

Abstract
BACKGROUND AND PURPOSE
Mucociliary malfunction occurs in chronic obstructive pulmonary disease (COPD) and compromised functions of ciliated bronchial epithelial cells may contribute to this. Cigarette smoke, a major risk factor for COPD, impairs ciliary beat frequency (CBF). cAMP augments CBF. This in vitro study addressed, in differentiated, primary human bronchial epithelial cells, whether roflumilast N-oxide, a PDE4 inhibitor, (i) augments CBF; (ii) prevents the reduction in CBF induced by cigarette smoke extract (CSE); and (iii) protects against the loss of the ciliated phenotype following long-term CSE exposure.
EXPERIMENTAL APPROACH
Air-liquid interface cultured human bronchial epithelial cells were incubated with roflumilast N-oxide and exposed to CSE. CBF was assessed by digital high speed video microscopy (DHSV). Ciliated cells were characterized by β-tubulin IV staining and analyses of Foxj1 and Dnai2 mRNA and protein (real-time quantitative PCR, Western blotting).
KEY RESULTS
Roflumilast N-oxide concentration-dependently triggered a rapid and persistent increase in CBF and reversed the decrease in CBF following CSE. Long-term incubation of bronchial epithelial cells with CSE resulted in a loss in ciliated cells associated with reduced expression of the ciliated cell markers Foxj1 and Dnai2. The PDE4 inhibitor prevented this loss in the ciliated cell phenotype and the compromised Foxj1 and Dnai2 expression. The enhanced release of IL-13 following CSE, a cytokine that diminishes the proportion of ciliated cells and in parallel, reduces Foxj1 and Dnai2, was reversed by roflumilast N-oxide.
CONCLUSION AND IMPLICATIONS
Roflumilast N-oxide protected differentiated human bronchial epithelial cells from reduced CBF and loss of ciliated cells following CSE.
Keywords: bronchial epithelial cells, ciliary motility, phosphodiesterase-4, roflumilast
Introduction
Mucociliary clearance is a major host defence mechanism of the lungs and is governed by the bronchial epithelium. The coordinated secretion of airway mucus from goblet cells along with synchronized ciliary beating and maintenance of the periciliary lining fluid at an optimal height by ciliated epithelium provides an efficient defence from inhaled noxious particles and pathogens (Wanner et al., 1996).
The frequency of ciliary beating is controlled by numerous factors such as temperature, pH, noxious particles and pathogens, a number of endogenous mediators present in the periciliary lining fluid (e.g. ATP, adenosine, nitric oxide), stress and exercise or by therapeutic interventions (Olseni and Wollmer, 1990; Salathe, 2007). Most of these factors modify cellular levels of Ca2+, cAMP or cGMP. Indeed, activation of cAMP-dependent PKA enhances ciliary beat frequency (CBF) secondary to a specific phosphorylation of dynein light chains (Salathe, 2007).
Tobacco smoking is acknowledged as the major risk factor of chronic obstructive pulmonary disease (COPD). By anatomy, bronchial epithelial cells are the primary target of tobacco smoke. As a corollary, cigarette smoke (CS) was shown to impair CBF in vitro, in vivo and in patients (Ballenger, 1960; Elliott et al., 2007; Simet et al., 2010), and a role of an activated PKCε (Salathe et al., 1993; Elliott et al., 2007; Salathe, 2007; Simet et al., 2010) was discussed in this context. Such a compromised CBF adds to mucus hypersecretion, impaired functioning of Cl--secreting channels, loss of ciliated epithelial cells secondary to tobacco smoke altogether resulting in malfunction of the mucociliary apparatus that is considered to be a major disease mechanism in COPD (Sisson et al., 1994; Gensch et al., 2004; Tamashiro et al., 2009).
CBF is augmented by cAMP and one option to increase cellular cAMP is to prevent its degradation through inhibition of cyclic nucleotide hydrolysing PDEs. The PDE superfamily comprises 11 families (PDE1-11). Among these, the cAMP selectively hydrolysing PDE4 has attracted much interest as a therapeutic target in respiratory ailments such as COPD. The PDE4 family is composed of four subtypes (PDEA-D) encoded by different genes that by alternative splicing are expressed as multiple variants with sequence diversity in their N-terminal domains (Conti et al., 2003; Houslay et al., 2005). PDE4 was found and shown to be of functional relevance in virtually all cells related to COPD. Among them are airway epithelial cells where selective PDE4 inhibitors (i) suppress EGF-induced production of the MUC5AC mucus protein (Mata et al., 2005); (ii) enhance activity of the cystic fibrosis transmembrane conductance regulator Cl--secreting channel (Barnes et al., 2005); and (iii) increase CBF (Cervin and Lindgren, 1998; Wohlsen et al., 2010) that altogether may integrate into an improved mucociliary clearance. Specifically, the PDE4 inhibitor rolipram was reported to augment CBF of epithelial cells from rabbit maxillary sinus and trachea (Cervin and Lindgren, 1998) and in rat precision cut lung slices (Wohlsen et al., 2010). In the latter protocol, the PDE4 inhibitor roflumilast was more potent than rolipram at increasing CBF.
The first PDE4 inhibitor roflumilast is now available for the treatment of severe COPD. Indeed, in large clinical trials (over up to 1 year), roflumilast improved lung function and reduced the rate of acute exacerbations in severe COPD (Calverley et al., 2009; Fabbri et al., 2009). The clinical benefit of roflumilast is considered to emanate primarily from its anti-inflammatory effects, although results from cellular and animal studies indicate that the PDE4 inhibitor also has the potential to curb lung architectural remodelling, mucociliary malfunction and the burden of oxidative stress that are often associated with COPD (Hatzelmann et al., 2010).
The current study was mainly dedicated to explore the effects of the PDE4 inhibitor roflumilast N-oxide on ciliated human bronchial epithelial cells (in an air-liquid interface culture) compromised by exposure to cigarette smoke extracts (CSEs) in vitro. Roflumilast N-oxide is the active metabolite of roflumilast that largely accounts for its clinical efficacy (Hatzelmann and Schudt, 2001; Bethke et al., 2007; Hatzelmann et al., 2010). The culture of bronchial epithelial cells on porous membranes in an air-liquid interface protocol allows formation of a pseudostratified epithelium comprising ciliated cells, mucus producing cells and basal cells that is widely accepted as an appropriate model to reflect differentiated bronchial epithelium in situ.
Methods
Isolation of bronchial epithelial cells and air-liquid interface culture
Primary human bronchial epithelial cells were isolated and cultured from human lung tissue of patients undergoing surgery for lung cancer (approval of the local ethics committee and informed consent was obtained). Small pieces of bronchi were excised from microscopically normal lung areas, carefully dissected free from lung parenchyma and plated on collagen-coated culture dishes [10 µg·cm−2 rat type I collagen (Sigma) in bronchial epithelial growth medium (BEGM, comprising bronchial epithelial basal medium supplemented with bovine pituitary extract 52 µg·mL−1, hydrocortisone 0.5 µg·mL−1, human recombinant EGF 25 ng·mL−1, adrenaline 0.5 µg·mL−1, transferrin 10 µg·mL−1, insulin 5 µg·mL−1, retinoic acid 50 nM, triiodo-L-thyronine 6.5 ng·mL−1, gentamycin 40 µg·mL−1, amphotericin B 50 ng·mL−1, bovine serum albumin 1.5 µg·mL−1)]. Small bronchi were orientated with the epithelial layer to be in contact with the culture plate. After a period of approximately 2 weeks, bronchial epithelial cells were observed around bronchi. The identity of the monolayer as bronchial epithelial cells was affirmed by morphological criteria and immunofluorescence for cytokeratin 5. At about 75% confluency, bronchi were removed and cells were trypsinized and subpassaged on polyester Transwell inserts (Millipore, Billerica, MA, USA) at 82 500 cells per insert. Cells were left for 7 days submerged in BEGM/Dulbecco's modified Eagle's minimal essential medium 1:1. From day 7, the air-liquid interface culture was initiated by removing the medium from the upper well leaving the apical surface of the cells exposed to air and changing the final EGF concentration to 0.5 ng·mL−1 (differentiation medium). In general, air-liquid interface cultures were pursued until about 80–90% of cells were ciliated by microscopic inspection (about 3–4 weeks after initiation) before experiments were commenced. Cells were maintained at 37°C with 5% CO2 and medium changed every other day. At this stage, a pseudostratified bronchial epithelium comprising basal cells, ciliated cells and goblet cells was obtained and considered as ‘differentiated’.
Preparation of CSE and incubations
CSE solutions were prepared as previously described (Ortiz et al., 2010). Briefly, the smoke of a research cigarette (2R4F, from Tobacco Health Research, University of Kentucky, Orlando, KY, USA) was bubbled into a flask containing 25 mL of pre-warmed (37°C) differentiation medium using a respiratory pump model (Harvard Apparatus Rodent Respirator 680, Harvard Apparatus, Holliston, MA, USA) that operates through a puffing mechanism corresponding to the human smoking pattern (three puffs min−1; 35 mL per each puff of 2 s duration with a volume of 0.5 cm above the filter). The CS solution was then drawn into a syringe through a 0.22 µm pore size filter to remove particles and the tar phase and to obtain a sterile solution (Corning, NY, USA). The resulting solution was defined as CSE at 100% and used in the different experiments within 30 min of preparation following appropriate dilution as indicated. CSE at 10% reportedly corresponds to an exposure associated with smoking of approximately two packs per day (Su et al., 1998). Cytotoxicity possibly emanating from CSE was analysed by exposing differentiated bronchial epithelial cells to 10% CSE for up to 7 days (CSE and culture medium were replaced every 24 h) or to 20% CSE for up to 24 h based on the release of LDH in culture supernatants. No significant differences in LDH activities were observed in the culture supernatants between the CSE and control group (LDH cytotoxicity assay; Cayman, Spain, data not shown).
‘Acute’ effects of CSE to compromise CBF of differentiated human bronchial epithelial cells were explored in ‘short-term incubations’ (up to 24 h), while ‘chronic’ effects of CSE entailing a loss in cilia were investigated in long-term incubations (up to 7 days).
Roflumilast N-oxide was added at 0, 2 or 1000 nM at 30 min before CSE if not indicated otherwise; 0 nM corresponds to vehicle (0.1% DMSO), 2 nM corresponds to the free plasma concentrations (unbound to plasma protein) after repeated, oral, once-daily dosing of roflumilast at the clinical dose of 500 µg·day−1 (Bethke et al., 2007; Hatzelmann et al., 2010), and at 1000 nM roflumilast N-oxide completely and selectively inhibits PDE4 (Hatzelmann and Schudt, 2001). The half-maximum potency of roflumilast N-oxide to inhibit PDE4 amounts to 2 nM (Hatzelmann and Schudt, 2001).
In experiments exceeding a 24 h incubation period, both roflumilast N-oxide or vehicle and CSE were replaced every 24 h.
In some experiments, theophylline was added at 10 µM, forskolin at 10 µM, dbcAMP at 1 mM, apocynin at 100 µM or N-acetylcysteine at 1 mM.
Both test compounds and CSE were added to the basolateral media and at the apical surface. Because manipulations at the apical surface may affect cilia physiology, all incubations with vehicle controls were run under identical conditions as those of the CSE and test compounds (e.g. identical volumes of medium comprising test compounds/CSE of 25 µL were added to the apical surface throughout all conditions, for the basolateral compartment the volume was kept at 500 µL for all conditions). In control experiments where vehicles/medium (instead of test compounds/CSE) were added to the apical surface of differentiated human bronchial epithelial cells over a maximum of 7 days (including daily replacement procedures, as indicated), the number of ciliated cells and expression of cilia markers were found to be not different from cultures of differentiated human bronchial epithelial cells in the absence of the manipulations at the apical surface.
CBF and functional ciliated cell analysis
Measurement of CBF and functional ciliated cell analysis followed previously described protocols (Armengot et al., 2010). Briefly, cells were imaged with a digital high speed video (DHSV) imaging technique using a Nikon Eclipse TS100 microscope equipped with a 40× Nikon phase-contrast objective, providing a final optical gain of ×400. A CCD camera (Digital Quad High Speed Progressive Scan Camera, JAI CV-A33 CL) at 128 frames s−1 was used. Video signals were digitized and processed using appropriate software. Contrast-enhanced video images were selected and the light intensity of the selected pixels was recorded on a frame-by-frame basis. In addition, the magnitude spectrum from a fast Fourier transformation of the variation of pixel intensity signal was analysed in an average of six different regions of (3 × 3 pixels) the cilia in individual cells in a minimum of 10 cells per experiment. CBF experiments were performed with cells submerged in HBSS (Lonza, Cambridge, UK) at an apical volume of 100 µL at room temperature (26°C).
The average number of ciliated cells with cilia motility in relation to total cells was determined using the same technique as described above for CBF (Armengot et al., 2010). For each experimental condition, a minimum of 10 separate fields was captured (approximately 500 cells per field in ×400 augmentations). Cells with cilia motility were identified by the movement of cilia detected by DHSV. Data were calculated as the average number of cells with cilia motility per field and expressed as % of control.
Real time RT-PCR
Total RNA was isolated from differentiated bronchial epithelial cells using TriPure® Isolation Reagent (Roche, Indianapolis, IN, USA). Reverse transcription was performed in 300 ng of total RNA with TaqMan reverse transcription reagents kit (Applied Biosystems, Madrid, Spain); 1.5 µL of the resulting cDNA was amplified using specific primers and probes included in TaqMan Gene Expression Assays (Assay-On-Demand; Applied Biosystems) for IL-13 (IL-13, Hs99999038_m1), forkhead transcription factor 1 (Foxj1, Hs00230964_m1), axonemal dynein intermediate polypeptide 2 (Dnai2, Hs01001551_m11), β-tubulin IV (β-tubulin IV, Hs00893144_g1) the NADPH oxidases NOX2 (NOX2, Hs01553391_m1), DUOX1 (DUOX1, Hs00213694_m1) or DUOX2 (DUOX2, Hs00204187_m1) in a 7900HT Fast Real-Time PCR System (Applied Biosystems). Relative quantification of these different transcripts was determined with the 2−ΔΔCT method using GAPDH as an endogenous control (Applied Biosystems; cat no: 4352339E).
