformation, which facilitates formation of a thymine dimer, 5-thyminyl-5,6-dihydrothymine (Scheme 1), also called spore photoproduct or SP, as the exclusive UV damage product (1–7). SP is rapidly repaired by a metalloenzyme, spore photoproduct lyase (SPL), when spores start germinating (1, 8–16), thus posing little threat to spores' survival. However, despite the fact that SP was discovered nearly half a century ago (17) and the strong interest of the scientific community to understand its formation and repair (1), neither process is well understood at this point.
Scheme 1.
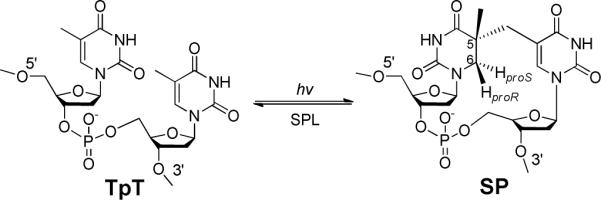
Our laboratory is interested in understanding both aspects of SP biochemistry. We recently employed deuterium labeled dinucleotide TpT and demonstrated that SP is formed via an intramolecular H atom transfer process between two thymine residues (18), answering a key question in SP photoformation (1, 17). By using a neutral formacetal linker instead of a negatively-charged phosphate between the two thymine residues, we successfully obtained the crystal structure of SP 50 years after its discovery (19). Analyzing the structure not only confirms SP to have a chiral center with an R configuration, but also reveals that both 2-deoxyriboses adopt the C3′-endo conformation, which is typically found in A-form DNA. Although SP formation needs the genomic DNA to adopt an A-like conformation (3, 4, 6), once formed, the methylene bridge between the thymine bases becomes dominant, forcing the two 2-deoxyriboses to maintain the A-like conformation even though the DNA binding proteins are absent.
SP is repaired by the radical SAM enzyme SPL at the bacterial early germination phase (1, 20, 21). All radical SAM enzymes use a tri-cysteinate motif to coordinate to three irons in the [4Fe-4S] cluster, and S-adenosylmethionine (SAM) to the fourth one (Scheme 2) (20–38). The cluster donates an electron to SAM to cleave its C-S bond, generating a 5′-deoxyadenosyl radical (5′-dA•). Utilizing dinucleotide SP TpTs with the two H6 atoms selectively labeled by deuterium, we proved that it is the H6proR atom that is taken by the 5′-dA•, confirming that the SPL reaction is highly stereo-selective (39). However, different from the prediction by the previously hypothesized mechanism (10, 11), the resulting TpT radical does not take the H-atom back from 5′-dA, but from an unknown protein residue, which is able to exchange a proton with the aqueous solution via acid-base chemistry, to produce the repaired TpT (39).
Scheme 2.

Which protein residue serves as the direct H atom donor to the TpT radical? The research to date suggests that a conserved cysteine residue, C141, in Bacillus subtilis SPL is the most likely candidate. An in vivo study found that a C→A mutation totally abolished the enzyme activity (40). An in vitro assay found that the C141A mutant produced a TpTSO2− species as the major product from the SP repair reaction, with the -SO2− group originating from the dithionite added as the reductant to the SAM [4Fe-4S]2+ cluster (14). These results suggest that the C141 residue plays a key role in enzyme catalysis.
The crystal structures of the WT SPL as well several SPL C140 mutants from Geobacillus thermodenitrificans were solved at high resolution very recently (41). The C140 mutants were found to have same structures as the WT SPL, suggesting that the conserved cysteine has no structural role. The enzyme contains a dinucleoside SP. As shown in Fig. 1, the bridging methylene carbon in SP was found to be 4.6 Å away from the C140 (equivalent to the C141 in B. Subtilis SPL) and 4.3 Å away from an active-site tyrosine (Y98). It was suggested that after the methylene bridge breaks during the repair process, the resulting thymine allylic radical moves toward C140, making the C140 perfectly positioned as the intrinsic H atom donor. To support this assumption, the activity of the C140A mutant was studied and the correspondent -SO2− adduct to DNA was found by MALDI spectroscopy. The yield of the -SO2− adduct however appeared to be low. Furthermore, the < 0.5 turnover observed also makes the mechanistic analysis complicated. In contrast, when dinucleotide SP was used as substrate for B. Subtilis C141A mutant, multiple turnovers were observed and > 70% of the repaired SPs yielded the TpTSO2− adduct, with TpT a minor species (14).
Figure 1.
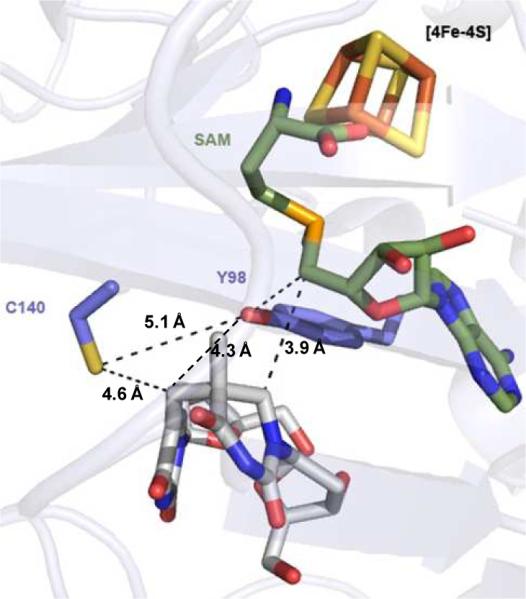
Crystal structure of the WT SPL from Geobacillus thermodenitrificans which contains a SAM and a dinucleoside SP in the enzyme active site. The distance between the SP methylene carbon to the conserved cysteine (C140) was found to be 4.6 Å, and that between the methylene carbon and a conserved tyrosine (Y98) was found to be 4.3 Å.
Although the structural data suggests that the conserved cysteine is likely the H atom donor, the possibility that a conserved tyrosine (Y98 in G. thermodenitrificans SPL and Y99 in B. subtilis SPL) is the donor cannot be excluded. This Y98 is closer to the SP methylene carbon in the SPL structure; it may be able to donate the H atom to the thymine allylic radical, which could explain why TpT also forms in the C141A mutant reaction (14). To address this problem, the B. subtilis C141A catalyzed SP TpT repair reaction was certainly worthy of re-visiting to reveal if the TpTSO2− and TpT form at the same time. Such information is of significance because if the Y99 in B. subtilis SPL, but not the C141, is the direct H atom donor, it is still perfectly positioned to donate the H atom in the C141A mutant, enabling the formation of TpT.
Here, we report the enzyme kinetics studies of the SPL C141A mutant. Our data confirm that C141 is the direct H atom donor to the TpT radical. Moreover, after removing this putative H atom donor from SPL, the SPL C141A mutant produces TpT likely via an unprecedented thymine radical cation reduction (proton coupled electron transfer) mechanism.
Experimental Section
Materials and Methods
Unless otherwise stated all solvents and chemicals used were of commercially available analytical grade and used without further purification. Purification of reaction products was carried out by flash chromatography using silica gel (Dynamic Adsorbents Inc, 32–63 μm). The 1H and 13C NMR spectra were obtained on a Bruker 500 MHz NMR Fourier transform spectrometer. UV–visible spectra were recorded using UV-Mini 1240 Spectrophotometer and the associated data manager software package. The photoreaction was carried out using a Spectroline germicidal UV lamp (Dual-tube, 15 w, intensity: 1550 uw/cm2) with samples ~5 cm from the lamp. The protein purification as well as the enzyme reactions were carried out under an inert atmosphere using a Coylab anaerobic chamber (Grass Lake, MI) with the H2 concentration around 3%. DNA sequencing was performed by the GENEWIZ Inc. at South Plainfield, NJ.
All DNA-modifying enzymes and reagents were purchased from Fermentas Life Sciences (Glen Burnie, MD). B. subtilis strain 168 chromosomal DNA was purchased from the ATCC (ATCC 23857D). Oligonucleotide primers were obtained from Integrated DNA Technologies (Coralville, IA). E. Coli BL21(DE3) and expression vector pET-28a was purchased from Novagen (Madison, WI). The construct containing the SPL gene was co-expressed with plasmid pDB1282, which was a generous gift from Prof. Squire Booker at the Pennsylvania State University. 5 Prime Perfectpro* Nickel nitrilotriacetic acid (Ni-NTA) resin was purchased from Fisher Scientific. And the SP Sepharose fast flow ion exchange resin was purchased from the GE Healthcare Bio-Sciences Corporation.
Construction of the SPL C141A expression vector
The splB gene was cloned from the B. subtilis chromosomal DNA (strain 168) into the in pET-28a vector with a N-terminal His6-tag as previously described (39). A site-directed mutagenesis was performed to change the cysteine 141 to an alanine using the synthetic oligonucleotide primers 5′-CAAGGTTCGAAGCATCAGCTACGTCAG ACATTG-3′ and 5′-CAATGTCTGACGTAGCTGATGCTTCGAACCTTG-3′ with the QuickChange site-directed mutagenesis kits from Stratagene under the manufacturer's instruction. The construct was transformed into E. coli 10 G chemically competent cells purchased from Lucigen Corporation (Middleton, WI) for isolation and amplification of the resulting plasmid DNA. The resulting vector was named SPL C141A-pET28 and co-transformed with a pDB 1282 vector into E. coli BL21(DE3) obtained from Stratagene (La Jolla, CA) for protein overexpression as described before (39).
Expression and purification of SPL C141A mutant
Both the WT SPL and the C141A mutant were expressed in LB medium containing the appropriate antibiotics as previously described.(39) The proteins were purified via Ni%NTA chromatography followed by an ion exchange chromatography using the SP Sepharose fast flow ion exchange resin (GE Healthcare Life Sciences, Piscataway, NJ). The bound protein was washed using a buffer containing 25 mM Tris, 250 mM NaCl and 10% glycerol (pH 7.0) for 10 column volumes. The protein was then eluted using the same buffer containing 500 mM NaCl instead. The resulting protein was diluted by 2-fold to reduce the salt concentration to 250 mM and saved for activity studies. In a separate purification process, phosphate buffer containing 25 mM sodium phosphate, 250 mM NaCl and 10% glycerol (pH 7.0) was used for the protein purification.
Protein, iron, and sulfide assays
Routine determinations of protein concentration were conducted by the Bradford method (42), using bovine gamma globulin as the protein standard. Protein concentrations were calibrated on the basis of the absorption of aromatic residues at 280 nm in the presence of 6 M guanidine hydrochloride using the method of Gill and von Hippel (43). Iron content was determined using obathophenanthroline (OBP) under reductive conditions after protein digestion in 0.8% KMnO4 and 1.2 M HCl as described by Fish (44). Iron standards were prepared from commercially available ferric chloride. Sulfide assays were carried out using the method described by Beinert (45).
Preparation of SP TpT and d4-SP TpT
These compounds were prepared via either chemical synthesis or solid state TpT photolysis as described before (39).
