

Abstract
BACKGROUND AND PURPOSE
So far, there is only limited information about the regulation of the endogenous synthesis of hydrogen sulfide (H2S), an important gaseous signalling molecule. This study was done to evaluate the redox-dependent signalling events that regulate the expression of the H2S synthesising enzyme cystathionine-γ-lyase (CSE) in rat mesangial cells.
EXPERIMENTAL APPROACH
The effects of platelet-derived growth factor (PDGF)-BB and antioxidants on CSE expression and activity in cultured rat renal mesangial cells were assessed. Activity of nuclear factor erythroid-2-related factor-2 (Nrf2) was measured as the binding capacity to a radiolabelled consensus element by electrophoretic mobility shift assay (EMSA). Furthermore, CSE and Nrf2 expression was analysed in a rat model of anti-Thy-1-induced glomerulonephritis by immunohistochemistry.
KEY RESULTS
Treatment of mesangial cells with PDGF-BB resulted in a marked time- and dose-dependent up-regulation of CSE mRNA and protein levels, as well as CSE activity accompanied with increased formation of reactive oxygen species. Remarkably, co-administration of antioxidants, such as N-acetylcysteine, ebselen or diphenylene iodonium chloride, drastically reduced PDGF-BB-induced CSE expression. PDGF-BB induced binding of Nrf2 to a corresponding consensus antioxidant element in a redox-dependent manner. Furthermore, PDGF-BB-induced CSE expression in mouse mesangial cells was completely abolished in Nrf2 knockout mice compared with wild-type mice. In a rat model of anti-Thy-1-induced proliferative glomerulonephritis, we observed a marked up-regulation of CSE protein paralleled by a stabilization of Nrf2 protein.
CONCLUSIONS AND IMPLICATIONS
PDGF-BB regulated CSE via a redox-mediated activation of Nrf2. Such action would aid the resolution of glomerular inflammatory diseases.
LINKED ARTICLE
This article is commented on by Gallyas, pp. 2228–2230 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2012.01976.x
Keywords: Mesangial cells, cystathionine-γ-lyase, reactive oxygen species, redox, platelet-derived growth factor, nuclear erythroid-2-related factor-2
Introduction
Hydrogen sulfide (H2S), a poisonous gas that affects the respiratory chain, has been recognized as an endogenous mediator of well-defined physiological processes, and, consequently, H2S has joined the family of so-called gasotransmitters which includes NO and carbon monoxide (CO) (Wang, 2002; Łowicka and Bełtowski, 2007; Gadalla and Snyder, 2010). H2S can exert antioxidant properties simply as a scavenger of reactive oxygen species (ROS), NO and peroxynitrite (Łowicka and Bełtowski, 2007). More recently, an elegant mechanism has been uncovered by which H2S enhances the enzymic activity of different proteins, such as glyceraldehyde 3-phosphatedehydrogenase (GAPDH) (Mustafa et al., 2009; Gadalla and Snyder, 2010). Obviously, the formation of a –SSH moiety at cysteine residues by H2S via a sulfhydration reaction is the crucial molecular mechanism of H2S action (Mustafa et al., 2009). One of the first targets found to be activated by H2S was the ATP-dependent K+ channel (KATP) (Zhao et al., 2001; channel and receptor nomenclature follow Alexander et al., 2011). More recently, Bucci et al. (2010) were able to demonstrate that, in isolated aortic rings, endogenously produced H2S inhibited phosphodiesterases leading to increased cGMP levels, thus suggesting a cross-talk between NO and H2S signalling in the cardiovascular system.
In mammalian cells, H2S is currently thought to be synthesized by two different enzymes, namely cystathionine γ-lyase (CSE, EC 4.4.1.1) and cystathionine β-synthase (CBS, EC 4.2.1.22), both involved in the metabolism of the amino acid cysteine. However, very recently, another enzyme, 3-mercaptopyruvate sulfurtransferase, in conjunction with cysteine aminotransferase (CAT), has been identified to produce considerable amounts of H2S in brain and vascular endothelium (Shibuya et al., 2009a, b). Regarding their localization, CBS is expressed predominantly in the brain, whereas CSE expression is found predominantly in vascular tissue. It is worth noting that in analogy to the discovery of the localization of the different forms of NO synthases, it seems to be too early to discriminate between a brain and a vascular form of H2S-generating enzymes.
The existence and the mechanisms of activity of H2S-generating enzymes have been known for a long time (Stipanuk and Beck, 1982). However, it was not known whether synthesis of H2S constitutes simply an adverse or non-essential by-product of the cysteine metabolic pathway or such synthesis had a distinct signalling purpose to modify physiological functions. An important answer to this question was provided by Yang et al. (2008), who found that CSE knockout mice had drastically reduced H2S levels accompanied with a clear elevation in blood pressure. These findings indicated a role of H2S as another endothelium-derived, vascular smooth muscle relaxing factor.
A further indication of a pathophysiological function of CSE would be that its expression was tightly regulated by cytokines or growth factors. So far, only a few reports describe the regulation of the CSE gene. For instance, LPS induced CSE expression in macrophages and this effect was inhibited by glucocorticoids, suggesting a role of CSE regulation in inflammatory processes (Zhu et al., 2010). Inflammatory cytokines, such as TNF-α, IL-1β and IL-6, induced the expression of CSE and enhanced H2S formation in primary human articular chondrocytes, as well as in mesenchymal progenitor cells (Fox et al., 2012). In addition, these authors demonstrated that genetic or pharmacological blockade of cytokine-induced H2S formation resulted in a higher susceptibility to oxidative stress, indicating a protective role of endogenously produced H2S in joint inflammation.
Mesangial cells are glomerular pericytes that resemble vascular smooth muscle cells. In a complex cross-talk with glomerular endothelial cells, glomerular podocytes and invading monocytes/macrophages, mesangial cells produce high amounts of inflammatory mediators, such as cytokines, ROS and NO (Baud and Ardaillou, 1993; Cattell, 2002; Pfeilschifter et al., 2002; Schlöndorff and Banas, 2009). In culture, rat mesangial cells can be stimulated with cytokines, such as IL-1β and TNF-α to form large amounts of NO via the induction of iNOS (Pfeilschifter and Schwarzenbach, 1990). This process results in a positive regulatory loop based on further activation of the iNOS transcriptional machinery by NO and ROS (Mühl and Pfeilschifter, 1995; Beck et al., 1998). Platelet-derived growth factor (PDGF) is a highly potent mitogen in mesangial cells (Abboud, 1995). We have shown that NO induced the expression of PDGF receptor α and this allowed several isoforms of PDGF to limit the effects of NO (Kunz et al., 1997; Beck et al., 2005). As the BB isoform of PDGF (PDGF-BB) recognizes both of the receptors for PDGF so far described, we have here examined the effects of PDGF-BB on CSE expression in rat mesangial cells.
Methods
Animals
All animal care and experimental procedures complied with the German Animal Protection Law and was approved by the Ethics Review Committee for laboratory animals of the District Government of Darmstadt, Germany. The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). In these experiments, a total of 123 rats and 6 mice were used.
Cell culture
Rat glomerular mesangial cells (from three 1 month old male Sprague-Dawley rats; 70-100 g; Charles River, Sulzfeld, Germany) were cultivated as described previously (Kurtz et al., 1982) and grown in RPMI 1640 medium supplemented with 10 % fetal calf serum (FCS), 2 mM glutamine, 5 ng·mL−1 insulin, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin (Pfeilschifter and Vosbeck, 1991). Quiescent cells were obtained by incubating mesangial cells for 24 h in serum-free Dulbecco's minimal essential medium (DMEM) supplemented with 0.1 mg·mL−1 of essentially fatty acid-free bovine serum albumin. Stimulation of mesangial cells with PDGF-BB was performed with fresh serum-free culture medium. Mesangial cells were used between passages 12 and 21.
Mouse spleen macrophages and glomerular mesangial cells were obtained from C57BL6 (wild-type) mice or mice lacking the transcription factor, nuclear factor erythroid-2-related factor-2 (Nrf2 knockout, on a C57BL6 background). These mice were a kind gift from Prof. Yamamoto (Tohoku University, Japan). For each strain, cells from 2 females and 1 male (3 months old, 22–28 g) were pooled. Spleen macrophages were isolated and differentiated as previously described (Jennewein et al., 2008). Mouse mesangial cells were isolated and cultured, as previously described (Hofmann et al., 2008).