To determine the relative expression of the PDE4A, PDE4B and PDE4D subtypes as well as of the NADPH oxidase NOX1, p47phox and p67phox SYBR Green real-time PCR was used. We designed the PDE4 primers between the exons 11 and 12 of the different PDE4 transcripts in order to recognize the different isoforms (Table 1). The percentage primer efficiency and correlation coefficients of the SYBR Green primers was calculated and accepted for efficiencies 100 ± 10%. The amplification specificity for each RT-PCR analysis was confirmed by melting curve analysis; 4 µL of cDNA was added to 19 µL of reaction mixture containing 7 µL H2O, 10 µL QuantiTect® SYBR® Green PCR Master Mix (Qiagen, Crawley, UK) and 1 µL each of forward and reverse primers (10 µM) (Table 1). Relative quantification of transcript levels (compared with control groups) was determined by evaluating the expression as 2−ΔΔCT as described above.
Table 1.
Primers for SYBR Green real-time quantitative reverse transcriptase polymerase chain reaction
| Name | Sequence (5′-3′) | GenBank accession no | Product size (bp) |
|---|---|---|---|
| PDE4A forward | TCATGGCCGAGTTCTTCCAG | NM_006202.2 | 193 |
| PDE4A reverse | TCCAAAGTGTCCAAGATCTCCTG | ||
| PDE4B forward | ATTGTATCGGCAATGGACAGAC | NM_002600.3 | 213 |
| PDE4B reverse | GTATCGAGAATGTCCTGAGCATC | ||
| PDE4D forward | AGTTCTTCCAGCAGGGAGACC | NM_000923.3 | 196 |
| PDE4D reverse | TCTCGATTGTCCTCCAGCG | ||
| NOX1 forward | TGGAACAGGAGATGGAGGAATT | NM_007052.4 | 174 |
| NOX1 reverse | CATTGTCCCACATTGGTCTCC | ||
| p47phox forward | TGAGCCATACGTCGCCATC | NM_000265.4 | 180 |
| p47phox reverse | GACACGTCTTGCCCTGACTTT | ||
| p67phox forward | ACTACTGCCTGACTCTGTGGTGTG | NM_000433.3 | 158 |
| p67phox reverse | GGTAGCCTCATAACTGAAGAGTGC |
DCF fluorescence measurement of reactive oxygen species
2′,7′-Dichlorodihydrofluorescein diacetate (H2DCF-DA; Molecular Probes, Manchester, UK) is a cell-permeable compound that following intracellular ester hydrolysis is oxidized to fluorescent 2′,7′-dichlorofluorescein (DCF) by O2·- and H2O2, and can therefore be used to monitor intracellular generation of reactive oxygen species (ROS) (Trayner et al., 1995). To quantify ROS levels, polarized human bronchial epithelial cells were washed twice with PBS and incubated for 30 min with 50 µM H2DCF-DA diluted in Opti-MEM with 10% fetal calf serum in the presence or absence of roflumilast N-oxide, dbcAMP or the antioxidant NAC. Then, cells were again washed twice with PBS to remove remaining H2DCF-DA and stimulated with CSE in the presence or absence of the same drugs indicated before for 30 min. Five randomly selected fields per condition were measured for fluorescent intensity using an epifluorescence microscope (Nikon Eclipse-TE-200, Tokyo, Japan) with filter set for fluorescein isothiocyanate (FITC). Subsequent image capture and analysis was performed using Metafluor® 5.0 software (Analytical Technologies, San Francisco, CA, USA). Results were expressed as DCF fluorescence in relative fluorescence units.
Immunofluorescence and ciliated cell count
Foxj1, Dnai2 and β-tubulin IV in differentiated human bronchial epithelial cells were analysed by immunofluorescence. To this end, transwell inserts of air-liquid interface-cultured human bronchial epithelial cells were fixed in paraformaldehyde (4%) for 45 min and embedded in Tissue-Tek® OCT™ cryosectioning compound (Sakura Finetek Europe BV, Leiden, the Netherlands). Blocks were cut into 10 µm thick sections, permeabilized in Triton X 100 (0.1% in PBS) for 5 min, blocked in 10% goat serum in PBS and immunostained with mouse anti-human monoclonal antibodies against either Foxj1 (1:50; ab95697; Abcam, Cambridge, UK), Dnai2 (1:50; ab72746; Abcam) or β-tubulin IV (1:100; Clone ONS.1A6; Sigma, Gillingham, UK) for 24 h at 4°C, followed by a secondary FITC (Foxj1, Dnai2) or rhodamine (β tubulin IV) conjugated anti-mouse IgG antibody (1:100; Molecular Probes, Manchester, UK), and finally 4′,6-diamidino-2-phenylindole (DAPI) (2 µg·mL−1) to mark nuclei (Molecular Probes, Leiden, the Netherlands). The cells were visualized at excitation/emission of 485/525 nm (FITC), or 570/590 nm (rhodamine) or 350/470 nm (DAPI) with a ×400 magnification in a TE-200 epifluorescence microscope (Nikon Eclipse-TE-200).
The antibodies against Foxj1, Dnai2 and β-tubulin IV showed a single band in Western blots of appropriate cell or tissue lysates within a large range from 25 to 250 kDa according to information given by the manufacturer. This was confirmed by our own studies wherein lysates of differentiated human bronchial epithelial cell single bands at the appropriate molecular weights between 25 and 250 kDa were found. No other bands were detected. The manufacturer recommended both antibodies as appropriate for immunohistochemistry/immunofluorescence.
The number of ciliated cells was quantified based on immunofluorescent labelling for β-tubulin IV, a commonly used cilia marker. To this end, whole transwell inserts of differentiated human bronchial epithelial cells were fixed in paraformaldehyde (4%) for 45 min, permeabilized with Triton X 100 (0.1% in PBS) for 5 min, blocked with 10% goat serum in PBS and immunostained with a primary mouse anti-human β-tubulin IV antibody for 24 h (1:100; Clone ONS.1A6; Sigma, Gillingham, UK). β-tubulin IV expressing cells in whole transwell inserts were then visualized with a secondary FITC-conjugated anti-mouse IgG antibody (1:100; Molecular Probes, Manchester, UK). The cells were visualized at excitation/emission of 485/525 nm with a ×400 magnification in a TE-200 epifluorescence microscope (Nikon Eclipse-TE-200).
To determine the percentage of ciliated cells present in untreated and treated bronchial epithelial air-liquid interface cultures, three transwell inserts each from control cells and treated cells were examined. Ciliated cells were identified by morphometric analysis of the β-tubulin IV positively staining cilia on the cell surface as previously outlined (Look et al., 2001). The percentages of airway luminal cells that expressed β-tubulin IV were calculated visually using 10 regions per Transwell insert. Results are expressed as % β-tubulin IV-positive cells. The specificity of the immunostaining was confirmed by substituting the primary antibody with an irrelevant mouse IgG or by omitting the primary antibody. No positive immunostaining was observed in these controls (data not shown).
Western blot
Western blot analysis was used to detect Foxj1, Dnai2, β-tubulin IV and PDE4B proteins in differentiated bronchial epithelial cells. Cells were scraped from air-liquid interface transwell inserts and lysed on ice with a lysis buffer consisting of 20 mM Tris, 1 mM EDTA, 150 mM NaCl, 0.1% Triton X-100, 1 mM dithiothreitol (DTT) and 1 µg·mL−1 pepstatin A supplemented by a complete protease inhibitor cocktail. The Bio-Rad assay (Bio-Rad Laboratories Ltd, Herts, UK) was used to quantify the level of protein in each sample to ensure equal protein loading. SDS-PAGE was used to separate the proteins according to their molecular weight. Briefly, 20 µg of protein (denatured) mixed with 2× loading buffer [comprising 160 mM Tris HCl (pH 6.8), 4% SDS, 20% glycerol, 1.4 mM β-mercaptoethanol, 0.04% bromophenol blue], along with a molecular weight protein marker (Bio-Rad Kaleidoscope marker; Bio-Rad Laboratories), was loaded onto an acrylamide gel consisting of a 5% acrylamide stacking gel on top of a 12% acrylamide resolving gel and run through the gel by application of 100 V for 1 h. Proteins were transferred to a PVDF membrane using a wet blotting method. The membrane was blocked with 5% Marvel in PBS containing 0.1% Tween20 and then probed with either mouse anti-human Foxj1 (1:1000; Abcam), or Dnai2 (1:1000; Abcam) or β-tubulin IV (1:1000; Sigma, Gillingham, UK) monoclonal antibodies, a goat anti-human PDE4B (1:1000; Sigma, Gillingham, UK) polyclonal antibody and a rabbit anti-human β-actin polyclonal antibody (1:1000; Sigma, Gillingham, UK) as housekeeping reference, followed by the corresponding peroxidase-conjugated secondary (1:10.000) antibody. The enhanced chemiluminescence method of protein detection using ECL-plus (GE Healthcare, Amersham Biosciences, Manchester, UK) was used to detect labelled proteins. Quantification of protein expression was performed by densitometry relative to β-actin expression using the software GeneSnap version 6.08 (Cambridge, UK).
cAMP assay
For measurements of cellular cAMP content following incubations with CSE or roflumilast N-oxide as indicated, culture medium was removed and the cells were washed with PBS. Then, cells were lysed and the intracellular cAMP content was determined with the cAMP Biotrak enzyme immunoassay system according to manufacturer's instructions (Amersham). Results are expressed as fmol cAMP mg−1 protein (per insert).
PKA activity
Differentiated human bronchial epithelial cells were incubated with roflumilast N-oxide or vehicle followed by CSE. After 24 h, cells were washed with PBS and lysed with a lysis buffer (20 mM MOPS, 50 mM β-glycerolphosphate, 50 mM sodium fluoride, 1 mM sodium vanadate, 5 mM EGTA, 2 mM EDTA, 1% NP40, 1 mM DTT, 1 mM benzamidine, 1 mM PMSF, and 10 µg·mL−1 leupeptin and aprotinin); 2 µg of total protein was used to determine PKA activity. PKA activity was measured using a colorimetric PKA Activity Assay Kit (Assay Designs, Farmingdale, NY, USA) according to manufacturer's instructions. Briefly, the non-radioactive, solid-phase ELISA is based on the PKA-dependent phosphorylation of a highly specific substrate attached to the assay plate that in its phosphorylated form is detected by a specific antibody. Results are expressed as x-fold of the PKA activity in untreated human differentiated bronchial epithelial cells.
PDE activity assay
Following air-liquid interface culture as described above, differentiated human bronchial epithelial cells were incubated with CSE (10%) for 24 h. Then cells were scraped, lysed and homogenized in a buffer containing 50 mM Tris-HCl (pH 7.8), 150 mM NaCl, 10 mM MgCl2, 10 mM 2-mercaptoethanol, 1 mM EGTA, 50 mM sodium fluoride, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.5% SDS, 50 mM benzamidine, 0.5 µg·mL−1 leupeptin, 0.7 µg·mL−1 pepstatin, 4 µg·mL−1 aprotinin, 10 µg·mL−1 soybean trypsin inhibitor and 2 mM PMSF. The homogenate was centrifuged at 14.000 g for 10 min. PDE activities were measured in supernatants using 15 µg of protein per condition (protein was determined using the Bio-Rad assay as described in the section on Western blotting).
Total PDE and PDE4 activities were measured as described previously (Thompson et al., 1974) with modifications (Cortijo et al., 1996; Singh et al., 2003). Briefly, a reaction mixture with a final volume of 200 µL consisting of 40 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 1.25 mM 2-mercaptoethanol, 20 µg bovine serum albumin, 1 µM [3H]-cAMP and 25 µL of the 14.000 g cell extract supernatant (comprising 15 µg of protein with PDE activity) was incubated at 37°C for 15 min. Then 200 µL of 40 mM Tris-HCl (pH 7.5) was added and reactions were terminated by boiling in a water bath for 3 min. Once the reaction mixture had returned to room temperature, 50 µL of Crotalus atrox venom (1 mg·mL−1) was added comprising 5′ nucleotidase. Following an incubation for 10 min at 37°C allowing all [3H]-5′-AMP being converted into [3H]-adenosine, an anion exchange resin was used to separate uncharged [3H]-adenosine from negatively charged [3H]-cAMP. To this end, 500 µL of a 1:3 slurry of Bio-Rad resin (AG 1-X2, 200–400 mesh) in de-ionized water was added to each one of the assay tubes. After centrifugation for 10 min, 250 µL aliquots of the supernatant were collected and counted in a liquid scintillation counter. To determine PDE4 activity, assays were run in the presence of roflumilast N-oxide at 1 µM (that completely and selectively blocks all PDE4) or vehicle (0.1% DMSO). PDE4 was calculated as the difference of PDE activity measured in the presence or absence of roflumilast N-oxide. PDE enzymatic activity and is expressed as pmol cAMP hydrolysed min−1 mg−1 protein.
Results were corrected for blank values (measured in the presence of denatured proteins) that remained below 2% of total radioactivity. cAMP degradation was <25% of the amount of substrate added and, thus, was in the linear range.
ELISA
IL-13 protein was analysed in culture supernatants of differentiated human bronchial epithelial cells by using a commercially available enzyme-linked immunosorbent assay kit (R&D Systems, Abingdon, Oxon, UK) according to the manufacturer's protocol.
