

Abstract
BACKGROUND AND PURPOSE
Diabetes is characterized by hyperglycaemia, which facilitates the formation of advanced glycation end-products (AGEs). Type 2 diabetes mellitus is commonly accompanied by non-alcoholic steatohepatitis, which could lead to hepatic fibrosis. Receptor for AGEs (RAGE) mediates effects of AGEs and is associated with increased oxidative stress, cell growth and inflammation. The phytochemical curcumin inhibits the activation of hepatic stellate cells (HSCs), the major effectors during hepatic fibrogenesis. The aim of this study was to explore the underlying mechanisms of curcumin in the elimination of the stimulating effects of AGEs on the activation of HSCs. We hypothesize that curcumin eliminates the effects of AGEs by suppressing gene expression of RAGE.
EXPERIMENTAL APPROACH
Gene promoter activities were evaluated by transient transfection assays. The expression of rage was silenced by short hairpin RNA. Gene expression was analysed by real-time PCR and Western blots. Oxidative stress was evaluated.
KEY RESULTS
AGEs induced rage expression in cultured HSCs, which played a critical role in the AGEs-induced activation of HSCs. Curcumin at 20 µM eliminated the AGE effects, which required the activation of PPARγ. In addition, curcumin attenuated AGEs-induced oxidative stress in HSCs by elevating the activity of glutamate-cysteine ligase and by stimulating de novo synthesis of glutathione, leading to the suppression of gene expression of RAGE.
CONCLUSION AND IMPLICATIONS
Curcumin suppressed gene expression of RAGE by elevating the activity of PPARγ and attenuating oxidative stress, leading to the elimination of the AGE effects on the activation of HSCs.
LINKED ARTICLE
This article is commented on by Stefanska, pp. 2209–2211 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2012.01959.x
Keywords: diabetes, hyperglycaemia, hepatic fibrosis, hepatic stellate cells, gene expression, phytochemical
Introduction
Elevated levels of blood glucose (i.e. hyperglycaemia) are a feature of diabetes. Non-alcoholic fatty liver disease (NAFLD) refers to a wide spectrum of liver damage, ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), advanced fibrosis and cirrhosis. Accumulating evidence has suggested that NAFLD/NASH patients are more likely to have impaired glucose metabolism, including insulin resistance and elevated levels of blood glucose (Hatziagelaki et al., 2012), as reviewed by Leclercq (2010). Approximately 15–40% NASH patients progress into hepatic fibrosis (Clark, 2006). Studies have demonstrated that hyperglycaemia is a key factor in the initiation and induction of renal fibrogenesis in in vitro and in vivo models with diabetic nephropathy (Lam et al., 2003; Chen et al., 2004). However, the role and underlying mechanisms of hyperglycaemia in the induction of hepatic fibrogenesis remain largely to be defined.
Exposure of cells to excess glucose induces inflammation and other pathological disorders, but the underlying mechanisms remain to be established. Most current hypotheses are based on the formation of advanced glycation end-products (AGEs), which are the non-enzymatic results of a chain of non-oxidative and oxidative reactions of excessive sugars with proteins and/or lipids (Bierhaus et al., 2005). AGEs deposit in tissues, causing cellular dysfunctions and even organ failures, such as cardiovascular diseases, neuropathy and blindness. The impairments caused by AGEs are largely associated with the interaction of AGEs and their receptors mediating many signalling transduction pathways and pro-inflammatory responses. Receptor for AGEs (RAGE) is a member of the immunoglobulin superfamily of cell surface molecules and is associated with increased oxidative stress, cell growth and inflammation (Schmidt et al., 2000). A dramatic increase in the abundance of RAGE was observed in diabetic patients with high levels of blood AGEs (Brett et al., 1993; Tanji et al., 2000). The interaction of AGEs and RAGE may stimulate the activation of a diverse array of signalling cascades, including MAPKs, Jak/STAT, PI3K and members of the Rho GTPase signalling pathway (Ramasamy et al., 2009). The interaction of AGEs–RAGE mediates the development of many chronic diseases, including diabetic complications, which was confirmed in studies with RAGE-/- mice (Bierhaus et al., 2005). Pharmacological blockade of RAGE actions or genetic deletion of RAGE imparts significant protection in murine models of diabetes (Ramasamy et al., 2009). RAGE is, therefore, a promising therapeutic target for the development of diabetic complications (Ramasamy et al., 2009).
Hepatic fibrosis is excessive accumulation of extracellular matrix proteins including type I collagen in the liver (Bataller and Brenner, 2005; Friedman, 2008). Hepatic stellate cells (HSCs) are the central mediators and major effectors during hepatic fibrogenesis. In response to hepatic injury, quiescent HSCs undergo a process of activation, rendering them becoming highly proliferative and fibrogenic cells (Bataller and Brenner, 2005; Friedman, 2008). The process of activation is coupled with the induction of oxidative stress and down-regulation of PPARγ (Tsukamoto et al., 2006; Friedman, 2008). It has been reported that RAGE is up-regulated in cultured HSCs (Fehrenbach et al., 2001), and AGEs induce cell proliferation of HSCs (Iwamoto et al., 2008). It is noteworthy that culturing quiescent HSCs on plastic plates causes spontaneous activation, mimicking the process seen in vivo, which provides a good model for elucidating underlying mechanisms of HSC activation and studying potential therapeutic intervention of the process (Friedman, 2008).
Few effective medicines are available for combating T2DM- and NASH-associated hepatic fibrosis (Calamita and Portincasa, 2007; Federico et al., 2008). It is, thus, of high priority to identify innocuous anti-fibrotic agents. Most evolving anti-fibrogenic therapies are aimed at inhibiting HSC activation. We and others have shown that curcumin, the yellow pigment in curry from turmeric, inhibits HSC activation in vitro (Xu et al., 2003; Zheng and Chen 2004; 2006; 2007; Zheng et al., 2007) and protects the liver from fibrogenesis in vivo (Park et al., 2000; Nanji et al., 2003; Fu et al., 2008). We recently reported that curcumin attenuated the effect of leptin on elevating glucose levels in HSCs by blocking translocation of glucose transporter-4 and increasing glucokinase (Tang and Chen, 2010). Curcumin has received attention as a promising dietary component in the protection of the liver against fibrogenic insults (O'Connell and Rushworth, 2008). In addition, we recently observed that AGEs stimulated the activation of HSCs in vitro by inducing cell proliferation and stimulating expression of genes relevant to HSC activation, which were dramatically eliminated by curcumin (Lin et al., 2012). However, the underlying mechanisms remain undefined.
The aims of this study were to evaluate the effect of AGEs on regulating gene expression of RAGE in HSCs, the effect of RAGE on the activation of HSCs and the role of curcumin in eliminating the AGE effects, and to further explore the underlying mechanisms. Results from this study support our original hypothesis that one of mechanisms of AGEs in the stimulation of HSC activation is to induce gene expression of RAGE. Curcumin eliminated the effects of AGEs and suppressed gene expression of RAGE in HSCs by elevating the activity of PPARγ and attenuating oxidative stress.
Methods
AGE preparation and chemicals
AGEs-BSA was prepared following the protocol described by others (Makita et al., 1992). In brief, 50 mg·mL−1 of BSA (USB Corp., Cleveland, OH) and 0.5 M of glucose (Sigma-Aldrich Corp. St. Louis, MO) were dissolved in 0.2 M of sodium phosphate buffer (pH 7.4). A BSA control and a glucose control were respectively prepared by dissolving BSA (50 mg·mL−1) alone, or glucose (0.5 M) alone, in sodium phosphate buffer (pH 7.4) (0.2 M). After sterilization with sterile Acrodisc® syringe filters, the solutions were incubated in the dark at 37°C for 60 days. Unbound materials were removed by extensive dialysis against PBS. The concentration of AGEs was determined as described previously by measuring AGEs-specific fluorescence with excitation at 360 nm and emissions at 440 nm (Monnier et al., 1984; Wrobel et al., 1997). The fluorescence of the BSA control was used as a base line. The fluorescence of qualified AGEs–BSA used in our experiments must be at least 70-fold higher than that of the BSA control. The quality and eligibility of AGEs were evaluated and confirmed as suggested previously (Miele et al., 2003). No contamination with insulin-like growth factor-1 and/or endotoxin was detected. l-Buthionine-sulfoximine (BSO), N-acetyl-l-cysteine (NAC) and curcumin (purity > 94%) were purchased from Sigma (St. Louis, MO). PD68235, a specific PPARγ antagonist, was kindly provided by Pfizer (Ann Arbor, MI) (Camp et al., 2001). 15-deoxy-Δ12,14-prostaglandin J2 (PGJ2) and rosiglitazone (BRL 49653), purchased from Cayman Chemical (Ann Arbor, MI), were dissolved in dimethyl sulfoxide (100 mM). Primary antibodies were purchased from Santa Cruz Biotech. Inc. (Santa Cruz, CA), unless otherwise noted, and were as previously described (Lin et al., 2009).
Isolation and culture of HSCs
Male Sprague–Dawley rats (200–250 g), or C57B/L6 mice at 4 weeks, purchased from the Harlan Laboratories, Inc. (Indianapolis, IN), were housed in a temperature-controlled animal facility (23°C) with a 12:12 h light–dark cycle and allowed free access to regular chew and water ad libitum. HSCs were isolated by the pronase–collagenase perfusion in situ before density gradient centrifugation, as we previously described (Xu et al., 2003). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010).The animal protocol for the use of rats was approved by Institutional Animal Care and Use Committee of Saint Louis University. Freshly isolated HSCs were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% of fetal bovine serum (FBS). Cells were passaged in DMEM with 10% of FBS. Semi-confluent HSCs with four to nine passages were used in experiments. In some of experiments, cells were cultured in serum-depleted DMEM for 24 h before treatment, which rendered HSCs more sensitive to exogenous stimuli (Lin et al., 2009). Cells were subsequently treated and cultured in serum-depleted media, which excluded the interference from other factors in FBS.
Western blotting analyses
Preparation of whole cell extracts, SDS-PAGE, transblotting and subsequent immunoreactions were conducted as we previously described (Xu et al., 2003). β-Tubulin was used as an invariant control for equal loading. Densities of bands in Western blotting analyses were normalized with the internal invariable control. Levels of target protein bands were densitometrically determined by using Quantity One® 4.4.1 (Bio-Rad, Hercules, CA). Variations in the density were expressed as fold changes compared with the control in the blot.