Analysis of CSE mRNA expression by semi-quantitative and quantitative PCR
For a preliminary analysis of CSE and CBS expression, semi-quantitative RT-PCR was performed using the oligonucleotides (sense: 5′ ACCAAATTGCTGGAGGCAGCGAT 3′ and antisense: 5′ TCATGATTGCTGGAAGCTCAGC 3′) for rat CSE (Acc.-Nr. NM 017074) and (sense: 5′ GTGAGCAGACCCGGAAATAGAGT 3′ and antisense: 5′ CTTGTCGGGAAAGGAGGGAGAT 3′) for rat CBS (Acc.-Nr. NM 012522).
Quantitative PCR was performed using a pre-developed TaqMan gene expression assay for CSE in a GeneAmp 7700 System according to the manufacturer's instructions (Applied Biosystems, Weiterstadt, Germany). The reactions were performed with 40 cycles (3 s at 95°C; 30 s at 60°C). Each sample was measured in duplicate. Quantification was performed using the ΔΔCt method. To correct for unequal amounts of RNA also mRNA levels for GAPDH were analysed using a primer set provided by Applied Biosystems.
Western blot analysis
Cells were harvested in a lysis buffer containing 20 mM Tris-HCl (pH 7.5), 1 mM EGTA, 1 mM EDTA, 1 % TritonX-100 and a complete protease Inhibitor Cocktail (Roche, Mannheim, Germany). Samples (30-80 µg protein) of the homogenates were subjected to SDS-PAGE (12% acrylamide gel) and separated for 2 h with 150V in SDS-PAGE buffer (25 mM Tris, 192 mM glycine, 0.1 % SDS). After transfer in 25 mM Tris, 192 mM glycine and 20 % methanol to a PVDF membrane, CSE protein was detected using either an polyclonal, affinity purified antibody raised against a CSE-derived peptide FKQDSPGQSSGFVYSC, representing amino acids 46–60 of the rat CSE protein, SwissProt Accession Number P18757 (Eurogentec, Köln, Germany) or an polyclonal CSE antibody (Proteintech, Chicago, IL, USA). The blot was then incubated with a secondary horseradish peroxidase-conjugated antibody (goat anti-rabbit IgG-HRP, Santa Cruz Biotechnology) and developed with the ECL detection system (Amersham, Braunschweig, Germany). As a control for equal loading, the blot was stripped and re-probed with an antibody for β-actin (Sigma).
Analysis of [3H]methyl-thymidine incorporation
Quiescent mouse or rat mesangial cells were treated with or without growth factors or antioxidants for 14 h. Thereafter, 1 µCi·mL−1[3H]methyl-thymidine was added, and the cells were incubated for a further 24 h. Thereafter, cells were washed twice with PBS and incubated in 5% trichloroacetic acid at 4°C for 30 min, and the DNA was solubilized in 0.5 M NaOH for 30 min at 37°C. [3H]thymidine incorporation was determined in a β-counter (Döll et al., 2005).
CSE activity assay
A method for the determination of CSE activity was described previously (Ogasawara et al., 2002). This method is based on the conversion of β-chloro-L-alanine to hydrogen peroxide via pyruvate and the oxidation of a colourless leuco-dye to produce Bindschedler's green. Cells were harvested in ice-cold 50 mM potassium phosphate buffer (pH 7.4) and lysed by sonification. To an 85 µL reaction mixture containing 100 mM Tris-phosphate (pH 8), 2.35 mM EDTA and 35 µM PLP, 15 µL of 0.1 M β-chloro-L-alanine and 60 µg of lysates were added and incubated at 37°C for 15 min. As a control for the specificity of CSE, 10 mM of β-cyano-L-alanine was added to inhibit CSE activity. The reaction is terminated by the addition of 100 µL of 6.5 mM 4,4-dithiopyridine to inactivate CSE. After a 5 min incubation step at room temperature, 700 µL of the colour-producing buffer were added to the reaction mixture (0.1 M PIPES, pH 6.4; 0.823 mM thiamine pyrophosphate; 6857 mM Mg2+; 0.274 U·mL−1 horseradish peroxidase type VI; 0.274 mM DA-64 dye (Wako, Neuss, Germany); 10.97 U·mL−1 pyruvate oxidase). The solution was mixed and incubated for 10 min at 37°C. The absorbance of the formed Bindschedler's green was measured at 727 nm. The sample blank was treated in the same way except the 4,4-dithiopyridine was added before the addition of β-chloro-L-alanine.
Analysis of ROS production
Mesangial cells were grown on 6-well plates. After stimulation with or without PDGF-BB, cells were washed and incubated in HEPES-modified Tyrode's solution containing dihydrodichlorofluorescein-DA (5 µmol·L−1) for 20 min in the dark. Subsequently, formation of the oxidation product dichlorofluorescein was determined using a micro-plate fluorimeter (excitation 488 nm, emission 515 nm).
Electrophoretic mobility shift (EMSA)
Quiescent mesangial cells were treated with PDGF (25 ng·mL−1) for different time periods and nuclear extracts were prepared according to the method of Schreiber et al. (1989). To allow binding of Nrf2 to its respective binding site, nuclear extracts were incubated with a double-stranded radioactively labelled Nrf2 consensus sequence (5′ TGGGGAACCTGTGCTGAG 3′, Santa Cruz, Heidelberg, Germany). Nrf2/ antioxidant responsive element (ARE) complexes were separated on a 4.5% acrylamide gel and visualized using an automated detector system BAS 1500 of Fujifilm (Raytest, Straubenhardt, Germany). Competition experiments were carried out by pre-incubation of the DNA-binding reaction for 30 min with a 100-fold excess of the unlabeled double stranded oligonucleotide.
In vivo model of anti-Thy-1 glomerulonephritis
Anti-Thy-1 glomerulonephritis was induced in adult male Wistar rats (n = 6; 180–200g; Charles River) by a single intravenous injection of mouse anti-rat Thy-1.1 IgG, clone OX-7 (BioTrend, Cologne, Germany) dissolved in 18 mM sodium phosphate, pH 7.4/0.15 M NaCl (PBS), at a dose of 1 mg·kg−1 b.w. Control animals (n = 6) received a single intravenous injection of PBS only. Animals were anaesthetized with pentobarbital (150 mg·kg−1). Kidneys were harvested at 2, 4 and 8 h, as well as at 1, 3, 7, 10, 15, 21 and 29 days following induction of anti-Thy-1 glomerulonephritis.
Immunohistochemistry
Serial sections (4 µm) of paraffin-embedded samples were treated with 3% H2O2 (10 min, room temperature) and processed for immunohistochemical analysis by immunoperoxidase technique using mouse anti-CSE (clone 1E4, Abnova) and rabbit anti-Nrf2 antibodies (Santa Cruz, Heidelberg, Germany) (Schaefer et al., 2002). After blocking with 4% non-fat dry milk/0.1% Triton X-100/TBS (30 min, room temperature) sections were treated with Avidin/Biotin Blocking Kit, followed by incubation with Vectastain ABC peroxidase kit, mouse or rabbit, respectively, applied according to the manufacturer's instructions (all from Vector Laboratories). Finally, DAB (3,3′-diaminobenzidine) peroxidase substrate (Vector Laboratories) and counterstaining with haematoxylin (AppliChem) were used for the visualization and counterstaining respectively. The specificity of immunostaining was ascertained by omitting the primary antibody, using non-immune serum and blocking peptide for Nrf2 (Santa Cruz, Heidelberg, Germany).
Statistical analyses
If not otherwise indicated, data are expressed as per cent of vehicle-treated controls (means ± SD). Significance was tested by Student's t-test or anova, and P values < 0.05 were considered to be statistically significant.