Synthesis of TpTSO2−
The TpTSO2− was prepared via a modified procedure as previously described in literature (14). As shown in Scheme 3, the synthesis took advantage of the weak strength of the C-S bond connected at the thymine methyl moiety. Photo-cleavage of this bond readily generates a thymine allylic radical, which subsequently reacts with dithionite to yield TpTSO2−. Although the yield for the photo-substitution is only 7%, the unreacted compound 5 can be readily recovered and re-used for the next round of photoreaction. Care has been taken to remove the dissolved O2 by purging with argon. The photosynthesis was then conducted in the Coy anaerobic chamber. Experimental details about the synthesis are available in supporting information (SI).
Scheme 3.

Synthesis of TpTOH
The synthesis of TpTOH was conducted via the reactions shown in Scheme 4. The hydroxyl group was introduced first to the methyl moiety of the thymine residue, which was subsequently protected before being cross-linked to another thymine residue via standard phosphoramidite chemistry. Detailed information regarding the synthesis is available in the SI.
Scheme 4.

SPL C141A activity assay
In a typical set of experiments, the reaction mixture contained 30 μM SPL C141A, 0.6 mM SP TpT substrate, 100 μM SAM in a final volume of 500 VL of buffer containing 25 mM Tris, 250 mM NaCl and 10% glycerol at pH 7.0. Freshly made sodium dithionite (final concentration 1 mM) was added as the reductant to initiate the enzyme reaction. The reaction was conducted as described before and analyzed by HPLC (39). In a separate set of experiments, the C141A mutant reaction was examined in phosphate buffer (containing 25 mM sodium phosphate instead of 25 mM Tris) to exclude the involvement of Tris in enzyme catalysis.
Enzyme activity with limited amount of SAM
To study the impact of SAM on the SP repair reaction, the WT SPL and the C141A mutant reactions were repeated using 1, 2, 5 and 10 equivalents of SAM (relative to protein) respectively. A typical reaction mixture contained 15 VM enzyme, 0.4 mM SP TpT substrate and differing amounts of SAM in a final volume of 800 VL of buffer containing 25 mM Tris, 250 mM NaCl and 10% glycerol at pH 7.0. Freshly made sodium dithionite (final concentration 1 mM) was added as reductant to initiate the reaction. At certain reaction time, 100 μL of the reaction solution was taken out, quenched by 3 μL of 3 M HCl and analyzed by HPLC.
Enzyme activity in the absence of external reductant
The C141A mutant protein was reduced by 1 mM of freshly prepared sodium dithionite for 30 min. Excess inorganic ions were removed via a Thermo Scientific Zeba Spin Desalting Column. SP TpT and SAM were then added to a final concentration of 0.3 mM and 50 μM (~ 3-fold of SPL) respectively to initiate the SP repair reaction. In another set of experiments, 1 mM or 10 mM DTT was added together with SP TpT and SAM. At different time points, 100 μL solution was taken out, quenched by HCl and analyzed by HPLC.
SPL C141A activity with limited dithionite
To understand the effect of dithionite to the C141A mutant reaction, the protein is pre-reduced and de-salted as described above. Dithionite or DTT was then re-supplemented to the de-salted enzyme. The reactions were quenched by HCl after 1 hr.
Examination of the TpTSPh (5) photoreaction
To reveal the reaction mechanism of the TpT formation, the TpTSPh photoreaction (final concentration of 1 mM) was studied in dd H2O or in a Tris buffer containing 25 mM Tris, 250 mM NaCl and 10% glycerol at pH 7.0 in the presence and absence of varying amounts of sodium dithionite respectively under an inert atmosphere. The reaction was allowed to proceed under 254 nm UV light for 10 minutes and the products analyzed by LC-MS. In another set of experiments, the reactions above were supplemented with DTT to a final concentration of 1 or 10 mM. To understand the origin of the proton incorporated into the formed TpT, the reactions were also repeated in D2O, methanol (CH3OH) and d1-methanol (CH3OD) respectively.
HPLC assay for product analysis
HPLC Chromatography was performed at room temperature with a Waters (Milford, MA) breeze HPLC system with a 2489 UV/Visible detector at 268 nm. An Agilent Zorbax reverse-phase C-18 column (3.5 μM, 4.6×50 mm) was equilibrated in 50 mM triethylammonium acetate, pH 6.5 (buffer A), and compounds were eluted with an ascending gradient (5% – 20%) of buffer B in 15 minutes which is composed of 50% buffer A and 50% acetonitrile at a flow rate of 1 mL/min. Under this gradient, SP TpT was eluted at 5.4 min, 5′-dA at 8.9 min, TpTSO 2− at 9.8 min, TpTOH at 12.9 min and TpT at 14.1 min. The identity of the products was confirmed by co-injection of respective authentic samples as well as by LC-MS spectrometry. The area of the product peak was determined after subtraction of the baseline from the t = 0 chromatograph and the amounts of 5′-dA, TpTSO2− and TpT formed were determined by reference to standard curves constructed with authentic samples.
The SAM analysis was conducted according to a modified literature procedure (46). Briefly, An Agilent Zorbax reverse-phase C-18 column (3.5 μM, 4.6×75 mm) was equilibrated in 0.1% formic acid supplemented by 1 mM heptanesulfonic acid (buffer A), and compounds were eluted from the HPLC column with an ascending gradient (0% – 30%) of buffer B in 14 minutes which is composed of 50% buffer A and 50% acetonitrile at a flow rate of 1 mL/min. Under this gradient, SP TpT was eluted at 3.3 min, TpTSO 2− and TpT at 6.5 min under a single peak, 5′-dA at 9.8 min, SAH (S-adenosyl-L-homocysteine) at 10.2 min, SAM at 11.4 min and MTA (methylthioadenosine) at 12.5 min (47).
LC MS assay for product analysis
LC-MS analyses were conducted via an Agilent 6130 Quadrupole LC/MS spectrometer coupled to an Agilent 1100 series chromatography system using a Waters X- bridgeTM OST C18 column (2.5 μM, 4.6×50 mm). The column was equilibrated in solvent A (5 mM ammonium acetate, pH 6.5), and compounds were eluted with an ascending gradient (0 – 17%) of solvent B (5 mM ammonium acetate in a 1:1 acetonitrile/methanol mixture) at a flow rate of 1 mL/min in 12 min. Under this gradient TpTSO2− elutes at 4.8min, TpTOH at 7.6 min and TpT at 8.2 min. The mass signals were monitored under positive and negative ion mode respectively.
The photoreaction of TpTSPh was analyzed with an Agilent Eclipse XDB reverse-phase C-18 column (5 μM, 4.6×150 mm). The buffers A and B are same as described above. The compounds were eluted with an ascending gradient (1–15%) in 20 minutes followed by another gradient (15–95%) in 10 minutes. Under this program, the TpTSO2− was eluted at 9.3 min, TpTOH at 16.3 min, TpT at 17.7 minutes, and TpTSPh at 25.2 minute.
Alkylation of Cysteine 141 and analyses
Cysteine alkylation of WT SPL or C141A mutant was carried out in 200 μL sodium phosphate buffer at pH 7.0 containing 250 mM NaCl and 10% glycerol. The protein (5 μM) was treated with 0.5 mM iodoacetamide in the dark for 1 hour. The reaction was conducted in anaerobic chamber, and the protein kept on ice through the treatment to minimize protein denaturation. The solution was then diluted by 2-fold using dd water, and 0.1 μL of the resulting solution injected into the Agilent 6520 Accurate-Mass Q-TOF LC/MS spectrometer. The data was acquired via Agilent MassHunter Workstation Data Acquisition (B.03.00) and analyzed via Qualitative Analysis of MassHunter Acquisition Data (B.03.00) software.
The remaining protein was digested by α-chymotrypsin and trypsin for overnight in 37 ° incubator. The resulting peptide fragments were injected into Agilent 6520 Accurate-Mass Q-TOF LC/MS and analyzed as described above. The activity of the alkylated protein was also studied under the standard procedure using 5-fold SAM and 10-fold SP and analyzed by HPLC.
EPR experiments
Continuous wave (CW) EPR spectra were recorded on a modified Varian spectrometer at 35 GHz (“Q”-band) and 2 K (48). The as-isolated SPL mutant (300 μM, 3.0 iron/protein) was reduced with 2 mM dithionite inside the anaerobic chamber for 60 min and placed into the EPR tube and immediately frozen in liquid N2. The EPR spectra were recorded as described previously (39). EPR simulations were performed using the program QPOW (49), as modified by J.Telser.
Results
SPL C141A expression, purification and characterization
The C141A-pET28 vector contains a his6-tag, which allows the protein to be purified to up to ~ 90% purity via Ni-NTA chromatography as judged by SDS-PAGE (47). As SPL protein is positively charged at pH 7 (pI = 8.5), it binds tightly to the SP sepharose ion exchange resin in the presence of 150 mM NaCl; while most of the E. coli proteins do not bind. In our hands, the ion exchange chromatography is absolutely needed. The protein purified only by Ni-NTA chromatography was found to be contaminated by an unknown enzyme, which consumes the 5′-dA generated from the SP repair process. This enzyme can be removed by a thorough washing process from the ion-exchange column (a minimum of 5-column volumes of washing is needed). After elution from the ion-exchange column, the SPL C141A was obtained in > 99% purity (47).
The purified protein possesses a dark-brown color, suggesting the presence of the [4Fe-4S] cluster. The presence of such a cluster is supported by the characteristic UV absorption band at 420 nm (47). The iron/sulfur content analysis of the as-isolated protein found that each protein molecule contains 3.2 Fe and 3.0 S. Upon dithionite reduction, the cluster exhibits an S = 1/2 signal (Figure S3) that is essentially indistinguishable from the EPR signal from the WT SPL cluster (47). Simulation yields g = [2.026(5), 1.928(5), 1.890(5)], which is equivalent to the g values reported for this center by other workers (11, 12, 50). These observations agree with the previous studies of this mutant enzyme (14), namely that the C141 residue is not involved in coordinating with the radical SAM FeS cluster.
Iodoacetamide treatment
As described by our previous work, 5′-dA is not the direct H-atom donor to the thymine allylic radical (39). The TpT radical takes an H atom from a source which is able to exchange proton with the buffer, making the cysteine 141 in B. subtilis SPL the most likely H donor. Such an assumption is further supported by an in vivo study (40), which showed that spores carrying the SPL C141A mutant are highly sensitive to UV irradiation as well as an in vitro study by Foncecave(12) that TpT is no longer the major SP repair product in the C141A mutant reaction. The G. thermodenitrificans SPL structure further suggests that the cysteine is close to the SP substrate (41), although a conserved tyrosine cannot be excluded as the H donor. If C141 is the direct H donor, we expect that (1) The C141 is solvent exposable so that the TpT radical can have a chance to accept the H atom; and (2) No other protein residues are involved in this H transfer process.
