

Abstract
BACKGROUND AND PURPOSE
The use of ±3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) is associated with cardiovascular complications and hyperthermia.
EXPERIMENTAL APPROACH
We assessed the effects of the α1- and β-adrenoceptor antagonist carvedilol on the cardiostimulant, thermogenic and subjective responses to MDMA in 16 healthy subjects. Carvedilol (50 mg) or placebo was administered 1 h before MDMA (125 mg) or placebo using a randomized, double-blind, placebo-controlled, four-period crossover design.
KEY RESULTS
Carvedilol reduced MDMA-induced elevations in blood pressure, heart rate and body temperature. Carvedilol did not affect the subjective effects of MDMA including MDMA-induced good drug effects, drug high, drug liking, stimulation or adverse effects. Carvedilol did not alter the plasma exposure to MDMA.
CONCLUSIONS AND IMPLICATIONS
α1- and β-Adrenoceptors contribute to the cardiostimulant and thermogenic effects of MDMA in humans but not to its psychotropic effects. Carvedilol could be useful in the treatment of cardiovascular and hyperthermic complications associated with ecstasy use.
Keywords: MDMA; 3,4-methylenedioxymethamphetamine; ecstasy; noradrenaline; carvedilol; α- and β-adrenoceptors
Introduction
±3,4-Methylenedioxymethamphetamine (MDMA, ‘ecstasy’) is widely abused for its euphoric effects. The use of ecstasy is associated with hyperthermia (Henry et al., 1992; Liechti et al., 2005; Halpern et al., 2011). MDMA-induced hyperthermia is a life-threatening disorder that may lead to rhabdomyolysis, disseminated intravascular coagulation, acute hepatic and renal failure and death (Henry et al., 1992; Liechti et al., 2005). Severe hyperthermia has typically been observed when ecstasy is used in crowded clubs, at high ambient temperatures or during physical activity (Henry et al., 1992; Parrott, 2012). In laboratory animals, crowding, high ambient temperature, reduced water consumption and repeated dosing similarly enhanced MDMA-induced hyperthermia (Dafters, 1995; Docherty and Green, 2010). However, MDMA also elevates body temperature under controlled laboratory conditions in humans in the absence of permissive factors (Liechti et al., 2001; Freedman et al., 2005; Dumont and Verkes, 2006; Parrott, 2012). The clinical treatment of sympathomimetic amphetamine toxicity is mainly supportive and includes volume repletion and sedation with benzodiazepines (Liechti et al., 2005; Halpern et al., 2011). The management of severe MDMA-related hyperpyrexia includes cooling and ventilation (Hall and Henry, 2006). Dantrolene, which acts peripherally at skeletal muscles to inhibit release of calcium from the sarcoplasmic reticulum, has also been used (Green et al., 1995; Hall and Henry, 2006; Grunau et al., 2010). However, dantrolene does not inhibit the thermogenic effects of MDMA (Rusyniak et al., 2004) and the drug does not specifically interfere with the presumed mechanism of MDMA-induced hyperthermia. MDMA mainly releases 5-HT, NA and dopamine (Rudnick and Wall, 1992; Liechti and Vollenweider, 2001; Verrico et al., 2007). Stimulation of both α1- and β3-adrenoceptors has been implicated in the thermogenic effects of MDMA (Sprague et al., 2004a; 2005). Specifically, increasing NA levels through the inhibition of phenylethanolamine N-methyltransferase potentiated the hyperthermic effects of MDMA in rats (Sprague et al., 2007). Combined pretreatment with the α1-adrenoceptor antagonist prazosin plus the β3-adrenoceptor antagonist SR59230A attenuated MDMA-induced elevations in core body temperature and creatine kinase levels in rats (Sprague et al., 2004a). The α1 and β1,2,3 antagonist carvedilol similarly prevented the hyperthermic response to MDMA in rats (Sprague et al., 2005). Moreover, carvedilol reversed established hyperthermia when it was administered 1 h after MDMA (Sprague et al., 2005). Selective inhibition of β3 receptors with low concentrations of SR59230A attenuated the slowly developing late hyperthermic response to MDMA, suggesting a role for β3 receptors in this late response in mice (Bexis and Docherty, 2008). In contrast, α1 blockade with prazosin induced an early hypothermic reaction to MDMA, consistent with a role for α1-receptors in this early response to MDMA in mice (Bexis and Docherty, 2008). Finally, mice deficient in uncoupling protein 3, which is regulated by NA, were protected against the hyperthermic effects of MDMA (Mills et al., 2003) and methamphetamine (Sprague et al., 2004b). Altogether, the preclinical data suggest that MDMA-induced hyperthermia results from noradrenergic activation of mitochondrial uncoupling that involves both α1- and β3-adrenoceptors (Mills et al., 2004; Rusyniak et al., 2005). Additionally, α1-receptors contribute to the vasoconstriction of skin blood vessels, impairing heat dissipation, which enhances hyperthermia induced by MDMA (Pedersen and Blessing, 2001).
Psychostimulants, including MDMA, also produce hypertension and tachycardia. Myocardial ischaemia and stroke are complications of the sympathomimetic action of cocaine and ecstasy (Brody et al., 1990; Liechti et al., 2005; Bruggisser et al., 2010; Halpern et al., 2011). Selective β-adrenoceptor blockers are commonly used in the treatment of myocardial infarction or acute hypertension but are not recommended if psychostimulants are involved because of the risk of unopposed α1-receptor stimulation (Hoffman, 2008). Indeed, propranolol potentiated cocaine-induced coronary vasoconstriction (Lange et al., 1990) and worsened cocaine-associated hypertension (Ramoska and Sacchetti, 1985). β blockade also did not affect the blood pressure response to MDMA (Hysek et al., 2010). In contrast, α- and β-adrenoceptor blockade with labetalol (Boehrer et al., 1993; Sofuoglu et al., 2000b) and carvedilol (Sofuoglu et al., 2000a) dose-dependently prevented the haemodynamic response to cocaine in humans. Labetalol also had no negative effect on cocaine-induced coronary vasoconstriction (Boehrer et al., 1993). Combined α- and β-blockers may therefore be the treatment of choice for stimulant-associated hypertension and myocardial ischaemia.