Materials
Human recombinant IL-1β was from Cell Concept (Umkirch, Germany), and TNF-α was a gift from Knoll AG (Ludwigshafen, Germany). Media were from Invitrogen (Karlsruhe, Germany), fetal calf serum from Biochrom AG (Berlin, Germany) and tissue culture plastic was from Greiner BioOne (Frickenhausen, Germany). Radioactive materials were obtained from GE Healthcare (München, Germany). Immobilon-P (PVDF) membranes were from Millipore (Eschborn, Germany). Double-stranded consensus oligonucleotides for Nrf2 and NFκB and Ebselen, as well as secondary horseradish peroxidase conjugated antibodies for goat, mouse or rabbit IgG, were purchased from Santa Cruz Biotechnologies (Heidelberg, Germany). N-acetylcysteine, tert-butyl hydroquinone (tBHQ) and PDGF-BB were purchased from Sigma (Deisenhofen, Germany). (E)-2-cyano-3-(3,4-dihydrophenyl)-N-(phenylmethyl)-2-propenamide (tyrphostin, AG-490), 4-methoxyphenyl(morpholino)phosphinodithioate morpholinium (GYY4137) and haem oxygenase 1 (HO-1) antibodies were from Enzo Life Sciences (Lörrach, Germany). Diphenylene iodonium (DPI), as well as Zn-protoporphyrin IX (ZnPP), was obtained from Axxora (Lörrach, Germany). The JAK2 inhibitor II was purchased from Merck, Darmstadt, Germany. Unless indicated otherwise, all other chemicals were obtained from Sigma.
Results
CSE but not CBS mRNA is detectable in rat mesangial cells
To evaluate whether CBS or CSE or both are expressed in rat mesangial cells, a RT-PCR analysis was performed using gene-specific primers for rat CBS and CSE respectively. To confirm the efficiency of the primers for CBS, total RNA from rat brain tissue was included in these experiments. Electrophoretic separation of the RT-PCR products yielded a clear band of the expected size of 645 bp for CSE in mesangial cells and a band of 520 bp for CBS in rat brain (Figure 1). The cDNA fragments were cloned into a TOPA-TA vector and were confirmed to encode CSE and CBS, respectively, by sequencing. Interestingly, no PCR product for CBS was detectable in mesangial cells and no product for CSE was visible in tissue from rat brain. Therefore, we considered CSE as the relevant H2S-generating enzyme in rat mesangial cells.
Figure 1.
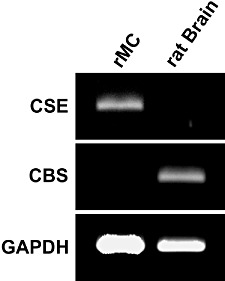
Analysis of CSE and CBS mRNA expression in rat mesangial cells and rat brain. Total RNA obtained from mesangial cells and rat brain tissue as indicated was reversed transcribed and subjected to semi-quantitative RT-PCR using gene-specific primers for CSE, CBS and GAPDH. Thereafter, the PCR products were separated by gel electrophoresis and visualized by staining with ethidium bromide.
PDGF-BB affects CSE mRNA levels in a time- and dose-dependent manner in mesangial cells
Quiescent mesangial cells were treated with PDGF-BB (25 ng·mL−1) for different time points, and CSE mRNA levels were analysed by qPCR. As depicted in Figure 2A, CSE mRNA levels were potently upregulated by PDGF-BB. CSE mRNA expression was about fourfold control levels at 8 h and declined nearly to basal levels after 24 h. As shown in Figure 2B, the upregulation of CSE mRNA by PDGF-BB after 8 h occurred in a dose-dependent manner. As a consequence of this result, a concentration of 25 ng·mL−1 PDGF-BB was chosen for further experiments.
Figure 2.
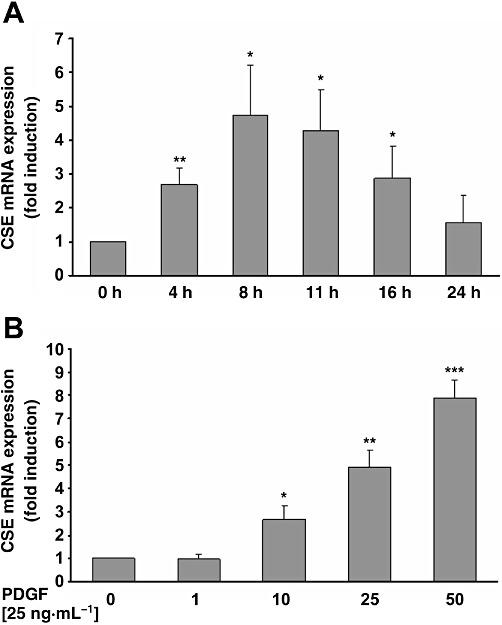
Time- and dose dependency of PDGF-induced CSE mRNA expression in rat mesangial cells. Quiescent mesangial cells were treated with or without PDGF-BB (25 ng·mL−1) for the indicated time-periods (A) or with the indicated concentrations of PDGF-BB (B) for 8 h. Thereafter, CSE mRNA levels were analysed by real-time RT-PCR. Data are means + SD, n= 3. ***P < 0.001, **P < 0.01, *P < 0.05 significantly different from vehicle-treated controls.
Effects of PDGF on CSE protein expression and activity
To test whether PDGF-BB-induced CSE mRNA expression is also transduced into protein levels, quiescent mesangial cells were treated for 4, 8, 10 and 16 h with PDGF-BB, and total protein was subjected to Western blotting. As illustrated in Figure 3A, PDGF-BB affected CSE protein levels in a manner comparable with CSE mRNA levels (Figure 2A), with a maximal effect after a 10 h incubation period. Furthermore, analysis of CSE activity after 10 h stimulation with PDGF-BB resulted in a nearly 2.5 fold increase of CSE enzymic activity (Figure 3B).
Figure 3.
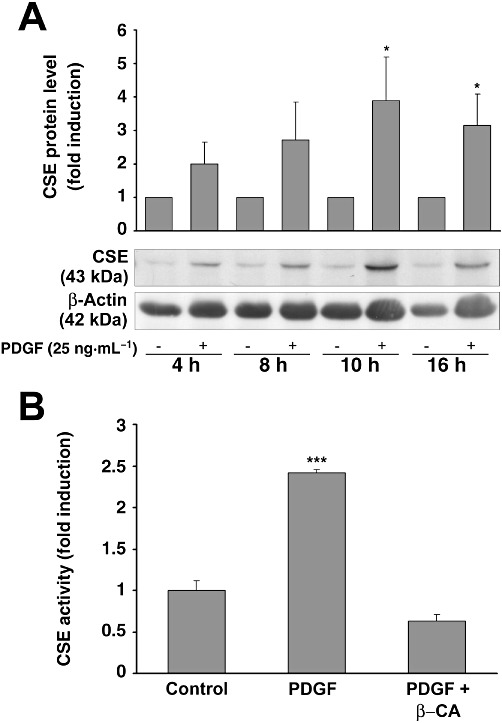
Analysis of PDGF-induced protein expression in mesangial cells. (A) Quiescent mesangial cells were treated with or without PDGF-BB (25 ng·mL−1) for the indicated time-periods. Thereafter, lysates were analysed by Western blotting. A representative Western blot experiment is shown in the lower panel. The bar graph above represents densitometric analysis of three independent experiments. Data are means + SD, *P < 0.05 versus vehicle-treated controls. (B) Quiescent mesangial cells were treated with or without PDGF-BB (50 ng·mL−1) for 10 h. Thereafter, lysates were analysed for CSE activity in the presence or absence of β-cyano-L-alanine, as described. Data are means + SD, ***P < 0.001 significantly different from vehicle-treated controls.
Involvement of the JAK/STAT pathway in PDGF-induced CSE expression
As PDGF-induced signalling may result in an activation of the JAK/STAT pathway (Vignais et al., 1996), we tested whether tyrphostin (AG 490), a potent JAK inhibitor, was able to alter PDGF-BB-induced CSE expression. To this end, mesangial cells were treated with PDGF-BB with or without different concentrations of tyrphostin AG 490 (20 and 30 µM) and CSE protein expression was analysed by Western blotting. Tyrphostin AG 490 treatment reduced CSE protein expression to basal levels when used in the higher concentration, indicating a role for the JAK/STAT signalling pathway in the increased CSE expression (Figure 4A). Similar results were obtained using another JAK inhibitor (JAK2 inhibitor II). As shown in Figure 4B, co-administration of JAK2 inhibitor II completely blocked the CSE mRNA levels induced by PDGF-BB.