Neutralization of IL-13
In some experiments, a neutralizing antibody against IL-13 was used to investigate the role of IL-13 in CSE-induced loss in Foxj1, Dnai2 and β tubulin IV protein expression. To this end, a rat anti-human IL-13 monoclonal antibody (mAb-IL-13; clone JES 10-5A2, affinity purified, azide free and low endotoxin, acquired from BD Pharmingen™, Madrid, Spain) was used at 150 ng·mL−1 that should result in > 95% neutralization of IL-13. This antibody or its respective isotype control (rat IgG1 at 150 ng·mL−1) was added to the basolateral and apical compartment of the differentiated human bronchial epithelial cell cultures from 30 min before initiation of CSE exposure over 3 days. Following cell lysis and protein extraction, Foxj1, Dnai2 and β tubulin IV were detected by Western blotting as described before.
Statistical analysis
Data are presented as mean ± SEM of n experiments. Statistical analysis of data was performed by anova followed by Bonferroni test (GraphPad Software Inc, San Diego, CA, USA). Significance was accepted when P < 0.05.
Materials
Roflumilast N-oxide (N-(3,5-dichloro-1-oxypyridin-4-yl)-3-cyclopropylmethoxy-4-difluoromethoxybenzamide) was synthesized by Nycomed GmbH, Konstanz, Germany. Unless indicated otherwise, all other reagents used were obtained from Sigma (Chemical Co, Madrid, Spain). Roflumilast N-oxide or forskolin was dissolved in dimethyl sulfoxide (DMSO) at 10 mM stock concentration. Theophylline was dissolved in HCl 0.1 M at 100 mM stock concentration. Several dilutions of the stocks were performed with cell culture medium. The final concentrations of DMSO (0.1%) or HCl (10 µM) in the cell culture did not affect cellular functions. Other chemicals [dibutyryl cAMP (dbcAMP), apocynin, N-acetyl cysteine (NAC)] were dissolved in medium.
Results
Short-term effects of roflumilast N-oxide and forskolin on CBF in differentiated human bronchial epithelial cells
Adding the adenylyl cyclase activator forskolin (10 µM) or the PDE4 inhibitor roflumilast N-oxide (2 nM, 1 µM) to well-differentiated cultures of ciliated human bronchial epithelial cells resulted in a rapid increase in CBF (Figure 1A,B). CBF peaked within 3 min after addition of forskolin but about 70% of this increment was lost after 30 min and CBF returned to baseline values another 90 min later. Roflumilast N-oxide at 1 µM (corresponding to complete and selective inhibition of PDE4) reproduced the rather immediate rise in CBF that, however, persisted when monitored over 3 h. Roflumilast N-oxide at 2 nM [corresponding to about half-maximum inhibition of PDE4 (Hatzelmann and Schudt, 2001) and free plasma concentrations in man following clinical dosing of roflumilast (Bethke et al., 2007)] resulted in a gradually increasing rise in CBF attaining a maximum at 60 min following addition of the PDE4 inhibitor that corresponded to about half the peak CBF found with 1 µM roflumilast N-oxide. Again, the increase in CBF was maintained over the remaining 2 h observation period.
Figure 1.
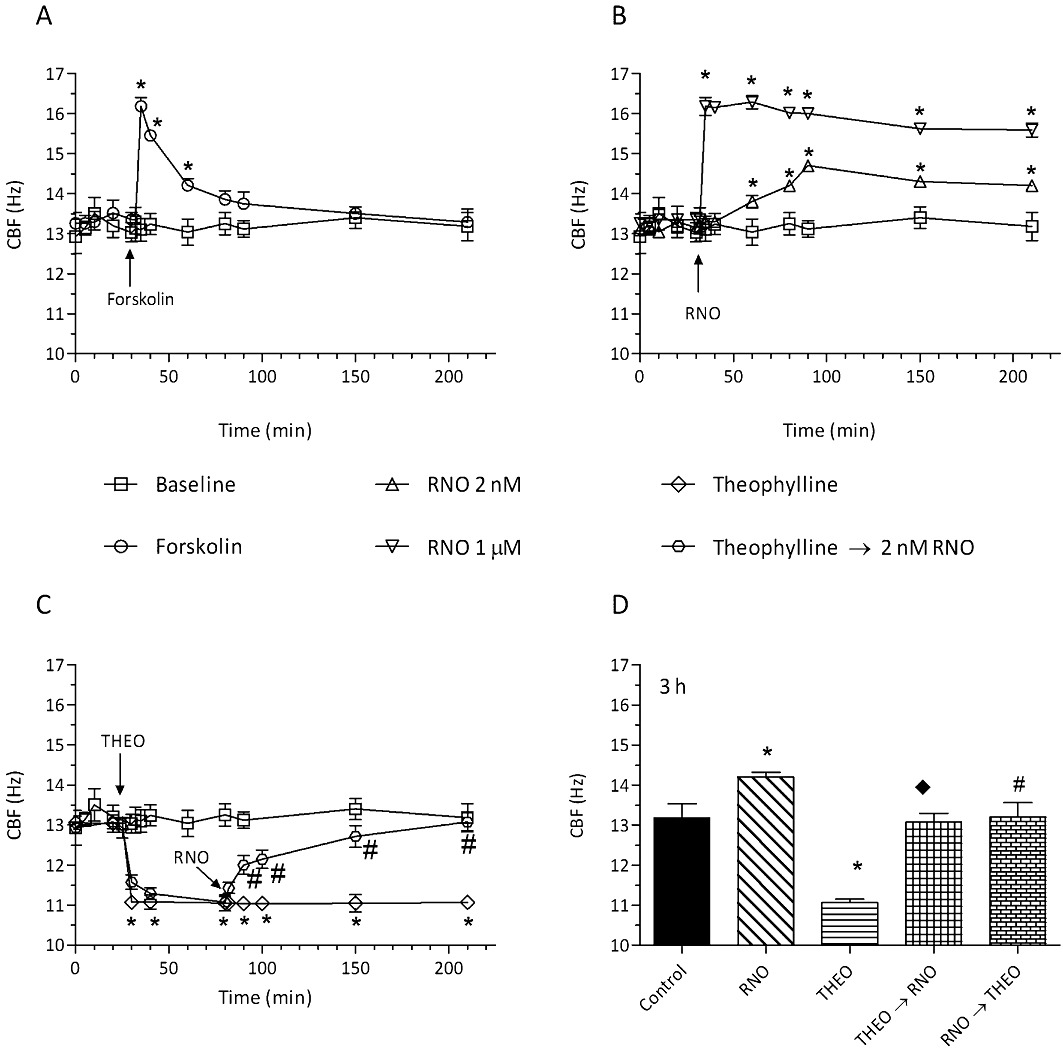
Forskolin and roflumilast N-oxide rapidly increased CBF of differentiated human bronchial epithelial cells, effects of theophylline. Following an initial 20 min baseline period, differentiated bronchial epithelial cells (D-BECs) were incubated with forskolin at 10 µM (A), or roflumilast N-oxide (RNO) at 2 nM or 1 µM (B), or theophylline (THEO) at 10 µM followed by roflumilast N-oxide (2 nM) (C) or vehicle (baseline), and ciliary beat frequency (CBF) was monitored for up to 3 h. (D) D-BECs were incubated with vehicle (control), or 2 nM RNO, or THEO at 10 µM, or THEO followed by RNO (from 80 min) (THEO→RNO), or RNO followed by THEO (from 80 min) (RNO→THEO), and the CBF assessed at 3 h is depicted. CBF was measured by DHSV microscopy and quantified by Fourier transformation as detailed in Methods. Results are shown as the means ± SEM of three experiments per donor based on cultures from two to four different donors per condition. Vehicle (0.1% DMSO for RNO or forskolin and 10 µM HCl for THEO) was included in the controls and did not affect CBF over the observation period. (A and B) *P < 0.05 versus baseline. (C) *P < 0.05 versus baseline; #P < 0.05 versus THEO. (D) *P < 0.05 versus control; #P < 0.05 versus RNO; ◆P < 0.05 versus THEO.
Because endogenously produced adenosine may regulate the homeostasis of bronchial epithelial cells (Rollins et al., 2008) and adenosine (by stimulating A2B receptors) was reported to augment CBF in airway epithelial cells (Lazarowski and Boucher, 2009; Allen-Gipson et al., 2011), the effects of theophylline at 10 µM, a concentration at which the xanthine is a non-specific antagonist to adenosine receptors including A2B but does not interfere with PDE activities, were studied. Strikingly, there was a rapid and persistent fall in CBF following theophylline (Figure 1C). Then roflumilast N-oxide (2 nM) was added to the incubations with theophylline once this fall in CBF had reached a robust plateau (1 h after theophylline). In the presence of theophylline, the PDE4 inhibitor restored CBF to baseline, but not above (Figure 1C,D). On the other hand, the increment in CBF secondary to roflumilast N-oxide at 2 nM was abolished by theophylline (Figure 1D).
Effects of roflumilast N-oxide and forskolin on the rapid loss of CBF following exposure of differentiated human bronchial epithelial cells to CSE
In order to determine the short-term effect of CSE on CBF, differentiated bronchial epithelial cells were incubated with CSE at 10% for up to 3 h and CBF was regularly monitored. CSE almost instantaneously impaired CBF and this reduction was maintained over at least 3 h. Forskolin, added from 60 min after CSE, resulted in a rapid, yet partly transient recovery in CBF (Figure 2A). Full and maintained recovery from the CSE-induced loss in CBF, however, emanated from roflumilast N-oxide at 1 µM while the PDE4 inhibitor at the concentration of 2 nM resulted in a more gradually improved CBF approaching fully restored CBF at the end of the 3 h monitoring period (Figure 2B).
Figure 2.
CSE triggered a rapid loss of CBF in differentiated human bronchial epithelial cells, an effect which was reversed by forskolin and roflumilast N-oxide (A, B). Following an initial 20 min baseline period, differentiated bronchial epithelial cells (D-BECs) were exposed to CSE at 10% for up to 3 h. From the time point the reduction in CBF was optimal (at 80 min), forskolin (FSK) at 10 µM (A) or roflumilast N-oxide (RNO) at 2 nM or 1 µM (B), or vehicle was added. CBF was monitored for 3 h by DHSV and quantified by Fourier transformation as detailed in Methods. Results are shown as the means ± SEM of three experiments per donor based on cultures from two to four different donors per condition. *P < 0.05 versus control, #P < 0.05 versus CSE. CSE rapidly reduced cAMP content in differentiated human bronchial epithelial cells (C). D-BECs were exposed to vehicle (baseline), RNO at 2 nM, CSE at 10% followed by vehicle or RNO (2 nM) at the indicated times and cellular cAMP content was measured at different time points. Results are shown as the means ± SEM from cultures of two donors in duplicates. *P < 0.05 versus control.
Because cAMP is a critical regulator of CBF, the potential effects of CSE on the cAMP content of differentiated human bronchial epithelial cells were explored next. Exposure to CSE at 10% reduced the initial cAMP content by 46% within 5 min and this had not recovered 1 h later, whereas cAMP content did not change over this time period in appropriate controls (Figure 2C). Roflumilast N-oxide (2 nM) when added at 15 min following initiation of CSE exposure rescued the cells from this loss in cAMP; the cAMP content was significantly elevated to almost control levels after 45 min (Figure 2C). This effect of was more or less in parallel to its effects on CBF (Figure 2B).
In the absence of CSE, roflumilast N-oxide at 2 nM enhanced the cellular cAMP content by about 1.7-fold within 15 min and this remained sustained over at least another 30 min (Figure 2C).
Long-term exposure (24 h) to CSE impaired CBF: effects of roflumilast N-oxide and role of PKA
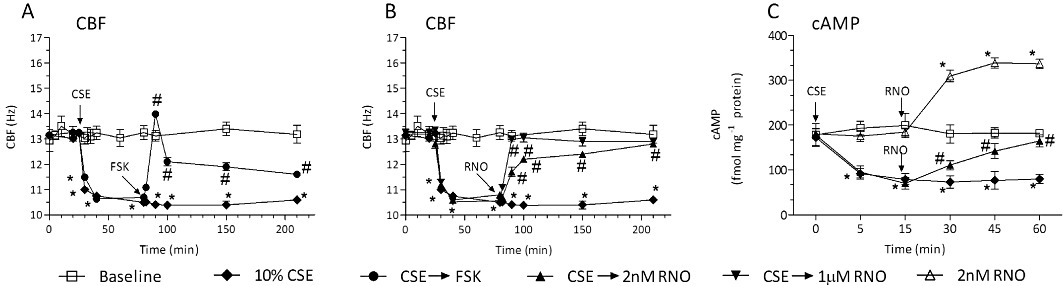
When differentiated human bronchial epithelial cells were exposed to CSE for 24 h, CBF was concentration- dependently compromised (Figure 3A) in the absence of cytotoxicity (assessed by LDH release in separate experiments). Apparently, CBF deteriorated more markedly when CSE was present for 24 h compared with 3 h (Figure 2A,B). The following experiments were then conducted with CSE at 10%. The concomitant presence of roflumilast N-oxide at 1 µM along with CSE for 24 h prevented the loss in CBF secondary to the smoke extract. The PDE4 inhibitor at 2 nM resulted in approximately 68% recovery of CBF (Figure 3B). Quenching ROS by adding apocynin (100 µM) or N-acetylcysteine (1 mM) reversed the loss in CBF following a 24 h exposure to CSE (10%) by 70–80% (Figure 3C).
Figure 3.