We used the iodoacetamide treatment assay to test if C141 is solvent exposable. Iodoacetamide treatment is widely used to label the cysteine residues in proteins at either the native form or the denatured form (51, 52). It was used to alkylate the free cysteine residues in iron-sulfur proteins such as APS reductase (53) and aconitase (54). Should the cysteine 141 in B. subtilis SPL be the direct H atom donor to the thymine allylic radical (39), it would be accessible from the aqueous solution and readily alkylated by the iodoacetamide treatment. As shown in Figure 2A, after treated by 100-fold of iodoacetamide under the native condition, ~ 50% of the resulting enzyme molecules carry one alkyl label and the rest carry 0, 2, 3 or 4 labels respectively. In contrast, ~ 80% of the C141A mutant remains unlabeled after the reaction. Among the labeled C141A species, the intensity of the mono-alkylated protein is comparable to the di-alkylated peak in the treated WT enzyme, indicating that the alkylation site is at one of the three cluster cysteines. Trypsin digestion experiment further proves that the C141 residue of the WT SPL carried the acetamide label. Such a label was missing in the same peptide fragment from the SPL C141A mutant, suggesting that the C141 in WT SPL is prone to the iodoacetamide treatment.
Figure 2.
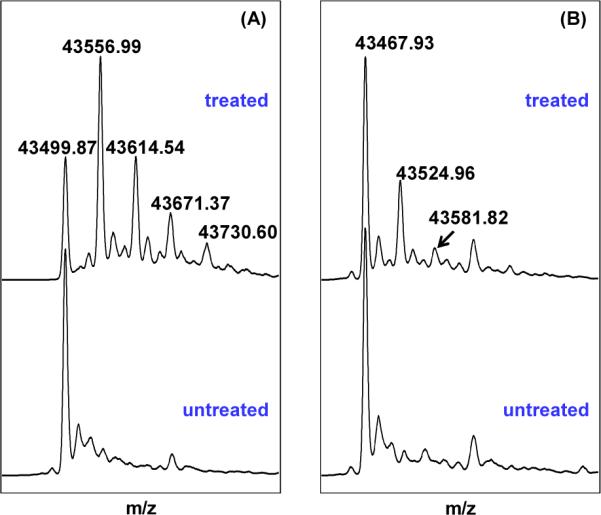
(A) Deconvoluted ESI-MS spectrum of the WT SPL enzyme treated by excess iodoacetamide under native conditions. Comparing with the untreated protein, the major species in treated enzyme exhibits a mass gain of 57.13. The intensity of this peak is much stronger than those corresponding to proteins which carry 2, 3 or 4 labels. This observation suggests that one of the four cysteines in WT SPL is prone to alkylation. (B) Deconvoluted ESI-MS spectrum of the SPL C141A mutant treated by excess iodoacetamide under an identical condition. The mono-labeled species in the treated C141A mutant exhibited a peak whose intensity is comparable with that of the treated WT enzyme carrying two alkyl labels, but much weaker than that corresponding to the mono-alkylated WT enzyme, suggesting that the alkylation site is at one of the three cluster cysteines. The different behavior toward the iodoacetamide treatment between these two proteins suggests that C141 residue in the WT SPL is accessible from the aqueous solution.
As shown by Cravatt et.al., a fully exposed cysteine in a native protein can be readily labeled by a stoichiometic amount of iodoacetamide (52). Even with 100-fold iodoacetamide, only 50% of the WT enzymes have their C141 alkylated, suggesting that although it is accessible from the aqueous buffer, the C141 is relatively well protected in the enzyme binding pocket. Prolonged incubation resulted in the alkylation of all cysteine residues in the WT SPL as well as C141A mutant (47). The fact that the three cysteines in the radical SAM motif were alkylated is surprising as the cysteinate residues are usually protected once chelated by the iron-sulfur cluster as revealed in iron-sulfur enzymes such as the APS reductase (53) and aconitase (54). On the other hand, the radical SAM motif typically resides on a flexible loop at the protein surface (24), making the three cysteine residues less likely to be effectively protected. Such a secondary structure may cause the cysteines to be attacked by the labeling reagent, as indicated by our observations here.
To exclude the possibility that the alkylation of the four cysteine residues in WT SPL are due to the SPL denaturation during the treatment, we tested the activity of the alkylated WT enzyme. In contrast to the WT enzyme, which yields the dinucleotide TpT as the SP repair product, the alkylated SPL should behave like the C141A mutant, producing TpTSO2− as the major product (14). Under the hypothesis that the mono-alkylated species shown in Figure 2A only had the C141 residue modified, the alkylated protein and unmodified WT SPL were thus present in a ~5:3 ratio. As described in the following section, the WT enzyme repairs SP at a rate which is ~3-fold faster than the C141A mutant. Therefore, TpT/TpTSO2− should be produced under a ~ 2: 1 ratio. Indeed, the isolated TpT/TpTSO2− exhibited a mole ratio of 2.5 : 1 (47), suggesting that the SPL carrying an alkylated cysteine 141 is still in its native state; the C141 in the WT SPL enzyme is accessible from the aqueous buffer.
SPL C141A reaction with regular SP TpT
The iodoacetamide treatment proves that C141 is accessible to the TpT radical, thus fulfilling the prerequisite to be the H atom donor. Next, we ask whether other protein residues are involved in this H atom transfer process. Should a protein residue other than the C141, for instance the Y99 in B. subtilis SPL as suggested by the structure shown in Figure 1, serve as the H donor, the residue would still be present in the C141A mutant, which is expected to produce TpT as the SP repair product at least for the first enzyme turnover. A lag phase for the TpTSO2− formation is thus expected. In our kinetic studies of the SPL C141A reaction, we found that in the presence of 1 mM sodium dithionite and the absence of DTT, TpTSO2− was isolated as the major reaction product and TpT as the minor one. The third product, TpTOH, was also detected although its yield was extremely low (< 0.1%). This is consistent with the earlier findings from Fontecave et.al. More importantly, using the calibration curves constructed with the authentic TpT, and TpTSO2− respectively, their formation rates were calculated and shown in Table 1. The formation rate of TpTOH cannot be determined due to its low reaction yield. Our studies revealed that the TpTSO2− and TpT formed under constant reaction rates from the very beginning of the reaction, no lag phase was observed for the TpTSO2− formation (Figure 3). These findings strongly suggest that no other protein residue locates between the TpT radical and the C141 in the WT SPL reaction pathway.
Table 1.
The formation rates (min−1) of TpTSO2− and TpT in C141A mutant reaction with 1 mM sodium dithionite, 0.1 mM of SAM and varying concentrations of DTT as the H atom donor.
| No DTT | 1 mM DTT | 10 mM DTT | |
|---|---|---|---|
| TpTSO2− | 0.11 ± 0.01 | 0.11 ± 0.01 | 0.05 ± 0.005 |
| TpT | 0.014 ± 0.002 | 0.019 ± 0.002 | 0.041 ± 0.005 |
| TpTSO2− + TpT | 0.12 ± 0.01 | 0.13 ± 0.01 | 0.09 ± 0.01 |
Figure 3.

HPLC chromatograph of the SP TpT repair process mediated by the B. subtilis SPL C141A mutant with 30 μM enzyme, 150 μM SAM and 1 mM dithionite. Under the HPLC program, the SP TpT was eluted at 5.4 min, 5'-dA at 8.9 min, TpTSO2− at 9.8 min, TpTOH at 12.9 min and TpT at 14.1 min. Linear formations of TpTSO2− and TpT were observed in the first 30 minutes of the reaction. The TpTOH peak overlapped with an uncharacterized compound in our HPLC chromatograph. The yield of TpTOH was too low to be determined.
TpT formation in SPL C141A reaction
Our data above suggest that C141 is the most likely intrinsic H atom donor in SPL. However, the C141A mutant still produces TpT during the SP repair reaction, which is puzzling since it does not contain such an H donor any more. One possibility is that the TpT radical obtains an H atom from the small molecules in the reaction buffer. We first excluded the possibility of Tris as the H atom donor, as same reaction pattern was obtained when the C141A reaction was conducted in the phosphate buffer. As thiol compounds are usually supplemented in radical SAM reactions, we next examined the SPL C141A reaction in the presence of various concentrations of DTT. As shown in Table 1, under equal amounts of dithionite and DTT (1 mM), little difference was observed in both TpT and TpTSO2− formation. In contrast, the presence of 10 mM DTT increased the TpT formation rate by 3-fold, however, the formation of TpTSO2− and the overall SP repair rate were found to be slower. Other small thiol compounds such as β-mercaptoethanol and methanethiol also facilitate TpT formation; however, they are not as effective as DTT probably due to the stronger S-H bond in these compounds.
The dithionite reduces the [4Fe-4S]2+ cluster via an outer-sphere electron transfer process; the excess DTT may block the enzyme binding pocket, preventing the dithionite from accessing the cluster and resulting in a partial inhibition of the SP repair reaction. A similar inhibition by concentrated DTT was also observed in the WT SPL reaction. Despite the inhibitory effect, the improved yield suggests that presence of an H atom donor could facilitate the TpT formation. However, this enhancement requires a large amount of DTT. Considering that no small molecule possesses a 10 mM concentration in our enzyme reaction, the H atom donation from a small molecule can be ruled out.
SPL C141A reaction with varying amount of dithionite
A second possibility for TpT formation in C141A mutant is that TpT radical obtains an H atom from the residues on the protein such as the tyrosine Y99. We hence studied the enzyme reaction under varying concentrations of dithionite. Dithionite possesses dual functions in the SPL C141A reaction: 1) reducing the [4Fe-4S]2+ cluster to initiate the SAM cleavage reaction, and 2) quenching the thymine radical to yield TpTSO2−. Should the protein provide the needed H atom, the availability of this H atom would be constant. Under a low dithionite supply, the TpTSO2− formation would be disfavored due to the lack of reactant; production of TpT should be subsequently increased.
To focus on the latter function, we pre-reduced the cluster using a large access of dithionite and removed the unreacted dithionite by desalting columns. We then added varying amounts of dithionite or 10 mM DTT as the radical quenching reagent, together with SP and SAM, to reveal the quenching effect of dithionite on the SP repair reaction.
Surprisingly, under low dithionite concentration, TpTSO2− becomes an even more dominating product (Table 2). Increasing the dithionite concentration however favors the formation of TpT, as indicated by the ascending TpTSO2−/TpT ratio found in Table 2. Under 10 mM dithionite, almost comparable amounts of TpTSO2− and TpT were obtained. Should H atom donor from the protein be responsible for the TpT formation, an opposite trend for the TpTSO2−/TpT vs dithionite concentration would be expected. We thus conclude that dithionite is directly responsible for the TpT formation; TpT found in the mutant reaction is unlikely generated via a direct H atom transfer process as that in the WT SPL reaction (39). Although supplementing 10 mM DTT to the pre-reduced mutant resulted in an almost exclusive formation of TpT (Table 2) (47), the TpT formed here is clearly via a different mechanism from that in the regular SPL C141A reaction.