RNA extraction and real-time PCR
Total RNA was treated with DNase I before the synthesis of the first strand of cDNA. Real-time PCR were performed as we previously described using SYBR Green Supermix (Xu et al., 2003). mRNA levels are expressed as fold changes after normalization with glyceraldehyde-3-phosphate dehydrogenase (GAPDH), as described by Schmittgen et al. (2000). The following primers were used in real-time PCR.
| RAGE: | (F) 5′-GAA TCC TCC CCA ATG GTT CA-3′, (R) 5′-GCC CGA CAC CGG AAA GT-3′; |
| GCLc: | (F) 5′-TGT GTG ATG AGC CCA AGG AC-3′, (R) 5′- AGT TGG CTC GCA TCA TAG TTG-3′; |
| GCLm: | (F) 5′-CTG CTA AAC TGT TCA TTG TAG-3′, (R) 5′-CTA TGG GTT TTA CCT GTG-3′; |
| GAPDH: | (F) 5′-GGC AAA TTC AAC GGC ACA GT-3′, (R) 5′-AGA TGG TGA TGG GCT TCC C-3′. |
Plasmids and transient transfection assays
The luciferase reporter plasmid pRAGE-Luc was generated by subcloning a fragment (∼2168 bp) of the mouse rage gene promoter at the NheI and HindIII sites of the pGL3-basic vector. The fragment was from mouse genomic DNA by PCR using the following primers: forward, 5′-ATT GCT AGC GGG TCA GAT ATA CAG TCC-3′ and reverse, 5′-ATT AAGCTT CCA TCT CCC ATC TCG TTC TGT C-3′. The luciferase report plasmids pPDGF-βR-Luc and pTGFβ-RII-Luc were gifts, respectively, from Dr Keiko Funa (Ballagi et al., 1995) and Seong-Jin Kim (Bae et al., 1995). pPDGF-βR-Luc contained a fragment (1366 bp) of the gene promoter of platelet-derived growth factor-β receptor (PDGF-β receptor) (Ballagi et al., 1995). pTGFβ-RII-Luc had a fragment (1670 bp) of the gene promoter of type II TGFβ receptor. The cDNA expression plasmid pRAGE, containing a full-length of rage cDNA in the plasmid pcDNA3, was kindly provided by Dr Ann Marie Schmidt (Kislinger et al., 1999).
For co-transfection, semi-confluent HSCs in six-well cell culture plates were transiently transfected with a total of 3–4.5 µg DNA per well, using the LipofectAMINE® reagent (Invitrogen Corp., Carlsbad, CA), as we previously described (Xu et al., 2003). Each sample was in triplicate in every experiment. Transfection efficiency was normalized by co-transfection of the β-galactosidase reporter plasmid pSV-β-gal (0.5 µg·per well) (Promega Corporation, Madison, WI, USA). β-Galactosidase activities were measured by using a chemiluminescence assay kit (Tropix, Bedford, MA). Luciferase activities were expressed as relative unit after normalization with β-galactosidase activities µg-1 protein. Results were combined from at least three independent experiments.
Knockdown of RAGE by short hairpin RNA (shRNA)
The RNAi sequence targeting mouse rage mRNA was selected using online RNAi design program from Thermo Fisher Scientific, Dharmacon RNAi Technologies (Lafayette, CO). The sequence of RAGE shRNA was 5′-GCT AGA ATG GAA ACT GAA CA-3′. Construction of shRNA expression cassettes and subsequent cloning in the lentiviral vector pFLRu were conducted as described (Feng et al., 2010; Fraley et al., 2010). Briefly, the hU6 promoter (f1) was obtained by PCR using pBShU6-1 as a template and the primers of (F) 5′-ACA GAA TTC TAG AAC CCC AGT GGA AAG ACG CGC AG-3′ and (R) 5′-GGT GTT TCG TCC TTT CCA CAA G-3′. The hairpin-containing fragment (f2) was obtained by PCR using the primers of (F) 5′-GTG GAA AGG ACG AAA CAC C GC TAG AAT GGA AAC TGA ACATTC AAG AGA tgttca and (R) 5′-TCC AGC TCG AGA AAA A GC TAG AAT GGA AAC TGA ACATCT CTT GAA tgttca. Overlapping PCR were carried out by using the hU6 forward primer, the respective shRNA reverse primer and mixed fragments of f1 and f2 as templates. The PCR products were purified, cut with XbaI/XhoI and subcloned into the lentiviral vector pFLRu. A construct targeting a firefly luciferase shRNA was also similarly created and used as a non-specific shRNA control. The sequence of shRNA for the firefly luciferase (i.e. Luc shRNA) was 5′-GCT TAC GCT GAG TAC TTC GA-3′.
To generate lentiviruses, sub-confluent Lenti-X™ 293T Cells, a lentiviral packaging cell line (Clontech Laboratories, Inc.. Mountain View, CA), were co-transfected with the pFLRu derived vectors and a mixture (8:1) of lentiviral packaging plasmids pHR8.2ΔR and pCMV-VSVG. Media were refreshed for virus production 24 h after transfection. Media containing viruses were collected and filtered with syringe filters (0.45 µm) in the following day. After mixing the virus-containing media with an equal volume of fresh media, the mixture was added to HSCs in 70–80% confluence. Protamine sulfate was added to a final concentration of 10 µg·mL−1. After overnight incubation, viral transduced cells were selected for three days in fresh media containing puromycin at 15 µg·mL−1, which was determined by a killing curve of non-transduced HSCs generated in pilot experiments. The knockdown efficiency of RAGE by RAGE shRNA was determined by Western blotting analyses.
Determination of levels of intracellular reactive oxygen species (ROS)
Levels of ROS in HSC were determined by analysing dichlorofluorescein fluorescence (DCF), as described previously (Zheng et al., 2007).
Analyses of lipid peroxidation (LPO)
LPO assays were performed by using the Lipid Hydroperoxide Assay Kit purchased from Cayman Chemical (Zheng et al., 2007).
Glutathione assays
Levels of reduced glutathione (GSH) and oxidized glutathione (GSSG) were determined by using the enzyme immune assay kit GSH-400® (Cayman Chemical) (Zheng et al., 2007).
Analyses of activities of glutamate-cysteine ligase (GCL)
Activities of GCL were spectrophotometrically determined using a coupled assay with pyruvate kinase and lactate dehydrogenase, as we previously described (Zheng et al., 2007).
Statistical analyses and nomenclatures
Differences between means were evaluated using Student's unpaired two-sided test (P < 0.05 was considered as significant). Nomenclatures used in this manuscript were checked to conform to British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2011).
Results
AGEs dose-dependently induced gene expression of RAGE in HSCs in vitro
To evaluate the effect of AGEs on gene expression of RAGE, passaged HSCs were transfected with the rage promoter luciferase reporter plasmid pRAGE-Luc. After overnight recovery, HSCs were deprived of serum for 4 h before the treatment with AGEs at different concentrations in serum-free media for an additional 24 h. Serum-deprivation rendered HSCs more sensitive to exogenous stimuli (Lin et al., 2009). The subsequent treatment in serum-free media excluded the influence from factors in sera. Luciferase activity assays in Figure 1A indicated that AGEs caused a dose-dependent increase in luciferase activities, suggesting that AGEs stimulated the rage promoter activity of in HSCs in vitro. To confirm the observations, serum-deprived HSCs were treated with AGEs at different concentrations in serum-free media for 24 h. Real-time PCR (Figure 1B) and Western blotting analyses (Figure 1C) demonstrated that AGEs elevated the mRNA level and the protein abundance of AGE receptors in a dose-dependent manner. These results collectively indicated that AGEs induced gene expression of AGE receptors in HSCs in vitro.
Figure 1.
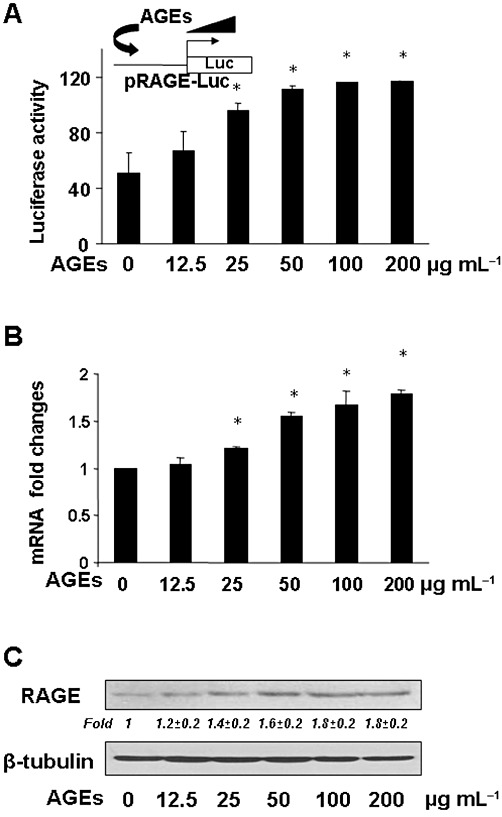
AGEs dose-dependently induced gene expression of RAGE in HSCs in vitro. (A) HSCs were transfected with the rage promoter luciferase reporter plasmid pRAGE-Luc. After overnight recovery, cells were serum-deprived for 4 h before the treatment with AGEs at different doses in serum-depleted media for an additional 24 h. Luciferase activity assays were conducted (n = 6). *P < 0.05, versus untreated control cells (first column). The floating inset denotes the pRAGE-Luc construct in use and the application of AGEs to the system. (B and C) HSCs were serum-deprived in serum-free DMEM for 24 h prior to the stimulation with AGEs at different doses in serum-depleted media for an additional 24 h. Total RNA and whole cell lysates were prepared. (B) Real- time PCR analyses. Values were presented as mRNA fold changes (mean ± SD, n ≥ 3). *P < 0.05, versus the untreated control (first column). (C) Western blotting analyses. Representatives were from three independent experiments. β-Tubulin was used as an internal control for equal loading. Italic numbers beneath the upper blot were fold changes (mean ± SD) in the densities of the bands compared with the untreated control in the blot (n = 3), after normalization with the internal invariable control.
Curcumin abolished the effect of AGEs on inducing gene expression of RAGE in HSCs in vitro
Curcumin has been shown to affect the regulation of expression of genes relevant to the activation of HSCs in vitro and in vivo (Xu et al., 2003; Zheng and Chen, 2004; 2006; Fu et al., 2008). To evaluate the effect of curcumin on the expression of RAGE, passaged HSCs were treated with curcumin at various concentrations in DMEM with 10% FBS for 24 h. Western blotting analyses indicated that curcumin by itself reduced the abundance of RAGE in HSCs in a dose-dependent manner (Figure 2A). To further determine the role of curcumin in the attenuation of the effects of AGEs on the activation of HSCs, passaged HSCs were transfected with the rage promoter luciferase reporter plasmid pRAGE-Luc. After recovery, cells were deprived of serum for 4 h before treatment with AGEs at 100 µg·mL−1 in the presence of curcumin at indicated concentrations in serum-free media for an additional 24 h. Luciferase activity assays in Figure 2B indicated that AGEs increased, as expected, the luciferase activity (second column), which was diminished by curcumin in a dose-dependent manner (third to fifth columns). These observations suggest that curcumin eliminated the effect of AGEs on stimulating the rage promoter activity in HSCs in vitro. To verify the observations, serum-deprived HSCs were treated with AGE at 100 µg·mL−1 plus curcumin at different concentrations in serum-depleted media for 24 h. Real-time PCR (Figure 2C) and Western blotting analyses (Figure 2D) demonstrated that curcumin eliminated the effect of AGEs and dose-dependently reduced the mRNA level and the protein abundance of RAGE in HSCs (the corresponding third to fifth columns or lanes). These results collectively demonstrated that curcumin dose-dependently abolished the effect of AGEs on inducing gene expression of RAGE in HSCs in vitro.