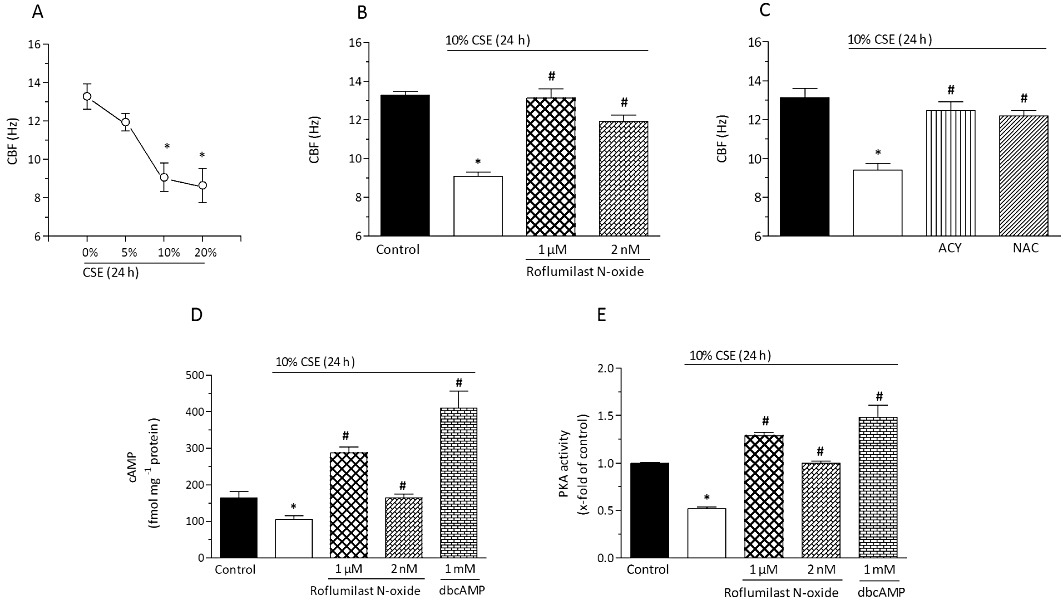
Roflumilast N-oxide reversed the CSE-induced persistent loss of CBF in differentiated human bronchial epithelial cells (A-C), role of cAMP (D), PKA (E). Differentiated human bronchial epithelial cells (D-BECs) were exposed to CSE at 0–20% for 24 h (A), or pre-incubated for 30 min with roflumilast N-oxide at 2 nM or 1 µM (B) or with apocynin (ACY) at 100 µM or with at NAC at 1 mM (C) or vehicle (B, C) and then exposed to CSE at 10% for 24 h. Then CBF was measured by DHSV and quantified by Fourier transformation as detailed in Methods. Results are shown as the means ± SEM of cultures from two to four donors and three experiments per donor. In (D) and (E), D-BECs were pre-incubated for 30 min with roflumilast N-oxide at 2 nM or 1 µM or with dbcAMP (1 mM) before exposure to 10% CSE. Cellular cAMP content (D) and PKA (E) were assessed after 24 h as described in Methods. Results are depicted as means ± SEM of cultures from three donors and three experiments per donor. *P < 0.05 versus control, #P < 0.05 versus CSE.
Next, the effects of CSE on cAMP and PKA were investigated under these experimental conditions. In differentiated human bronchial epithelial cells exposed to tobacco smoke for 24 h, the cAMP content (Figure 3D) and in parallel PKA activity (Figure 3E) were both reduced compared with controls, and for cAMP the loss was comparable to that observed after 5 min incubation with CSE (Figure 2C). The additional presence of roflumilast N-oxide at 2 nM almost restored the cAMP content (Figure 3D) and PKA activity (Figure 3E), while both cAMP and PKA activity recovered to higher than baseline with 1 µM roflumilast N-oxide; this also occurred with dbcAMP, a cAMP analogue serving as a positive control (Figure 3D,E).
Regulation of PDE4 expression by CSE: effects of roflumilast N-oxide and role of oxidative stress
The loss in cAMP content secondary to CSE exposure for 24 h prompted us to analyse the activity and expression of PDE4, well-known as the prominent cAMP hydrolysing PDE in human bronchial epithelial cells. Firstly, exposure of differentiated bronchial epithelial cells to CSE at 10% for 24 h induced about a twofold increase in PDE4 activity (Figure 4A), probably as a result of an up-regulation in the expression of PDE4B. Indeed, of the PDE4 subtypes analysed, mRNA transcripts for PDE4B were most markedly increased by 2.8-fold over control (P < 0.05), while the 1.4-fold and 1.3-fold increment in PDE4A and D transcripts, respectively, remained below significance (Figure 4B). Strikingly, quenching ROS by apocynin (100 µM) or N-acetylcysteine (1 mM) reversed the CSE-induced increment in PDE4B transcripts (Figure 4C). As an enhancement in the levels of cAMP may prevent CSE-induced oxidative stress (as exemplified in Figure 5), next the effects of dbcAMP (1 mM) on PDE4B expression were investigated. The cAMP analogue prevented the CSE-induced increment in PDE4B transcripts (Figure 4D). In line with this result, inhibiting PDE4 (roflumilast N-oxide at 1 µM) abolished the CSE-induced increment in PDE4B transcripts (Figure 4E) and protein (Figure 4F). Roflumilast N-oxide at 2 nM modestly reduced the increment in PDE4B transcripts secondary to CSE (Figure 4E). At this concentration, the PDE4 inhibitor was not found to significantly reduce PDE4B protein based on the densitometric analyses (Figure 4F).
Figure 4.
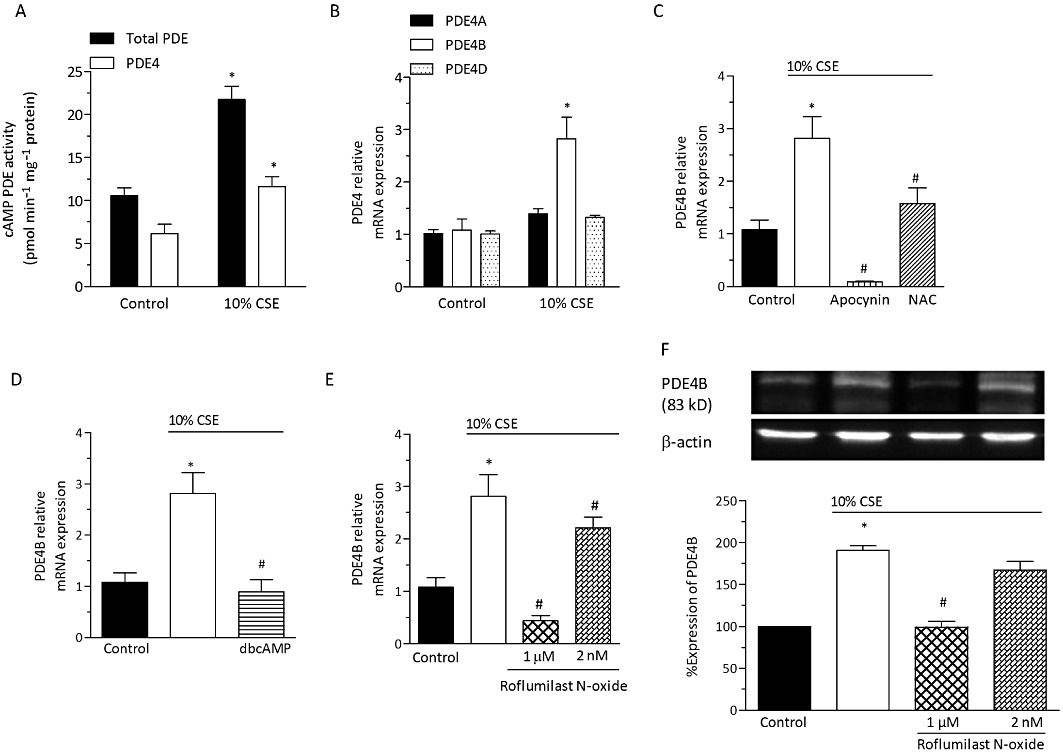
CSE up-regulates PDE4B expression and PDE4 activity in differentiated human bronchial epithelial cells: Effects of roflumilast N-oxide and role of ROS. Differentiated bronchial epithelial cells (D-BECs) were exposed to CSE (10%) or medium (control) for 24 h. (A) Total cAMP hydrolysing PDE and PDE 4 activity following incubation of differentiated bronchial epithelial cells were measured in cell lysates. PDE4 activity was calculated as the difference in PDE activity measured in the presence and absence of roflumilast N-oxide (1 µM). Data are the mean ± SEM (n= 3 per condition; *P < 0.05 versus control). (B) Relative expression levels of mRNA transcripts for PDE4A, B and D subtypes were determined by real-time quantitative RT-PCR using the SYBR Green protocol as described in Methods. In (C–F), D-BECs were pre-incubated (30 min) with either apocynin at 100 µM (C), or NAC at 1 mM (C), or dbcAMP at 1 mM (D), or roflumilast N-oxide at 2 nM or 1 µM (E, F), or vehicle before exposure to CSE at 10% for 24 h. In (C, D, E), relative expression levels of PDE4B mRNA transcripts are shown. In (F), expression of PDE4B protein is depicted. Cells were lysed and proteins separated by SDS-PAGE followed by blotting and detection using a PDE4B-specific antibody as detailed in Methods. A representative Western blot and results from densitometric evaluations are illustrated. Means ± SEM of D-BEC cultures from three donors and two experiments per donor are shown (B–F). *P < 0.05 versus control, #P < 0.05 versus CSE.
Figure 5.
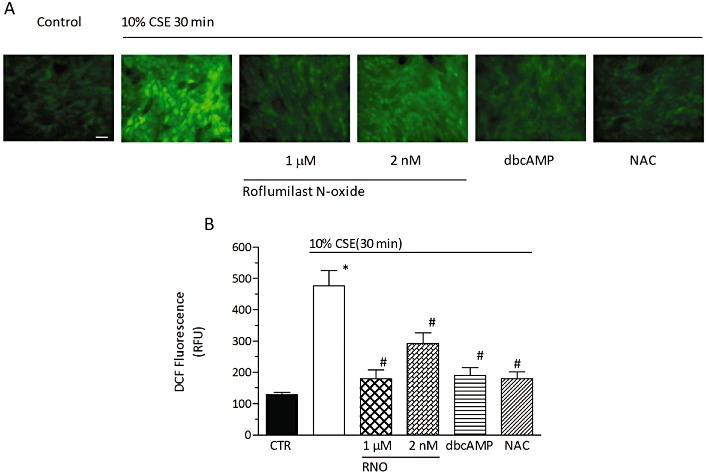
Roflumilast N-oxide attenuates CSE-induced intracellular ROS generation in differentiated human bronchial epithelial cells. Differentiated human bronchial epithelial cells (D-BECs) were loaded with H2DCF-DA in the presence of roflumilast N-oxide (RNO) at 2 nM or 1 µM, or dbcAMP at 1 mM, or NAC at 1 mM, or vehicle for 30 min. Excess H2DCF-DA was removed by washing with PBS. RNO, dbcAMP, NAC or vehicle was appropriately replenished before cells were exposed to CSE at 10%. Following a 30 min incubation period, accumulation of fluorescent DCF was assessed with fluorescence microscopy in a total of five fields per condition as detailed in Methods. (A) Representative microphotographs (scale bar corresponds to 20 µm). (B) Quantitative evaluation of DCF fluorescence. Results in (B) represent the means ± SEM of cultures from two donors and three experiments per donor. *P < 0.05 versus control, #P < 0.05 versus CSE. CTR, control.
Effects of roflumilast N-oxide on CSE-induced intracellular ROS and NADPH oxidases in differentiated human bronchial epithelial cells
The role of ROS and its possible regulation by roflumilast N-oxide was further addressed. Intracellular ROS was quantified as DCF following oxidation from H2DCF-DA. Exposure of differentiated human bronchial epithelial cells to CSE at 10% for 30 min resulted in an approx. fourfold increase in DCF accumulation over control. Pre-incubation with roflumilast N-oxide concentration-dependently reduced the increment in DCF accumulation by about 53 and 86% at 2 nM and 1 µM, respectively (Figure 5). dbcAMP (1 mM) reproduced the reduction in DCF accumulation observed with the PDE4 inhibitor. Finally, NAC (1 mM) serving as a control prevented DCF accumulation triggered by CSE (Figure 5).
Exposure of differentiated human bronchial epithelial cells to CSE at 10% for 24 h up-regulated the expression of the NADPH oxidase isoenzymes NOX1, NOX2 (gp91phox) and DUOX2 (Figure 6) while DUOX1 was not affected (data not shown). This up-regulation was reduced by roflumilast N-oxide at 2 nM and 1 µM and dbcAMP at 1 mM. Quenching of ROS by apocynin (100 µM) or NAC (1 mM) also suppressed CSE-triggered expression of the NADPH oxidase isoenzymes (Figure 6). Finally, CSE (10%) for 24 h enhanced the expression of the p47phox (2.5-fold) and p67phox (threefold) cytosolic components required for assembling the active NOX2 NADPH oxidase complex that again was reversed by the PDE4 inhibitor or the ROS quenchers (data not shown).
Figure 6.
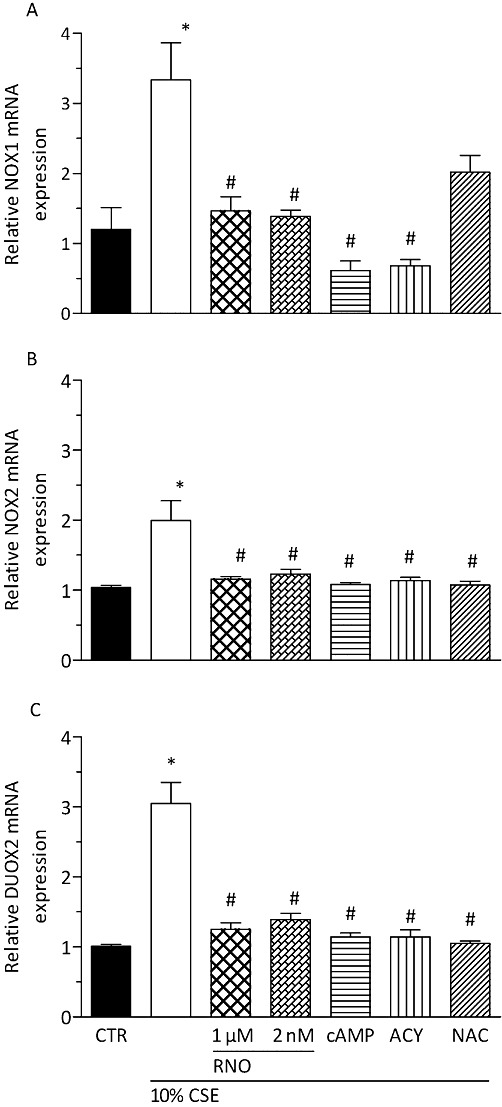
CSE up-regulates mRNA expression of NADPH oxidases NOX1, NOX2 and DUOX2 in differentiated human bronchial epithelial cells (D-BECs) that was reversed by roflumilast N-oxide (RNO). D-BECs were pre-incubated with RNO at 2 nM or 1 µM, or dibutyryl cAMP at 1 mM (cAMP), or apocynin (ACY) at 100 µM, or NAC at 1 mM, or vehicle for 30 min and then exposed to CSE (10%) for 24 h. mRNA transcripts of NOX1, NOX2 and DUOX2 were quantified by real-time quantitative RT-PCR as described in Methods. Results are given as means ± SEM of cultures from two donors and three experiments per donor. *P < 0.05 versus control, #P < 0.05 versus CSE. CTR, control.