Table 2.
TpTSCO2−/TpT ratio found in (a) the SP repair reaction mediated by a pre-reduced C141A mutant (10 μM) supplemented by varying amount of dithionite or DTT, and (b) photoreaction of TpTSPh (1 mM) supplemented by varying amount of dithionite.
| Dithionite (mM) | 0.02 | 0.05 | 0.2 | 1 | 10 | 10mM DTT |
| TpTSO2−/TpT a | 7.1±1 | 5.8±0.6 | 3.2±0.3 | 2.8±0.3 | 1.8±0.3 | <0.1 |
| TpTSO2−/TpT b | 0.08 ± 0.01 | 0.14 ± 0.01 | 0.18 ± 0.02 | 0.21 ± 0.02 | 0.44 ± 0.03 | 0.22 ± 0.02 c |
The yield of the TpTSPh photoreaction is ~ 7%, making the concentration of the TpT radical generated from the photoreaction to be < 7 μM. This concentration should be comparable to the TpT radical concentration produced from the SPL C141A mutant reaction.
The reaction was conducted in the presence of both 10 mM dithionite and 10 mM DTT.
TpT is not produced via TpTSO2− decomposition
A third possibility for TpT formation in C141A mutant is via a TpTSO2− degradation reaction as indicated in Scheme 5. This reaction resembles the reduction of the pyridinium salts by dithionite in the coenzyme NAD+ (55–57). To test this hypothesis, we incubated the TpTSO2− with the enzyme in a pH 7.0 buffer for 1 hr. No TpT was found as analyzed by HPLC. Moreover, we repeated the experiments in the pH 7.0 and 9.0 Tris buffer in the presence or absence of UV light respectively, as the neutral or mildly basic condition is suggested to favor the SO2 elimination (55–57). Analysis of the reaction products revealed that TpTSO2− is stable under these conditions and no TpT was observed either. These observations suggest that the TpT found in the C141A reaction originates directly from the TpT radical; it is not a secondary product resulting from the TpTSO2− degradation process.
Scheme 5.

TpT formation in TpTSPh (5) photoreaction
The TpT formation in the SPL C141A mutant reaction is neither due to the quenching of TpT radical from the small molecules in the solution or from the residues on the protein, nor due to the TpTSO2− degradation. To reveal the mechanism for the TpT formation, we examined the TpTSPh photoreaction shown in Scheme 3 under an inert atmosphere using roughly identical conditions to the enzyme reaction. Surprisingly, TpTSO2− becomes a minor species even with dithionite concentration approaches 10 mM. Additionally, as shown in Table 2, as the dithionite concentration increased from 0.02 mM to 10 mM, the TpTSO2−/TpT ratio also increased, which is totally opposite to those observed in the SPL C141A reaction. As discussed below, the TpTSO2− formation is likely via a radical quenching mechanism and it should be fairly favorable for the TpT radical to recombine with the •SO2−, yielding TpTSO2−. If TpT forms via an H atom abstraction process, the reaction is relatively unfavorable unless a very weak X-H bond is available from the environment. The absence of such an X-H bond thus implies that TpT must form via a mechanism other than the radical mediated H atom abstraction reaction.
Further analysis of the TpTSPh (5) photoreaction revealed that in the absence of dithionite, irradiating 1 mM 5 in H2O still produces TpT (Figure 4). Addition of dithionite as a reductant improves the TpT yield by ~3 folds, suggesting that TpT forms via a radical reduction process. Due to the strong bond dissociation energy (BDE) for the O-H bond in water, it is highly unlikely for the TpT formation to proceed via an H atom abstraction mechanism. Moreover, neither thiophenol nor diphenol disulfide was observed in the reaction, suggesting that they are unlikely to be involved in the reaction here. Thus, the reaction route that the thiophenol first exchanges a proton with the solvent followed by an S-H bond homolytic cleavage to donate the H atom to the TpT radical is unlikely to be involved here. The presence of H atom donor (1 mM DTT) has little impact to the reaction, as reflected by the similar reaction patterns exhibited by Figure 4A and 4C, further supporting this conclusion.
Figure 4.
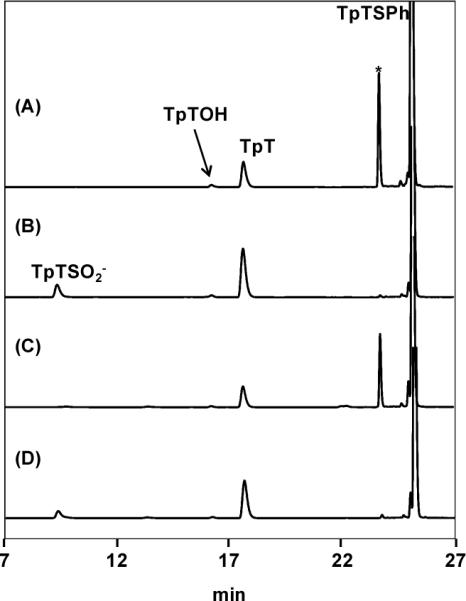
The HPLC chromatograph of the TpTSPh (5) photoreaction conducted under the 254 nm UV light in the anaerobic chamber. (A) Photoreaction in H2O with 1 mM 5; (B) reaction A + 1 mM sodium dithionite; (C) reaction A + 1 mM DTT. (D) reaction A + 1 mM sodium dithionite + 1 mM DTT. The peak with * is likely due to the addition of TpT cation to 5 as indicated by ESI-MS, the nature of which is characterized in reference 74.
Consumption of SAM and formation of 5'-dA
After elucidating the role of C141 in SPL catalysis and the mechanism behind the TpT formation in the SPL C141A mutant reaction, we next attempted to further characterize the SPL C141A reaction. In radical SAM enzyme reactions, the ratio between 5'-dA and product is of significance. A 1 : 1 ratio suggests that SAM is a co-substrate, being consumed after each catalytic turnover. While a 1 : X (X > 1) ratio suggests that SAM is a co-factor, being regenerated after each catalytic cycle (58). As shown in Figure 6B (also in the SI), SAM was consumed as the reaction progressed; the ratio between the consumed SAM (formed 5'-dA) and reacted SP was determined to be 1.08 ± 0.1, suggesting that in SPL C141A reaction, SAM serves as a co-substrate. As a control, analyzing the amounts of 5'-dA and TpT formed in the reaction using the carefully purified WT enzyme resulted in a ratio of 1.5 ± 0.2, which equals the ratio shown in the first several minutes of the WT SPL reaction in our previous report (39). As shown in Figure 5, when plotted together, the difference between the C141A and WT SPL reactions is obvious, suggesting that SAM plays a partially catalytic role in the WT SPL reaction, but a non-catalytic role in the C141A mutant.
Figure 6.

The [M - H]− signals of the TpT, TpTSO2− and TpTOH isolated from the SP TpT and d4-SP TpT repair reactions mediated by SPL C141A mutant. The deuterium abstracted from the d4-SP TpT during the repair process is not returned to the products, the corresponding d3-species were isolated for all three products. (* in the ESI-MS spectrum of d3-TpTSO2− donates an impurity from the background)
Figure 5.

(A) The formation of TpT, TpTSO2 −, 5'-dA as well as the consumption of SAM in the WT SPL reaction. 5 equivalents of SAM were added to initiate the reaction. A background level of TpTSO2− was observed along with the formation of TpT. The ratio between the SP repair products (TpT +TpTSO2−) and 5'-dA is found to be 1.5 : 1 for the WT SPL reaction, suggesting that SAM is partially catalytic here. (B) The formation of TpT, TpTSO2−2, 5'-dA as well as the consumption of SAM in the SPL C141A reaction with 5 equivalents of SAM supplemented. The ratio between the SP repair products (TpT +TpTSO2−) and 5'-dA was found to be 1.08 : 1 for the SPL C141A reaction, suggesting that SAM plays a non-catalytic role in the C141A reaction and is a co-substrate. Under the 5 equivalents of SAM, the SP TpT repair rate by WT SPL was determined to be 0.41 ± 0.03 min−1, and by SPL C141A mutant was determined to be 0.14 ± 0.02 min−1.
As mentioned earlier, the protein purified from the Ni-NTA column contains some uncharacterized enzyme(s), which is very difficult to remove without a thorough washing on the ion-exchange column, even though the SDS-PAGE suggests that the isolated protein is >99% pure. Different from the well characterized S-adenosylhomocysteine nucleosidase (SAH nucleosidase) which is known to hydrolyze the 5'-dA to adenine (59–62), the contaminant protein not only hydrolyzes the 5'-dA to adenine; but also further degrades the adenine to an unknown product, which cannot be detected by the HPLC UV detector at 260 nm. Presence of this contaminant protein drastically disturbs the SPL activity studies, as the 5'-dA can no longer be used as a marker to reflect the SAM regeneration process. Therefore, the disappearance of the 5'-dA in our previous report is not due to the SAM regeneration, but due to the presence of this contaminant enzyme. The Figure 9B in our previous report is an error (39).
Carefully analyzing the amount of SAM left in the solution, we found that SAM could be slightly increased by 0 ~ 0.2 equivalent as the SP repair reaction slowed down (47), which may indicate that a small portion of SAM was regenerated in the WT SPL reaction. No increase in SAM is found in the SPL C141A mutant reaction. However, we are hesitant to draw any conclusion here as the SAM increase can only be observed in ~ 50% of the WT SPL reactions we conducted (7 out of 13 experiments). Despite of the inconsistency, our findings could still be useful to other researchers as few experiments in characterizing the stoichiometry of SAM involved in a radical SAM reaction have been reported.
SPL C141A reaction in the absence of excess reductant
Being able to reliably measure the amount of yielded 5'-dA provides us a marker to accurately determine the reaction turnover numbers. In our previous report, after using desalting column to remove the excess dithionite, the pre-reduced cluster of WT SPL was found to support 12 turnovers under ~3 equivalents of SAM (39). Using our latest calibration curve, the number is now determined to be 5.4. This number still suggests that the WT SPL reaction has a closed catalytic cycle and is consistent with our latest results here.
We performed similar experiment with the C141A mutant to test if the C→A mutation disrupts such a cycle. After pre-reduction with dithionite for 30 min, the resulting [4Fe-4S]+ cluster in SPL C141A mutant supported only ~0.4 turnover, with TpT the dominant product when 10 mM of DTT was supplemented. No turnover was observed in the absence of DTT, suggesting that being able to quench the TpT radical is the pre-requisite for the occurrence of the SP repair reaction. Other thiol compounds can also rescue the SPL C141A reaction, but the yield of TpT would be even lower. The low concentration of DTT (< 1 mM) has little effect to stimulate the reaction, suggesting that the thymine radical is well protected by the enzyme against external H donors. This finding is consistent with the result obtained in the C141 labeling studies described above that C141 appears to be protected in the enzyme binding pocket. As participants for the key H atom transfer process during the SP repair process, both the TpT radical and the C141 are protected by the SPL enzyme.