Figure 4.
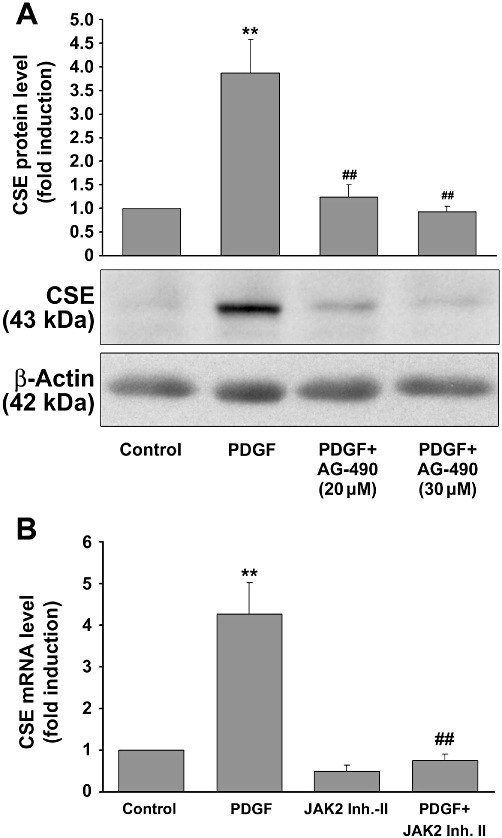
Effects of the STAT inhibitor tyrphostin AG-490 on PDGF-induced CSE expression. Quiescent mesangial cells were stimulated with or without PDGF-BB (25 ng·mL−1) and additionally treated with tyrphostin (AG-490) or the JAK2 inhibitor II, as indicated, for 8 h. CSE protein or mRNA expression were analysed by Western blotting or qPCR respectively. A representative Western blot experiment is shown in Figure 4A (lower panel). The bar graph above represents densitometric analysis of three independent experiments. Data are means + SD, **P < 0.01 significantly different from vehicle-treated controls, ##P < 0.01 significantly different from PDGF-BB (25 ng·mL−1) alone. The effects of JAK2 inhibitor-II (50 µM) on PDGF-BB-induced mRNA expression as determined by qPCR are summarised in Figure 4B. Data are means + SD, **P < 0.01 significantly different from vehicle-treated controls, ##P < 0.01 significantly different from PDGF-BB (25 ng·mL−1) alone.
Effects of redox modulators on PDGF-induced CSE expression, ROS formation and proliferation
We have shown recently that PDGF-BB induces ROS production in mesangial cells via the NADPH oxidase isoform Nox1 (Pleskováet al., 2006). Therefore, we tested whether PDGF-BB-induced CSE expression was dependent on ROS formation. To this end, mesangial cells were treated with PDGF-BB and additionally with the NADPH oxidase inhibitor, diphenylene iodonium (DPI), and the ROS scavenger N-acetylcysteine (NAC) for 16 h, and total RNA and protein levels were assessed by qPCR and Western blotting respectively. As shown in Figure 5A and B, respectively, this antioxidant treatment resulted in a significant decrease in CSE mRNA and protein expression, indicating that the ROS formation, induced by PDGF-BB (Figure 5C), subsequently triggered CSE expression. However, DPI reduced basal and PDGF-BB-induced CSE mRNA expression, as well as PDGF-BB-induced ROS formation to below control levels, implying that another constitutive source of ROS, such as Nox4, contributed to the triggering of basal CSE expression. To evaluate a role for ROS in mediating PDGF-BB-induced mesangial cell proliferation, we next analysed the effects of NAC and the slowly releasing H2S compound GYY4137 on PDGF-BB-induced proliferation. We observed that NAC, as well as GYY4137, markedly reduced PDGF-BB-induced mesangial cell proliferation almost to control levels, indicating an antioxidant action not only for NAC but also for the H2S released from GYY4137 (Figure 5D).
Figure 5.
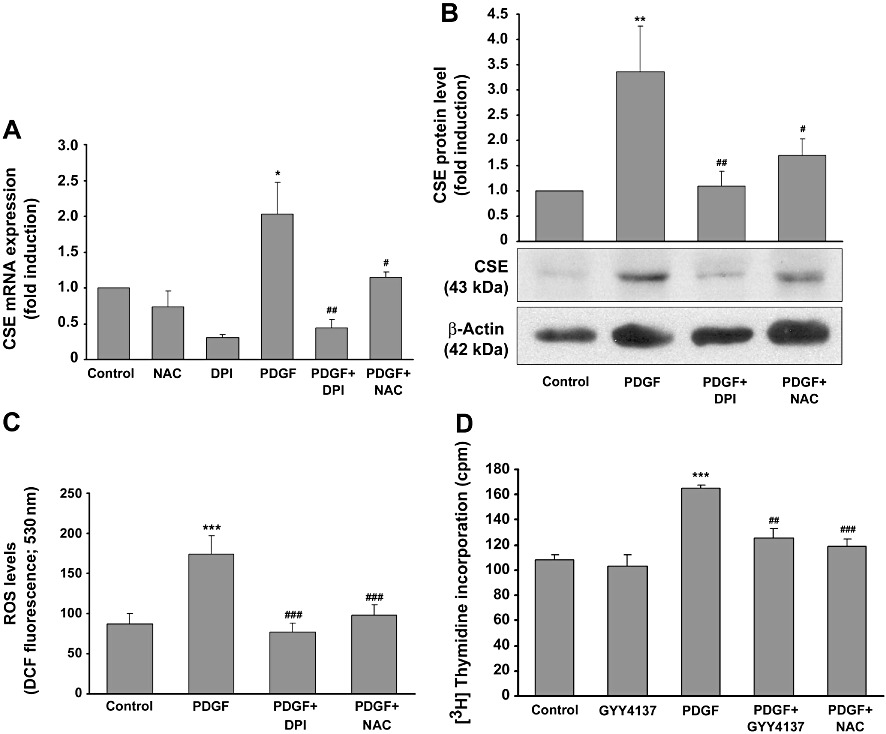
Effects of antioxidant treatment on PDGF-induced CSE expression, ROS production and proliferation. Quiescent mesangial cells were pre-treated with the NADPH oxidase inhibitor DPI (10 µM) or the ROS scavenger NAC (10 mM) or vehicle for 30 min. Thereafter, PDGF-BB (25 ng·mL−1) was added for a further 8 h, and CSE mRNA and protein levels, as well as ROS formation, were analysed by RT-PCR and Western blotting or DCF fluorescence respectively. CSE mRNA levels are shown in panel A, protein levels in panel B. Data are means + SD (n= 3 for mRNA and protein), *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from vehicle-treated controls; #P < 0.05, ##P < 0.01, ###P < 0.001 significantly different from PDGF-BB alone. The effects of PDGF-BB (8 h) as well as NAC and DPI on ROS formation measured as DCF fluorescence is shown in panel C, ***P < 0.001significantly different from vehicle treated controls; ###P < 0.001 significantly different from PDGF-BB. (D) Quiescent rat mesangial cells were treated with or without PDG-BB (25 ng·mL−1) in the presence or absence of GYY4137 (500 µM) and NAC (10 mM) for 14 h. Thereafter, 1 µCi·mL−1[3H]methyl-thymidine was added, and the cells were incubated for a further 24 h. Incorporation of [3H]methyl-thymidine was assessed as described. Data are means + SD (n= 3), ***P < 0.001 significantly different from vehicle treated controls; ##P < 0.01, ###P < 0.001 significantly different from PDGF-BB.