Figure 2.
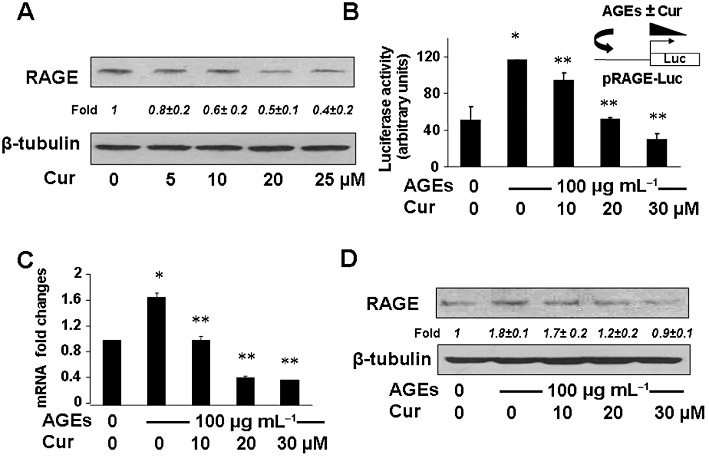
Curcumin abolished the effect of AGEs on inducing gene expression of RAGE in HSCs in vitro. (A). Passaged HSCs were treated with curcumin at various concentrations in DMEM with 10% FBS for 24 h. The abundance of RAGE in cells was analysed by Western blotting. Representatives were from three independent experiments. β-Tubulin was used as an internal control for equal loading. Italic numbers beneath the upper blot were fold changes (mean ± SD) in the densities of the bands compared with the untreated control in the blot (n = 3), after normalization with the internal invariable control. (B) HSCs were transfected with the rage promoter luciferase reporter plasmid pRAGE-Luc. After overnight recovery and following serum deprivation for 4 h, the cells were treated w/wt AGEs (100 µg·mL−1) in the presence of curcumin at indicated concentrations in serum-depleted media for an additional 24 h. Luciferase activity assays were conducted (n = 6). *P < 0.05, versus untreated control cells (first column). **P < 0.05, versus cells treated with AGEs only (second column). The floating inset denoted the pRAGE-Luc construct in use and the application of AGEs w/wt curcumin to the system. (C and D) Serum-deprived HSCs were treated w/wt AGEs (100 µg·mL−1) in the presence of curcumin at indicated concentrations in serum-depleted media for 24 h. Total RNA and whole cell lysates were prepared. (C) Real-time PCR analyses. Values were presented as mRNA fold changes (mean ± s. d., n ≥ 3). *P < 0.05, versus untreated control cells (first column); **P < 0.05, versus cells treated with AGEs only (second column). (D) Western blotting analyses. Representatives were from three independent experiments. β-Tubulin was used as an internal control for equal loading. Italic numbers beneath the upper blot were fold changes (mean ± SD) in the densities of the bands compared with the untreated control in the blot (n = 3), after normalization with the internal invariable control.
Forced expression of exogenous RAGE diminished the role of curcumin in the inhibition of the promoter activity of genes critically relevant to HSC activation
Previous studies have shown that the process of HSC activation is coupled with the expression of PDGF-β receptors and type II TGFβ receptors, whose signalling pathways played critical roles in stimulating HSC activation (Friedman, 2008). To evaluate the role of RAGE in the AGEs-induced activation of HSCs, passaged HSCs in six-well plates were co-transfected with a total of 4.5 µg of DNA mixture per well, including 2 µg of the PDGF-β receptor promoter luciferase reporter plasmid pPDGF-βR-Luc or the TGFβ-receptor II promoter luciferase reporter plasmid pTGFβ-RII-Luc, 0.5 µg of pSV-β-gal and 2 µg of the cDNA expression plasmid pRAGE at various doses plus the empty vector pcDNA. The latter was used to ensure an equal amount of total DNA in transfection assays. After overnight recovery, cells were serum-deprived for 4 h before the treatment with or without (w/wt) AGEs (100 µg·mL−1) in the presence or absence of curcumin (20 µM) in serum-depleted media for an additional 24 h. Results from luciferase activity assays, illustrated in Figure 3, demonstrated that, compared with the untreated control (the corresponding first column), AGEs significantly increased the luciferase activity in cells transfected with pPDGF-βR-Luc or pTGFβ-RII-Luc (the corresponding second column), confirming the role of AGEs in stimulating the promoter activity of PDGF-β receptor and TGFβ-receptorII genes in cultured HSCs. In further studies it was observed that curcumin (20 µM) significantly eliminated the stimulating role of AGEs and reduced luciferase activities in the cells (the corresponding third column). It was of interest to observe that forced expression of exogenous RAGE attenuated the inhibitory effect of curcumin and dose-dependently increased luciferase activities in HSCs (the corresponding fourth to sixth columns). These results indicate that forced expression of exogenous RAGE dose-dependently diminishes the role of curcumin (20 µM) in the inhibition of the promoter activity of genes critically relevant to HSC activation.
Figure 3.
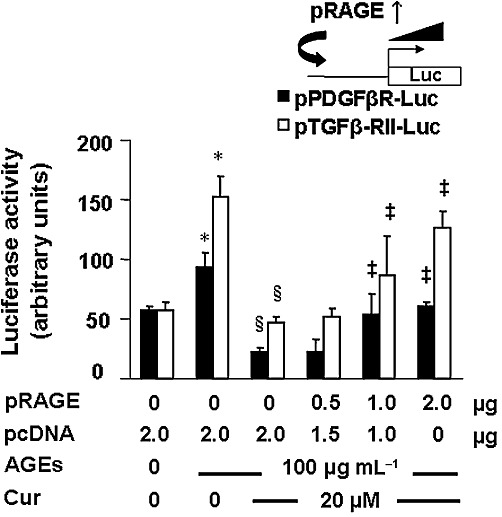
Forced expression of exogenous RAGE diminished the role of curcumin in the inhibition of the promoter activity of genes critically relevant to HSC activation. Passaged HSCs in six-well plates were co-transfected with a total of 4.5 µg of a DNA mixture per well, including 2 µg of the PDGF-βR promoter luciferase reporter plasmid pPDGF-βR-Luc, or the TGFβ-RII promoter luciferase reporter plasmid pTGFβ-RII-Luc, 0.5 µg of pSV-β-gal and 2 µg of the cDNA expression plasmid pRAGE at various doses plus the empty vector pcDNA. The latter was used to ensure an equal amount of total DNA in transfection assays. After recovery, cells were serum-deprived for 4 h before the treatment w/wt AGEs (100 µg·mL−1) in the presence or absence of curcumin (20 µM) in serum-depleted media for an additional 24 h. Luciferase activity assays were conducted (n = 6). *P < 0.05, versus untreated control cells (the corresponding first column); §P < 0.05, versus cells treated with AGEs only (the corresponding second column); ‡P < 0.05, versus control cells treated with AGEs plus curcumin (the corresponding third column). The floating inset denoted the plasmids pPDGF-βR-Luc, or pTGFβ-RII-Luc, and pRAGE in use for co-transfection.
The knockdown of RAGE by RAGE shRNA dramatically diminished the stimulant effects of AGEs, suggesting a critical role of the induction of gene expression of RAGE in the AGEs-induced activation of HSCs in vitro
To verify the role of RAGE in mediating the stimulating effects of AGEs on the activation of HSCs, gene expression of RAGE in mouse HSCs was silenced by shRNA. Pilot experiments of Western blotting analyses in Figure 4A demonstrated that compared with that in HSCs without transduction (no shRNA) (first lane), the abundance of RAGE in cells transduced with lentiviruses containing a non-specific shRNA targeting a firefly luciferase (Luc shRNA) (second lane) showed no apparent difference. In great contrast, compared with that in the two control HSCs (first and second lanes), the abundance of RAGE was significantly reduced in HSCs transduced with shRNA targeting RAGE (RAGE shRNA) by more than 95% (third lane). These results indicate that gene expression of RAGE in passaged HSCs was efficiently and significantly silenced by RAGE shRNA.
Figure 4.
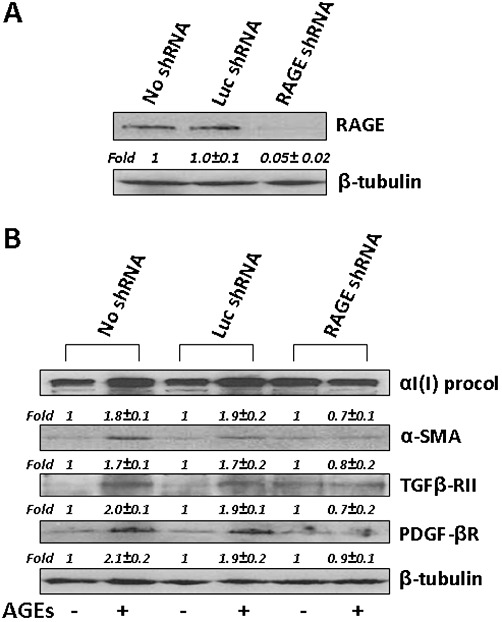
The knockdown of RAGE by RAGE shRNA diminished the stimulant effect of AGEs, suggesting a critical role for the induction of gene expression of RAGE in the AGEs-induced activation of HSCs in vitro. Passaged mouse HSCs were transduced w/wt lentiviruses containing RAGE shRNA or Luc shRNA as a non-specific shRNA control. After selection with puromycin, viral transduced HSCs were treated w/wt AGEs at 100 µg·mL−1 in serum-depleted media for 24 h. Whole cell lysates were prepared for Western blotting analyses. Italic numbers beneath the upper blot were fold changes (mean ± SD) in the densities of the bands compared with the untreated control in the blot (n = 3), after normalization with the internal invariable control. Representatives were from three independent experiments. β-Tubulin was used as an internal control for equal loading. (A) Determination of the knockdown efficiency of shRNA targeting RAGE. (B) Evaluation of the effect of AGEs on the abundance of proteins critically relevant to, or associated with, the activation of HSCs after knockdown of RAGE.