Loss of ciliated bronchial epithelial cells by CSE: effects of roflumilast N-oxide
Differentiated human bronchial epithelial cells were exposed to CSE at different concentrations (0–10%) and time periods (1, 3, 5 and 7 days). Then, the average number of cells with cilia motility was determined by DHSV followed by Fourier transformation. Under control conditions (0% CSE), the average number of cells with cilia motility remained unchanged over 7 days. Exposure to CSE concentration- and time-dependently reduced the average number of cells with cilia motility, which was significant after 3 days of incubation with CSE at 2.5% (about 30% inhibition), and reached a maximum of about 75% inhibition versus control after 7 days of incubation with CSE at 10% (Figure 7A, left panel). Morphologically, differentiated bronchial epithelial cells exposed to CSE adopted a flattened and elongated phenotype with fragmented cilia.
Figure 7.
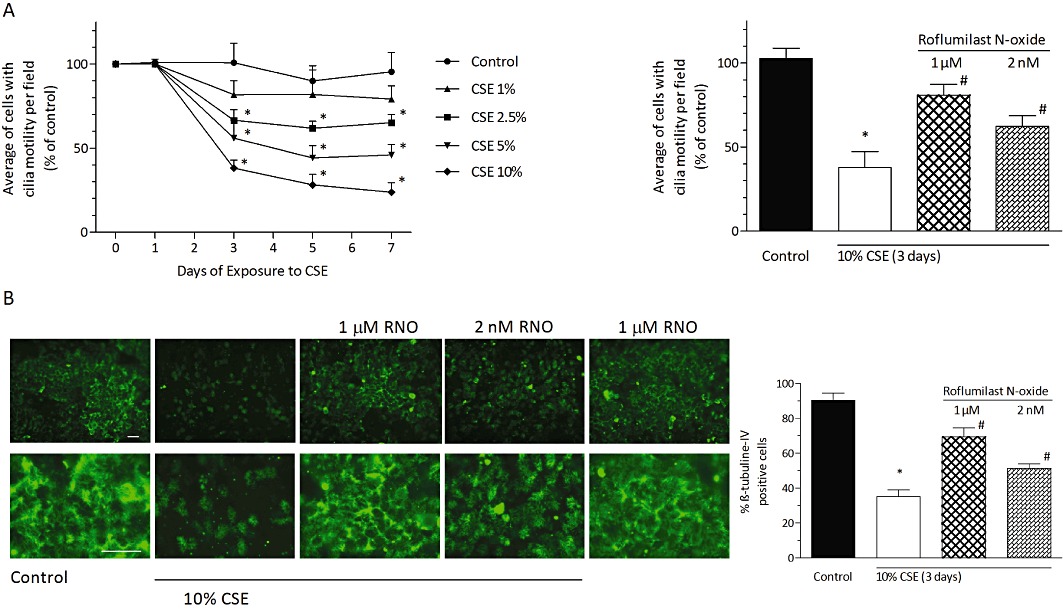
Roflumilast N-oxide (RNO) protected from loss of ciliated bronchial epithelial cells induced by CSE. In (A, left panel), differentiated bronchial epithelial cells (D-BECs) were exposed to CSE at different concentrations (0–10%) and times (0–7 days). Medium was replenished with fresh CSE every 24 h. Cells with cilia motility were identified based on DHSV as described in Methods. At each of the indicated times and concentrations of CSE exposure, the average number of cells with cilia motility per field was then calculated based on captures from a minimum of 10 separate fields and expressed as % of corresponding cilia motility obtained for non-treated cells at day 0 (= 100%) (control). In (A, right panel), D-BECs were pre-incubated with RNO at 2 nM or 1 µM or vehicle for 30 min and then exposed to CSE at 10% for 3 days. Culture medium with fresh CSE and RNO was replaced every 24 h. The average number of cells with cilia motility per field was determined as in (A), left panel. In (B), D-BECs were pre-incubated with RNO and then exposed to CSE at 10% for 3 days as described in (A). β-tubulin IV was identified by immunofluorescent labelling with a primary anti-β-tubulin IV antibody followed by a secondary FITC-conjugated antibody as detailed in Methods. Representative microphotographs are depicted in the upper, left panel (scale bar 20 µm, original magnification × 400) with zoomed pictures in the lower, left panel. Results of the morphometric evaluation are shown in the right panel as the percentage of cells that expressed β-tubulin IV of total cells. Results are given as means ± SEM of cultures from two donors and three experiments per donor. *P < 0.05 versus control, #P < 0.05 versus CSE.
The additional presence of roflumilast N-oxide at 2 nM or 1 µM partially prevented the loss in bronchial epithelial cells endowed with motile cilia elicited by incubation with 10% CSE for 3 days. Indeed, the approx. 67% reduction of cells with cilia motility following CSE compared with control was diminished by 38 and 67% with roflumilast N-oxide at 2 nM and 1 µM, respectively (Figure 7A, right panel).
In parallel, immunostaining against the cilia marker β-tubulin IV was performed to monitor the occurrence of cilia on bronchial epithelial cells. Incubation with 10% CSE for 3 days reduced the percentage of cells positively staining for β-tubulin IV by about 60%. Roflumilast N-oxide at 2 nM and 1 µM concentration-dependently prevented the loss of this cilia marker by 34 and 62%, respectively (Figure 7B), which is in the range comparable to findings from acquisition of the average of cells with cilia motility.
Effects of CSE and roflumilast N-oxide on Foxj1 and Dnai2 expression in human bronchial epithelial cells
Foxj1 is a transcription factor that is critical for and a regulator of the motile ciliogenic programme (Yu et al., 2008). Therefore, the expression of Foxj1 along with the dynein axonemal intermediate chain-2 (Dnai2) protein, required to assemble the outer dynein arm of the motile cilia and β-tubulin IV to mark cilia, was studied in human bronchial epithelial cells. First, expression levels of Foxj1 and Dnai2 along with β-tubulin IV were monitored over the time of transition from undifferentiated bronchial epithelial cells into the differentiated phenotype comprising ciliated cells under in vitro air-liquid interface culture conditions. Immunofluorescence stainings in sections of bronchial epithelial cells revealed a time-dependent increase in ciliated cells based on apical staining for β-tubulin IV over 28 days of air-liquid interface culture (Figure 8A). This time course for β-tubulin IV staining was closely paralleled by immunofluorescent labelling of Dnai2 and Foxj1. Indeed, while at day 7 after instigating the differentiating culture conditions, immunofluorescence for β-tubulin IV, Foxj1 and Dnai2 remained virtually absent, a moderate level of cilia markers β-tubulin IV and Dnai2 emerged apically located along with some Foxj1 in the nuclear region at day 14 (Figure 8A). Four weeks of air-liquid interface culture resulted in an almost complete covering of the cell layer by apical β-tubulin IV and Dnai2 staining corresponding to the high fraction of ciliated cells in these cultures as depicted in Figure 7B. In parallel, the expression of Foxj1 in the nuclear region was further enhanced at day 28 (Figure 8A). These progressive increases in the markers of the ciliated cell phenotype were reflected in analyses of total protein (Western blot) (Figure 8B) and mRNA (Figure 8C). For example, transcripts for Foxj1, Dnai2 and β-tubulin IV (Figure 8C) were significantly enhanced at day 14 and further increased to day 28 after initiation of the air-liquid interface culture. That enhanced expression of Foxj1 over time was paralleled by increased expression of the cilia constituents Dnai2 and β-tubulin IV, and by the morphological observation of cells equipped with motile cilia is in line with the critical role this transcription factor has in the process of differentiation towards ciliated human bronchial epithelial cells.
Figure 8.
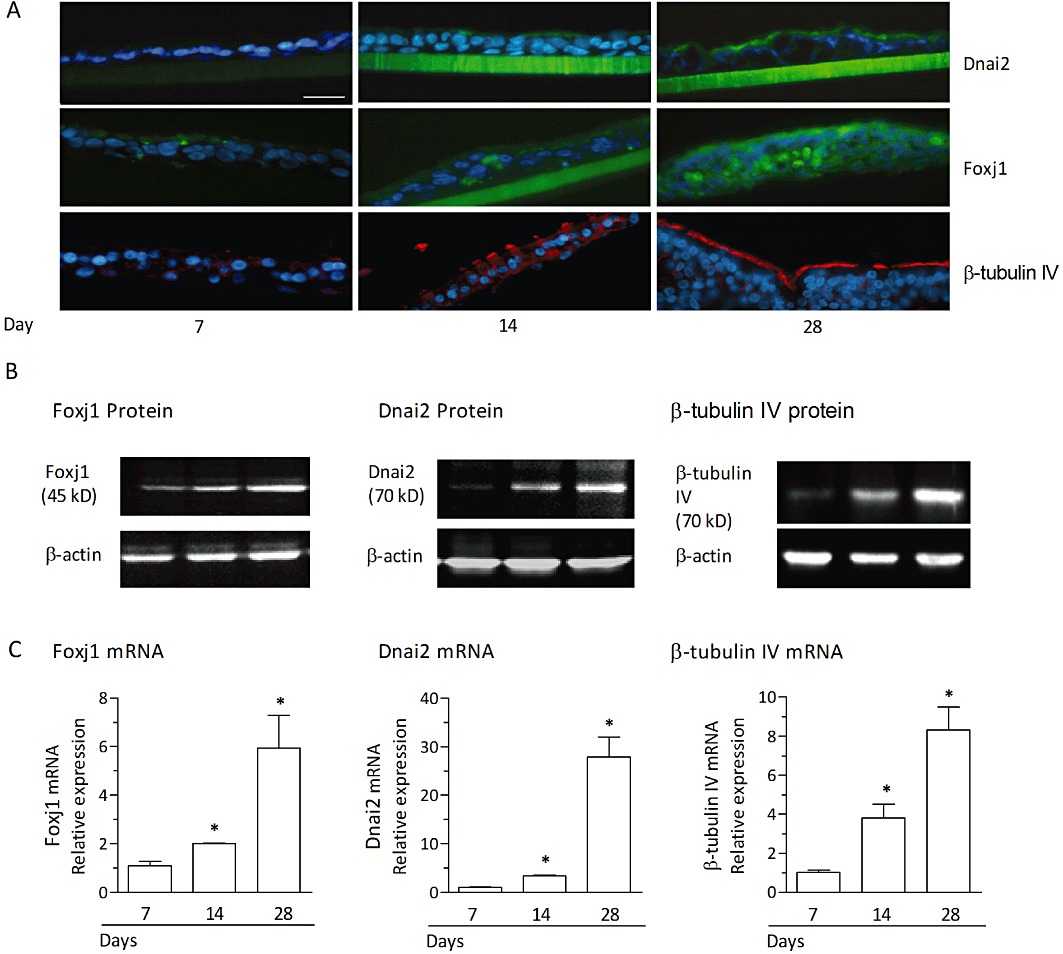
Differentiation of human bronchial epithelial cells in air-liquid interface culture is associated with an increase in Foxj1, Dnai2 and β-tubulin IV. Undifferentiated bronchial epithelial cells from monolayer cultures were plated on porous transwell inserts and differentiated in an air-liquid interface culture for 28 days. (A) Immunofluorescence detection of Foxj1, Dnai2 and β-tubulin IV in sections prepared at days 7, 14 and 28 after initiation of the air-liquid interface culture. Green staining (FITC) indicates the presence of Foxj1 or Dnai2, red staining (rhodamine) reflects β-tubulin IV. Blue staining indicates cell nuclei (DAPI). Scale bar 30 µm. Representative sections are shown and no detectable staining was observed for corresponding isotype controls. In (B) and (C), Western blots (B) and relative mRNA expression levels (C) for Foxj1, Dnai2 or β tubulin IV at day 7, 14 and 28 following initiation of the air-liquid interface culture are shown. Western blots are a representative of three independent experiments. The mRNA expression levels were quantified by real-time RT-PCR and evaluated as the x-fold change versus control (immediately before initiation of the air-liquid interface culture) using the 2−ΔΔCt formula with GAPDH as endogenous standard. Results from mRNA analyses represent the means ± SEM of cultures from three donors and three experiments per donor. *P < 0.05 versus control.
Next, differentiated human bronchial epithelial cells were exposed to CSE (at 10%) over 24–72 h and the expression of Foxj1 and Dnai2 was assessed. Compared with baseline, CSE resulted in a comparable decrease in Foxj1 (Figure 9A) and Dnai2 (Figure 9B) mRNA transcript levels by about 40% at 24 h and 70% at 72 h exposure. This fall in mRNA levels of two proteins required for motile cilia integrity precedes the loss in cells with cilia motility following CSE exposure (Figure 7A).
Figure 9.