The transcription factor Nrf2 is involved in PDGF-induced CSE expression
Considering the redox-dependent regulation of CSE expression, it was tempting to speculate that PDGF-BB-induced CSE expression was triggered at the transcriptional level by a redox-sensitive transcription factor. An in silico analysis of the DNA sequences flanking the 5′ untranslated region of the rat CSE gene revealed a putative binding site for Nrf2 (GTGACTCAG), also referred to as ‘antioxidant responsive element’ (ARE), 355 bp upstream from the transcriptional start site derived from the published sequence of the murine CSE promoter (Ishii et al., 2004). This prompted us to analyse the effects of PDGF-BB on Nrf2 activity in mesangial cells. Indeed, treatment of mesangial cells with PDGF-BB (25 ng·mL−1) induced a marked binding to an ARE consensus element after 4 h, that was completely reversed when a 100-fold excess of a ‘cold’ double-stranded ARE consensus element was added to the reaction mixture. In contrast, adding a 100-fold excess of a consensus element for NFκB as a control did not affect PDGF-BB-induced Nrf2 binding (Figure 6A). As shown in Figure 6B, PDGF-induced binding of Nrf2 was abolished when mesangial cells were co-stimulated with the antioxidants DPI (10 µM), NAC (10 mM) or ebselen (40 µM), clearly indicating a role for ROS in activation of Nrf2.
Figure 6.
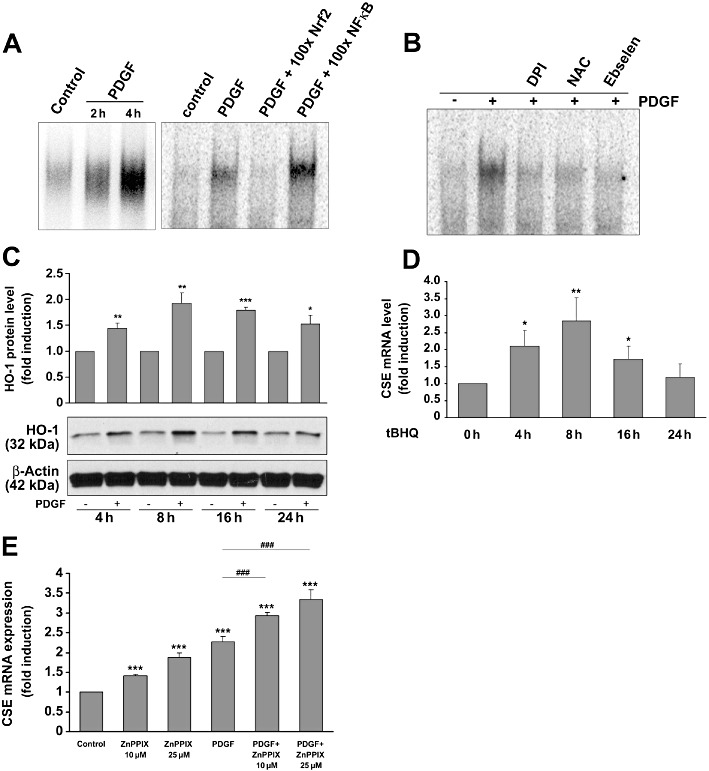
The role of the transcription factor Nrf2 in PDGF-induced CSE expression. Mesangial cells were stimulated without or with PDGF-BB (25 ng·mL−1) with or without pre-stimulation (30 min) with DPI, NAC or ebselen as indicated, and nuclear protein was analysed by EMSA using a radiolabeled ARE consensus motif. Representative results of at least three similar experiments are illustrated in panels A and B. The Nrf2-specific complex formation was diminished by a 100-fold excess of double-stranded ‘cold’ Nrf2-specific consensus oligonucleotides but not by NFκB-specific oligonucleotides (A) and by the antioxidants DPI (10 µM), NAC (10 mM) or ebselen (40 µM) (B). (C) Mesangial cells were stimulated with PDGF-BB (25 ng·mL−1) as indicated, and HO-1 protein expression was analysed by immunoblotting. Data analysis was performed after re-probing the blot with antibodies specific for β-actin. The bar graph above represents densitometric analysis of three independent experiments. Data are means + SD; *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from controls. (D) Mesangial cells were stimulated with tBHQ (10 µM) for the indicated time-periods. Total RNA was subjected to RT-PCR analysis for CSE mRNA determination. *P < 0.05, **P < 0.01 significantly different from controls (n= 3). (E) Mesangial cells were stimulated with or without PDGF-BB (25 ng·mL−1) in the presence or absence of ZnPP at the indicated concentrations for 8 h. Thereafter, total RNA was subjected to reverse transcription and qPCR. ***P < 0.001 significantly different from controls (n= 3), ###P < 0.001 significantly different from PDGF-BB (n= 3).
To test whether PDGF-BB triggers putative Nrf2-targeted gene expression, we analysed the effects of PDGF-BB on haem oxygenase 1 (HO-1), a prototypical Nrf2-regulated gene (Alam et al., 1999), and found that PDGF-BB triggered HO-1 protein expression in a time-dependent manner, comparable with the induction of CSE expression by PDGF-BB (Figures 3 and 6C). As shown in Figure 6D, tBHQ, a commonly accepted activator of Nrf2 and inducer of HO-1 expression (Li et al., 2000), also enhanced CSE mRNA expression in a time-dependent manner, which further supported a role of Nrf2 in CSE expression. It is worth noting that the CO formed by the activity of HO-1 inhibits CSE expression in aortic smooth muscle cells (Jin et al., 2006) and this would imply that activation of Nrf2 by PDGF-BB could limit CSE production. This hypothesis was corroborated by the fact that inhibition of PDGF-BB-induced HO-1 activity by Zn-protoporphyrin (ZnPP) resulted in a further increase in CSE mRNA expression in mesangial cells (Figure 6E).
To analyse the consequence of a genetic deletion of Nrf2, we studied PDGF-BB-induced expression of CSE in mesangial cells from Nrf2 knockout mice. To this end, cultures of mesangial cells from Nrf2 knockout and wild-type mice were treated with or without PDGF-BB (50 ng·mL−1) and CSE expression was analysed by immunoblotting. PDGF-BB induced CSE protein expression in cells from wild-type, but not in those from Nrf2 knockout mice (Figure 7A). Remarkably, basal levels of CSE were clearly reduced in Nrf2 knockout mice, suggesting that Nrf2 also played a role in constitutive CSE expression.
Figure 7.
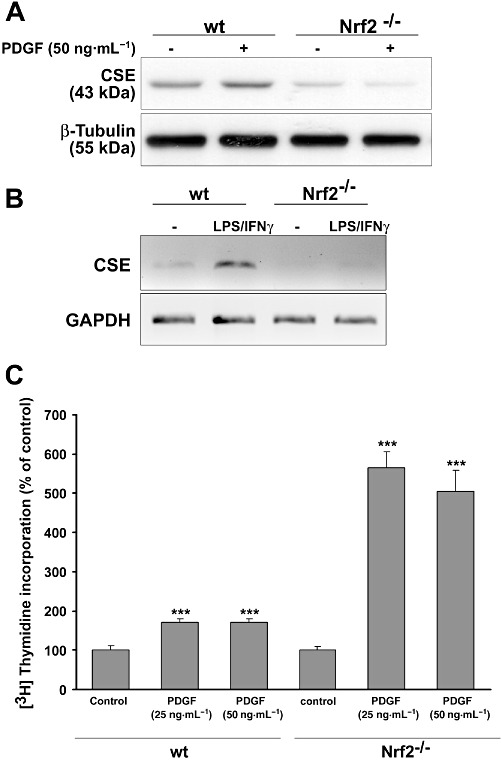
Analysis of CSE expression in Nrf2 knockout mice. Mouse mesangial cells and mouse spleen macrophages from three wild-type and three Nrf2 knockout mice were pooled and further processed for the evaluation of Nrf2-dependent CSE expression. Mesangial cells were treated with PDGF-BB (50 ng·mL−1) for 8 h and CSE protein expression was assessed by immunoblotting. A representative blot out of three similar experiments is depicted in (A). Mouse spleen macrophages were treated with or without LPS (100 U·mL−1) + IFN-γ (1 µg·mL−1) for 18 h. Total RNA was subjected to semi-quantitative RT-PCR. The PCR products were separated on a 1% agarose gel and CSE mRNA expression (B) was analysed. (C): Mesangial cells derived from Nrf2 knockout and wild-type mice were stimulated as indicated for 14 h. Thereafter, 1 µCi·mL−1[3H]methyl-thymidine was added, and the cells were incubated for a further 24 h. Incorporation of [3H]methyl-thymidine was assessed. Data are means + SD; *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from controls.