Because carvedilol has been shown to prevent MDMA-induced hyperthermia and rhabdomyolysis in rats (Sprague et al., 2005) and the cardiostimulant response to cocaine in humans (Sofuoglu et al., 2000a), we evaluated the effects of carvedilol on the cardiovascular and hyperthermic response to MDMA in healthy subjects.
Methods
Study design
We used a double-blind, double-dummy placebo-controlled, randomized, crossover study design with four experiential conditions (placebo-placebo, carvedilol-placebo, placebo-MDMA and carvedilol-MDMA) in a balanced order. The washout periods between the sessions were at least 10 days. The study was conducted at the University Hospital of Basel in accordance with the Declaration of Helsinki and International Conference on Harmonization Guidelines on Good Clinical Practice and approved by the Ethics Committee of the Canton of Basel, Switzerland, and Swiss Agency for Therapeutic Products (Swissmedic). The use of MDMA in healthy subjects was authorized by the Swiss Federal Office of Public Health. The study was registered at ClinicalTrials.gov (NCT01270672). The reduction in the MDMA-induced increase in blood pressure by carvedilol was the predefined primary outcome of this clinical trial.
Study procedures
The subjects completed a screening visit, four test sessions and an end-of-study visit. The test sessions were conducted in a quiet hospital research ward with no more than two research subjects present per session. The mean (SD) room temperature was 23.3°C (0.7°C). At the beginning of each test session, an indwelling i.v. catheter was placed in the antecubital vein for blood sampling. Carvedilol (50 mg) or placebo was administered at 8 h 00 min. MDMA (125 mg) or placebo was administered at 9 h 00 min. A standardized lunch was served at 12 h 00 min, and the subjects were sent home at 15 h 00 min.
Subjects
Sixteen healthy subjects (eight men, eight women) with a mean (SD) age of 24.2 (2.2) years and a mean body weight of 67 (13) kg were recruited from the university campus. The allocation to treatment order was performed by drawing from blocks of eight different balanced drug treatment sequences by two pharmacists not involved in the study. Each code was stored in a sealed envelope until the termination of the study. Data from all 16 subjects were available for the final analysis. The exclusion criteria included the following: (i) age <18 or >45 years; (ii) pregnancy determined by a urine test before each test session; (iii) body mass index <18.5 kg·m−2 or >25 kg·m−2; (iv) personal or family (first-degree relative) history of psychiatric disorder [determined by the structured clinical interview for Axis I and Axis II disorders according to the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (Wittchen et al., 1997) supplemented by the SCL-90-R Symptom Checklist (Derogatis et al., 1976; Schmitz et al., 2000)]; (v) regular use of medications; (vi) chronic or acute physical illness assessed by physical examination, electrocardiogram, standard haematology and chemical blood analyses; (vii) smoking more than seven cigarettes per day; (viii) a lifetime history of using illicit drugs more than five times, with the exception of cannabis; (ix) illicit drug use within the last 2 months; and (x) illicit drug use during the study, determined by urine tests conducted before the test sessions using TRIAGE 8 (Biosite, San Diego, CA, USA). The subjects were asked to abstain from excessive alcohol consumption between test sessions and limit alcohol use to one glass on the day before each test session. All of the subjects were non-smokers. All of the subjects, with the exception of one, had previously used cannabis. Four subjects reported using illicit drugs, in which three subjects had tried amphetamine once and one had tried ecstasy once and amphetamine three times. All of the subjects were phenotyped for cytochrome P450 (CYP) 2D6 activity using dextromethorphan as the probe drug. Nine extensive, six intermediate and one poor CYP2D6 metabolizer were identified in the study. The female subjects were investigated during the follicular phase (day 2–14) of their menstrual cycle when the reactivity to amphetamines is expected to be similar to men (White et al., 2002). All of the subjects provided their written informed consent before participating in the study, and they were paid for their participation.
Drugs
±MDMA hydrochloride (Lipomed AG, Arlesheim, Switzerland) was prepared as gelatine capsules (100 and 25 mg of the salt). Identical placebo (lactose) capsules were prepared. MDMA was administered in a single oral dose of 125 mg, corresponding to a dose of 1.93 ± 0.36 mg·kg−1 body weight. Carvedilol tablets (50 mg, Dilatrend, Roche Pharma AG, Basel, Switzerland) were encapsulated within opaque gelatine capsules, and identical placebo (lactose) capsules were prepared. An oral dose of carvedilol (50 mg) was used that has previously been shown to attenuate the smoked cocaine-induced increases in heart rate and blood pressure in humans (Sofuoglu et al., 2000a). At this dose, carvedilol is expected to inhibit both α1- and β-adrenoceptors (Tham et al., 1995; Sofuoglu et al., 2000a). Carvedilol or placebo was administered 1 h before MDMA or placebo administration so that the maximal plasma concentration (Cmax) of carvedilol was reached (Morgan, 1994) shortly before the Cmax of MDMA occurred. Oral medication administration was supervised by study personnel.
Pharmacodynamic measurements
Vital signs
Vital signs were assessed repeatedly 1 h before and 0, 0.33, 0.66, 1, 1.5, 2, 2.5, 3, 4, 5 and 6 h after MDMA or placebo administration. Heart rate, systolic blood pressure and diastolic blood pressure were measured using an OMRON M7 blood pressure monitor (Omron Healthcare Europe, Hoofddorp, The Netherlands) in the dominant arm after a resting time of 5 min. Measures were taken twice per time point with an interval of 1 min, and the average was used for analysis. Core (tympanic) temperature was assessed using a GENIUS 2 ear thermometer (Tyco Healthcare Group, Watertown, NY, USA).