In further experiments, gene expression of RAGE in mouse HSCs was silenced by RAGE shRNA, or by Luc shRNA as a non-specific shRNA control. After selection with puromycin, viral transduced HSCs were treated w/wt AGEs at 100 µg·mL−1 in serum-depleted media for 24 h. The abundance of proteins critically relevant to, or associated with, the activation of HSCs was analysed by Western blotting analyses (Figure 4B). Compared with untreated HSCs transduced with no shRNA (first lane), or with the Luc shRNA (third lane), HSCs treated with AGEs in the second or fourth lane showed significantly higher levels of αI(I) procollagen, α-SMA, PDGF-β receptors and TGFβ- receptor II. It was of interest to observe that there was no significant difference in the abundance of the proteins in HSCs treated with or without AGEs in HSCs transduced with RAGE shRNA (fifth and sixth lanes). These results indicate that the knockdown of RAGE by RAGE shRNA dramatically diminished the stimulating effects of AGEs, suggesting a critical role for the induction of the gene expression of RAGE in the AGEs-induced activation of HSCs in vitro.
The activation of PPARγ eliminated the effect of AGEs on inducing gene expression of RAGE in HSCs in vitro
Our previous studies indicated that curcumin induced gene expression of PPARγ and enhanced its activity in cultured HSCs, which was a prerequisite for curcumin to inhibit HSC activation in vitro (Xu et al., 2003; Zheng and Chen, 2004). It was, therefore, plausible to postulate that the inhibition of gene expression of RAGE by curcumin might be mediated by the stimulation of PPARγ activity. To test this hypothesis, serum-deprived HSCs were treated with AGEs at 100 µg·mL−1 in the presence or absence of PGJ2, a natural PPARγ agonist, or rosiglitazone, a synthetic PPARγ agonist, at various doses in serum-depleted media for 24 h. Real-time PCR (Figure 5A) and Western blotting analysis (Figure 5B) showed that the PPARγ agonists, mimicking curcumin observed in Figure 2B and C, dose-dependently reduced the mRNA levels and the protein abundance of RAGE, which was induced by AGEs. These results support our hypothesis and suggest that the activation of PPARγ eliminated the effect of AGEs on inducing gene expression of RAGE in HSCs in vitro.
Figure 5.
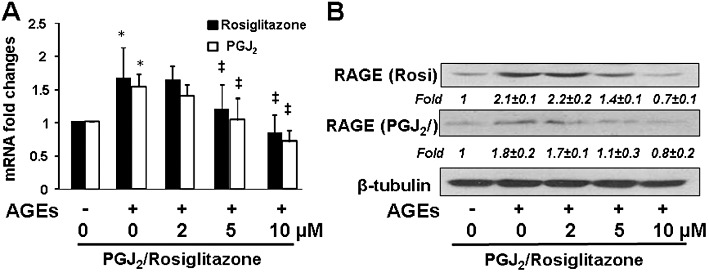
The activation of PPARγ eliminated the effect of AGEs on inducing gene expression of RAGE in HSCs in vitro. Serum-deprived HSCs were treated with AGEs (100 µg·mL−1) in the presence or absence of PGJ2 or rosiglitazone at various doses in serum-depleted media for 24 h. Total RNA and whole cell lysates were prepared for evaluating gene expression of RAGE. (A) Real-time PCR analyses. Values are presented as mRNA fold changes (mean ± SD, n ≥ 3). *P < 0.05, versus untreated control cells (the corresponding first column); ‡P < 0.05, versus cells treated with AGEs only (the corresponding second column); (B) Western blotting analyses. Representatives were from three independent experiments. β-Tubulin was used as an internal control for equal loading. Italic numbers beneath blots were fold changes (mean ± SD) in the densities of the bands compared with the untreated control in the blot (n = 3), after normalization with the internal invariable control.
The activation of PPARγ played a critical role in he suppression by curcumin of the gene expression of RAGE in HSCs
To further test our hypothesis, HSCs were transfected with the plasmid pRAGE-Luc. After recovery, cells were deprived of serum for 4 h before the treatment w/wt AGEs (100 µg·mL−1) in the presence or absence of curcumin (20 µM) and PD68235 (20 µM), a specific PPARγ antagonist, in serum-depleted media with PGJ2 (5 µM) for an additional 24 h. The exogenous PPARγ agonist PGJ2 was added because there was no PPARγ agonist in serum-depleted media. Luciferase activity assays in Figure 6A showed that compared with the untreated control (first column), AGEs significantly increased, as expected, the luciferase activity (second column), which was markedly diminished by curcumin (third column). Compared with the cells treated with curcumin plus AGEs (third column), the presence of the PPARγ antagonist PD68235 suppressed the inhibitory role of curcumin and significantly increased the luciferase activity (the last column). This result indicates that the blockade of PPARγ activation reduces the inhibitory effect of curcumin on rage promoter activity in cultured HSCs. This result was confirmed in HSCs with the aforesaid treatment by real-time PCR (Figure 6B) and Western blotting analyses (Figure 6C). It was demonstrated that the inhibition of PPARγ activation by the PPARγ antagonist PD68235 eliminated the inhibitory role of curcumin and significantly increased the levels of mRNA and protein of RAGE (Figure 6B and C). Taken together, the results demonstrate that the activation of PPARγ plays a critical role in the suppression of the gene expression of RAGE in HSCs induced by curcumin (20 µM).
Figure 6.
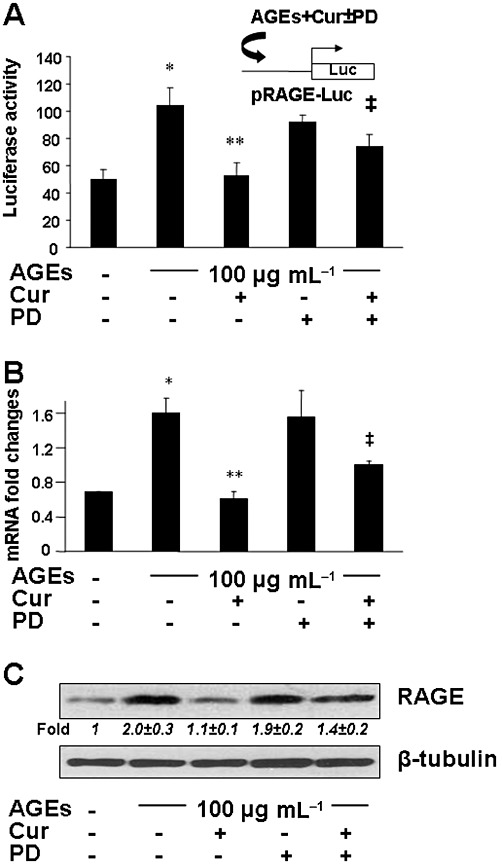
The activation of PPARγ played a critical role in the ability of curcumin to suppress gene expression of RAGE in HSCs. (A) HSCs were transfected with the plasmid pRAGE-Luc. After overnight recovery, cells were serum-deprived for 4 h before the treatment w/wt AGEs (100 µg·mL−1) in the presence or absence of curcumin (20 µM) and PD68235 (PD) (20 µM), a specific PPARγ antagonist, in serum-depleted media with PGJ2 (5 µM) for an additional 24 h. Luciferase activity assays were conducted (n = 6). *P < 0.05, versus untreated control cells (first column); **P < 0.05, versus cells treated with AGEs only (second column); ‡P < 0.05, versus cells treated with curcumin and AGEs (third column). The floating inset denoted the pRAGE-Luc construct in use and the application of AGEs and curcumin w/wt PD68235 to the system. (B and C) After pretreatment w/wt PD68235 at 20 µM for 1 h, serum-deprived HSCs were treated w/wt AGEs (100 µg·mL−1) in the presence or absence of curcumin (20 µM) in serum-depleted media with PGJ2 (5 µM) for an additional 24 h. (B) Real-time PCR analyses. Values are presented as mRNA fold changes (mean ± SD, n ≥ 3). *P < 0.05, versus untreated control cells (first column); **P < 0.05, versus cells treated with AGEs only (second column); ‡P < 0.05, versus cells treated with curcumin and AGEs (third column). (C) Western blotting analyses. Representatives are from three independent experiments. β-Tubulin was used as an internal control for equal loading. Italic numbers beneath the upper blot were fold changes (mean ± SD) in the densities of the bands compared with the untreated control in the blot (n = 3), after normalization with the internal invariable control.
However, it is noteworthy that compared with AGEs plus curcumin (third column or lane in Figure 6B and C), the addition of PD68235 to the system to inhibit the activity of PPARγ did not completely eliminate the inhibitory effect of curcumin on the expression of RAGE (fifth column or lane in Figure 6B and C). This observation suggested that other PPARγ-independent mechanism(s) might be involved in the process. Further experiments were designed to explore the additional mechanism(s).
AGEs induced oxidative stress in cultured HSCs, which was attenuated by curcumin
AGEs are well known to induce oxidative stress (Sahin et al., 2008; Yamagishi and Matsui, 2010). Curcumin is a potent antioxidant (Ruby et al., 1995). To further explore the underlying mechanisms by which curcumin eliminated the stimulating effects of AGEs on the activation of HSCs and inhibited gene expression of RAGE in HSCs, we further presumed that curcumin attenuated the AGEs-induced oxidative stress, leading to the inhibition of gene expression of RAGE. To test this presumption, serum-deprived HSCs were treated with AGEs in the presence or absence of curcumin at indicated doses in serum-depleted media for 24 h. As shown in Figure 7A and B, AGEs dose-dependently elevated, as expected, levels of cellular ROS and LPO (the corresponding first to fourth columns). Compared with cells treated with AGEs at 100 µg·mL−1 (the corresponding third column), curcumin diminished the stimulant effects of AGEs on the levels of cellular ROS and LPO in a dose-dependent manner (the corresponding fifth to seventh columns). These observations were verified by analyses of the level of cellular GSH (Figure 7C), a most abundant and effective thiol antioxidant in eukaryotic cells (Wu et al., 2004), and by determining the ratio of reduced GSH to oxidative GSH (GSSG) (Figure 7D), a sensitive indicator of oxidative stress in cells (Fridovich, 1978). Taken together, these results indicate that AGEs induce oxidative stress in cultured HSCs, which was attenuated by curcumin, at least partially, by increasing the level of intracellular GSH and improving the ratio of GSH/GSSG.
Figure 7.