The 0.4 turnover could be due to the incomplete reduction of the radical SAM cluster upon the 30-min dithionite treatment. As shown by our previous EPR experiment, a 30-minute reduction led to only ~ 40% of SPL cluster reduction; 1 hr reduction resulted in ~ 70% of the cluster being converted to the 1+ oxidation state (39). The observation is in line with the 20% [4Fe-4S]+ cluster reduction observed by Broderick (63) and the 40% cluster reduction by Knappe (64) in PFL-AE studies upon prolonged reduction. More importantly, the difficulty for the externally added thiol to be correctly positioned to quench the TpT radical may play a major role for the low yield of TpT as under an identical condition, > 5 turnovers were observed for the WT SPL reaction (39). Nevertheless, the < 1 turnover in the C141A mutant suggests that the C141A mutant no longer possesses a closed catalytic cycle.
SPL C141A reaction with d4-SP TpT as substrate
As shown in Scheme 2, the WT SPL enzyme takes the H6proR atom to initiate the SP repair reaction, the abstracted H atom however is not returned to the TpT product. In the aqueous buffer, the repair of d4-SP TpT by WT SPL leads to the formation of d3-TpT. As shown in Figure 6, all three products, TpTSO2−, TpT, and TpTOH, from the d4-SP TpT repair reaction mediated by the C141A mutant contain only three deuteriums, suggesting that the abstracted deuterium by the 5′-dA• was not returned as expected. The C→A mutation disturbs the H-atom back-donation step and the steps after, but likely leaves the previous steps intact.
Linear formations against reaction time can be obtained for TpTSO2− and TpT species under a saturated substrate concentration; comparing the correspondent rates using d0- and d4-SP TpT as substrates reveals the DVmax KIE to be 1.7 ± 0.2 for TpTSO2− and TpT respectively (Table 3). The identical KIEs for TpT and TpTSO2− suggest that these species share the same rate determining step. Thus the DVmax KIE for the C141A mutant reaction can be reported as 1.7 ± 0.2, which is smaller than the KIE of 2.8 ± 0.3 exhibited by the WT enzyme (39).
Table 3.
Formation rates (min−1) of TpTSO2− and TpT and Vmax KIE in SPL C141A reaction.
| Without DTT | With 1 mM DTT | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Substrate used | SP TpT | d4-SP TpT | Vmax KIE | SP TpT | d4-SP TpT | Vmax KIE |
| TpTSO2− | 0.1 ± 0.01 | 0.055 ± 0.005 | 1.78 | 0.1 ± 0.01 | 0.055 ± 0.005 | 1.70 |
| TpT | 0.02 ± 0.002 | 0.012 ± 0.001 | 1.67 | 0.026 ± 0.003 | 0.015 ± 0.002 | 1.73 |
| TpTSO2− + TpT | 0.12 ± 0.01 | 0.067 ± 0.006 | 1.76 | 0.126 ± 0.013 | 0.07 ± 0.007 | 1.71 |
Discussion
Photosynthesis of TpTSO2−
As shown in Scheme 3, the synthesis of TpTSO2−2 took advantage of the weak C-S bond which is readily cleaved upon photo-excitation. The dithionite dianion has a small dissociation constant in water with a Kd ≈ 10−6 mM (65–68); auto-dissociation of S2O42− results in the formation of 2•SO2−. The •SO2− radical readily recombines with the thymine allylic radical after photocleavage of the C-S bond in TpTSPh to yield the TpTSO2− (Scheme 6A). The •SO2− produced by the dithionite auto-dissociation reaction was used to activate the initiator and trigger the radical polymerization of the vinyl chloride (69), further supporting the mechanism in Scheme 6A.
Scheme 6.

Although the radical recombination is the most plausible mechanism, the radical induced S-S bond replacement reaction as shown in Scheme 6B cannot be excluded. The thymine allylic radical attacks the disulfide bond in dithionite, replacing a –SO2− moiety of the dithionite to form the TpTSO2−. Thiyl radical based S-S replacement is a common reaction encountered in biochemistry (70); other organic radicals are implied to be able induce the homolytic cleavage of the S-S bond as well (71). Although the vast majority of the TpTSO2− is believed to form via the radical recombination reaction, considering the millimolar dithionite concentration used in our synthesis, we cannot exclude the involvement of the radical replacement mechanism to produce a very minor amount of TpTSO2−.
It is worth pointing out in TpTSO2− synthesis that the reaction should be conducted under an inert atmosphere. Should the photoreaction be conducted in the air, the product would contain ~ 30% of TpTSO3−, the two-electron oxidation product of TpTSO2−. As the TpTSO2− is reasonably stable and exposing its aqueous solution to air for 24 h did not result in obvious oxidation (as monitored by LC-MS), we conclude that the oxidation likely occurred during the photosynthesis, where the UV light excited the O2, which then oxidized the sulfinic acid to yield the sulfonate product TpTSO3−.
C141 is likely the H atom donor
To donate the H atom directly to the thymine allylic radical, the C141 –SH moiety must be accessible to the substrate. Our observation that the C141 residue can be alkylated by iodoacetamide under the native condition suggests that this cysteine is solvent accessible and should be readily available to the thymine radical. Such a conclusion is also supported by the crystal structures of the G. thermodenitrificans SPL (41). The similar behaviors of WT SPL and SPL C141A mutant under the iodoacetamide treatment suggest that the C→A mutation is unlikely to change the protein structure, which is also supported by the structural data (41). Should the C141 not be the direct H atom donor, but pass the H atom to another protein residue (Y99, for instance) during the course of the catalysis, the residue would also possess a weak X-H bond, which is perfectly positioned for the H atom transfer to the TpT radical. Different from the class I ribonucleotide reductase, where the radical species has to be passed to the substrate binding pocket before the ribonucleotide modification reaction is initiated (72, 73), the radical chain in the SPL reaction is initiated from the substrate. Given the weak BDE for the protein residues which are known to be involved in the radical relay process, should a protein residue other than C141 (For instance, the Y99) serve as the direct H atom donor, an H atom would be readily available to the TpT radical, producing TpT during the first turnover. Such a hypothesis is against our observations here as the formations of TpT and TpTSO2− are linear from the very beginning of the SP repair reaction, suggesting that both species form at the same time.
Furthermore, should the putative residue be responsible for H atom donation, an X• radical will be left behind on this residue. Consequently, the enzyme is likely to be modified (formation of a SO2− adduct, for instance) in order to quench this X• as the electron transfer chain is broken after the C→A mutation. However, the C141A protein is not modified after the reaction, as proved by the ESI-MS analysis (47). Thus, even though our data tell us nothing about the proximity between the C141 and the TpT radical, it is extremely unlikely for a residue other than the C141 to directly donate an H atom to the TpT radical.
Formation of three SP repair products
Among the three products TpTSO2−, TpT, and TpTOH formed in the SPL C141A reaction, the TpTSO2− likely forms via a radical recombination reaction as discussed above. The radical replacement mechanism may also play a very minor role in its formation.
The formation of TpT is more interesting. Although TpT is produced in both SPL WT and C141A mutant reactions, it likely forms via different mechanisms. In WT SPL reaction, the –SH moiety on C141 likely donates an H atom to the thymine radical to form TpT. Such a low energy H donor is no longer present in the C141A mutant unless the reaction solution is supplemented by a large amount of thiols. As revealed by our thymine allylic radical activity studies, such a radical readily undergoes disproportionation and reduction reactions, with reaction rates likely comparable to the radical recombination reaction (74). Actually, the fact that the presence of large excess of thiol compounds has little impact to the TpT formation further suggests that the H atom abstraction is an unfavorable reaction due to the relatively slow reaction rate (74). A reduction of thymine radical is the most likely thymine formation mechanism here, which is likely involved in the TpT formation in C141A reaction as well.
However, a direct reduction of the thymine allylic radical is unfavored as the resulting thymine methyl anion is extremely unstable, which makes the thymine radical cation reduction the most favorable mechanism (74). Since the reaction is conducted in water, the thymine C4=O bond must readily associate with a proton through the hydrogen bonding interaction with the aqueous solvent. Reducing the resulting TpT radical cation would generate TpT (Scheme 7). Such a radical cation reduction mechanism (proton coupled electron transfer) has not received much attention before in the DNA biochemical studies (75–78). Our data here argue that the thymine cation radical reduction pathway may play an important role in the thymine damage repair, which has been largely overlooked in the past.
Scheme 7.

As shown in Table 2, in the SPL C141A mutant reaction, the TpTSO2− is the dominating product. Contrastingly, the TpT prevailed during the photoreaction of TpTSPh. The trend of TpTSO2−/TpT vs dithionite concentration is also different in these two reactions. We believe that the protein network plays a major role here in changing the reaction pattern. In the TpTSPh photoreaction, all reactants can freely interact in the solution. The cation radical reduction to produce TpT is relatively unaffected when the dithionite concentration increases while the formation of TpTSO2− becomes more competitive due to the availability of more reactants. Contrastingly, the TpT radical is well protected in the C141A binding pocket, as suggested by the observation that a large amount of externally added DTT (10 mM) is needed in order to quench the TpT radical. As shown by the CPD photolyase structure, the thymine C4=O bond is recognized by a protein residue via the hydrogen bonding interaction (79). A similar recognition pattern is expected here. The pre-organized enzyme–substrate complex likely hinders a proton from the solvent (or protein) to adopt the right position to associate with the allylic radical as shown in Scheme 7, thus slowing down the TpT formation and making the TpTSO2− formation the favorable reaction.
Similar to that in the TpTSPh photoreaction, the dithionite is likely to serve as the electron donor in SPL C141A mutant reaction to facilitate the formation of TpT as well. However, we cannot exclude the possibility that the protein harbored [4Fe-4S]+ cluster donates the needed electron. Nevertheless, the TpT likely forms via a proton coupled electron transfer process in the SPL C141A mutant, which is different from the H atom transfer mechanism in the WT SPL reaction.
TpTOH was isolated in previous thymine oxidation studies, its formation is typically induced by the oxidation of TpT allylic radical by either addition of O2(80) or radical recombination with a •OH radical (81). Our latest data suggests that it can also be produced via a thymine radical disproportionation reaction (74). As the enzyme reaction was conducted anaerobically, O2 can be ruled out, making the radical disproportionation the most likely mechanism. The thymine cation then reacts with water to produce TpTOH (Scheme 6). Under the reducing conditions adopted in the enzyme reaction, the thymine cation must be reductively quenched. Consequently, only a tiny amount of TpTOH was produced.