Plasma catecholamines
Blood samples to determine the concentrations of NA and adrenaline were taken 1 h before and 1 and 2 h after MDMA or placebo administration. All of the blood samples were collected on ice and centrifuged within 10 min at 4°C. The plasma was then stored at −20°C until analysis. The plasma levels of free catecholamines (NA and adrenaline) were determined by HPLC with an electrochemical detector as described previously (Hysek et al., 2011).
Psychometric scales
Subjective measures were repeatedly assessed using Visual Analogue Scales (VASs; (Hysek et al., 2011) 1 h before and 0, 0.33, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5 and 6 h after MDMA or placebo administration. The VASs included ‘any drug effect’, ‘good drug effect’, ‘bad drug effect’, ‘drug liking’, ‘drug high’ and ‘stimulated’ (Farre et al., 2007; Kolbrich et al., 2008; Hysek et al., 2011). The VASs were presented as 100-mm horizontal lines marked ‘not at all’ on the left and ‘extremely’ on the right. Additionally, the 5-Dimensions of Altered States of Consciousness Scale [5D-ASC; (Dittrich, 1998; Studerus et al., 2010)] was applied 4 h after MDMA or placebo administration. The 5D-ASC rating scale measures alterations in mood, perception and experience of self in relation to the environment and thought disorder (Studerus et al., 2010). The 5D-ASC dimension ‘oceanic boundlessness’ (27 items) measures derealization and depersonalization associated with positive mood. The dimension ‘anxious ego dissolution’ (21 items) summarizes ego disintegration and loss of self-control, phenomena associated with anxiety. The dimension ‘visionary restructuralization’ (18 items) describes perceptual alterations. Two other dimensions of the scale were not used in our study. The total ASC score was determined by adding the scores of the three dimensions.
Adverse effects
Adverse effects were assessed 1 h before and 3 and 24 h after MDMA or placebo administration using the List of Complaints (Zerssen, 1976; Hysek et al., 2011). The scale consists of 66 items that yield a total adverse effects score, reliably measuring physical and general discomfort.
Pharmacokinetic measurements
Samples of plasma for the determination of MDMA and ±3,4-methylenedioxyamphetamine (MDA), the active metabolite of MDMA, were collected 1 h before and 0 (just before), 0.33, 0.66, 1, 1.5, 2, 2.5, 3, 4 and 6 h after MDMA or placebo administration. The plasma concentrations of MDMA and MDA were determined using HPLC coupled to tandem MS as described previously (Hysek et al., 2012).
Data analysis
Pharmacokinetic analysis
The data for the plasma concentrations of MDMA and MDA were analysed using non-compartmental methods. Cmax and time to Cmax were obtained directly from the concentration–time curves of the observed values. The area under the plasma concentration–time curve (AUC)0–6 h was calculated using the linear trapezoidal rule. Plasma concentrations were only determined up to 6 h after MDMA administration because the aim of the study was to assess potential changes in plasma levels of MDMA during the time of the pharmacodynamic effects of MDMA.
Statistical analysis
Values were transformed to differences from baseline. The maximal effect (Emax) values were determined for repeated measures and analysed by two-way General Linear Models repeated-measures anova with the two drug factors MDMA (MDMA vs. placebo) and carvedilol (carvedilol vs. placebo) using STATISTICA 6.0 software (StatSoft, Tulsa, OK, USA). Tukey's post hoc comparisons were performed based on significant main effects or interactions. Additional anovas were performed, with drug order as an additional factor, to exclude carry-over effects. The criterion for significance was P < 0.05. A sample-size estimation based on previous data (Hysek et al., 2011; 2012) showed that eight subjects would be needed to detect a relevant change in the primary study outcome with 80% power using a within-subjects study design.
Results
Vital signs and circulating catecholamines
MDMA significantly increased blood pressure, heart rate and body temperature compared with placebo (Table 1 and Figure 1). Carvedilol significantly inhibited the MDMA-induced increases in blood pressure, heart rate and body temperature (Table 1 and Figure 1). Carvedilol alone also moderately lowered blood pressure and heart rate compared with placebo. The effect of carvedilol on the pressure and hyperthermic response to MDMA was more pronounced than the effect of carvedilol alone compared with placebo, corroborated by the significant carvedilol × MDMA interaction in the two-way anova. Carvedilol alone increased the plasma concentration of NA compared with placebo. MDMA also tended to increase circulating NA compared with placebo, but the effect was not significant. The co-administration of carvedilol and MDMA significantly increased both circulating adrenaline and NA (Table 1 and Figure 2).
Table 1.