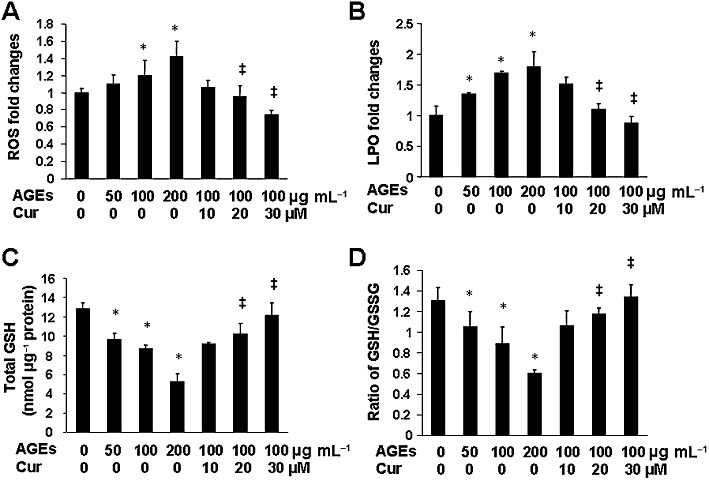
AGEs induced oxidative stress in cultured HSCs, which was attenuated by curcumin. Serum-deprived HSCs were treated with AGEs at indicated doses in the presence or absence of curcumin at various concentrations in serum-depleted media for 24 h. Levels of intracellular ROS (A), LPO (B), total GSH (C) and the ratio of GSH/GSSG (D) were determined. Values are expressed as mean ± SD (n ≥ 3). *P < 0.05, versus untreated control cells (the corresponding first column); ‡P < 0.05, versus cells treated with AGEs at 100 µg·mL−1 (the corresponding third column).
AGEs dose-dependently reduced GCL activity and inhibited expression of GCL genes in HSCs in vitro, which were eliminated by curcumin
De novo synthesis of GSH is mainly regulated by GCL, a rate-limiting enzyme in the process of GSH synthesis. To elucidate the mechanism by which curcumin eliminated the inhibitory effect of AGEs on the level of cellular GSH, serum-deprived HSCs were treated with AGEs in the presence or absence of curcumin at indicated doses in serum-depleted media for 24 h. As demonstrated in Figure 8A by GCL activity assays, compared with the untreated control (first column), AGEs dose-dependently reduced the activity of GCL (second to fourth columns), whereas curcumin diminished the inhibitory effect and increased the activity of GCL in a dose-dependent manner (fifth to seventh columns). In addition, real-time PCR and Western blotting analyses were conducted in HSCs with the aforesaid treatment to elucidate the effect of AGEs w/wt curcumin on the gene expression of GCL, composed of a catalytic subunit GCLc and a modifier subunit GCLm. It was observed that AGEs dose-dependently reduced the mRNA levels (Figure 8B) and the protein abundance (Figure 8C) of GCLc and GCLm, which were suppressed by curcumin (the corresponding fifth to seventh columns or lanes). Taken together, these results indicate that AGEs reduced the GCL activity by inhibiting gene expressions of GCL in HSCs in vitro, which were suppressed by curcumin in dose-dependent manner.
Figure 8.
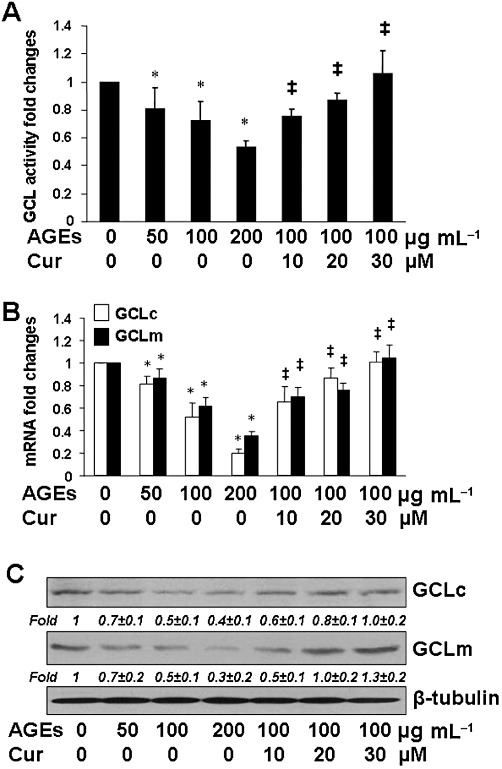
AGEs dose-dependently reduced GCL activity and inhibited expression of GCL genes in HSCs in vitro, which were eliminated by curcumin. Serum-deprived HSCs were treated with AGEs at indicated doses in the presence or absence of curcumin at various concentrations in serum-depleted media for 24 h. *P < 0.05, versus untreated control cells (the corresponding first column); ‡P < 0.05, versus cells treated with AGEs at 100 µg·mL−1 (the corresponding third column). (A) Analyses of GCL activities. Values are expressed as mean ± SD (n ≥ 6). (B) Real-time PCR analyses. Values are presented as mRNA fold changes (mean ± SD, n ≥ 3). (C) Western blotting analyses. Representatives were from three independent experiments. β-Tubulin was used as an internal control for equal loading. Italic numbers beneath blots were fold changes (mean ± SD) in the densities of the bands compared with the untreated control in the blot (n = 3), after normalization with the internal invariable control.
De novo synthesis of GSH played a pivotal role in the suppression of AGEs-induced gene expression of RAGE in HSCs by curcumin in vitro
Additional experiments were conducted to verify the role of the synthesis of GSH and the attenuation of oxidative stress in the curcumin-induced suppression of AGEs-induced gene expression of RAGE in HSCs. In the following experiments, cellular GSH contents were altered by two well-known manipulators of GSH synthesis. NAC is a precursor of GSH, increasing GSH contents by supplying cysteine (Cotgreave, 1997). BSO is a specific inhibitor of GCL, depleting cellular GSH (Anderson and Luo, 1998). Serum-deprived HSCs were divided into two groups. One was treated w/wt AGEs (100 µg·mL−1) in the presence of curcumin (20 µM), or NAC (5 mM), in serum-depleted media for 24 h. The other group was pretreated with BSO (0.25 mM) for 1 h before the treatment w/wt AGEs (100 µg·mL−1) in the presence of curcumin (20 µM), or NAC (5 mM), in serum-depleted media for 24 h. Real-time PCR (Figure 9A) and Western blotting analyses (Figure 9B) demonstrated that compared with the no treatment control (first column and lane), AGEs elevated, as expected, the mRNA level and the protein abundance of RAGE in the cells (second column and lane). NAC (fourth column and lane), mimicking curcumin (third column and lane), significantly eliminated the stimulant effect of AGEs. The inhibition of GSH synthesis by the pre-exposure to BSO apparently abolished the inhibitory effect of both NAC and curcumin (the last two columns and lanes), suggesting a critical role of de novo synthesis of GSH in the suppression of the AGEs-induced gene expression of RAGE in HSCs by curcumin, as well as by NAC. Taken together, these results indicate that de novo synthesis of GSH plays a pivotal role in the suppression of AGEs-induced gene expression of RAGE in HSCs by curcumin (20 µM) in vitro.
Figure 9.
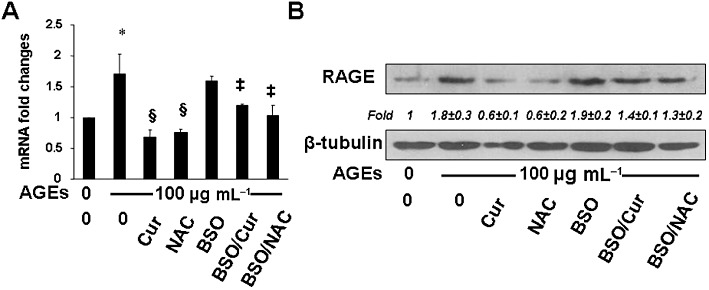
De novo synthesis of GSH played a pivotal role in the ability of curcumin to suppress the AGEs-induced gene expression of RAGE in HSCs in vitro. Serum-deprived HSCs were divided into two groups. One was treated w/wt AGEs (100 µg·mL−1) in the presence of curcumin (20 µM), or NAC (5 mM), in serum-depleted media for 24 h. The other group was pretreated with BSO (0.25 mM) for 1 h before the treatment w/wt AGEs (100 µg·mL−1) in the presence of curcumin (20 µM), or NAC (5 mM), in serum-depleted media for 24 h. (A) Real-time PCR analyses. Values were presented as mRNA fold changes (mean ± SD, n ≥ 3). *P < 0.05, versus untreated control cells (first column); **P < 0.05, versus cells treated with AGEs only (second column); ‡P < 0.05, versus cells treated with AGEs plus curcumin or NAC (third and fourth column respectively). (B) Western blotting analyses. Representatives were from three independent experiments. β-Tubulin was used as an internal control for equal loading. Italic numbers beneath the upper blot were fold changes (mean ± SD) in the densities of the bands compared with the untreated control in the blot (n = 3), after normalization with the internal invariable control.
Discussion
Factors associated with T2DM, including hyperglycaemia, hyperinsulinaemia, hyperleptinaemia and hypercholesterolemia, have been implicated in stimulating HSC activation and hepatic fibrogenesis (Kang and Chen, 2009c; Lin et al., 2009; Tang et al., 2009; Lin and Chen, 2011). It is unlikely that any single factor is solely responsible for T2DM- and NASH-associated hepatic pathogenesis. Few breakthroughs occur in therapeutic intervention of the disease. Establishing causal mechanisms is an important and first step to identify innocuous anti-fibrotic agents (Wells, 2009). Most evolving anti-fibrotic therapies are aimed at inhibiting HSC activation. We have shown that curcumin eliminates the effects of hypercholesterolaemia (Kang and Chen, 2009a, b, c), hyperinsulinaemia (Lin et al., 2009), hyperleptinaemia (Tang et al., 2009) and hyperglycaemia (Lin and Chen, 2011) on inducing HSC activation in vitro. Our recent studies demonstrated that AGEs stimulated HSC activation by stimulating cell proliferation and inducing expression of genes closely relevant to HSC activation (Lin et al., 2012). Our present study focused on exploring the underlying mechanisms. We observed in this report that the AGEs-induced stimulation of HSC activation was associated with an increase in gene expression of RAGE, which played a critical role in the process. The phytochemical curcumin eliminated the stimulating effects of AGEs and inhibited the gene expression of RAGE by attenuating oxidative stress and stimulating the activity of PPARγ. It should be emphasized that the current study focused on the effects of AGEs and curcumin on regulating gene expression of RAGE in HSCs. However, our results do not exclude the roles of AGEs and curcumin in regulating gene expression of AGE clearance receptors, including AGE receptor-1 (Lu et al., 2004). For the AGE preparation used for these experiments, a carrier protein (i.e. BSA), was incubated with glucose. The result is a crude mixture of different AGE structures. We are cognizant that it is not known to what extent AGEs created in vitro could represent ‘native’ AGEs generated in vivo. Although AGE adducts generated in vivo were detected in AGEs formed in vitro (Horiuchi et al., 1991; Makita et al., 1992), the extent of AGE modifications formed in vitro are unlikely to be as high as those generated in vivo.