Enzyme activity and Kinetic isotope effect
The bond dissociation energy (BDE) of the cysteine S-H bond is suggested to be ~3–4 kcal/mol lower than the allylic C-H bond (82–85). Assuming that the SPL configuration is such that the C141 is at the vicinity of the thymine allylic radical, the H atom back transfer step must be heavily favored and should not affect the overall SP repair rate. In contrast, the C→A mutation removes this natural H atom transfer process; the TpT allylic radical has to either be released before it is quenched or wait until a quenching reagent diffuses into the enzyme binding pocket to react with the radical and generate a stable product. Such a quenching step must be slow, resulting in the ~ 3-fold rate reduction exhibited by the C141A mutant relative to that of the WT enzyme (Figure 5).
Theoretical calculations suggest that the two steps involved in the H-atom exchange with 5′-dA possess the highest energy barriers (82, 83, 86, 87). Of these two steps, the H-atom back-donation step has the higher barrier due to the energy difference between the allylic radical and the C-H bond associated with the methyl moiety of 5′-dA. Although our data suggests that it is the –SH moiety of C141, not the –CH3 of 5′-dA, that serves as the H atom donor for the thymine allylic radical, the resulting protein radical(s) are likely to be stable as well. The regeneration of the 5′-dA• is expected to be slow (72, 88). Thus, it is reasonable to hypothesize that the rate-determining step in SPL reaction is the regeneration of 5′-dA•; all steps before, including the H atom abstraction by the 5′-dA• from the C6 carbon of SP to obtain the SP radical, belong to the “rate-determining zone” (89). The rate constants from all steps in this zone will contribute to the Vmax KIE (89–91). The intrinsic isotope effect caused by the reaction between the 5′-dA• and the deuterated SP substrate is likely to be “diluted” by other steps in this zone, resulting in a moderate Vmax KIE of 2.8 for the WT SPL reaction (39).
The rationale above assumes that SAM is regenerated after every catalytic cycle. However, the observed TpT/5′-dA ratio of 1.5 suggests that the roughly two thirds of the formed 5′-dA and methionine molecules exchange with SAM from the environment during the SPL catalysis. Currently, no data is available to help us determine how fast this exchange step is. However, it is clear that the reaction rates we observed here seem to be the average of two reactions: one results in the SAM regeneration, the other results in the exchange of 5′-dA and methionine with SAM. Therefore, the Vmax KIE here is unlikely to represent the true Vmax KIE occurring in vivo. To determine the Vmax KIE for the in vivo reaction, a method has to be found to prevent the SAM/5′dA-methionine exchange from occurring.
Such a moderate Vmax KIE is further reduced in the C141A mutant reaction due to the slower thymine radical quenching process. Although changing the SP TpT to deuterated d4-SP TpT slows down the SP repair reaction, the repair rate was reduced to a less extent, resulting in a Vmax KIE of 1.7 ± 0.2 for both TpTSO2− and TpT formations. The almost identical KIE exhibited by these two products indicates that despite the different quenching mechanism, the reaction rates for this step must be similar. Comparing with the WT SPL (39), the reduced Vmax KIE in the mutant reaction suggests that the H abstraction step by 5'-dA• becomes even less rate-determining (92). The radical quenching step likely becomes the new rate-limiting step, as the SAM regeneration process is no longer possible. However, in C141A reaction, SAM exchanges with the formed 5′-dA and methionine at the end of each catalytic cycle. Should this step be rate-determining, a slower TpT radical quenching step shown in Figure 7B can still lead to the reduced Vmax KIE as observed here.
SAM regeneration
To recycle SAM after each turnover is probably the most efficient choice for the germinated spores. UV irradiation can convert as much as 8% of the total thymine in bacterial genomic DNA to SP (4, 93); these SPs must be repaired by SPL for the bacteria to survive (7, 94, 95). Germinated spores have many urgent needs in resuming their normal life cycle with limited resources, to make good use of every SAM molecule must be important. The in vitro reactions under a large excess of SAM reported here are unlikely to happen in vivo.
Despite the importance of SAM regeneration for in vivo reaction, such a process is not fully established by in vitro experiments. Previous efforts to establish the SAM regeneration were conducted via a tritium labeling experiment in SPL and lysine-2,3-aminomutase (LAM) (10, 20, 37). In both cases, the percentage of label transfer from the substrate into SAM was suggested to be very low (39, 96, 97). Recent proposals by Eguchi in the studies of BtrN (98) and by Liu in the studies of DesII (27), both of which are radical SAM enzymes, suggested that SAM cleavage, as well as the subsequent H atom abstraction by the 5'-dA• from enzyme substrates, may be reversible. Such reversible processes could explain the tritium incorporation into SAM as previously observed in SPL reaction (10).
SAM becomes a co-substrate in the C141A reaction. This is reasonable considering that C141 is a part of the catalytic pathway; disrupting this pathway makes the closed catalytic cycle open. In WT SPL reaction, the TpT/5′-dA ratio of 1.5 ± 0.1 suggests that only one third of SAM behaves like a co-factor. We'd like to observe the SAM regeneration in a more significant scale; however, this seems unlikely using the dinucleotide SP TpT as the enzyme substrate.
The small regeneration scale predicted here highlights the difficulty to directly detect the SAM regeneration process. In B-12 enzymes, where the no excess B-12 cofactor is needed, the 5′-dA recycling process can be readily observed. In contrast, the binding of SAM to radical SAM enzyme is weak; to achieve enough enzyme activity, 2–3 equivalents of SAM is needed (10, 39). This then determines that there are always excess SAM molecules available in the reaction buffer, which can readily exchange with the yielded 5'-dA and methionine, making the SAM recyclization unnecessary. To study the SAM regeneration process, a limited SAM supply is required. In addition, the enzyme has to bind SAM (5'-dA and methionine) tightly. Such a condition may be achievable by using a tightly bound SP substrate. As shown in Broderick's experiments, using a SP containing plasmid DNA, one equivalent SAM can support > 500 turnovers (10). How a tighter substrate binding enhances the SAM recyclization is currently unclear.
How is SAM regenerated in the WT enzyme? Cysteine based thiyl radical is known to abstract an H atom from 5′-dA in class II ribonucleotide reductase (RNR) to produce the 5′-dA• before it recombines with cobalamin to regenerate the adenosylcobalamin at the end of each catalytic cycle (99). However, in class II RNR, the 5′-dA• interacts only with the active-site cysteine and does not associate with the substrate at any point of the catalytic cycle. In contrast, in SPL, the 5′-dA• abstracts the H6proR atom of the 5'-T of SP to initiate the repair reaction (39); while the C141 likely provides the needed H atom to the methyl radical at the 3'-T to generate the repaired TpT. Should the thiyl radical on C141 be directly responsible for H atom abstraction from 5′-dA to regenerate SAM, the TpT would be released first, followed by a major protein conformational change before the thiyl radical can interact with the 5′-dA. On the other hand, it is reasonable for the thiyl radical to oxidize a neighboring protein residue before the reaction with 5'-dA occurs. In that case, SPL must harbor an electron transfer pathway with the C141 a key element. As shown in the G. thermodenitrificans SPL structure, a conserved tyrosine is found between the conserved cysteine and SAM (41). Although protein conformational change is expected after the methylene bridge in SP and SAM C-S bond are cleaved, the conserved tyrosine is still likely to be involved in SPL catalysis/SAM regeneration. The role of this tyrosine residue in SPL reaction is to be investigated in the future.
Alternative route for SAM regeneration?
Although the SAM regeneration is the most reasonable hypothesis, we cannot rule out other possibilities for the SPL catalyzed SP repair reaction. For instance, what if the 5′-dA generated from the SAM reductive reaction is responsible only for the first catalytic cycle and the protein radical generated takes over and catalyzes the rest of the turnovers? Under in vitro conditions, the presence of excess SAM and reductant determines that both catalytic routes may occur.
The mechanistic studies of SPL thus far suggest that the enzyme reaction can be divided into two half reactions: SP repair/TpT formation and SAM regeneration. These two half reactions are tightly coupled under a low SAM concentration in vivo; but are uncoupled under sufficient SAM supply. Progress has been made in elucidating the half reaction involved in SP repair and TpT formation; however, the other half - SAM regeneration, remains largely unclear. Future work should be focused on the second half reaction before a clear mechanistic understanding of SPL catalysis can be achieved.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the reviewers for their insightful comments to help improve this manuscript. We thank the National Institute of Environmental Health Sciences (R00ES017177) as well as IUPUI startup funds for financial support. The NMR and MS facilities at IUPUI are supported by National Science Foundation MRI grants CHE-0619254 and DBI-0821661, respectively. We thank Professor Brian M. Hoffman, Northwestern University, for use of the 35 GHz EPR spectrometer, which is funded by NSF grant MCB-0316038. We thank Prof. Squire J. Booker, the Pennsylvania State University, for the generous gift of plasmid pDB1282, the E. coli SAH nucleosidase construct as well as all the helpful discussions. We thank Tyler Grove at the Pennsylvania State University for sharing protocols on SAM separation by HPLC. We thank Professor Steven Rokita at the Johns Hopkins University and Prof. Haibo Ge at IUPUI for the helpful discussions on the TpT radical reduction. We thank Professor Neil Marsh at the University of Michigan for the helpful discussions on the SAM and B12 regeneration processes. We thank Dr. Alhosna Benjdia at the Department of Biomolecular Mechanisms, Max-Planck Institute for Medical Research for her help in preparing Figure 1.