Values and statistics of pharmacodynamic changes
| Placebo-placebo | Carvedilol-placebo | Placebo-MDMA | Carvedilol-MDMA | MDMA | Carvedilol | Carvedilol × MDMA | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean (SEM) | F1,15 | P | F1,15 | P | F1,15 | P | ||||
| Physiological effects | ||||||||||
| Systolic blood pressure (mmHg) | 4.7 (1.8) | −8.1 (2.0)***### | 28.1 (3.2)* | 6.5 (2.2)### | 59.30 | <0.001 | 72.33 | <0.001 | 5.02 | <0.05 |
| Diastolic blood pressure (mmHg) | −1.0 (1.4) | −8.1 (1.6)*### | 15.3 (1.6)* | 9.3 (1.9)***# | 151.10 | <0.001 | 16.40 | <0.001 | 1.70 | NS |
| Heart rate (beats min−1) | 5.8 (3.0) | −5.0 (2.5)*### | 26.2 (3.9)* | 5.5 (3.0)### | 15.44 | <0.001 | 38.84 | <0.001 | 18.64 | <0.001 |
| Body temperature (°C) | 0.24 (0.06) | 0.32 (0.06)## | 0.69 (0.10)* | 0.40 (0.06)# | 13.78 | <0.01 | 3.29 | NS | 7.65 | <0.05 |
| Circulating catecholamines | ||||||||||
| Adrenaline (nM) | −0.03 (0.03) | 0.08 (0.05) | 0.23 (0.06) | 0.70 (0.15)***### | 19.63 | <0.001 | 14.04 | <0.01 | 14.20 | <0.01 |
| Noradrenaline (nM) | −0.34 (0.14) | 1.85 (0.36)***## | 0.29 (0.14) | 2.58 (0.40)***### | 4.33 | 0.055 | 59.86 | <0.001 | 0.04 | NS |
| Visual Analogue Scale (%max) | ||||||||||
| Any drug effect | 2.4 (1.4) | 7.1 (3.4)### | 64.8 (7.5)* | 69.6 (7.6)* | 94.67 | <0.001 | 1.50 | NS | 0.00 | NS |
| Good drug effect | 1.4 (1.4) | 0.0 (0.0)### | 71.1 (7.6)* | 76.8 (7.2)* | 112.69 | <0.001 | 0.40 | NS | 1.01 | NS |
| Bad drug effect | 0.3 (0.3) | 2.5 (1.1) | 13.6 (5.1) | 25.3 (9.1)* | 13.70 | <0.01 | 1.51 | NS | 0.92 | NS |
| Drug liking | 1.6 (1.4) | 0.0 (0.0)### | 74.8 (7.1)* | 75.9 (7.9)* | 106.20 | <0.001 | 0.01 | NS | 0.20 | NS |
| Drug high | 1.7 (1.7) | 0.0 (0.0)### | 59.4 (9.0)* | 66.3 (8.7)* | 56.46 | <0.001 | 0.49 | NS | 1.15 | NS |
| Stimulated | 2.0 (2.0) | 0.4 (0.4)### | 57.8 (9.2)* | 61.7 (8.9)* | 47.78 | <0.001 | 0.10 | NS | 0.38 | NS |
| 5D-ASC Scale | ||||||||||
| Total ASC score | 8.7 (8.7) | 0.0 (0.0)## | 747 (177)* | 894 (227)* | 20.06 | <0.001 | 0.55 | NS | 0.68 | NS |
| Oceanic boundlessness | 7.6 (7.6) | 0.0 (0.0)### | 436 (119)* | 531 (152)* | 13.91 | <0.01 | 0.86 | NS | 1.12 | NS |
| Anxious ego dissolution | 0.7 (0.7) | 0.0 (0.0)# | 192 (75)* | 161 (55) | 13.53 | <0.01 | 0.12 | NS | 0.11 | NS |
| Visionary restructuralization | 0.4 (0.4) | 0.0 (0.0)# | 119 (33)* | 202 (54)* | 16.39 | <0.001 | 3.88 | NS | 3.94 | 0.07 |
| List of complaints (total score) | ||||||||||
| Acute adverse effects (at 3 h) | −0.2 (0.3) | 0.9 (0.4)### | 8.4 (1.5)* | 9.9 (2.0)* | 46.96 | <0.001 | 0.37 | NS | 0.08 | NS |
| Subacute adverse effects (at 24 h) | 0.1 (0.3) | 1.1 (0.8) | 5.3 (1.6)* | 4.9 (1.5)* | 25.96 | <0.001 | 0.08 | NS | 0.47 | NS |
Values are expressed as mean (SEM) changes from baseline of 16 subjects. ASC, Altered States of Consciousness; NS, not significant.
*P < 0.05, **P < 0.01, ***P < 0.001, compared with placebo-placebo. #P < 0.05, ##P < 0.01, ###P < 0.001, compared with placebo-MDMA.
Figure 1.
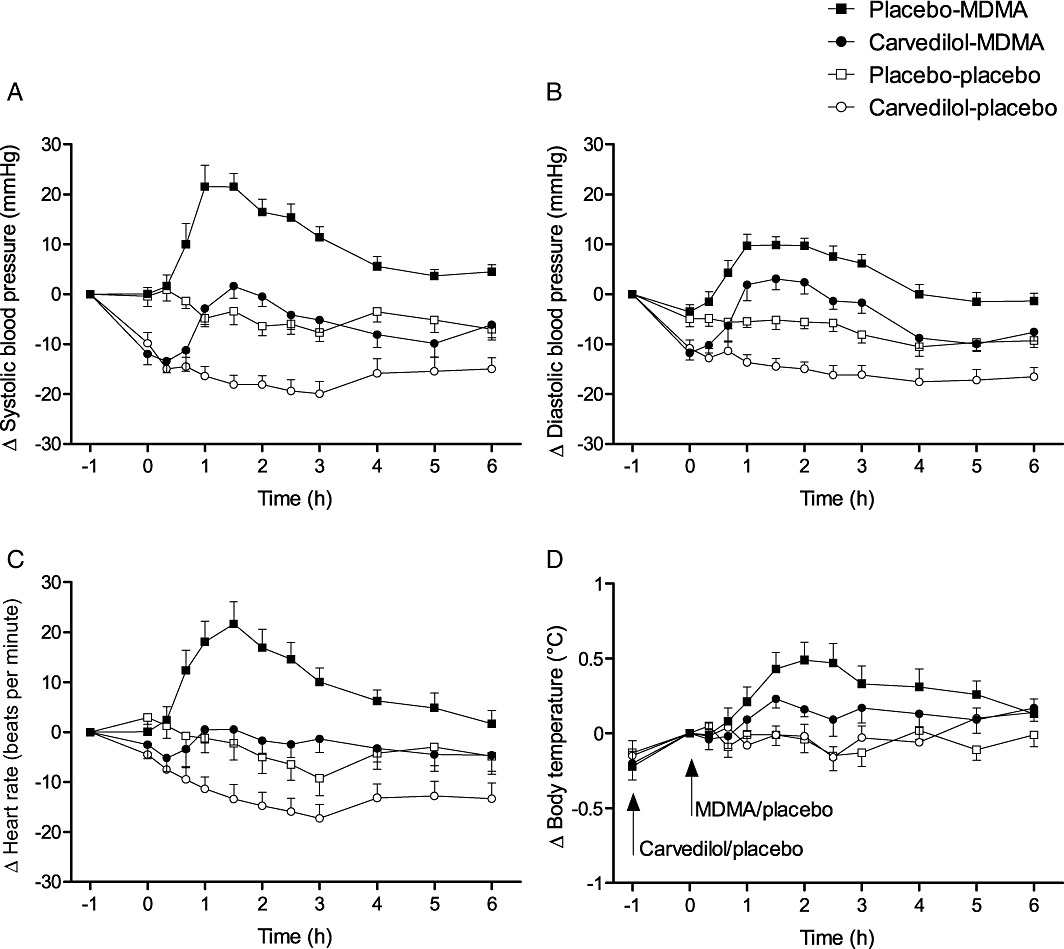
Physiological effects of carvedilol and MDMA. Carvedilol reduced MDMA-induced elevations in systolic (A) and diastolic (B) blood pressure, heart rate (C) and body temperature (D). Carvedilol was administered at t =−1 h. MDMA was administered at t = 0 h. The values are expressed as mean ± SEM changes from baseline in 16 subjects.