PPARγ is one of the nuclear receptor proteins that function as transcription factors regulating expression of genes (Michalik et al., 2006). Transcriptional regulation by PPARγ requires heterodimerization with retinoid X receptor (RXR). When activated by a ligand, the dimer modulates transcription via binding to a specific DNA sequence element, called peroxisome proliferator response element (PPRE), in the promoter of target genes (Michalik et al., 2006). Endogenous PPARγ is highly expressed in quiescent HSC and is functionally active (Galli et al., 2000; Marra et al., 2000; Miyahara et al., 2000). During the process of HSC activation, the abundance of PPARγ is markedly reduced. However, PPARγ was still detectable in activated HSCs in vitro by Western blotting analyses and responded to the agonist PGJ2 (Xu et al., 2003; Zheng and Chen, 2006). Our previous studies showed that curcumin dramatically induced gene expression of endogenous PPARγ and elevated its activity in cultured HSCs, which was required for curcumin to inhibit HSC activation (Xu et al., 2003; Zheng and Chen, 2004). No results have suggested curcumin to be an agonist of PPARγ or RXR. To evaluate the role of the activation of PPARγ, the agonist PGJ2 or rosiglitazone was shown to significantly inhibit gene expression of RAGE in HSCs in vitro. In addition, the blockade of PPARγ activation by its specific antagonist PD68235 apparently eliminated the ability of curcumin to inhibit gene expression of RAGE in cultured HSCs. These results collectively demonstrate that the induction of PPARγ activation plays a critical role in the curcumin-induced inhibition of gene expression of RAGE in HSCs. Our observations were consistent with previous reports that the activation of PPARγ suppressed gene expression of RAGE in other cells (Yamagishi et al., 2008a; Ihm et al., 2010; Matsui et al., 2010; Zhang et al., 2010). However, a brief computer-aided search did not find a consensus sequence of a PPARγ DNA binding site (i.e. the peroxisome proliferator response element) in the proximal promoter (∼2000 bp) of murine rage gene. Preliminary results from promoter deletion assays and site-directed mutagenesis suggested that a putative NF-κB binding site and a putative TCF/LEF1 binding site in the promoter might have synergistic effects on regulating the rage promoter activity in HSCs. A role for NF-κB in regulating rage expression has been demonstrated in human cells (Tanaka et al., 2000). AGEs activate NF-κB (Tanaka et al., 2000). TCF and LEF-1 are transcription factors and bind to a TCF/LEF1 site to provide a docking position for the transcription factor β-catenin. Upon activation of the canonical Wnt signalling pathway, β-catenin translocates to the nucleus to promote the transcription of target genes (Brantjes et al., 2002). We observed the activation of Wnt signalling in HSCs treated with AGEs (data not shown). Curcumin interrupted Wnt signalling and reduced the level of nuclear β-catenin in HSCs (Kang and Chen, 2009a). The activation of PPARγ inhibited the NF-κB activity (Chen and Zheng, 2008) and interrupted Wnt signalling (Kang and Chen, 2009a) in HSCs. We previously showed that curcumin enhanced the trans-activity of PPARγ in HSCs, which was required for curcumin to inhibit HSC activation (Xu et al., 2003; Zheng and Chen, 2004). It is plausible to assume that curcumin attenuates the effects of AGEs by inducing gene expression of PPARγ and enhancing its activity. The latter leads to the inhibition of NF-κB activity and the blockade of the Wnt signalling pathway, which collectively suppress gene expression of RAGE in HSCs. Further experiments are ongoing in our lab to test this assumption. Our results do not exclude possible effects of AGEs w/wt curcumin on the stability of RAGE mRNA or protein, or on any post-translational regulation.
Oxidative stress is a mediator in the AGEs-induced gene expression of RAGE (Yamagishi and Matsui, 2010). Oxidative stress is defined as the imbalance between the production and the elimination of free radicals and reactive metabolites. The imbalance leads to injury of important biomolecules and cells and to negative effects on the whole organism. GSH is the most abundant thiol antioxidant in cells. It reacts with ROS or functions as a cofactor of antioxidant enzymes, leading to the protection of functions of redox-sensitive molecules, including enzymes and transcription factors (Wu et al., 2004). Oxidative stress plays a critical role in HSC activation and in hepatic fibrogenesis, regardless of aetiology (Di Sario et al., 2007; De Minicis and Brenner, 2008). It is well-known that AGEs induce oxidative stress and show a negative relationship with the level of GSH in patients (Sahin et al., 2008; Yamagishi and Matsui, 2010). Curcumin is anti-proliferative and a potent antioxidant (O'Connell and Rushworth, 2008). Its antioxidant capacity is 100-fold stronger than that of vitamin E/C (Ruby et al., 1995). We have shown that oxidative stress negatively regulates gene expression of PPARγ in HSCs (Zheng et al., 2007). On the other hand, the induction of PPARγ activity attenuates oxidative stress in cultured HSCs (Lin and Chen, 2008), and de novo synthesis of GSH is a prerequisite for curcumin to inhibit HSC activation (Zheng et al., 2007). In this report, we demonstrated that curcumin elevated the level of cellular GSH and attenuated the AGEs-induced oxidative stress by inducing the expression of GCL genes, which was a prerequisite for curcumin to inhibit gene expression of RAGE. Our observations are supported by those from previous studies (Yamagishi, 2009; Yamagishi and Matsui, 2010). The blockade of AGE-RAGE-oxidative stress system has been proposed to be a novel therapeutic strategy for diabetic complications (Yamagishi et al., 2008b).
The interaction of AGEs with RAGE has been shown to activate multiple cellular signalling cascades in cells, including ERK, JNK, p38 and NF-κB, etc (Cortizo et al., 2003; Dukic-Stefanovic et al., 2003; Shanmugam et al., 2003). The activation of the signalling cascades might facilitate the induction of oxidative stress and the inhibition of PPARγ gene expression in HSCs. We previously showed that curcumin interrupted MAPK signalling cascades and inhibited NF-κB activity in HSCs in vitro (Xu et al., 2003; Zhou et al., 2007; Lin and Chen, 2008). In the current report, we observed that curcumin suppressed gene expression of RAGE, which might lead to further interruption of its downstream signalling cascades and to the inhibition of HSC activation. Curcumin showed unique mechanisms in attenuating oxidative stress by inducing expression of GCL genes and elevating the level of cellular GSH. Studies have suggested curcumin to be an innocuous and dietary supplemental anti-fibrotic candidate for the treatment and/or the prevention of T2DM- and NASH-associated hepatic fibrosis (O'Connell and Rushworth, 2008).
The toxicity of curcumin was previously evaluated in cultured HSCs (Xu et al., 2003). Based on results from lactate dehydrogenase release assays, trypan blue exclusion assays and a rapid recovery of cell proliferation after withdrawal of curcumin, it was concluded that curcumin up to 100 µM was not toxic to cultured HSCs. Curcumin at 20 µM was used in most of our in vitro experiments. The systemic bioavailability of curcumin is relatively low (Sharma et al., 2001). Curcumin concentrations in human plasma can reach up to 2 µM following oral intake of very high amounts of curcumin (Garcea et al., 2004). Few reports could be found regarding serum levels of the AGE-proteins in human populations with or without diabetes. Among these limited studies, the levels of serum AGEs in human were contradictory (Schiel et al., 2003; Basta et al., 2006; Butscheid et al., 2007; Scharnagl et al., 2007; Ghanem et al., 2011). AGEs at 100 µg·mL−1 were used in most of the experiments in this project. The same dose of human glycated albumin was also used to examine its effects on insulin signalling in L6 skeletal muscle cells (Miele et al., 2003). The concentration of AGEs used in our experiments was determined by measuring AGEs-specific fluorescence with excitation at 360 nm and emissions at 440 nm (Monnier et al., 1984; Wrobel et al., 1997). N-Carboxymethyl-lysine (CML) was a major AGE among them (Monnier et al., 1984; Wrobel et al., 1997). The level of CML was previously observed to be higher in diabetic patients than non-diabetic controls (Basta et al., 2006). It is noteworthy that because the in vivo system is multifactorial, directly extrapolating in vitro conditions and results (e.g. effective concentrations), to the in vivo system, or vice versa, might be misleading.
Curcumin is naturally occurring yellow-orange pigment. The maximum absorption spectrum of curcumin in aqueous buffer is located at 425 nm (Kunwar et al., 2006). Curcumin in aqueous buffer shows weak fluorescence at 550 nm (Kunwar et al., 2006). Whereas the absorption spectrum of curcumin is red-shifted, the fluorescence spectrum of curcumin is blue-shifted in the presence of BSA (Barik et al., 2003). Therefore, curcumin probably has the ability to bind to some proteins, including BSA, probably at the hydrophobic cavities inside the protein (Barik et al., 2003). In our experience, cell extracts showed a little yellowish colour, if cells were treated with curcumin at 20 µM for less than 2 h (e.g. experiments for studying the effect of curcumin on the level of protein phosphorylation; not presented in this report). However, cell extracts showed no apparent variation in colour if cells were treated with curcumin (20 µM) for 24 h, such as in most of our experiments used to evaluate levels of proteins by Western blotting analyses. In our ROS-DCF assays, fluorescence was excited at 480 nm and detected for emission at 530 nm. SYBR Green I bound to DNA for real-time PCR assays. The resulting DNA–dye complex absorbed blue light (λmax = 497 nm) and emitted green light (λmax = 520 nm). Protein concentration was detected at the absorption of 570 nm using BCA reagents. Because of the photophysical properties of curcumin, when possible, a proper control with curcumin was used in our assays to eliminate potential photophysical interference from curcumin. In addition, internal controls, including β-tubulin for Western blotting analyses and GAPDH for real-time PCR assays, were used for equal loading and normalization. It was, thus, concluded that the treatment with curcumin did not significantly interfere with our assays and results.
A simplified model is proposed (Figure 10) to demonstrate the effects of AGEs on the induction of gene expression of RAGE and the stimulation of HSC activation, as well as the role of curcumin in the attenuation of the stimulating effects of AGEs. It is suggested that the underlying mechanisms of curcumin in the elimination of the stimulant effects of AGEs are, at least partially, mediated by attenuating oxidative stress, inducing gene expression of PPARγ and stimulating its trans-activity. Our results in this report provide a novel insight into mechanisms by which curcumin inhibits the AGE-induced activation of HSCs. Additional experiments are necessary to verify these in vitro observations in vivo and further evaluate the role of curcumin as an anti-fibrotic agent for the therapeutic treatment of T2DM- and NASH-associated hepatic fibrogenesis.
Figure 10.
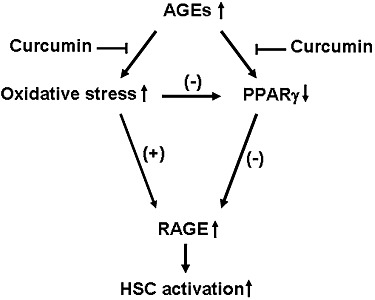
A simplified mechanistic model of AGEs in the induction of gene expression of RAGE, leading to the activation of HSCs, which is attenuated by curcumin. AGEs induces gene expression of RAGE in HSCs, which facilitates HSC activation. Curcumin eliminates the effects of AGEs probably by attenuating oxidative stress, inducing gene expression of PPARγ and stimulating its trans-activity. ‘↑’ or ‘↓’ indicates the effects of AGEs.