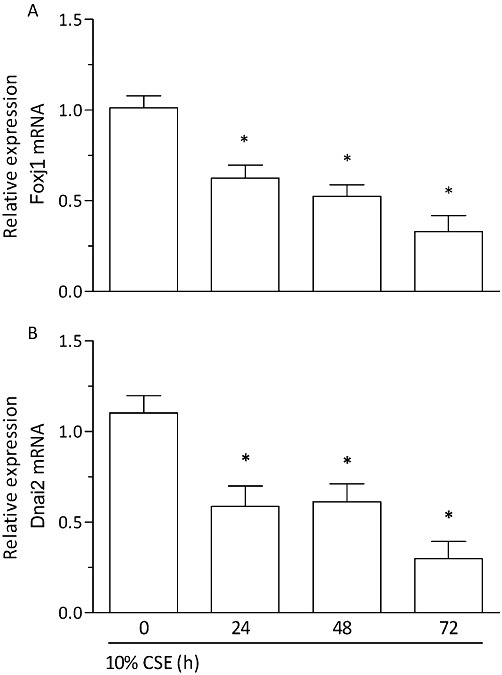
CSE provokes a loss of Foxj1 and Dnai2 expression in differentiated human bronchial epithelial cells (D-BECs). D-BECs were exposed to CSE at 10% or medium (control) for 24, 48 and 72 h and the levels of Foxj1 (A) and Dnai2 (B) mRNA transcripts were quantified by real-time RT-PCR. mRNA expression is evaluated as the x-fold change versus control (medium) at the corresponding time points using the 2−ΔΔCt formula with GAPDH as endogenous standard. Results are shown as the means ± SEM of cultures from two donors and three experiments per donor. *P < 0.05 versus control.
Addition of the PDE4 inhibitor roflumilast N-oxide at 2 nM or 1 µM concentration-dependently prevented the decrease in the expression levels of Foxj1 (Figure 10A) and Dnai2 (Figure 10B) following 3 days of exposure of differentiated bronchial epithelial cells to CSE at 10% with respect to mRNA transcripts and protein. These observations are in line with earlier findings that roflumilast N-oxide partly prevented the loss in cells with cilia motility (Figure 7A) and those labelled with β-tubulin IV (Figure 7B) secondary to exposure of differentiated bronchial epithelial cells to CSE at 10% for 3 days. Roflumilast N-oxide (1 µM) did not affect baseline Foxj1 and Dnai2 in cultures not exposed to tobacco smoke.
Figure 10.
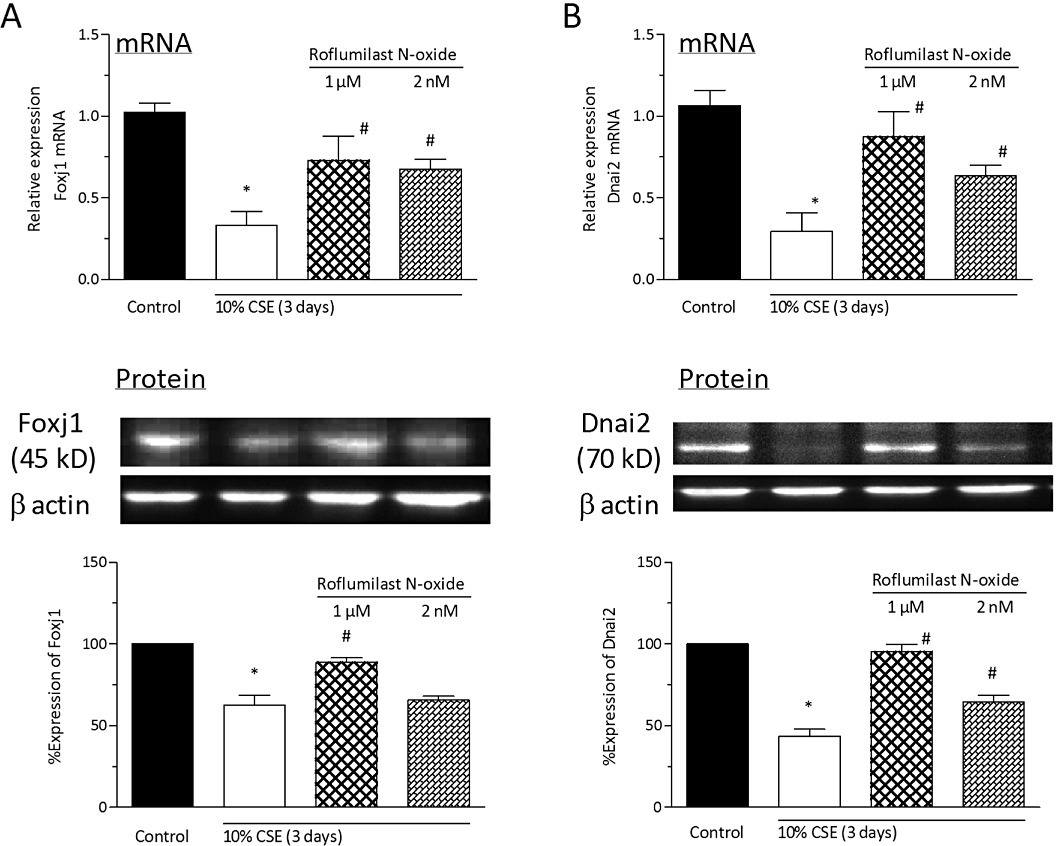
Roflumilast N-oxide reverses the CSE-induced loss in Foxj1 and Dnai2 of ciliated human bronchial epithelial cells. Differentiated human bronchial epithelial cells (D-BECs) were pre-incubated with roflumilast N-oxide at 2 nM or 1 µM, or vehicle for 30 min before being exposed to CSE at 10% for 3 days. Culture medium with fresh CSE and roflumilast N-oxide was replaced every 24 h. Then, the levels of Foxj1 (A) and Dnai2 (B) mRNA transcripts (upper panels) and protein (lower panels) were determined by quantitative real-time RT-PCR or densitometric evaluation of Western blots, respectively. A representative Western blot illustrating the corresponding incubations is depicted as well. Results are expressed relative to control levels (= 1 for mRNA, = 100% for densitometric protein analyses) and shown as the means ± SEM of cultures from two donors and three experiments per donor. *P < 0.05 versus control, #P < 0.05 versus CSE.
Given the documented role of IL-13 in regulating the number of ciliated cells and Foxj1 expression in differentiated bronchial epithelial cells (Turner et al., 2011), the effects of CSE and roflumilast N-oxide on the release of this cytokine were analysed. Exposure of differentiated human bronchial epithelial cells to CSE at 10% for 3 days resulted in a strong increase in IL-13 mRNA and protein. Roflumilast N-oxide at 2 nM and 1 µM partly reduced these effects. Specifically, roflumilast N-oxide diminished the approx. 3.8-fold increment in the release of IL-13 induced by CSE by 34% at 2 nM and by 60% at 1 µM (Figure 11).
Figure 11.
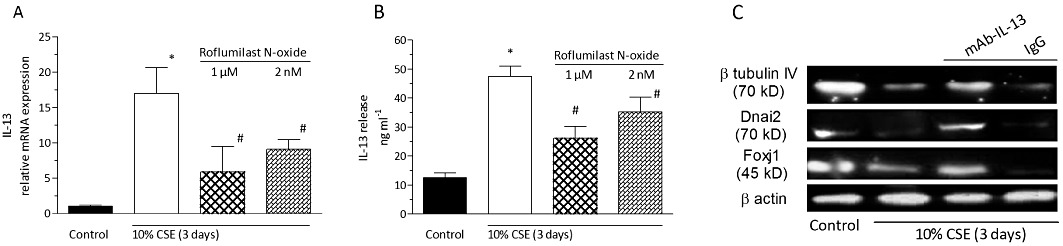
Roflumilast N-oxide inhibits the expression and release of IL-13 provoked by CSE in differentiated human bronchial epithelial cells (D-BECs). D-BECs were pre-incubated with roflumilast N-oxide at 2 nM or 1 µM, or vehicle for 30 min before being exposed to CSE at 10% for 3 days. (A) RNA was isolated from cell extracts to determine the levels of IL-13 transcripts by real-time RT-PCR. The mRNA expression is given as x-fold versus control using the 2−ΔΔCt formula with GAPDH as endogenous standard. (B) IL-13 release in culture supernatants was measured by ELISA. Results are shown as the means ± SEM of cultures from two donors and three experiments per donor. *P < 0.05 versus control, #P < 0.05 versus CSE. An IL-13 neutralizing antibody reversed the CSE-induced loss in β-tubulin IV, Dnai2 and Foxj1. (C) D-BECs were pre-incubated with a human IL-13 neutralizing monoclonal antibody generated in rats (mAb-IL-13) or the isotype control (rat IgG1) both at 150 ng·mL−1 for 30 min before cells were exposed to CSE (10%) for 3 days. At the end of the incubation period, cells were lysed and total protein extracted for Western blot detection of Foxj1, Dnai2 and β tubulin IV with β-actin serving as an internal control. A representative of three independent blots is shown.
In order to further elucidate a potentially critical role of IL-13 in CSE-induced down-regulation of ciliated cell markers, the effects of a monoclonal antibody specifically neutralizing human IL-13 (mAb-IL-13) were explored. The neutralizing antibody partially prevented the reduced expression of Foxj1, Dnai2 and β-tubulin IV following CSE while the isotype control was without effect (Figure 11C).
Discussion
Mucociliary malfunction may contribute to the progressive decline of lung function in COPD (Agusti, 2005). Ciliary beating and the fraction of ciliated cells within the bronchial epithelial cells are among the critical determinants of mucociliary clearance. Tobacco smoking is a major risk factor in COPD.
The current in vitro study in air-liquid cultured, polarized human bronchial epithelial cells reports that CSE triggers a rapid and sustained reduction in CBF and a loss of ciliated cells following long-term exposure. This compromised CBF and ciliated cell phenotype was prevented by roflumilast N-oxide, a PDE4 inhibitor. Indeed, roflumilast N-oxide (2 nM, 1 µM) (i) triggered a rapid (≤10 min) and sustained (at least 3 h) increase in CBF; (ii) reversed the decline in CBF secondary to CSE (10%); and (iii) alleviated the loss in ciliated cells caused by long-term (3 days) exposure to CSE.
In ciliated cells, an increase in cAMP translates into enhanced CBF attributed to PKA-dependent phosphorylation of dynein light chain (Schmid et al., 2006; 2007; Salathe, 2007). Consequently, complete and selective inhibition of PDE4 (1 µM roflumilast N-oxide) acting to enhance cAMP triggered an immediate and persistent rise in CBF. Roflumilast N-oxide at 2 nM [corresponding to the range of therapeutic plasma levels (unbound to plasma proteins) following repeated dosing of roflumilast at the clinical dose of 500 µg·day−1 in man (Bethke et al., 2007) and about 50% inhibition of PDE4 (Hatzelmann and Schudt, 2001)] reproduced the persistent increase in CBF over 3 h; however, to a lesser, about half-maximum extent, and with delayed time course. The reason for this delayed time course is not clear but may be attributed to a time-dependent concentration gradient until equilibrium is attained at the axonemal compartment with the lower concentration.
That PDE4 inhibitors augment airway epithelial CBF has been described previously (Cervin and Lindgren, 1998; Wohlsen et al., 2010), although apparently not in air-liquid-cultured, differentiated human bronchial epithelial cells. In rat precision cut lung slices, the PDE4 inhibitors roflumilast and rolipram increase CBF (Wohlsen et al., 2010) in line with the results from the current study.
Cellular effects emanating from PDE4 inhibition require prior cAMP synthesis. A candidate cAMP increasing molecule released by bronchial epithelial cells into the periciliary lining fluid is adenosine that enhances CBF (Lazarowski and Boucher, 2009; Allen-Gipson et al., 2011). Conversely, antagonism at adenosine receptors (10 µM theophylline) reduced baseline CBF and compromised the increase in CBF by roflumilast N-oxide (2 nM). On the other hand, the PDE4 inhibitor reversed the loss in CBF secondary to theophylline. Therefore, aside from adenosine, other factors maintaining cAMP synthesis may operate and in this context, a role of the soluble, bicarbonate sensitive adenylyl cyclase cannot be excluded (Schmid et al., 2007).
Theophylline (10 µM) is a non-specific antagonist at all adenosine receptors, and in the current study, which of the receptors was involved in the reduction of CBF was not investigated. However, published evidence indicates that of the four adenosine receptors, the A2B receptor is apparently exclusively involved in CBF regulation by adenosine. Firstly, in ciliated bronchial epithelial cells, transcripts of the A2B receptor were more abundant than of A2A or A3 receptors while mRNA encoding for the A1 receptor was virtually absent (Rollins et al., 2008). Secondly, in human nasal epithelial cells, CBF is enhanced by 5′-(N-ethylcarboxamide)adenosine (NECA) but not a selective A2A adenosine receptor agonist (Morse et al., 2001). Thirdly, CBF in tracheal rings from mice deficient in A2B receptors failed to respond to NECA while mice deficient in either A1, A2A or A3 receptors displayed the expected increase in CBF following the non-selective adenosine receptor agonist (Allen-Gipson et al., 2011).
Patients with COPD display compromised CBF (Piatti et al., 2005). CS deteriorates CBF (Ballenger, 1960; Cohen et al., 2009) probably as a result of enhanced axonemal PKCε (Wyatt et al., 2000; Simet et al., 2010). In line with this, PKC activators slow ciliary beating (Salathe, 2007).
In the current study, reduced CBF following CSE (5 min) appeared to decline in parallel with cAMP. Therefore, the notion is raised that aside from PKC activation, a decreased cAMP content may contribute to the compromised CBF following CSE. The CSE-induced rapid loss in cAMP may originate from reduced synthesis, enhanced degradation or efflux. Currently, the exact mechanism remains elusive. For example, cAMP-degrading PDE activities (specifically PDE4) may have been enhanced. Hydrogen peroxide, a component of tobacco smoke (Nakayama et al., 1989), swiftly elevates the activity of PDE4D3 attributed to PI3K and ERK-dependent phosphorylations (Hill et al., 2006). In the current study, the rapid reversal of CSE-induced loss in CBF by forskolin and roflumilast N-oxide, shown to replenish intracellular cAMP (as depicted in Figure 2C), may underpin the role of this cyclic nucleotide in the regulation of CBF in the presence of CSE.