Figure 2.
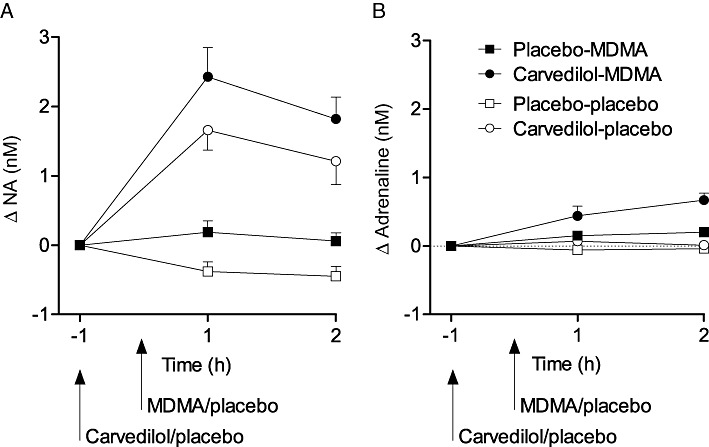
Effects of carvedilol and MDMA on circulating catecholamines. Carvedilol alone increased the plasma levels of noradrenaline (A) compared with placebo. MDMA alone produced a similar non-significant increase in noradrenaline. Co-administration of carvedilol and MDMA increased the concentrations of circulating noradrenaline (A) and adrenaline (B) compared with placebo. The values are expressed as mean ± SEM changes from baseline in 16 subjects.
Subjective effects
Carvedilol did not affect the psychotropic response to MDMA. It did not alter the pronounced MDMA-induced increases in the VAS (Table 1 and Figure 3) or 5D-ASC ratings of subjective drug effects (Table 1). Carvedilol alone had no subjective effects.
Figure 3.
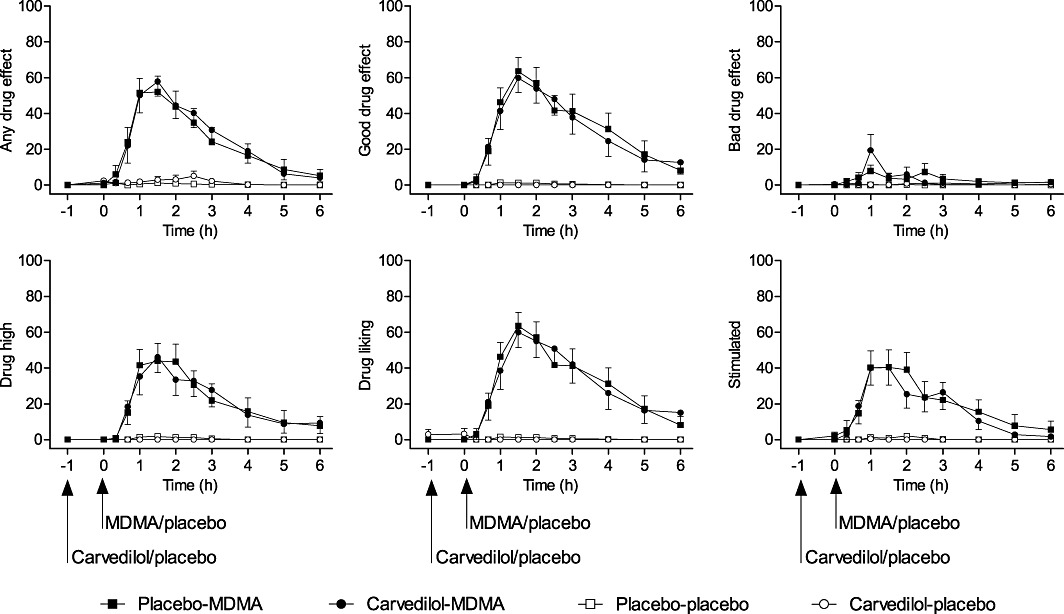
Time course of subjective drug effects on Visual Analogue Scale ratings. MDMA increased scores on all scales. Carvedilol did not affect any of the MDMA-induced increases in Visual Analogue Scale ratings. Carvedilol was administered at t =−1 h. MDMA was administered at t = 0 h. The values are expressed as mean ± SEM percentage of maximal values in 16 subjects.
Adverse effects
MDMA increased the total adverse effect score on the List of Complaints, both 3 and 24 h after drug administration compared with placebo (Table 1). Carvedilol had no effect on the MDMA-induced increase in the total score. However, fewer subjects reported palpitations and hot flushes after carvedilol and MDMA co-treatment (n= 2 and n= 2, respectively) compared with MDMA treatment alone (n= 6 and n= 5, respectively). Frequent adverse effects of MDMA and carvedilol-MDMA were thirst (n= 10 and n= 11, respectively), lack of appetite (n= 9 and n= 7, respectively), sweating (n= 8 and n= 7, respectively), restlessness (n= 7 and n= 5, respectively) and bruxism (n= 7 and n= 7, respectively). No severe adverse effects were reported.