Acknowledgments
The work was supported by the grant DK 47995 from NIH/NIDDK to AC.
Glossary
- AGEs
advanced glycation end-products
- BSO
l-buthionine-sulfoximine
- DMEM
Dulbecco's modified Eagle's Medium
- FBS
fetal bovine serum
- GAPDH
glyceraldehydes-3-phosphate dehydrogenase
- GCL
glutamate-cysteine ligase
- GSH
glutathione
- HSCs
hepatic stellate cells
- LPO
lipid peroxidation
- NAC
N-acetyl-l-cysteine
- NASH
non-alcohol steatohepatitis
- PDGF-β
platelet-derived growth factor-β
- PGJ2
deoxy-Δ12,14-prostaglandin J2
- RAGE
receptor for AGEs
- ROS
reactive oxygen species
- shRNA
short or small hairpin RNA
- T2DM
type 2 diabetes mellitus
Conflict of interest
None.
References
- Alexande SPH, Mathi A, Peter JA. Guide to receptors and channels (GRAC) Br J Pharmacol. (5th edn.) 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson ME, Luo JL. Glutathione therapy: from prodrugs to genes. Semin Liver Dis. 1998;18:415–424. doi: 10.1055/s-2007-1007174. [DOI] [PubMed] [Google Scholar]
- Bae HW, Geiser AG, Kim DH, Chung MT, Burmester JK, Sporn MB, et al. Characterization of the promoter region of the human transforming growth factor-beta type II receptor gene. J Biol Chem. 1995;270:29460–29468. doi: 10.1074/jbc.270.49.29460. [DOI] [PubMed] [Google Scholar]
- Ballagi AE, Ishizaki A, Nehlin JO, Funa K. Isolation and characterization of the mouse PDGF beta-receptor promoter. Biochem Biophys Res Commun. 1995;210:165–173. doi: 10.1006/bbrc.1995.1642. [DOI] [PubMed] [Google Scholar]
- Barik A, Priyadarsini KI, Mohan H. Photophysical studies on binding of curcumin to bovine serum albumins. Photochem Photobiol. 2003;77:597–603. doi: 10.1562/0031-8655(2003)077<0597:psoboc>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Basta G, Sironi AM, Lazzerini G, Del Turco S, Buzzigoli E, Casolaro A, et al. Circulating soluble receptor for advanced glycation end products is inversely associated with glycemic control and S100A12 protein. J Clin Endocrinol Metab. 2006;91:4628–4634. doi: 10.1210/jc.2005-2559. [DOI] [PubMed] [Google Scholar]
- Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83:876–886. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- Brantjes H, Barker N, van Es J, Clevers H. TCF: Lady Justice casting the final verdict on the outcome of Wnt signalling. Biol Chem. 2002;383:255–261. doi: 10.1515/BC.2002.027. [DOI] [PubMed] [Google Scholar]
- Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- Butscheid M, Schafer C, Brenner S, Alscher D, Murdter T, Niwa T, et al. Unchanged serum levels of advanced glycation endproducts in patients with liver disease. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:401–406. doi: 10.1007/s00210-007-0171-9. [DOI] [PubMed] [Google Scholar]
- Calamita G, Portincasa P. Present and future therapeutic strategies in non-alcoholic fatty liver disease. Expert Opin Ther Targets. 2007;11:1231–1249. doi: 10.1517/14728222.11.9.1231. [DOI] [PubMed] [Google Scholar]
- Camp HS, Chaudhry A, Leff T. A novel potent antagonist of peroxisome proliferator-activated receptor gamma blocks adipocyte differentiation but does not revert the phenotype of terminally differentiated adipocytes. Endocrinology. 2001;142:3207–3213. doi: 10.1210/endo.142.7.8254. [DOI] [PubMed] [Google Scholar]
- Chen A, Zheng S. Curcumin inhibits connective tissue growth factor gene expression in activated hepatic stellate cells in vitro by blocking NF-kappaB and ERK signalling. Br J Pharmacol. 2008;153:557–567. doi: 10.1038/sj.bjp.0707542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Hoffman BB, Lee JS, Kasama Y, Jim B, Kopp JB, et al. Cultured tubule cells from TGF-beta1 null mice exhibit impaired hypertrophy and fibronectin expression in high glucose. Kidney Int. 2004;65:1191–1204. doi: 10.1111/j.1523-1755.2004.00492.x. [DOI] [PubMed] [Google Scholar]
- Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40(Suppl. 1):S5–S10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- Cortizo AM, Lettieri MG, Barrio DA, Mercer N, Etcheverry SB, McCarthy AD. Advanced glycation end-products (AGEs) induce concerted changes in the osteoblastic expression of their receptor RAGE and in the activation of extracellular signal-regulated kinases (ERK) Mol Cell Biochem. 2003;250:1–10. doi: 10.1023/a:1024934008982. [DOI] [PubMed] [Google Scholar]
- Cotgreave IA. N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv Pharmacol. 1997;38:205–227. [PubMed] [Google Scholar]
- De Minicis S, Brenner DA. Oxidative stress in alcoholic liver disease: role of NADPH oxidase complex. J Gastroenterol Hepatol. 2008;23(Suppl. 1):S98–S103. doi: 10.1111/j.1440-1746.2007.05277.x. [DOI] [PubMed] [Google Scholar]
- Di Sario A, Candelaresi C, Omenetti A, Benedetti A. Vitamin E in chronic liver diseases and liver fibrosis. Vitam Horm. 2007;76:551–573. doi: 10.1016/S0083-6729(07)76021-1. [DOI] [PubMed] [Google Scholar]
- Dukic-Stefanovic S, Gasic-Milenkovic J, Deuther-Conrad W, Munch G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs) J Neurochem. 2003;87:44–55. doi: 10.1046/j.1471-4159.2003.01988.x. [DOI] [PubMed] [Google Scholar]
- Federico A, Niosi M, Vecchio Blanco CD, Loguercio C. Emerging drugs for non-alcoholic fatty liver disease. Expert Opin Emerg Drugs. 2008;13:145–158. doi: 10.1517/14728214.13.1.145. [DOI] [PubMed] [Google Scholar]
- Fehrenbach H, Weiskirchen R, Kasper M, Gressner AM. Up-regulated expression of the receptor for advanced glycation end products in cultured rat hepatic stellate cells during transdifferentiation to myofibroblasts. Hepatology. 2001;34:943–952. doi: 10.1053/jhep.2001.28788. [DOI] [PubMed] [Google Scholar]
- Feng Y, Nie L, Thakur MD, Su Q, Chi Z, Zhao Y, et al. A multifunctional lentiviral-based gene knockdown with concurrent rescue that controls for off-target effects of RNAi. Genomics Proteomics Bioinformatics. 2010;8:238–245. doi: 10.1016/S1672-0229(10)60025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley SI, Feng Y, Krishnamurthy R, Kim DH, Celedon A, Longmore GD, et al. A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat Cell Biol. 2010;12:598–604. doi: 10.1038/ncb2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridovich I. The biology of oxygen radicals. Science. 1978;201:875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zheng S, Lin J, Ryerse J, Chen A. Curcumin protects the rat liver from CCl4-caused injury and fibrogenesis by attenuating oxidative stress and suppressing inflammation. Mol Pharmacol. 2008;73:399–409. doi: 10.1124/mol.107.039818. [DOI] [PubMed] [Google Scholar]
- Galli A, Crabb D, Price D, Ceni E, Salzano R, Surrenti C, et al. Peroxisome proliferator-activated receptor gamma transcriptional regulation is involved in platelet-derived growth factor-induced proliferation of human hepatic stellate cells. Hepatology. 2000;31:101–108. doi: 10.1002/hep.510310117. [DOI] [PubMed] [Google Scholar]
- Garcea G, Jones DJ, Singh R, Dennison AR, Farmer PB, Sharma RA, et al. Detection of curcumin and its metabolites in hepatic tissue and portal blood of patients following oral administration. Br J Cancer. 2004;90:1011–1015. doi: 10.1038/sj.bjc.6601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem AA, Elewa A, Arafa LF. Pentosidine and N-carboxymethyl-lysine: biomarkers for type 2 diabetic retinopathy. Eur J Ophthalmol. 2011;21:48–54. doi: 10.5301/ejo.2010.4447. [DOI] [PubMed] [Google Scholar]
- Hatziagelaki E, Karageorgopoulos DE, Chounta A, Tsiavou A, Falagas ME, Dimitriadis G. Predictors of impaired glucose regulation in patients with non-alcoholic fatty liver disease. Exp Diabetes Res. 2012;2012:351974. doi: 10.1155/2012/351974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi S, Araki N, Morino Y. Immunochemical approach to characterize advanced glycation end products of the Maillard reaction. Evidence for the presence of a common structure. J Biol Chem. 1991;266:7329–7332. [PubMed] [Google Scholar]
- Ihm SH, Chang K, Kim HY, Baek SH, Youn HJ, Seung KB, et al. Peroxisome proliferator-activated receptor-gamma activation attenuates cardiac fibrosis in type 2 diabetic rats: the effect of rosiglitazone on myocardial expression of receptor for advanced glycation end products and of connective tissue growth factor. Basic Res Cardiol. 2010;105:399–407. doi: 10.1007/s00395-009-0071-x. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Kanno K, Hyogo H, Yamagishi S, Takeuchi M, Tazuma S, et al. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J Gastroenterol. 2008;43:298–304. doi: 10.1007/s00535-007-2152-7. [DOI] [PubMed] [Google Scholar]
- Kang Q, Chen A. Curcumin eliminates oxidized LDL roles in activating hepatic stellate cells by suppressing gene expression of lectin-like oxidized LDL receptor-1. Lab Invest. 2009a;89:1275–1290. doi: 10.1038/labinvest.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Q, Chen A. Curcumin inhibits srebp-2 expression in activated hepatic stellate cells in vitro by reducing the activity of specificity protein-1. Endocrinology. 2009b;150:5384–5394. doi: 10.1210/en.2009-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Q, Chen A. Curcumin suppresses expression of low-density lipoprotein (LDL) receptor, leading to the inhibition of LDL-induced activation of hepatic stellate cells. Br J Pharmacol. 2009c;157:1354–1367. doi: 10.1111/j.1476-5381.2009.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem. 1999;274:31740–31749. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- Kunwar A, Barik A, Pandey R, Priyadarsini KI. Transport of liposomal and albumin loaded curcumin to living cells: an absorption and fluorescence spectroscopic study. Biochim Biophys Acta. 2006;1760:1513–1520. doi: 10.1016/j.bbagen.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Lam S, van der Geest RN, Verhagen NA, van Nieuwenhoven FA, Blom IE, Aten J, et al. Connective tissue growth factor and igf-I are produced by human renal fibroblasts and cooperate in the induction of collagen production by high glucose. Diabetes. 2003;52:2975–2983. doi: 10.2337/diabetes.52.12.2975. [DOI] [PubMed] [Google Scholar]
- Leclercq I. Non-alcoholic fatty liver disease. Bull Mem Acad R Med Belg. 2010;165:147–155. Discussion 155–158. [PubMed] [Google Scholar]