Recently, Zhu et al., 2010 reported that LPS induced CSE expression in murine peritoneal macrophages. To evaluate whether macrophages from Nrf2 knockout mice were still able to express CSE, we obtained spleen macrophages from wild-type and Nrf2 knockout mice and stimulated them with LPS for 18 h (Itoh et al., 1997). LPS strongly induced CSE expression in wild-type mice, whereas CSE expression was abolished in the Nrf2 knockout mice (Figure 7B), clearly demonstrating an essential role of Nrf2 in LPS-induced CSE expression. To evaluate whether knockout of Nrf2 affected PDGF-BB-induced cell proliferation, mesangial cells derived from Nrf2 knockout and wild-type mice were stimulated as indicated with PDGF-BB, and [3H]-thymidine incorporation was analysed. We found that PDGF-BB affected proliferation of mesangial cells lacking Nrf2 much more than that of wild-type cells (Figure 7C). Interestingly, this behaviour of mesangial cells depleted of Nrf2 is very similar to that of CSE-deficient smooth muscle cells (Yang et al., 2010). Therefore, it is tempting to speculate whether reduced CSE expression (Figure 7A) is responsible for enhanced proliferation in Nrf2 knockout cells. However, the reduced expression, in Nrf2 knockout mice, of other Nrf2-regulated genes that exert anti-proliferative properties, such as HO-1, may also contribute to our observations.
CSE expression is strongly upregulated in the course of anti-Thy-1 glomerulonephritis
The rat model of anti-Thy-1 glomerulonephritis serves as a classical model for an acute mesangioproliferative glomerulonephritis. The growth factor PDGF is regarded as the most potent mitogen that triggers mesangial cell proliferation (Iida et al., 1991). This prompted us to analyse whether the expression of CSE is affected in this model of glomerular disease.
The immunostaining pattern of CSE and Nrf2 was analysed in kidneys from healthy (control) rats and from animals with anti-Thy-1 glomerulonephritis at 2, 4 and 8 h, as well as at 1, 3, 7, 10, 15, 21 and 29 days following induction of disease. There was no immunostaining for CSE in healthy glomeruli (Figure 8, upper panel). In nephritic glomeruli CSE was not detectable until day 3 of disease. Three days after induction of anti-Thy-1 glomerulonephritis, CSE became detectable in the mesangium with increasing expression up to day 7 (Figure 8, upper panel). Nrf2 was present only in trace amounts in control glomeruli (Figure 8, lower panel). Induction of anti-Thy-1 glomerulonephritis resulted in a progressively increasing intensity of Nrf2 immunostaining, starting at day 1 and peaking at day 7 (Figure 8, lower panel). With the resolution of the glomerular disease, the mesangial expression of CSE and Nrf2 started to decline around day 10 and returned to baseline levels at day 29 (data not shown). The time course of Nrf2 and CSE expression nicely corresponded to the kinetics of PDGF-driven mesangial cell expansion in anti-Thy-1 glomerulonephritis (Iida et al., 1991; Johnson et al., 1992; 1993).
Figure 8.
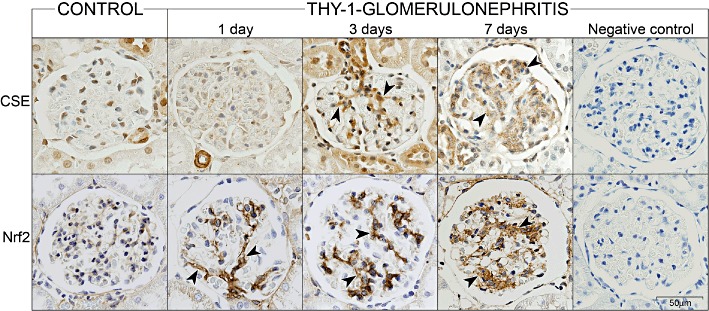
Glomerular immunostaining for CSE and Nrf2 in kidneys from control and anti-Thy-1 nephritic rats. Immunostaining (brown) of CSE (upper panel) and of Nrf2 (lower panel) in glomeruli from kidneys of healthy (control) rats and from animals with anti-Thy-1 glomerulonephritis at 1, 3 and 7 days following induction of disease. The mesangial immunostaining pattern is indicated by arrowheads. Negative controls (7 days of anti-Thy-1 glomerulonephritis shown here as an example) were performed by omitting the primary antibodies. The bar indicates the degree of magnification.
Discussion
PDGF is one of the main mediators in the progression of several inflammatory kidney diseases (Iida et al., 1991; Johnson et al., 1992; 1993). In particular, PDGF is an important player in a complex cross-talk with inflammatory autocoids, such as NO and ROS. In renal mesangial cells, PDGF-BB inhibits cytokine-induced iNOS expression and NO formation and induces ROS formation via the NADPH oxidase Nox1 (Kunz et al., 1997; Pleskováet al., 2006). Interestingly, ROS and NO enhance PDGF signalling by inhibition of PDGF receptor phosphotyrosine phosphatases (Callsen et al., 1999). Moreover, NO induces PDGF receptor α expression in rat renal mesangial cells and in a rat model of anti-Thy-1 glomerulonephritis (Beck et al., 2005). This results in a increased potential for PDGF to trigger PKB/Akt activation in mesangial cells. The property of PDGF to actively influence the redox state of a cell by affecting the NO/ROS equilibrium in an inflammatory setting and its sensitivity to the redox state prompted us to evaluate a role for PDGF on the synthesis of H2S, recognized recently as an important gaseous redox mediator.
We found that PDGF-BB induced mRNA and protein expression as well as enzymic activity of CSE in mesangial cells in a time- and dose-dependent manner (Figures 1–3), and this effect was markedly reduced by co-administration of antioxidants (Figure 5), suggesting a role for a redox-sensitive transcription factor, such as nuclear factor-κB (NFκB), activating protein-1 (AP-1) or Nrf2, in CSE transcription. Nrf2 is a redox-sensitive transcription factor that is activated by oxidative or electrophilic stress (Kaspar et al., 2009; Maher and Yamamoto, 2010). Under normal conditions, Nrf2 is bound to its cytosolic inhibitor INrf2, also referred to as Kelch-like ECH-associating protein 1 (Keap1) that directs Nrf2 to proteasomal degradation (Itoh et al., 1999). Oxidation of INrf2 allows Nrf2 to enter the nucleus and to bind to an antioxidant element (ARE) to mediate gene transcription of protective genes, such as glutamate cysteine ligase, glutathionine S-transferase or HO-1 (Kaspar et al., 2009). Moderate synthesis of H2S is commonly regarded as cytoprotective and, therefore, it was possible that PDGF-BB triggered protective CSE expression by a redox-dependent activation of Nrf2, possibly by an endogenous generation of ROS via the NADPH-oxidase Nox1 in mesangial cells (Pleskováet al., 2006).
To this end, we performed several experiments, first to analyse a role for PDGF-BB in inducing Nrf2 activity, and second to evaluate whether CSE expression is triggered at least in part by Nrf2. We found that PDGF-BB induced Nrf2 activity in a redox-dependent manner, shown as binding to a synthetic Nrf2 response element in Figure 6A and B. Furthermore, we demonstrated that HO-1, a prototypical Nrf2-regulated gene (Alam et al., 1999; Paine et al., 2010), was induced by PDGF-BB by a manner strikingly similar to the induction of CSE (Figure 6C) and that tBHQ, a commonly used synthetic activator of the Nrf2 machinery, also triggered CSE expression in rat mesangial cells (Figure 6D). In addition, the PDGF-BB-induced, as well as basal, CSE expression, was nearly abolished in mesangial cells derived from Nrf2 knockout, compared with wild-type mice (Figure 7A). Remarkably similar results were obtained from the analysis of LPS-induced CSE expression in spleen macrophages (Figure 7B). From these data, we have concluded that Nrf2 is involved in mediating CSE expression.