Pharmacokinetics and pharmacokinetic–pharmacodynamic relationship
The decrease in the cardiovascular and thermogenic response to MDMA after carvedilol pretreatment was not attributable to a pharmacokinetic interaction between carvedilol and MDMA. Carvedilol did not affect the Cmax or AUC0–6 h of MDMA or MDA (Table 2 and Figure 4A). The effect of MDMA on blood pressure in relation to the plasma concentration of MDMA is illustrated by the hysteresis curves in Figure 4B. Carvedilol produced a pronounced downward shift in the Emax of the systolic pressure response to MDMA and a rightward shift in the Cmax of MDMA in the concentration-effect curve (Figure 4B). The pharmacokinetic parameters of MDMA did not depend on CYP2D6 phenotype or the dextromethorphan : dextrorphan ratio in our small study sample.
Table 2.
Pharmacokinetic parameters of MDMA and MDA
| Cmax (ng·mL−1) | Tmax (h) | AUC0–6 h (h·ng·mL−1) | |
|---|---|---|---|
| MDMA | |||
| Placebo-MDMA | 214 (12) | 2.9 (0.3) | 866 (47) |
| Carvedilol-MDMA | 224 (12) | 2.8 (0.2) | 921 (45) |
| MDA | |||
| Placebo-MDMA | 12.3 (1.0) | 5.5 (0.2) | 46.3 (3.5) |
| Carvedilol-MDMA | 12.5 (0.9) | 5.0 (0.4) | 49.3 (2.8) |
Values are mean (SEM) of 16 healthy subjects. AUC, area under concentration–time curve; Cmax, maximum plasma concentration; Tmax, time to maximum plasma concentration.
Figure 4.
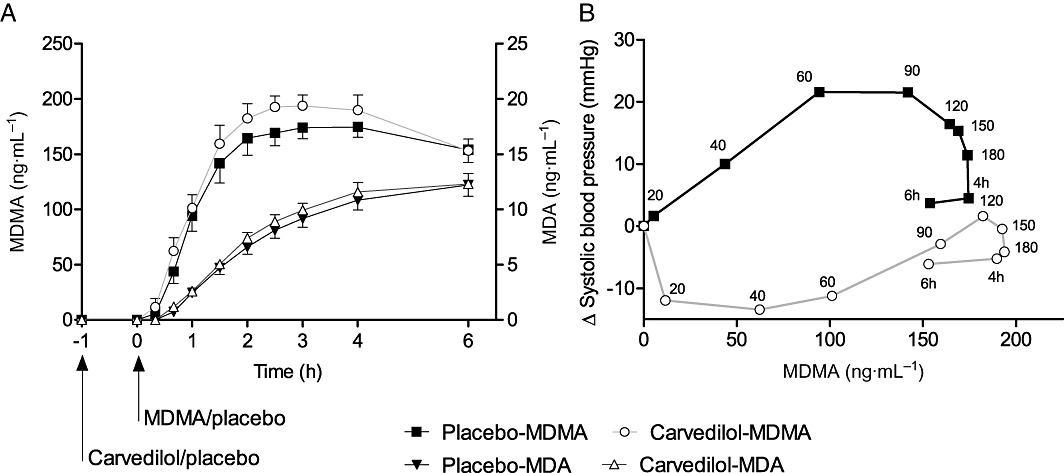
Pharmacokinetics (A) and pharmacokinetic–pharmacodynamic relationship (B). Carvedilol non-significantly increased the exposure to MDMA and MDA (A). The values are expressed as mean ± SEM in 16 subjects. Carvedilol was administered at t =−1 h. MDMA was administered at t = 0 h. MDMA effects on systolic blood pressure plotted against MDMA plasma concentration (B). The values are expressed as means of the changes from baseline in 16 subjects. The time of sampling is noted next to each point in min or h after MDMA administration. Carvedilol produced a downward and rightward shift of the concentration-blood pressure response curve of MDMA (B).
Discussion
The α1- and β1,2,3-adrenoceptor antagonist carvedilol reduced the cardiostimulant and hyperthermic response to MDMA in healthy subjects. Carvedilol similarly reduced MDMA-induced hyperthermia in rats (Sprague et al., 2004a; 2005). Additional studies in rats and mice showed that the transient and early hypothermic effect of MDMA are enhanced by blocking α1-receptors (Bexis and Docherty, 2008), whereas the late hyperthermic response to MDMA is blunted by blocking β3-receptors (Sprague et al., 2004a; Bexis and Docherty, 2008). Moreover, α1-receptors mediate peripheral vasoconstriction and heat dissipation, which are impaired by MDMA (Pedersen and Blessing, 2001). Administration of β1,2-receptor antagonists had no effect on the thermogenic response to MDMA in rats (Sprague et al., 2005) or humans (Hysek et al., 2010). These data suggest a role for both α1- and β3-receptors in MDMA-induced hyperthermia. Carvedilol should be considered for the treatment of hyperthermia associated with ecstasy use because it effectively reduced MDMA-induced hyperthermia in both animals and humans and reversed established hyperthermia in rats (Sprague et al., 2005).
In addition to adrenoceptors, other sites have been implicated in stimulant-induced hyperthermia. MDMA primarily induces the release of 5-HT, NA and dopamine through their respective presynaptic monoamine transporters (Rudnick and Wall, 1992; Rothman et al., 2001; Verrico et al., 2007). MDMA binds to α2-adrenoceptors, 5-HT2A-receptors, H1-histamine and trace amine-1 receptors (Battaglia et al., 1988; Bunzow et al., 2001). The 5-HT2A-receptor antagonist ketanserin inhibited the thermogenic effects of MDMA in rats (Shioda et al., 2008), mice (Di Cara et al., 2011) and humans (Liechti et al., 2000). In both mice and humans, ketanserin administered alone lowered body temperature compared with vehicle and placebo, respectively (Liechti et al., 2000; Di Cara et al., 2011). Thus, no interactive effect of ketanserin and MDMA on body temperature was observed, in contrast to carvedilol and MDMA in the present study. Furthermore, ketanserin has α1-adrenoceptor-blocking properties (Brogden and Sorkin, 1990), and its ability to reduce MDMA-associated hyperthermia may be explained, at least partially, by α1-receptor antagonism. A recent study showed that mice that lack trace amine-1 receptors did not exhibit the early hypothermic response to MDMA, indicating a role for this receptor in the early hypothermic effects of MDMA (Di Cara et al., 2011). D1- and D2-dopamine receptors, α2-adrenoceptors and 5-HT1-receptors do not appear to be involved in the effects of MDMA on body temperature, demonstrated by preclinical (Docherty and Green, 2010; Di Cara et al., 2011) and clinical (Liechti and Vollenweider, 2000; Hysek et al., 2010; 2012) studies.