Exposure of differentiated bronchial epithelial cells to CSE for 24 h reduced CBF more markedly than after 3 h. Over this period, CSE enhanced the expression of the NADPH oxidase isoenzymes NOX1, NOX2 and DUOX2, but not DUOX1. Hence, (long-term) up-regulation of NOXs (Figure 6) in addition to the (short-term) increase of ROS (probably as a result of elevated NOX activity) following CSE (Figure 5) may account for the more accentuated reduction of CBF after a 24 h exposure to CSE. Quenching intracellular ROS by NAC or apocynin reversed the CSE (24 h)-induced loss in CBF, underpinning a critical role for ROS in the compromised CBF. Expression of NOX1 (Schneider et al., 2010), NOX2 (Schwarzer et al., 2004; Fink et al., 2008) and DUOX2 (Petry et al., 2010) has been described in airway epithelial cells. Bronchial epithelial NOX1 is up-regulated in COPD (Schneider et al., 2010) and the expression of DUOX2 is increased in smokers (Nagai et al., 2008). In vitro, in polarized human bronchial epithelial cells, DUOX1 transcripts are not affected by exposure to CSE (Lavigne and Eppihimer, 2005) as found here. These authors further demonstrated enhanced intracellular ROS after CSE exposure of bronchial epithelial cells, confirming the current observations.
Roflumilast N-oxide prevented the suppression of CBF induced by 24 h exposure to CSE. As the PDE4 inhibitor diminished the short-term ROS formation (Figure 5) and the expression of NOX isoforms (Figure 6) induced by CSE and given the potential role of ROS to induce the loss in CBF following CSE, one may postulate that inhibition of ROS accounts for the potential of roflumilast N-oxide to reverse the decrease in CBF following CSE.
It is likely that roflumilast N-oxide and dbcAMP diminished the rapid rise in CSE-induced ROS in differentiated bronchial epithelial cells by inhibiting NOX activities. Such an effect has been well documented in neutrophils (Hatzelmann and Schudt, 2001) and was recently reproduced in human pulmonary artery smooth muscle cells where roflumilast N-oxide reduced U46619-triggered rapid ROS release by curbing Rac1 activation, involved in the induction of NOX1- and NOX2-dependent NADPH oxidase activities (Muzaffar et al., 2008). In the same cells, roflumilast N-oxide attenuates the enhanced expression of NOX1 induced by a TXA2 analogue (Muzaffar et al., 2008). Essentially, the ability of this PDE4 inhibitor to suppress ROS formation secondary to CSE in differentiated human bronchial epithelial cells may represent a critical mechanism potentially accounting for the observed reversal of the CSE-induced enhanced expression of NOX isoenzymes. Indeed, this up-regulation was prevented when intracellular ROS was quenched by apocynin or NAC, and earlier others suggested that superoxide itself may elevate the expression of NOX2 in porcine pulmonary artery endothelial cells (Muzaffar et al., 2006).
Earlier studies have shown that in smokers with COPD, there was a rise in PDE4 in alveolar macrophages (Barber et al., 2004). In the current study CSE (24 h) enhanced PDE4 activity and PDE4B expression. Roflumilast N-oxide and dbcAMP reversed this up-regulated PDE4B expression. The latter observation may allude to another level of PDE4 inhibition but appears inconsistent with the view that cAMP elevates PDE4B expression (Conti and Beavo, 2007). However, processes in the current study are probably controlled by ROS because apocynin and NAC prevented the CSE-induced PDE4B up-regulation and roflumilast N-oxide and dbcAMP suppressed ROS secondary to CSE. In support of this notion, a ROS-dependent enhanced expression and activity of PDE4 has recently been unveiled in human umbilical vein endothelial cells (Muzaffar et al., 2012).
The F-box forkhead transcription factor Foxj1 (HFH-4) is a master regulator of the motile ciliogenic programme (Yu et al., 2008). In line with a previous study (Turner et al., 2011), there was a time-dependent increase in Foxj1 mRNA and protein over a 4 week period of air-liquid interface culture from non-differentiated to ciliated bronchial epithelial cells. In parallel, the expression levels of Dnai2 contributing to the outer dynein arm of cilia as well as of the cilia marker β-tubulin IV were gradually enhanced. Mutants of Dnai2 may account for primary ciliary dyskinesia (Loges et al., 2008) supporting the critical role of this axonemal protein for cilia motility.
On the other hand, exposure of the differentiated air-liquid interface cultures to CSE resulted in a time-dependent loss in Foxj1 transcripts that was paralleled by reduced Dnai2 and followed by compromised cilia motility and cells exhibiting the cilia marker β-tubulin IV. Previous work demonstrates an enhanced degree of axonemal ultrastructural abnormalities in smokers and ex-smokers compared with non-smokers (Verra et al., 1995), and CSE was shown to impede the development of ciliated cells (ciliogenesis) in air-liquid interface-cultured mouse nasal epithelial cells (Tamashiro et al., 2009). In mice exposed to tobacco smoke for up to a year, a progressive reduction in ciliated airway epithelial cells over time is observed (Simet et al., 2010). In the current study, roflumilast N-oxide (2 nM, corresponding to therapeutic plasma levels unbound to plasma proteins, and 1 µM) concentration-dependently reversed the CSE-induced loss in Foxj1, Dnai2 and ciliated cells in the air-liquid interface cultures. What might have caused these effects of the PDE4 inhibitor to protect the ciliated cell phenotype from being jeopardized by CSE? In mice, chronic smoke exposure results in increased IL-13 in the airway epithelium associated with deciliated areas (Bartalesi et al., 2005). It was further disclosed that exposure of rats to tobacco smoke over 2–3 months enhances whole lung IL-13 (Cooper et al., 2010). Strikingly, IL-13 transcripts and protein are augmented in lungs of patients afflicted with COPD (Kim et al., 2008) or in the bronchial submucosa of patients with chronic bronchitis (Miotto et al., 2003). In the current study, it was observed that exposure of the differentiated bronchial epithelial cells to CSE up-regulated the expression of IL-13 transcripts and protein, and this effect was gradually decreased by roflumilast N-oxide. Complementing these findings, neutralization of endogenously formed IL-13 using mAb-IL-13 partially prevented the reduced expression of ciliated cell markers (Foxj1, Dnai2, β-tubulin IV) secondary to CSE. On the other hand, there is firm evidence that IL-13 reduces Foxj1 in differentiated human bronchial epithelial cells associated with a loss in ciliated cells (Laoukili et al., 2001; Gomperts et al., 2007; Thavagnanam et al., 2011; Turner et al., 2011). Collectively, these observations suggest that roflumilast N-oxide may have prevented CSE-induced ciliated cell loss and the associated reduction in Foxj1 and Dnai2 by curbing the increased IL-13 release. That PDE4 inhibitors may reduce IL-13 has been demonstrated before. In vivo, roflumilast attenuates the increase in lung and BAL IL-13 following bleomycin in mice that presumably was driven by oxidative stress (Cortijo et al., 2009). In vitro, PDE4 inhibitors such as rolipram decrease the release of IL-13 from human T-cells (Essayan et al., 1997) and basophils (Eskandari et al., 2004).
Finally, one limitation of the current study is that the fate of the ciliated cells undergoing CSE-induced cilia loss has not yet been explored. In the reported experiments, differentiated bronchial epithelial cells exposed to CSE adopted a flattened and elongated phenotype with fragmented cilia. Shortening and shedding of cilia was earlier observed following exposure of mouse nasal epithelial cells to CSE in vitro (Tamashiro et al., 2009). While ciliated cells are usually considered as terminally differentiated (Rawlins et al., 2007), in at least one report it was shown that there may be exceptions, as IL-13 triggers transdifferentiation of ciliated cells into Goblet cells (Turner et al., 2011). This finding may be of interest in the context of the current study given the proposed role of IL-13 in the CSE-induced cilia loss and Foxj1 down-regulation. In any case, the technique employed by Turner & colleagues, that is Cre recombinase-mediated in vitro ciliated cell lineage tagging, may be instrumental in future studies addressing the fate of air-liquid interface-cultured human bronchial epithelial cell subpopulations following their exposure to tobacco smoke extract in the presence or absence of current or future therapeutic agents.
Taken together, the current in vitro study showed that a PDE4 inhibitor, roflumilast N-oxide, was able to partly protect differentiated human bronchial epithelial cells from a reduction in CBF and loss of ciliated cells following exposure to tobacco smoke extract. More studies are required to determine whether these findings may translate in vivo or in patients with COPD.
Acknowledgments
This work was supported by grants SAF2008-03113 (JC) and FIS CP11/00293 (JM) from CICYT (Ministry of Education and Science, Spanish Government) and co-financed by FEDER (European Funds for Regional Development), and by CIBER CB06/06/0027 from Health Institute ‘Carlos III’ of Ministry of Health (Spain), and research grants as Prometeo/2008/045, and AP-178/11 (JM) from Regional Government (‘Generalitat Valenciana’). This work was further supported by a grant from Nycomed GmbH, Konstanz, Germany.
Glossary
- CBF
ciliary beat frequency
- COPD
chronic obstructive pulmonary disease
- CSE
cigarette smoke extract
- DHSV
digital high speed video imaging technique
- Dnai2
axonemal dynein intermediate polypeptide 2
- DUOX
dual oxidase
- Foxj1
forkhead transcription factor 1
- NAC
N-acetyl cysteine
- NOX
NADPH oxidase
- ROS
reactive oxygen species
Conflict of interest
At the time of manuscript submission, RB and HT were employees of Nycomed GmbH: a Takeda Company, Konstanz, Germany. The other authors did not declare a conflict of interest.