Footnotes
Supporting Information Available: Syntheses and characterizations of the TpTSO2−2 and TpTOH. Protein purification and characterization. Enzyme activity assays. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Desnous CL, Guillaume D, Clivio P. Spore photoproduct: A key to bacterial eternal life. Chem. Rev. 2010;110:1213–1232. doi: 10.1021/cr0781972. [DOI] [PubMed] [Google Scholar]
- 2.Nicholson WL, Setlow B, Setlow P. Binding of DNA in vitro by a small, acid-soluble spore protein from Bacillus subtilis and the effect of this binding on DNA topology. J. Bacteriol. 1990;172:6900–6906. doi: 10.1128/jb.172.12.6900-6906.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohr SC, Sokolov NV, He CM, Setlow P. Binding of small acid-soluble spore proteins from Bacillus subtilis changes the conformation of DNA from B to A. Proc. Natl. Acad. Sci. USA. 1991;88:77–81. doi: 10.1073/pnas.88.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicholson WL, Setlow B, Setlow P. Ultraviolet irradiation of DNA complexed with alpha/beta-type small, acid-soluble proteins from spores of Bacillus or Clostridium species makes spore photoproduct but not thymine dimers. Proc. Natl. Acad. Sci. USA. 1991;88:8288–8292. doi: 10.1073/pnas.88.19.8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bumbaca D, Kosman J, Setlow P, Henderson RK, Jedrzejas MJ. Crystallization and preliminary x-ray analysis of the complex between a Bacillus subtilis [alpha]/[beta]-type small acid-soluble spore protein and DNA. Acta Crystallogr. 2007;F 63:503–506. doi: 10.1107/S1744309107022750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee KS, Bumbaca D, Kosman J, Setlow P, Jedrzejas MJ. Structure of a protein-DNA complex essential for DNA protection in spores of Bacillus species. Proc. Natl. Acad. Sci. USA. 2008;105:2806–2811. doi: 10.1073/pnas.0708244105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L. Mechanistic studies of the radical SAM enzyme spore photoproduct lyase (SPL) BBA - Proteins and Proteomics. 2012 doi: 10.1016/j.bbapap.2011.11.008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rebeil R, Sun Y, Chooback L, Pedraza-Reyes M, Kinsland C, Begley TP, Nicholson WL. Spore photoproduct lyase from Bacillus subtilis spores is a novel iron-sulfur DNA repair enzyme which shares features with proteins such as class III anaerobic ribonucleotide reductases and pyruvate-formate lyases. J. Bacteriol. 1998;180:4879–4885. doi: 10.1128/jb.180.18.4879-4885.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rebeil R, Nicholson WL. The subunit structure and catalytic mechanism of the Bacillus subtilis DNA repair enzyme spore photoproduct lyase. Proc. Natl. Acad. Sci. USA. 2001;98:9038–9043. doi: 10.1073/pnas.161278998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheek J, Broderick J. Direct H atom abstraction from spore photoproduct C-6 initiates DNA repair in the reaction catalyzed by spore photoproduct lyase: Evidence for a reversibly generated adenosyl radical intermediate. J. Am. Chem. Soc. 2002;124:2860–2861. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- 11.Buis J, Cheek J, Kalliri E, Broderick J. Characterization of an active spore photoproduct lyase, a DNA repair enzyme in the radical S-adenosylmethionine superfamily. J. Biol. Chem. 2006;281:25994–26003. doi: 10.1074/jbc.M603931200. [DOI] [PubMed] [Google Scholar]
- 12.Chandor A, Berteau O, Douki T, Gasparutto D, Sanakis Y, Ollagnier-De-Choudens S, Atta M, Fontecave M. Dinucleotide spore photoproduct, a minimal substrate of the DNA repair spore photoproduct lyase enzyme from Bacillus subtilis. J. Biol. Chem. 2006;281:26922–26931. doi: 10.1074/jbc.M602297200. [DOI] [PubMed] [Google Scholar]
- 13.Friedel M, Berteau O, Pieck J, Atta M, Ollagnier-De-Choudens S, Fontecave M, Carell T. The spore photoproduct lyase repairs the 5S- and not the 5R-configured spore photoproduct DNA lesion. Chem. Commun. 2006:445–447. doi: 10.1039/b514103f. [DOI] [PubMed] [Google Scholar]
- 14.Chandor-Proust A, Berteau O, Douki T, Gasparutto D, Ollagnier-De-Choudens S, Fontecave M, Atta M. DNA repair and free radicals, new insights into the mechanism of spore photoproduct lyase revealed by single amino acid substitution. J. Biol. Chem. 2008;283:36361–36368. doi: 10.1074/jbc.M806503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandra T, Silver SC, Zilinskas E, Shepard EM, Broderick WE, Broderick JB. Spore photoproduct lyase catalyzes specific repair of the 5R but not the 5S spore photoproduct. J. Am. Chem. Soc. 2009;131:2420–2421. doi: 10.1021/ja807375c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silver S, Chandra T, Zilinskas E, Ghose S, Broderick W, Broderick J. Complete stereospecific repair of a synthetic dinucleotide spore photoproduct by spore photoproduct lyase. J. Biol. Inorg. Chem. 2010;15:943–955. doi: 10.1007/s00775-010-0656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donnellan JE, Jr., Setlow RB. Thymine photoproducts but not thymine dimers found in ultraviolet-irradiated bacterial spores. Science. 1965;149:308–310. doi: 10.1126/science.149.3681.308. [DOI] [PubMed] [Google Scholar]
- 18.Lin G, Li L. Elucidation of spore-photoproduct formation by isotope labeling. Angew. Chem. Int. Ed. 2010;49:9926–9929. doi: 10.1002/anie.201005228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin G, Chen C-H, Pink M, Pu J, Li L. Chemical synthesis, crystal structure and enzymatic evaluation of a dinucleotide spore photoproduct analogue containing a formacetal linker. Chem. Eur. J. 2011;17:9658–9668. doi: 10.1002/chem.201101821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marsh ENG, Patterson DP, Li L. Adenosyl radical: Reagent and catalyst in enzyme reactions. ChemBioChem. 2010;11:604–621. doi: 10.1002/cbic.200900777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frey PA, Hegeman AD, Ruzicka FJ. The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 22.Chatterjee A, Li Y, Zhang Y, Grove TL, Lee M, Krebs C, Booker SJ, Begley TP, Ealick SE. Reconstitution of thic in thiamine pyrimidine biosynthesis expands the radical SAM superfamily. Nature Chem. Biol. 2008;4:758–765. doi: 10.1038/nchembio.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paraskevopoulou C, Fairhurst SA, Lowe DJ, Brick P, Onesti S. The elongator subunit Elp3 contains a Fe4S4 cluster and binds S-adenosylmethionine. Mol. Microbiol. 2006;59:795–806. doi: 10.1111/j.1365-2958.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- 24.Vey JL, Drennan CL. Structural insights into radical generation by the radical SAM superfamily. Chem. Rev. 2011;111:2487–2506. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: Functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruszczycky MW, Choi S-H, Liu H-W. Stoichiometry of the redox neutral deamination and oxidative dehydrogenation reactions catalyzed by the radical SAM enzyme DesII. J. Am. Chem. Soc. 2010;132:2359–2369. doi: 10.1021/ja909451a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szu P-H, Ruszczycky MW, Choi S-H, Yan F, Liu H-W. Characterization and mechanistic studies of DesII: A radical S-adenosyl-L-methionine enzyme involved in the biosynthesis of TDP-D-desosamine. J. Am. Chem. Soc. 2009;131:14030–14042. doi: 10.1021/ja903354k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulder DW, Boyd ES, Sarma R, Lange RK, Endrizzi JA, Broderick JB, Peters JW. Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydAΔEFG. Nature. 2010;465:248–251. doi: 10.1038/nature08993. [DOI] [PubMed] [Google Scholar]
- 29.Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ. A radically different mechanism for S-adenosylmethionine–dependent methyltransferases. Science. 2011;332:604–607. doi: 10.1126/science.1200877. [DOI] [PubMed] [Google Scholar]
- 30.Yan F, Lamarre JM, Roì^Hrich R, Wiesner J, Jomaa H, Mankin AS, Fujimori DG. RlmN and Cfr are radical SAM enzymes involved in methylation of ribosomal RNA. J. Am. Chem. Soc. 2010;132:3953–3964. doi: 10.1021/ja910850y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okada Y, Yamagata K, Hong K, Wakayama T, Zhang Y. A role for the elongator complex in zygotic paternal genome demethylation. Nature. 2010;463:554–558. doi: 10.1038/nature08732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ. A consensus mechanism for radical SAM-dependent dehydrogenation? BtrN contains two [4Fe-4S] clusters. Biochemistry. 2010;49:3783–3785. doi: 10.1021/bi9022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farrar CE, Siu KKW, Howell PL, Jarrett JT. Biotin synthase exhibits burst kinetics and multiple turnovers in the absence of inhibition by products and product-related biomolecules. Biochemistry. 2010;49:9985–9996. doi: 10.1021/bi101023c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Driesener R, Challand M, Mcglynn S, Shepard E, Boyd E, Broderick J, Peters J, Roach P. [FeFe]-hydrogenase cyanide ligands derived from S-adenosylmethionine-dependent cleavage of tyrosine. Angew. Chem. Int. Ed. 2010;49:1687–1690. doi: 10.1002/anie.200907047. [DOI] [PubMed] [Google Scholar]
- 35.Wecksler SR, Stoll S, Tran H, Magnusson OT, Wu S-P, King D, Britt RD, Klinman JP. Pyrroloquinoline quinone biogenesis: Demonstration that PqqE from Klebsiella pneumoniae is a radical S-adenosyl-L-methionine enzyme. Biochemistry. 2009;48:10151–10161. doi: 10.1021/bi900918b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grove TL, Lee K-H, St. Clair J, Krebs C, Booker SJ. In vitro characterization of Atsb, a radical SAM formylglycine-generating enzyme that contains three [4Fe-4S] clusters. Biochemistry. 2008;47:7523–7538. doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frey P, Magnusson O. S-adenosylmethionine: A wolf in sheep's clothing, or a rich man's adenosylcobalamin? Chem. Rev. 2003;103:2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 38.Mccusker KP, Fujimori DG. The chemistry of peptidyltransferase center-targeted antibiotics: Enzymatic resistance and approaches to countering resistance. ACS Chem. Biol. 2012;7:64–72. doi: 10.1021/cb200418f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang L, Lin G, Liu D, Dria KJ, Telser J, Li L. Probing the reaction mechanism of spore photoproduct lyase (SPL) via diastereoselectively labeled dinucleotide SP TpT substrates. J. Am. Chem. Soc. 2011;133:10434–10447. doi: 10.1021/ja110196d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fajardo-Cavazos P, Rebeil R, Nicholson W. Essential cysteine residues in Bacillus subtilis spore photoproduct lyase identified by alanine scanning mutagenesis. Curr. Microbiol. 2005;51:331–335. doi: 10.1007/s00284-005-0052-8. [DOI] [PubMed] [Google Scholar]
- 41.Benjdia A, Heil K, Barends TRM, Carell T, Schlichting I. Structural insights into recognition and repair of UV-DNA damage by spore photoproduct lyase, a radical SAM enzyme. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks603. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 43.Gill SC, Von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 44.Fish WW. Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol. 1988;158:357–364. doi: 10.1016/0076-6879(88)58067-9. [DOI] [PubMed] [Google Scholar]
- 45.Beinert H. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal. Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 46.She QB, Nagao I, Hayakawa T, Tsuge H. A simple HPLC method for the determination of S-adenosylmethionine and S-adenosylhomocysteine in rat tissues: The effect of vitamin B6 deficiency on these concentrations in rat liver. Biochem. Biophys. Res. Commu. 1994;205:1748–1754. doi: 10.1006/bbrc.1994.2871. [DOI] [PubMed] [Google Scholar]
- 47. See supporting information.