Recreational users of ecstasy report subjective increases in body temperature, sweating and hot flushes (Parrott et al., 2008). Hot flushes and sweating were also reported after administration of MDMA in the present and in previous studies (Liechti et al., 2001; Freedman et al., 2005). Carvedilol did not reduce the number of subjects who reported MDMA-induced subjective sweating but reduced the number of subjects reporting flushes. Interestingly, in another laboratory study, MDMA did not influence the perceptions of warmth and cold but delayed the onset of sweating at a warm ambient temperature along with an MDMA-induced increase in body temperature (Freedman et al., 2005).
Carvedilol also reduced the cardiostimulant response to MDMA, including blood pressure and heart rate. The α- and β-blockers carvedilol and labetalol have similarly been shown to inhibit the blood pressure response to cocaine in humans (Boehrer et al., 1993; Sofuoglu et al., 2000a, b). Blockade of β-receptors alone did not reduce the pressure response to cocaine (Ramoska and Sacchetti, 1985) or MDMA (Hysek et al., 2010) in humans and enhanced cocaine-induced coronary vasoconstriction (Lange et al., 1990). In rats, the blockade of α1-receptors inhibited both the pressure response and vasoconstriction in isolated vessels in response to cocaine (Mo et al., 1999). The data indicate that dual α,β-blockers, but not selective β-blockers, should be used in the treatment of psychostimulant-associated hypertension and myocardial ischaemia. The data indicate that carvedilol could be useful in the treatment of both psychostimulant-induced hypertension and hyperthermia.
Circulating catecholamine levels were increased by both MDMA and carvedilol. Plasma adrenaline is mainly derived from the adrenals, whereas plasma NA stems largely from transmitters released by sympathetic nerves and the escape of NA into the circulation (Esler et al., 1990; Eisenhofer et al., 1995). Circulating NA is therefore considered an indicator of sympathetic system activation. We observed a marked increase in plasma NA concentrations after carvedilol administration. This compensatory sympathoadrenal response with enhanced levels of catecholamines has previously been documented after α1- or α- and β-adrenoceptor blockade (Omvik et al., 1992; Mazzeo et al., 2001). The MDMA-induced increase in circulating NA in the present study did not reach statistical significance compared with previous work (Dumont et al., 2009; Hysek et al., 2011; 2012). It is possible that the peak effect was missed because we took only two samples. The catecholamine response was enhanced when MDMA was administered following carvedilol. A similar potentiation of the exercise-induced increases in plasma catecholamines has been shown following blockade of α1-adrenoceptors or α- and β-adrenoceptors (Berlin et al., 1993).
Preclinical and clinical studies suggest that NA contributes to the mediation of the subjective effects of MDMA and other psychostimulants (Sofuoglu and Sewell, 2009; Hysek et al., 2011; Newton, 2011). For example, MDMA is more potent in releasing NA than 5-HT or dopamine from monoamine-preloaded human embryonic kidney cells transfected with the corresponding human monoamine transporters (Verrico et al., 2007). Additionally, doses of stimulants that produce amphetamine-type subjective effects in humans correlated with their potency to release NA (Rothman et al., 2001). Furthermore, the NA transporter inhibitor reboxetine attenuated the cardiovascular and subjective response to MDMA in humans, indicating a role for MDMA-induced transporter-mediated NA release in the psychostimulant effects of MDMA (Hysek et al., 2011). Similarly, atomoxetine attenuated the effects of amphetamine in humans (Sofuoglu et al., 2009). Clonidine, which blocks the vesicular release of NA, did not affect the psychological effects of MDMA in humans (Hysek et al., 2012). Although these data suggest a role for transporter-mediated NA release in the psychotropic effects of psychostimulants, how and which postsynaptic adrenoceptors are involved are still unclear. Carvedilol did not alter the subjective effects of MDMA in the present study. Similar to our results, carvedilol and labetalol did not affect the subjective responses to cocaine in humans at doses of cocaine that effectively inhibited the cardiostimulant effects of the drug (Sofuoglu et al., 2000a, b). The available clinical data do not support a critical role for α1- and β1,2,3-receptors in the subjective effects of psychostimulants. Alternatively, the carvedilol concentrations in humans may not have been high enough to produce sufficient adrenoceptor occupancy in the brain. Carvedilol is lipophilic and enters the brain (Elsinga et al., 2005). However, carvedilol is a substrate of the efflux transporter P-glycoprotein in the blood-brain barrier (Elsinga et al., 2005; Bachmakov et al., 2006), and P-glycoprotein activity is known to limit brain exposure to carvedilol (Elsinga et al., 2005).