The rat model of anti-Thy-1 nephritis is a classical model for a mesangio-proliferative kidney disease. After the onset of the disease by injection of an antibody directed to the Thy-1 antigen on mesangial cells, a rapid and complement-dependent loss of glomerular mesangial cells (mesangiolysis) can be observed that is followed by a proliferative phase that is mediated mainly by the PDGF/PDGFR system (Iida et al., 1991; Johnson et al., 1992; 1993). The proliferation of mesangial cells is most prominent around day 3. We found that protein expression of CSE was maximal on day 3 in the course of anti-Thy-1 glomerulonephritis (Figure 8, upper panel), supporting a role of PDGF-BB on CSE expression also in vivo. In the same animal model, the antioxidant lipoic acid reduced ERK-dependent proliferation of mesangial cells in vivo and in vitro, resulting in a marked decrease of the severity of the symptoms of anti-Thy-1 glomerulonephritis (Budisavljevic et al., 2003). In addition, ROS formed in the course of anti-Thy-1 glomerulonephritis supported production of NO, which in turn aggravated acute glomerulonephritis (Narita et al., 1995; Mosley et al., 1999; Walpen et al., 2001). In this setting, endogenous production of H2S by the induction of CSE could limit PDGF-BB-induced mesangial cell growth and could subsequently inhibit the matrix expansion and profibrotic effects that accompany proliferation of mesangial cells (Li et al., 2011). This hypothesis is supported by a very recent report that described anti-proliferative and possibly anti-fibrotic effects of exogenously administered H2S in rat mesangial cells kept under high glucose conditions (Yuan et al., 2011). Interestingly, enhanced CSE expression in the course of glomerulonephritis was accompanied by a marked increase in the expression of Nrf2 protein. However, a clear increase of Nrf2 can be observed already at day 1 (Figure 8, lower panel), probably due to an enhanced ROS formation that may stabilize Nrf2 via oxidation of its inhibitor INrf2 on a post-translational level. We hypothesize that formation of active Nrf2 in the mesangiolytic phase of nephritis is a prerequisite to facilitate CSE expression in the later stages of the disease. Taking into account that PDGF-BB-induced activation of Nrf2 occurred earlier than that of CSE expression in cultured mesangial cells, we propose that the presence of active Nrf2 may be essential for CSE expression, but probably other transcription factors are needed to regulate CSE expression in a coordinated manner. Notably, the DNA recognition sites for Nrf2 and AP-1 are very similar and a direct interaction of c-Jun and Nrf2 to mediate gene expression has been already demonstrated (Levy et al., 2009; Paine et al., 2010). Therefore, an interaction of redox-sensitive signalling pathways that results in the activation of Nrf2 and/or AP-1 to trigger CSE expression should be considered in future experiments.
So far, there is only limited information about a role of CSE and H2S in mesangial cells. Besides the report demonstrating anti-proliferative effects under high glucose conditions (Yuan et al., 2011), there is a further study describing an inflammatory action of H2S in mouse mesangial cells Sen et al. (2011), in which overexpression of CSE and CBS antagonised homocysteine-mediated mesangial inflammation processes. These authors clearly demonstrated that exogenously administered NaHS or endogenously produced H2S by CBS and/or CSE gene transfer, reversed the effects of homocysteine on expression of the chemokines CXCL2 (MIP-2) and CCL2 (MCP-1), as well as the NADPH oxidase subunit p47phox and that these effects were accompanied by a H2S-mediated reduction of ERK1/2 and JNK1/2 pathways. Both studies clearly demonstrated an anti-proliferative and anti-inflammatory role of H2S in mesangial cells. We show here that PDGF-BB induced CSE expression in a redox-dependent manner, most probably via an activation of the transcription factor Nrf2 in vitro and in vivo. We suggest that limited gene expression of CSE, HO-1 and many other protective Nrf2-regulated genes should be considered in an endogenous antioxidant strategy that might also be targeted pharmacologically for the treatment of inflammatory glomerular diseases.
Acknowledgments
The authors thank Ute Schmidt and Riad Haceni for the valuable technical support. The authors thank Prof. Masayuki Yamamoto (Department of Medical Biochemistry, Tohoku University, Sendai, Japan) for providing us with Nrf2 knockout mice. Total RNA from rat brain was kindly provided by Dr. Ellen Niederberger (Institute of Clinical Pharmacology, Frankfurt am Main, Germany). The work was supported by the German Research Foundation (SFB 815, FOG 784, GRK 757, GRK 880/1, GRK 1172, EXC 147, PF361/7-1) and by the European Union (Grant No. LSHM-CT-2004–005033, EICOSANOX). MH was supported by a grant of the Ministry of Education of the Arab Republic of Egypt.
Glossary
- AP-1
activating protein 1
- ARE
antioxidant responsive element
- CBS
cystathionine β-synthase
- CSE
cystathionine-γ-lyase
- DPI
diphenylene iodonium
- FCS
fetal calf serum
- HO-1
haem oxygenase 1
- NAC
N-acetylcysteine
- Nox1
NADPH oxidase 1
- Nox4
NADPH oxidase 4
- Nrf2
nuclear factor erythroid-2-related factor-2
- PDGF
platelet-derived growth factor
- PLP
pyridoxal 5′-phosphate
- ROS
reactive oxygen species
- tBHQ
tert-butylhydroquinone
- ZnPP
Zn-protoporphyrin
Conflicts of interest
None.
References
- Abboud HE. Role of platelet-derived growth factor in renal injury. Annu Rev Physiol. 1995;57:297–309. doi: 10.1146/annurev.ph.57.030195.001501. [DOI] [PubMed] [Google Scholar]
- Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud L, Ardaillou R. Involvement of reactive oxygen species in kidney damage. Br Med Bull. 1993;49:621–629. doi: 10.1093/oxfordjournals.bmb.a072635. [DOI] [PubMed] [Google Scholar]
- Beck KF, Eberhardt W, Walpen S, Apel M, Pfeilschifter J. Potentiation of nitric oxide synthase expression by superoxide in interleukin 1 beta-stimulated rat mesangial cells. FEBS Lett. 1998;435:35–38. doi: 10.1016/s0014-5793(98)01035-7. [DOI] [PubMed] [Google Scholar]
- Beck KF, Güder G, Schaefer L, Pleskova M, Babelova A, Behrens MH, et al. Nitric oxide upregulates induction of PDGF receptor-alpha expression in rat renal mesangial cells and in anti-Thy-1 glomerulonephritis. J Am Soc Nephrol. 2005;16:1948–1957. doi: 10.1681/ASN.2004080638. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, et al. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- Budisavljevic MN, Hodge L, Barber K, Fulmer JR, Durazo-Arvizu RA, Self SE, et al. Oxidative stress in the pathogenesis of experimental mesangial proliferative glomerulonephritis. Am J Physiol Renal Physiol. 2003;285:F1138–F1148. doi: 10.1152/ajprenal.00397.2002. [DOI] [PubMed] [Google Scholar]
- Callsen D, Sandau KB, Brüne B. Nitric oxide and superoxide inhibit platelet-derived growth factor receptor. Free Radic Biol Med. 1999;26:1544–1553. doi: 10.1016/s0891-5849(99)00015-5. [DOI] [PubMed] [Google Scholar]
- Cattell V. Nitric oxide and glomerulonephritis. Kidney Int. 2002;61:816–821. doi: 10.1046/j.1523-1755.2002.00226.x. [DOI] [PubMed] [Google Scholar]
- Döll F, Pfeilschifter J, Huwiler A. The epidermal growth factor stimulates sphingosine kinase-1 expression and activity in the human mammary carcinoma cell line MCF7. Biochim Biophys Acta. 2005;1738:72–81. doi: 10.1016/j.bbalip.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Fox B, Schantz JT, Haigh R, Wood ME, Moore PK, Viner N, et al. Inducible hydrogen sulfide synthesis in chondrocytes and mesenchymal progenitor cells: is H2S a novel cytoprotective mediator in the inflamed joint? J Cell Mol Med. 2012;16:896–910. doi: 10.1111/j.1582-4934.2011.01357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla MM, Snyder SH. Hydrogen sulfide as a gasotransmitter. J Neurochem. 2010;113:14–26. doi: 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann L, Ren S, Schwalm S, Pfeilschifter J, Huwiler A. Sphingosine kinase 1 and 2 regulate the capacity of mesangial cells to resist apoptotic stimuli in an opposing manner. Biol Chem. 2008;389:1399–1407. doi: 10.1515/BC.2008.160. [DOI] [PubMed] [Google Scholar]