- 48.Werst MM, Davoust CE, Hoffman BM. Ligand spin densities in blue copper proteins by Q-band 1H and 14N ENDOR spectroscopy. J. Am. Chem. Soc. 1991:1533–1538. [Google Scholar]
- 49.Belford RL, Nilges MJ. Computer simulations of powder spectra. EPR Symposium, 21st Rocky Mountain Conference; Denver, Colorado. August,1979. [Google Scholar]
- 50.Pieck J, Hennecke U, Pierik A, Friedel M, Carell T. Characterization of a new thermophilic spore photoproduct lyase from Geobacillus stearothermophilus (Splg) with defined lesion containing DNA substrates. J. Biol. Chem. 2006;281:36317–36326. doi: 10.1074/jbc.M607053200. [DOI] [PubMed] [Google Scholar]
- 51.Shiio Y, Aebersold R. Quantitative proteome analysis using isotope-coded affinity tags and mass spectrometry. Nat. Protoc. 2006;1:139–145. doi: 10.1038/nprot.2006.22. [DOI] [PubMed] [Google Scholar]
- 52.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carroll KS, Gao H, Chen HY, Leary JA, Bertozzi CR. Investigation of the iron-sulfur cluster in Mycobacterium tuberculosis APS reductase: Implications for substrate binding and catalysis. Biochemistry. 2005;44:14647–14657. doi: 10.1021/bi051344a. [DOI] [PubMed] [Google Scholar]
- 54.Kennedy MC, Spoto G, Emptage MH, Beinert H. The active-site sulfhydryl of aconitase is not required for catalytic activity. J. Biol. Chem. 1988;263:8190–8193. [PubMed] [Google Scholar]
- 55.Carelli V, Liberatore F, Scipione L, Musio R, Sciacovelli O. On the structure of intermediate adducts arising from dithionite reduction of pyridinium salts: A novel class of derivatives of the parent sulfinic acid. Tetrahedron Lett. 2000;41:1235–1240. [Google Scholar]
- 56.Carelli V, Liberatore F, Scipione L, Di Rienzo B, Tortorella S. Dithionite adducts of pyridinium salts: Regioselectivity of formation and mechanisms of decomposition. Tetrahedron. 2005;61:10331–10337. [Google Scholar]
- 57.Fox JL. Sodium dithionite reduction of flavin. FEBS Lett. 1974;39:53–55. doi: 10.1016/0014-5793(74)80015-3. [DOI] [PubMed] [Google Scholar]
- 58.Marsh E, Patwardhan A, Huhta M. S-adenosylmethionine radical enzymes. Bioorg. Chem. 2004;32:326–340. doi: 10.1016/j.bioorg.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 59.Parveen N, Cornell KA. Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol. 2011;79:7–20. doi: 10.1111/j.1365-2958.2010.07455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi-Rhee E, Cronan JE. A nucleosidase required for in vivo function of the S-adenosyl-L-methionine radical enzyme, biotin synthase. Chem. Biol. 2005;12:589–593. doi: 10.1016/j.chembiol.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 61.Lee JE, Cornell KA, Riscoe MK, Howell PL. Structure of Escherichia coli 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase inhibitor complexes provide insight into the conformational changes required for substrate binding and catalysis. J. Biol. Chem. 2003;278:8761–8770. doi: 10.1074/jbc.M210836200. [DOI] [PubMed] [Google Scholar]
- 62.Lee JE, Cornell KA, Riscoe MK, Howell PL. Structure of E. Coli 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase reveals similarity to the purine nucleoside phosphorylases. Structure. 2001;9:941–953. doi: 10.1016/s0969-2126(01)00656-6. [DOI] [PubMed] [Google Scholar]
- 63.Henshaw TF, Cheek J, Broderick JB. The [4Fe-4S](1+) cluster of pyruvate formate-lyase activating enzyme generates the glycyl radical on pyruvate formate-lyase: EPR-detected single turnover. J. Am. Chem. Soc. 2000;122:8331–8332. [Google Scholar]
- 64.Kulzer R, Pils T, Kappl R, Huttermann J, Knappe J. Reconstitution and characterization of the polynuclear iron-sulfur cluster in pyruvate formate-lyase-activating enzyme - molecular properties of the holoenzyme form. J. Biol. Chem. 1998;273:4897–4903. doi: 10.1074/jbc.273.9.4897. [DOI] [PubMed] [Google Scholar]
- 65.Lynn S, Rinker RG, Corcoran WH. The monomerization rate of dithionite ion in aqueous solution. J. Phys. Chem. 1964;68:2363–2363. [Google Scholar]
- 66.Rinker RG, Gordon TP, Mason DM, Corcoran WH. The presence of the SO2 radical ion in aqueous solutions of sodium dithionite. J. Phys. Chem. 1959;63:302–302. [Google Scholar]
- 67.Rinker RG, Lynn S, Mason DM, Corcoran WH. Kinetics and mechanism of thermal decomposition of sodium dithionite in aqueous solution. Ind. Eng. Chem. Fund. 1965;4:282–288. [Google Scholar]
- 68.Lambeth DO, Palmer G. The kinetics and mechanism of reduction of electron transfer proteins and other compounds of biological interest by dithionite. J. Biol. Chem. 1973;248:6095–6103. [PubMed] [Google Scholar]
- 69.Rosen BM, Percec V. Single-electron transfer and single-electron transfer degenerative chain transfer living radical polymerization. Chem. Rev. 2009;109:5069–5119. doi: 10.1021/cr900024j. [DOI] [PubMed] [Google Scholar]
- 70.Bonifacic M, Asmus KD. Adduct formation and absolute rate constants in the displacement reaction of thiyl radicals with disulfides. J. Phys. Chem. 1984;88:6286–6290. [Google Scholar]
- 71.Schlosberg RH. Chemistry of coal conversion. Plenum Press; New York: 1985. [Google Scholar]
- 72.Stubbe J, Van Der Donk WA. Protein radicals in enzyme catalysis. Chem. Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 73.Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem. Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 74.Lin G, Li L. Disproportionation and reduction of 5-(2'-deoxyuridinyl)methyl radical. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hong IS, Greenberg MM. Efficient DNA interstrand cross-link formation from a nucleotide radical. J. Am. Chem. Soc. 2005;127:3692–3693. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]
- 76.Ding H, Greenberg MM. γ-radiolysis and hydroxyl radical produce interstrand cross-links in DNA involving thymidine. Chem. Res. Toxicol. 2007;20:1623–1628. doi: 10.1021/tx7002307. [DOI] [PubMed] [Google Scholar]
- 77.Ding H, Majumdar A, Tolman JR, Greenberg MM. Multinuclear NMR and kinetic analysis of DNA interstrand cross-link formation. J. Am. Chem. Soc. 2008;130:17981–17987. doi: 10.1021/ja807845n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peng XH, Pigli YZ, Rice PA, Greenberg MM. Protein binding has a large effect on radical mediated DNA damage. J. Am. Chem. Soc. 2008;130:12890–12891. doi: 10.1021/ja805440v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mees A, Klar T, Gnau P, Hennecke U, Eker APM, Carell T, Essen L-O. Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science. 2004;306:1789–1793. doi: 10.1126/science.1101598. [DOI] [PubMed] [Google Scholar]
- 80.Steenken S. Electron-transfer-induced acidity/basicity and reactivity changes of purine and pyrimidine bases. Consequences of redox processes for DNA base pairs. Free Radical Res. 1992;16:349–379. doi: 10.3109/10715769209049187. [DOI] [PubMed] [Google Scholar]
- 81.Teebor GW, Frenkel K, Goldstein MS. Ionizing radiation and tritium transmutation both cause formation of 5-hydroxymethyl-2'-deoxyuridine in cellular DNA. Proc. Natl. Acad. Sci. USA. 1984;81:318–321. doi: 10.1073/pnas.81.2.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo J, Luo Y, Himo F. DNA repair by spore photoproduct lyase: A density functional theory study. J. Phys. Chem. B. 2003;107:11188–11192. [Google Scholar]
- 83.Himo F. C-C bond formation and cleavage in radical enzymes, a theoretical perspective. BBA Bioenergetic. 2005;1707:24–33. doi: 10.1016/j.bbabio.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 84.S. Jursic B. Reliability of hybrid density theory-semiempirical approach for evaluation of bond dissociation energies. Journal of the Chemical Society, Perkin Transactions. 1999;2:369–372. [Google Scholar]
- 85.Blanksby SJ, Ellison GB. Bond dissociation energies of organic molecules. Acc. Chem. Res. 2003;36:255–263. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- 86.Hioe J, Zipse H. Radicals in enzymatic catalysis-A thermodynamic perspective. Faraday Discuss. 2010;145:301–313. [Google Scholar]
- 87.Hioe J, Zipse H. Radical stability and its role in synthesis and catalysis. Org. Biomol. Chem. 2010;8:3609–3617. doi: 10.1039/c004166a. [DOI] [PubMed] [Google Scholar]
- 88.Cotruvo JA, Stubbe J. Class I ribonucleotide reductases: Metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem. 2011;80:733–767. doi: 10.1146/annurev-biochem-061408-095817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Northrop DB. Steady-state analysis of kinetic isotope effects in enzymic reactions. Biochemistry. 1975;14:2644–2651. doi: 10.1021/bi00683a013. [DOI] [PubMed] [Google Scholar]
- 90.Cleland WW. Partition analysis and concept of net rate constants as tools in enzyme kinetics. Biochemistry. 1975;14:3220–3224. doi: 10.1021/bi00685a029. [DOI] [PubMed] [Google Scholar]
- 91.Li L, Marsh ENG. Deuterium isotope effects in the unusual addition of toluene to fumarate catalyzed by benzylsuccinate synthase. Biochemistry. 2006;45:13932–13938. doi: 10.1021/bi061117o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Madhavapeddi P, Marsh EN. The role of the active site glutamate in the rearrangement of glutamate to 3-methylaspartate catalyzed by adenosylcobalamin-dependent glutamate mutase. Chem. Biol. 2001;8:1143–1149. doi: 10.1016/s1074-5521(01)00081-3. [DOI] [PubMed] [Google Scholar]
- 93.Setlow B, Setlow P. Dipicolinic acid greatly enhances production of spore photoproduct in bacterial spores upon UV irradiation. Appl. Environ. Microbiol. 1993;59:640–643. doi: 10.1128/aem.59.2.640-643.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Setlow P. Mechanisms for the prevention of damage to DNA in spores of Bacillus species. Ann. Rev. Microbiol. 1995;49:29–54. doi: 10.1146/annurev.mi.49.100195.000333. [DOI] [PubMed] [Google Scholar]
- 95.Donnellan JE, Stafford RS. The ultraviolet photochemistry and photobiology of vegetative cells and spores of Bacillus megaterium. Biophys. J. 1968;8:17–28. doi: 10.1016/S0006-3495(68)86471-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kilgore JL, Aberhart DJ. Lysine 2,3-aminomutase - role of S-adenosyl-L-methionine in the mechanism - demonstration of tritium transfer from (2RS,3RS)-[3-3H]lysine to S-adenosyl-L-methionine. J. Chem. Soc. Perkin Trans. 1991;1:79–84. [Google Scholar]
- 97.Aberhart DJ. Studies on the mechanism of lysine 2,3-aminomutase. J. Chem. Soc. Perkin Trans. 1988;1:343–350. [Google Scholar]
- 98.Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. Characterization and mechanistic study of a radical SAM dehydrogenase in the biosynthesis of butirosin. J. Am. Chem. Soc. 2007;129:15147–15155. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- 99.Licht S, Gerfen GJ, Stubbe J. Thiyl radicals in ribonucleotide reductases. Science. 1996;271:477–481. doi: 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.