References
- Agusti AG. COPD, a multicomponent disease: implications for management. Respir Med. 2005;99:670–682. doi: 10.1016/j.rmed.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Allen-Gipson DS, Blackburn MR, Schneider DJ, Zhang H, Bluitt DL, Jarrell JC, et al. Adenosine activation of A(2B) receptor(s) is essential for stimulated epithelial ciliary motility and clearance. Am J Physiol Lung Cell Mol Physiol. 2011;301:L171–L180. doi: 10.1152/ajplung.00203.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armengot M, Milara J, Mata M, Carda C, Cortijo J. Cilia motility and structure in primary and secondary ciliary dyskinesia. Am J Rhinol Allergy. 2010;24:175–180. doi: 10.2500/ajra.2010.24.3448. [DOI] [PubMed] [Google Scholar]
- Ballenger JJ. Experimental effect of cigarette smoke on human respiratory cilia. N Engl J Med. 1960;263:832–835. doi: 10.1056/NEJM196010272631704. [DOI] [PubMed] [Google Scholar]
- Barber R, Baillie GS, Bergmann R, Shepherd MC, Sepper R, Houslay MD, et al. Differential expression of PDE4 cAMP phosphodiesterase isoforms in inflammatory cells of smokers with COPD, smokers without COPD, and nonsmokers. Am J Physiol Lung Cell Mol Physiol. 2004;287:L332–L343. doi: 10.1152/ajplung.00384.2003. [DOI] [PubMed] [Google Scholar]
- Barnes AP, Livera G, Huang P, Sun C, O'Neal WK, Conti M, et al. Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J Biol Chem. 2005;280:7997–8003. doi: 10.1074/jbc.M407521200. [DOI] [PubMed] [Google Scholar]
- Bartalesi B, Cavarra E, Fineschi S, Lucattelli M, Lunghi B, Martorana PA, et al. Different lung responses to cigarette smoke in two strains of mice sensitive to oxidants. Eur Respir J. 2005;25:15–22. doi: 10.1183/09031936.04.00067204. [DOI] [PubMed] [Google Scholar]
- Bethke TD, Bohmer GM, Hermann R, Hauns B, Fux R, Morike K, et al. Dose-proportional intraindividual single- and repeated-dose pharmacokinetics of roflumilast, an oral, once-daily phosphodiesterase 4 inhibitor. J Clin Pharmacol. 2007;47:26–36. doi: 10.1177/0091270006294529. [DOI] [PubMed] [Google Scholar]
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374:685–694. doi: 10.1016/S0140-6736(09)61255-1. [DOI] [PubMed] [Google Scholar]
- Cervin A, Lindgren S. The effect of selective phosphodiesterase inhibitors on mucociliary activity in the upper and lower airways in vitro. Auris Nasus Larynx. 1998;25:269–276. doi: 10.1016/s0385-8146(98)00010-8. [DOI] [PubMed] [Google Scholar]
- Cohen NA, Zhang S, Sharp DB, Tamashiro E, Chen B, Sorscher EJ, et al. Cigarette smoke condensate inhibits transepithelial chloride transport and ciliary beat frequency. Laryngoscope. 2009;119:2269–2274. doi: 10.1002/lary.20223. [DOI] [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem. 2003;278:5493–5496. doi: 10.1074/jbc.R200029200. [DOI] [PubMed] [Google Scholar]
- Cooper PR, Poll CT, Barnes PJ, Sturton RG. Involvement of IL-13 in tobacco smoke-induced changes in the structure and function of rat intrapulmonary airways. Am J Respir Cell Mol Biol. 2010;43:220–226. doi: 10.1165/rcmb.2009-0117OC. [DOI] [PubMed] [Google Scholar]
- Cortijo J, Villagrasa V, Navarrete C, Sanz C, Berto L, Michel A, et al. Effects of SCA40 on human isolated bronchus and human polymorphonuclear leukocytes: comparison with rolipram, SKF94120 and levcromakalim. Br J Pharmacol. 1996;119:99–106. doi: 10.1111/j.1476-5381.1996.tb15682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortijo J, Iranzo A, Milara X, Mata M, Cerda-Nicolas M, Ruiz-Sauri A, et al. Roflumilast, a phosphodiesterase 4 inhibitor, alleviates bleomycin-induced lung injury. Br J Pharmacol. 2009;156:534–544. doi: 10.1111/j.1476-5381.2008.00041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MK, Sisson JH, Wyatt TA. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am J Respir Cell Mol Biol. 2007;36:452–459. doi: 10.1165/rcmb.2005-0440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandari N, Wickramasinghe T, Peachell PT. Effects of phosphodiesterase inhibitors on interleukin-4 and interleukin-13 generation from human basophils. Br J Pharmacol. 2004;142:1265–1272. doi: 10.1038/sj.bjp.0705892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essayan DM, Kagey-Sobotka A, Lichtenstein LM, Huang SK. Regulation of interleukin-13 by type 4 cyclic nucleotide phosphodiesterase (PDE) inhibitors in allergen-specific human T lymphocyte clones. Biochem Pharmacol. 1997;53:1055–1060. doi: 10.1016/s0006-2952(97)00102-0. [DOI] [PubMed] [Google Scholar]
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, Bundschuh DS, Brose M, Martinez FJ, et al. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet. 2009;374:695–703. doi: 10.1016/S0140-6736(09)61252-6. [DOI] [PubMed] [Google Scholar]
- Fink K, Duval A, Martel A, Soucy-Faulkner A, Grandvaux N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-kappaB in airway epithelial cells. J Immunol. 2008;180:6911–6922. doi: 10.4049/jimmunol.180.10.6911. [DOI] [PubMed] [Google Scholar]
- Gensch E, Gallup M, Sucher A, Li D, Gebremichael A, Lemjabbar H, et al. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNK. J Biol Chem. 2004;279:39085–39093. doi: 10.1074/jbc.M406866200. [DOI] [PubMed] [Google Scholar]
- Gomperts BN, Kim LJ, Flaherty SA, Hackett BP. IL-13 regulates cilia loss and foxj1 expression in human airway epithelium. Am J Respir Cell Mol Biol. 2007;37:339–346. doi: 10.1165/rcmb.2006-0400OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzelmann A, Schudt C. Anti-inflammatory and immunomodulatory potential of the novel PDE4 inhibitor roflumilast in vitro. J Pharmacol Exp Ther. 2001;297:267–279. [PubMed] [Google Scholar]
- Hatzelmann A, Morcillo EJ, Lungarella G, Adnot S, Sanjar S, Beume R, et al. The preclinical pharmacology of roflumilast – a selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2010;23:235–256. doi: 10.1016/j.pupt.2010.03.011. [DOI] [PubMed] [Google Scholar]
- Hill EV, Sheppard CL, Cheung YF, Gall I, Krause E, Houslay MD. Oxidative stress employs phosphatidyl inositol 3-kinase and ERK signalling pathways to activate cAMP phosphodiesterase-4D3 (PDE4D3) through multi-site phosphorylation at Ser239 and Ser579. Cell Signal. 2006;18:2056–2069. doi: 10.1016/j.cellsig.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10:1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14:633–640. doi: 10.1038/nm1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoukili J, Perret E, Willems T, Minty A, Parthoens E, Houcine O, et al. IL-13 alters mucociliary differentiation and ciliary beating of human respiratory epithelial cells. J Clin Invest. 2001;108:1817–1824. doi: 10.1172/JCI13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavigne MC, Eppihimer MJ. Cigarette smoke condensate induces MMP-12 gene expression in airway-like epithelia. Biochem Biophys Res Commun. 2005;330:194–203. doi: 10.1016/j.bbrc.2005.02.144. [DOI] [PubMed] [Google Scholar]
- Lazarowski ER, Boucher RC. Purinergic receptors in airway epithelia. Curr Opin Pharmacol. 2009;9:262–267. doi: 10.1016/j.coph.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loges NT, Olbrich H, Fenske L, Mussaffi H, Horvath J, Fliegauf M, et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am J Hum Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Look DC, Walter MJ, Williamson MR, Pang L, You Y, Sreshta JN, et al. Effects of paramyxoviral infection on airway epithelial cell Foxj1 expression, ciliogenesis, and mucociliary function. Am J Pathol. 2001;159:2055–2069. doi: 10.1016/S0002-9440(10)63057-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata M, Sarria B, Buenestado A, Cortijo J, Cerda M, Morcillo EJ. Phosphodiesterase 4 inhibition decreases MUC5AC expression induced by epidermal growth factor in human airway epithelial cells. Thorax. 2005;60:144–152. doi: 10.1136/thx.2004.025692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto D, Ruggieri MP, Boschetto P, Cavallesco G, Papi A, Bononi I, et al. Interleukin-13 and -4 expression in the central airways of smokers with chronic bronchitis. Eur Respir J. 2003;22:602–608. doi: 10.1183/09031936.03.00046402. [DOI] [PubMed] [Google Scholar]
- Morse DM, Smullen JL, Davis CW. Differential effects of UTP, ATP, and adenosine on ciliary activity of human nasal epithelial cells. Am J Physiol Cell Physiol. 2001;280:C1485–C1497. doi: 10.1152/ajpcell.2001.280.6.C1485. [DOI] [PubMed] [Google Scholar]
- Muzaffar S, Shukla N, Angelini GD, Jeremy JY. Superoxide auto-augments superoxide formation and upregulates gp91(phox) expression in porcine pulmonary artery endothelial cells: inhibition by iloprost. Eur J Pharmacol. 2006;538:108–114. doi: 10.1016/j.ejphar.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Muzaffar S, Shukla N, Angelini GD, Jeremy JY. Roflumilast N-oxide inhibits NADPH oxidase expression and activity in human pulmonary artery smooth muscle cells. Proc Br Pharmacol Soc. 2008;6:027P. [Google Scholar]
- Muzaffar S, Shukla N, Angelini GD, Jeremy JY. NADPH oxidase 4 mediates upregulation of type 4 phosphodiesterases in human endothelial cells. J Cell Physiol. 2012;227:1941–1950. doi: 10.1002/jcp.22922. [DOI] [PubMed] [Google Scholar]
- Nagai K, Betsuyaku T, Suzuki M, Nasuhara Y, Kaga K, Kondo S, et al. Dual oxidase 1 and 2 expression in airway epithelium of smokers and patients with mild/moderate chronic obstructive pulmonary disease. Antioxid Redox Signal. 2008;10:705–714. doi: 10.1089/ars.2007.1941. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Church DF, Pryor WA. Quantitative analysis of the hydrogen peroxide formed in aqueous cigarette tar extracts. Free Radic Biol Med. 1989;7:9–15. doi: 10.1016/0891-5849(89)90094-4. [DOI] [PubMed] [Google Scholar]
- Olseni L, Wollmer P. Mucociliary clearance in healthy men at rest and during exercise. Clin Physiol. 1990;10:381–387. doi: 10.1111/j.1475-097x.1990.tb00798.x. [DOI] [PubMed] [Google Scholar]
- Ortiz JL, Milara J, Juan G, Montesinos JL, Mata M, Ramon M, et al. Direct effect of cigarette smoke on human pulmonary artery tension. Pulm Pharmacol Ther. 2010;23:222–228. doi: 10.1016/j.pupt.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Petry A, Weitnauer M, Gorlach A. Receptor activation of NADPH oxidases. Antioxid Redox Signal. 2010;13:467–487. doi: 10.1089/ars.2009.3026. [DOI] [PubMed] [Google Scholar]
- Piatti G, Ambrosetti U, Santus P, Allegra L. Effects of salmeterol on cilia and mucus in COPD and pneumonia patients. Pharmacol Res. 2005;51:165–168. doi: 10.1016/j.phrs.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Rawlins EL, Ostrowski LE, Randell SH, Hogan BL. Lung development and repair: contribution of the ciliated lineage. Proc Natl Acad Sci U S A. 2007;104:410–417. doi: 10.1073/pnas.0610770104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins BM, Burn M, Coakley RD, Chambers LA, Hirsh AJ, Clunes MT, et al. A2B adenosine receptors regulate the mucus clearance component of the lung's innate defense system. Am J Respir Cell Mol Biol. 2008;39:190–197. doi: 10.1165/rcmb.2007-0450OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salathe M. Regulation of mammalian ciliary beating. Annu Rev Physiol. 2007;69:401–422. doi: 10.1146/annurev.physiol.69.040705.141253. [DOI] [PubMed] [Google Scholar]
- Salathe M, Pratt MM, Wanner A. Protein kinase C-dependent phosphorylation of a ciliary membrane protein and inhibition of ciliary beating. J Cell Sci. 1993;106((Pt 4)):1211–1220. doi: 10.1242/jcs.106.4.1211. [DOI] [PubMed] [Google Scholar]
- Schmid A, Bai G, Schmid N, Zaccolo M, Ostrowski LE, Conner GE, et al. Real-time analysis of cAMP-mediated regulation of ciliary motility in single primary human airway epithelial cells. J Cell Sci. 2006;119((Pt 20)):4176–4186. doi: 10.1242/jcs.03181. [DOI] [PubMed] [Google Scholar]
- Schmid A, Sutto Z, Nlend MC, Horvath G, Schmid N, Buck J, et al. Soluble adenylyl cyclase is localized to cilia and contributes to ciliary beat frequency regulation via production of cAMP. J Gen Physiol. 2007;130:99–109. doi: 10.1085/jgp.200709784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D, Ganesan S, Comstock AT, Meldrum CA, Mahidhara R, Goldsmith AM, et al. Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:332–340. doi: 10.1164/rccm.200911-1673OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer C, Machen TE, Illek B, Fischer H. NADPH oxidase-dependent acid production in airway epithelial cells. J Biol Chem. 2004;279:36454–36461. doi: 10.1074/jbc.M404983200. [DOI] [PubMed] [Google Scholar]
- Simet SM, Sisson JH, Pavlik JA, Devasure JM, Boyer C, Liu X, et al. Long-term cigarette smoke exposure in a mouse model of ciliated epithelial cell function. Am J Respir Cell Mol Biol. 2010;43:635–640. doi: 10.1165/rcmb.2009-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SP, Barrett EG, Kalra R, Razani-Boroujerdi S, Langley RJ, Kurup V, et al. Prenatal cigarette smoke decreases lung cAMP and increases airway hyperresponsiveness. Am J Respir Crit Care Med. 2003;168:342–347. doi: 10.1164/rccm.200211-1262OC. [DOI] [PubMed] [Google Scholar]
- Sisson JH, Papi A, Beckmann JD, Leise KL, Wisecarver J, Brodersen BW, et al. Smoke and viral infection cause cilia loss detectable by bronchoalveolar lavage cytology and dynein ELISA. Am J Respir Crit Care Med. 1994;149:205–213. doi: 10.1164/ajrccm.149.1.8111584. [DOI] [PubMed] [Google Scholar]
- Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1998;19:819–825. doi: 10.1165/ajrcmb.19.5.3091. [DOI] [PubMed] [Google Scholar]
- Tamashiro E, Xiong G, Anselmo-Lima WT, Kreindler JL, Palmer JN, Cohen NA. Cigarette smoke exposure impairs respiratory epithelial ciliogenesis. Am J Rhinol Allergy. 2009;23:117–122. doi: 10.2500/ajra.2009.23.3280. [DOI] [PubMed] [Google Scholar]
- Thavagnanam S, Parker JC, McBrien ME, Skibinski G, Heaney LG, Shields MD. Effects of IL-13 on mucociliary differentiation of pediatric asthmatic bronchial epithelial cells. Pediatr Res. 2011;69:95–100. doi: 10.1203/PDR.0b013e318204edb5. [DOI] [PubMed] [Google Scholar]
- Thompson WJ, Brooker G, Appleman MM. Assay of cyclic nucleotide phosphodiesterases with radioactive substrates. Methods Enzymol. 1974;38:205–212. doi: 10.1016/0076-6879(74)38033-0. [DOI] [PubMed] [Google Scholar]
- Trayner ID, Rayner AP, Freeman GE, Farzaneh F. Quantitative multiwell myeloid differentiation assay using dichlorodihydrofluorescein diacetate (H2DCF-DA) or dihydrorhodamine 123 (H2R123) J Immunol Methods. 1995;186:275–284. doi: 10.1016/0022-1759(95)00152-z. [DOI] [PubMed] [Google Scholar]
- Turner J, Roger J, Fitau J, Combe D, Giddings J, Heeke GV, et al. Goblet cells are derived from a FOXJ1-expressing progenitor in a human airway epithelium. Am J Respir Cell Mol Biol. 2011;44:276–284. doi: 10.1165/rcmb.2009-0304OC. [DOI] [PubMed] [Google Scholar]
- Verra F, Escudier E, Lebargy F, Bernaudin JF, De Cremoux H, Bignon J. Ciliary abnormalities in bronchial epithelium of smokers, ex-smokers, and nonsmokers. Am J Respir Crit Care Med. 1995;151((3 Pt 1)):630–634. doi: 10.1164/ajrccm/151.3_Pt_1.630. [DOI] [PubMed] [Google Scholar]
- Wanner A, Salathe M, O'Riordan TG. Mucociliary clearance in the airways. Am J Respir Crit Care Med. 1996;154((6 Pt 1)):1868–1902. doi: 10.1164/ajrccm.154.6.8970383. [DOI] [PubMed] [Google Scholar]
- Wohlsen A, Hirrle A, Tenor H, Marx D, Beume R. Effect of cyclic AMP-elevating agents on airway ciliary beat frequency in central and lateral airways in rat precision-cut lung slices. Eur J Pharmacol. 2010;635:177–183. doi: 10.1016/j.ejphar.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Schmidt SC, Rennard SI, Tuma DJ, Sisson JH. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc Soc Exp Biol Med. 2000;225:91–97. doi: 10.1046/j.1525-1373.2000.22511.x. [DOI] [PubMed] [Google Scholar]
- Yu X, Ng CP, Habacher H, Roy S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat Genet. 2008;40:1445–1453. doi: 10.1038/ng.263. [DOI] [PubMed] [Google Scholar]