- Iida H, Seifert R, Alpers CE, Gronwald RG, Phillips PE, Pritzl P, et al. Platelet-derived growth factor (PDGF) and PDGF receptor are induced in mesangial proliferative nephritis in the rat. Proc Natl Acad Sci U S A. 1991;88:6560–6564. doi: 10.1073/pnas.88.15.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, et al. Murine cystathionine gamma-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J. 2004;381:113–123. doi: 10.1042/BJ20040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennewein C, Kuhn AM, Schmidt MV, Meilladec-Jullig V, von Knethen A, Gonzalez FJ, et al. Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J Immunol. 2008;181:5646–5652. doi: 10.4049/jimmunol.181.8.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin HF, Du JB, Li XH, Wang YF, Liang YF, Tang CS. Interaction between hydrogen sulfide/cystathionine gamma-lyase and carbon monoxide/heme oxygenase pathways in aortic smooth muscle cells. Acta Pharmacol Sin. 2006;27:1561–1566. doi: 10.1111/j.1745-7254.2006.00425.x. [DOI] [PubMed] [Google Scholar]
- Johnson RJ, Raines EW, Floege J, Yoshimura A, Pritzl P, Alpers C, et al. Inhibition of mesangial cell proliferation and matrix expansion in glomerulonephritis in the rat by antibody to platelet-derived growth factor. J Exp Med. 1992;175:1413–1416. doi: 10.1084/jem.175.5.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RJ, Floege J, Couser WG, Alpers CE. Role of platelet-derived growth factor in glomerular disease. J Am Soc Nephrol. 1993;4:119–128. doi: 10.1681/ASN.V42119. [DOI] [PubMed] [Google Scholar]
- Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz D, Walker G, Eberhardt W, Messmer UK, Huwiler A, Pfeilschifter J. Platelet-derived growth factor and fibroblast growth factor differentially regulate interleukin 1beta- and cAMP-induced nitric oxide synthase expression in rat renal mesangial cells. J Clin Invest. 1997;100:2800–2809. doi: 10.1172/JCI119827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz A, Jelkmann W, Bauer C. Mesangial cells derived from rat glomeruli produce an erythropoiesis stimulating factor in cell culture. FEBS Lett. 1982;137:129–132. doi: 10.1016/0014-5793(82)80330-x. [DOI] [PubMed] [Google Scholar]
- Levy S, Jaiswal AK, Forman HJ. The role of c-Jun phosphorylation in EpRE activation of phase II genes. Free Radic Biol Med. 2009;47:1172–1179. doi: 10.1016/j.freeradbiomed.2009.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Rose P, Moore PK. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- Li N, Venkatesan MI, Miguel A, Kaplan R, Gujuluva C, Alam J, et al. Induction of heme oxygenase-1 expression in macrophages by diesel exhaust particle chemicals and quinones via the antioxidant-responsive element. J Immunol. 2000;165:3393–3401. doi: 10.4049/jimmunol.165.6.3393. [DOI] [PubMed] [Google Scholar]
- Łowicka E, Bełtowski J. Hydrogen sulfide (H2S) – the third gas of interest for pharmacologists. Pharmacol Rep. 2007;59:4–24. [PubMed] [Google Scholar]
- Maher J, Yamamoto M. The rise of antioxidant signaling – the evolution and hormetic actions of Nrf2. Toxicol Appl Pharmacol. 2010;244:14–15. doi: 10.1016/j.taap.2010.01.011. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley K, Waddington SN, Ebrahim H, Cook T, Cattell V. Inducible nitric oxide synthase induction in Thy 1 glomerulonephritis is complement and reactive oxygen species dependent. Exp Nephrol. 1999;7:26–34. doi: 10.1159/000020581. [DOI] [PubMed] [Google Scholar]
- Mühl H, Pfeilschifter J. Amplification of nitric oxide synthase expression by nitric oxide in interleukin 1 beta-stimulated rat mesangial cells. J Clin Invest. 1995;95:1941–1946. doi: 10.1172/JCI117876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita I, Border WA, Ketteler M, Noble NA. Nitric oxide mediates immunologic injury to kidney mesangium in experimental glomerulonephritis. Lab Invest. 1995;72:17–24. [PubMed] [Google Scholar]
- Ogasawara Y, Ishii K, Tanabe S. Enzymatic assay of γ-cystathionase activity using pyruvate oxidase-peroxidase sequential reaction. J Biochem Methods. 2002;51:139–150. doi: 10.1016/s0165-022x(02)00010-6. [DOI] [PubMed] [Google Scholar]
- Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol. 2010;80:1895–1903. doi: 10.1016/j.bcp.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J, Schwarzenbach H. Interleukin 1 and tumor necrosis factor stimulate cGMP formation in rat renal mesangial cells. FEBS Lett. 1990;273:185–187. doi: 10.1016/0014-5793(90)81080-8. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J, Vosbeck K. Transforming growth factor β2 inhibits interleukin 1β- and tumour necrosis factor α-induction of nitric oxide synthase in rat renal mesangial cells. Biochem Biophys Res Commun. 1991;175:372–379. doi: 10.1016/0006-291x(91)91574-v. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J, Beck KF, Eberhardt W, Huwiler A. Changing gears in the course of glomerulonephritis by shifting superoxide to nitric oxide-dominated chemistry. Kidney Int. 2002;61:809–815. doi: 10.1046/j.1523-1755.2002.00225.x. [DOI] [PubMed] [Google Scholar]
- Plesková M, Beck KF, Behrens MH, Huwiler A, Fichtlscherer B, Wingerter O, et al. Nitric oxide down-regulates the expression of the catalytic NADPH oxidase subunit Nox1 in rat renal mesangial cells. FASEB J. 2006;20:139–141. doi: 10.1096/fj.05-3791fje. [DOI] [PubMed] [Google Scholar]
- Schaefer L, Macakova K, Raslik I, Micegova M, Gröne HJ, Schönherr E, et al. Absence of decorin adversely influences tubulointerstitial fibrosis of the obstructed kidney by enhanced apoptosis and increased inflammatory reaction. Am J Pathol. 2002;160:1181–1191. doi: 10.1016/S0002-9440(10)64937-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlöndorff D, Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol. 2009;20:1179–1187. doi: 10.1681/ASN.2008050549. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Müller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen U, Givvimani S, Abe OA, Lederer ED, Tyagi SC. Cystathionine β-synthase and cystathionine γ-lyase double gene transfer ameliorate homocysteine-mediated mesangial inflammation through hydrogen sulfide generation. Am J Physiol Cell Physiol. 2011;300:C155–C163. doi: 10.1152/ajpcell.00143.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009a;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulphide. J Biochem. 2009b;146:623–626. doi: 10.1093/jb/mvp111. [DOI] [PubMed] [Google Scholar]
- Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignais ML, Sadowski B, Watling D, Rogers NL, Gilman M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol Cell Biol. 1996;16:1759–1769. doi: 10.1128/mcb.16.4.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walpen S, Beck KF, Schaefer L, Raslik I, Eberhardt W, Schaefer RM, et al. Nitric oxide induces MIP-2 transcription in rat renal mesangial cells and in a rat model of glomerulonephritis. FASEB J. 2001;15:571–573. doi: 10.1096/fj.00-0518fje. [DOI] [PubMed] [Google Scholar]
- Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Wu L, Bryan S, Khaper N, Mani S, Wang R. Cystathionine gamma-lyase deficiency and overproliferation of smooth muscle cells. Cardiovasc Res. 2010;86:487–495. doi: 10.1093/cvr/cvp420. [DOI] [PubMed] [Google Scholar]
- Yuan P, Xue H, Zhou L, Qu L, Li C, Wang Z, et al. Rescue of mesangial cells from high glucose-induced over-proliferation and extracellular matrix secretion by hydrogen sulfide. Nephrol Dial Transplant. 2011;26:2119–2126. doi: 10.1093/ndt/gfq749. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XY, Liu SJ, Liu YJ, Wang S, Ni X. Glucocorticoids suppress cystathionine gamma-lyase expression and H2S production in lipopolysaccharide-treated macrophages. Cell Mol Life Sci. 2010;67:1119–1132. doi: 10.1007/s00018-009-0250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]