- Lin J, Chen A. Activation of peroxisome proliferator-activated receptor-gamma by curcumin blocks the signaling pathways for PDGF and EGF in hepatic stellate cells. Lab Invest. 2008;88:529–540. doi: 10.1038/labinvest.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Chen A. Curcumin diminishes the impacts of hyperglycemia on the activation of hepatic stellate cells by suppressing membrane translocation and gene expression of glucose transporter-2. Mol Cell Endocrinol. 2011;333:160–171. doi: 10.1016/j.mce.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Zheng S, Chen A. Curcumin attenuates the effects of insulin on stimulating hepatic stellate cell activation by interrupting insulin signaling and attenuating oxidative stress. Lab Invest. 2009;89:1397–1409. doi: 10.1038/labinvest.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Tang Y, Kang Q, Chen A. Curcumin eliminates the inhibitory effect of advanced glycation end-products (AGEs) on gene expression of AGE receptor-1 in hepatic stellate cells in vitro. Lab Invest. 2012 doi: 10.1038/labinvest.2012.53. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, He JC, Cai W, Liu H, Zhu L, Vlassara H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc Natl Acad Sci U S A. 2004;101:11767–11772. doi: 10.1073/pnas.0401588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita Z, Vlassara H, Cerami A, Bucala R. Immunochemical detection of advanced glycosylation end products in vivo. J Biol Chem. 1992;267:5133–5138. [PubMed] [Google Scholar]
- Marra F, Efsen E, Romanelli RG, Caligiuri A, Pastacaldi S, Batignani G, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466–478. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- Matsui T, Yamagishi S, Takeuchi M, Ueda S, Fukami K, Okuda S. Nifedipine inhibits advanced glycation end products (AGEs) and their receptor (RAGE) interaction-mediated proximal tubular cell injury via peroxisome proliferator-activated receptor-gamma activation. Biochem Biophys Res Commun. 2010;398:326–330. doi: 10.1016/j.bbrc.2010.06.093. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- Miele C, Riboulet A, Maitan MA, Oriente F, Romano C, Formisano P, et al. Human glycated albumin affects glucose metabolism in L6 skeletal muscle cells by impairing insulin-induced insulin receptor substrate (IRS) signaling through a protein kinase C alpha-mediated mechanism. J Biol Chem. 2003;278:47376–47387. doi: 10.1074/jbc.M301088200. [DOI] [PubMed] [Google Scholar]
- Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF, Jr, Motomura K, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- Monnier VM, Kohn RR, Cerami A. Accelerated age-related browning of human collagen in diabetes mellitus. Proc Natl Acad Sci U S A. 1984;81:583–587. doi: 10.1073/pnas.81.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanji AA, Jokelainen K, Tipoe GL, Rahemtulla A, Thomas P, Dannenberg AJ. Curcumin prevents alcohol-induced liver disease in rats by inhibiting the expression of NF-kappa B-dependent genes. Am J Physiol Gastrointest Liver Physiol. 2003;284:G321–G327. doi: 10.1152/ajpgi.00230.2002. [DOI] [PubMed] [Google Scholar]
- O'Connell MA, Rushworth SA. Curcumin: potential for hepatic fibrosis therapy? Br J Pharmacol. 2008;153:403–405. doi: 10.1038/sj.bjp.0707580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Jeon CH, Ko G, Kim J, Sohn DH. Protective effect of curcumin in rat liver injury induced by carbon tetrachloride. J Pharm Pharmacol. 2000;52:437–440. doi: 10.1211/0022357001774048. [DOI] [PubMed] [Google Scholar]
- Ramasamy R, Yan SF, Schmidt AM. RAGE: therapeutic target and biomarker of the inflammatory response–the evidence mounts. J Leukoc Biol. 2009;86:505–512. doi: 10.1189/jlb.0409230. [DOI] [PubMed] [Google Scholar]
- Ruby AJ, Kuttan G, Babu KD, Rajasekharan KN, Kuttan R. Anti-tumour and antioxidant activity of natural curcuminoids. Cancer Lett. 1995;94:79–83. doi: 10.1016/0304-3835(95)03827-j. [DOI] [PubMed] [Google Scholar]
- Sahin E, Gocmen AY, Kocak H, Tuncer M, Gumuslu S. The association of advanced glycation end-products with glutathione status. Ann Clin Biochem. 2008;45:369–374. doi: 10.1258/acb.2007.007186. [DOI] [PubMed] [Google Scholar]
- Scharnagl H, Stojakovic T, Winkler K, Rosinger S, Marz W, Boehm BO. The HMG-CoA reductase inhibitor cerivastatin lowers advanced glycation end products in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2007;115:372–375. doi: 10.1055/s-2007-973830. [DOI] [PubMed] [Google Scholar]
- Schiel R, Franke S, Appel T, Voigt U, Ross IS, Kientsch-Engel R, et al. Improvement in quality of diabetes control and concentrations of AGE-products in patients with type 1 and insulin-treated type 2 diabetes mellitus studied over a period of 10 years (JEVIN) J Diabetes Complications. 2003;17:90–97. doi: 10.1016/s1056-8727(02)00203-9. [DOI] [PubMed] [Google Scholar]
- Schmidt AM, Yan SD, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta. 2000;1498:99–111. doi: 10.1016/s0167-4889(00)00087-2. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- Shanmugam N, Kim YS, Lanting L, Natarajan R. Regulation of cyclooxygenase-2 expression in monocytes by ligation of the receptor for advanced glycation end products. J Biol Chem. 2003;278:34834–34844. doi: 10.1074/jbc.M302828200. [DOI] [PubMed] [Google Scholar]
- Sharma RA, McLelland HR, Hill KA, Ireson CR, Euden SA, Manson MM, et al. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin Cancer Res. 2001;7:1894–1900. [PubMed] [Google Scholar]
- Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem. 2000;275:25781–25790. doi: 10.1074/jbc.M001235200. [DOI] [PubMed] [Google Scholar]
- Tang Y, Chen A. Curcumin prevents leptin raising glucose levels in hepatic stellate cells by blocking translocation of glucose transporter-4 and increasing glucokinase. Br J Pharmacol. 2010;161:1137–1149. doi: 10.1111/j.1476-5381.2010.00956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zheng S, Chen A. Curcumin eliminates leptin's effects on hepatic stellate cell activation via interrupting leptin signaling. Endocrinology. 2009;150:3011–3020. doi: 10.1210/en.2008-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, et al. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11:1656–1666. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, She H, Hazra S, Cheng J, Miyahara T. Anti-adipogenic regulation underlies hepatic stellate cell transdifferentiation. J Gastroenterol Hepatol. 2006;21(Suppl. 3):S102–S105. doi: 10.1111/j.1440-1746.2006.04573.x. [DOI] [PubMed] [Google Scholar]
- Wells RG. Liver fibrosis: challenges of the new era. Gastroenterology. 2009;136:387–388. doi: 10.1053/j.gastro.2008.12.028. [DOI] [PubMed] [Google Scholar]
- Wrobel K, Wrobel K, Garay-Sevilla ME, Nava LE, Malacara JM. Novel analytical approach to monitoring advanced glycosylation end products in human serum with on-line spectrophotometric and spectrofluorometric detection in a flow system. Clin Chem. 1997;43:1563–1569. [PubMed] [Google Scholar]
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- Xu J, Fu Y, Chen A. Activation of peroxisome proliferator-activated receptor-gamma contributes to the inhibitory effects of curcumin on rat hepatic stellate cell growth. Am J Physiol Gastrointest Liver Physiol. 2003;285:G20–G30. doi: 10.1152/ajpgi.00474.2002. [DOI] [PubMed] [Google Scholar]
- Yamagishi S. Advanced glycation end products and receptor-oxidative stress system in diabetic vascular complications. Ther Apher Dial. 2009;13:534–539. doi: 10.1111/j.1744-9987.2009.00775.x. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Matsui T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid Med Cell Longev. 2010;3:101–108. doi: 10.4161/oxim.3.2.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi S, Matsui T, Nakamura K, Takeuchi M, Inoue H. Telmisartan inhibits advanced glycation end products (AGEs)-elicited endothelial cell injury by suppressing AGE receptor (RAGE) expression via peroxisome proliferator-activated receptor-gammaactivation. Protein Pept Lett. 2008a;15:850–853. doi: 10.2174/092986608785203746. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Nakamura K, Matsui T, Ueda S, Fukami K, Okuda S. Agents that block advanced glycation end product (AGE)-RAGE (receptor for AGEs)-oxidative stress system: a novel therapeutic strategy for diabetic vascular complications. Expert Opin Investig Drugs. 2008b;17:983–996. doi: 10.1517/13543784.17.7.983. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Luo Z, Ma L, Xu Q, Yang Q, Si L. Resveratrol prevents the impairment of advanced glycosylation end products (AGE) on macrophage lipid homeostasis by suppressing the receptor for AGE via peroxisome proliferator-activated receptor gamma activation. Int J Mol Med. 2010;25:729–734. doi: 10.3892/ijmm_00000398. [DOI] [PubMed] [Google Scholar]
- Zheng S, Chen A. Activation of PPARgamma is required for curcumin to induce apoptosis and to inhibit the expression of extracellular matrix genes in hepatic stellate cells in vitro. Biochem J. 2004;384:149–157. doi: 10.1042/BJ20040928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Chen A. Curcumin suppresses the expression of extracellular matrix genes in activated hepatic stellate cells by inhibiting gene expression of connective tissue growth factor. Am J Physiol Gastrointest Liver Physiol. 2006;290:G883–G893. doi: 10.1152/ajpgi.00450.2005. [DOI] [PubMed] [Google Scholar]
- Zheng S, Chen A. Disruption of transforming growth factor-beta signaling by curcumin induces gene expression of peroxisome proliferator-activated receptor-gamma in rat hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G113–G123. doi: 10.1152/ajpgi.00200.2006. [DOI] [PubMed] [Google Scholar]
- Zheng S, Yumei F, Chen A. De novo synthesis of glutathione is a prerequisite for curcumin to inhibit hepatic stellate cell (HSC) activation. Free Radic Biol Med. 2007;43:444–453. doi: 10.1016/j.freeradbiomed.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zheng S, Lin J, Zhang QJ, Chen A. The interruption of the PDGF and EGF signaling pathways by curcumin stimulates gene expression of PPARgamma in rat activated hepatic stellate cell in vitro. Lab Invest. 2007;87:488–498. doi: 10.1038/labinvest.3700532. [DOI] [PubMed] [Google Scholar]