Preclinical studies indicate that α1-receptors are involved in the mechanism of action of psychostimulants, including MDMA. For example, pretreatment with the α1-receptor antagonist prazosin inhibited locomotor stimulation induced by cocaine (Wellman et al., 2002), amphetamine (Vanderschuren et al., 2003) and MDMA (Fantegrossi et al., 2004; Selken and Nichols, 2007) in rats and mice. Additionally, α1-receptor activation in the ventral tegmental area contributed to the amphetamine-induced release of dopamine in the nucleus accumbens (Pan et al., 1996). Injection of prazosin directly into the ventral tegmental area also blocked the locomotor response to MDMA in rats (Selken and Nichols, 2007). Furthermore, administration of prazosin in the rat prefrontal cortex also blocked amphetamine-induced dopamine release in the nucleus accumbens and hyperactivity (Forget et al., 2011). Finally, α1-adrenoceptor knockout mice do not show increased amphetamine-induced dopamine release in the nucleus accumbens (Auclair et al., 2002) or behavioural sensitization to amphetamine or cocaine (Drouin et al., 2002). In contrast to α1-antagonism, the β-blocker propranolol enhanced both cocaine-induced locomotion and the cocaine-induced increase in dopamine in the nucleus accumbens (Harris et al., 1996). Altogether, the preclinical studies indicate that α1-adrenoceptors, but not β-receptors, play a role in the hyperlocomotion and dopaminergic neurochemical response to psychostimulants. However, the role of adrenoceptors in the reinforcing effects of psychostimulants is unclear. For example, prazosin reduced the self-administration of cocaine (Wee et al., 2008) and nicotine (Forget et al., 2011) in rats. In contrast, prazosin had no effect on cocaine self-administration in rhesus monkeys (Woolverton, 1987). The β-blocker propranolol also inhibited cocaine self-administration in rats (Harris et al., 1996). Carvedilol lowered the number of cocaine self-administrations in humans at a low but not high dose (Sofuoglu et al., 2000a). At low doses, carvedilol preferentially blocks β-receptors (Tham et al., 1995; Sofuoglu et al., 2000a) and active metabolites of carvedilol may contribute to the β- but not the α-adrenoceptor blocking effects of the drug (Spahn-Langguth and Schloos, 1996). The antagonism of α1-adrenoceptors by carvedilol may not have been sufficient in the brain to attenuate the subjective effects of MDMA and we cannot exclude a role for these receptors. The efficacy of carvedilol to reduce cocaine use or abstinence in addicted patients is currently being investigated in ongoing clinical trials [(Sofuoglu and Sewell, 2009) clinicaltrials.gov identifier: NCT00566969 and NCT01171183]. Further trials have investigated the effects of selective α1-blockers on the acute response to MDMA (NCT01386177) and cocaine (NCT01062945) and abstinence from cocaine use (NCT00880997).
Pharmacokinetic interactions between carvedilol and MDMA need to be considered in the interpretation of the present findings, because both drugs are metabolized by CYP2D6 (Graff et al., 2001; O'Mathuna et al., 2008). We therefore assessed the potential effects of carvedilol on the pharmacokinetics of MDMA. We found that carvedilol non-significantly increased the plasma exposure to MDMA or MDA. Thus, the reduced haemodynamic and thermogenic effects of MDMA after carvedilol pretreatment did not result from lower plasma levels of MDMA or MDA. We did not assess the plasma concentrations of carvedilol. MDMA inhibits CYP2D6 (O'Mathuna et al., 2008). CYP2D6 inhibition has been shown to increase the exposure to carvedilol but not its pharmacodynamic or adverse effects in humans (Graff et al., 2001).
Our laboratory study has a few limitations. The study design is limited by the use of single doses. We did not use a dose–response study because we did not want to expose the subjects to more than two doses of MDMA in a within-subject design. However, moderate to highly effective doses of both drugs were selected. The primary goal of the study was to investigate the role of adrenoceptors in the mechanism of action of MDMA in humans. Therefore, the study provides only indirect support for the use of carvedilol in the treatment of stimulant toxicity, in which carvedilol would be administered following the ingestion of ecstasy or other stimulants. Furthermore, the MDMA-induced increase in body temperature in our study was moderate, and we do not know whether carvedilol would also be effective in cases of severe hyperthermia following ecstasy use. Finally, thyroid function may modulate the thermogenic effects of MDMA (Martin et al., 2007; Sprague et al., 2007) and thyroid function parameters were not assessed in this study.
In conclusion, carvedilol inhibited the MDMA-induced increase in blood pressure and body temperature under controlled laboratory conditions. The results demonstrate that α1- and/or β1,2,3-adrenoceptors contribute to the cardiostimulant and thermogenic effects of MDMA in humans. The absence of an effect of carvedilol on the psychotropic response to MDMA does not support a role for α- and β-adrenoceptors in the mediation of the subjective effects of MDMA in humans. Combined α- and β-blockers could be useful in the treatment of intoxications with MDMA or other psychostimulants including other amphetamine derivatives or cocaine.
Acknowledgments
The authors acknowledge the assistance of C. Bläsi and L. Baseglia in study management and thank M. Arends for editorial assistance. This study was supported by the Swiss National Science Foundation (No. 323230_126231).
Glossary
- AUC
area under the concentration–time curve
- Cmax
maximal plasma concentration
- CYP
cytochrome P450
- 5D-ASC
5-Dimensions of Altered States of Consciousness
- Emax
maximal effect
- MDA
±3,4-methylenedioxyamphetamine
- MDMA
±3,4-methylenedioxymethamphetamine
- VAS
Visual Analogue Scale
Conflict of interest
None.
References
- Auclair A, Cotecchia S, Glowinski J, Tassin JP. D-amphetamine fails to increase extracellular dopamine levels in mice lacking alpha α1b-adrenergic receptors: relationship between functional and nonfunctional dopamine release. J Neurosci. 2002;22:9150–9154. doi: 10.1523/JNEUROSCI.22-21-09150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmakov I, Werner U, Endress B, Auge D, Fromm MF. Characterization of β-adrenoceptor antagonists as substrates and inhibitors of the drug transporter P-glycoprotein. Fundam Clin Pharmacol. 2006;20:273–282. doi: 10.1111/j.1472-8206.2006.00408.x. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Brooks BP, Kulsakdinun C, De Souza EB. Pharmacologic profile of MDMA (3,4-methylenedioxymethamphetamine) at various brain recognition sites. Eur J Pharmacol. 1988;149:159–163. doi: 10.1016/0014-2999(88)90056-8. [DOI] [PubMed] [Google Scholar]
- Berlin I, Lechat P, Deray G, Landault C, Maistre G, Chermat V, et al. Beta-adrenoceptor blockade potentiates acute exercise-induced release of atrial natriuretic peptide by increasing atrial diameter in normotensive healthy subjects. Eur J Clin Pharmacol. 1993;44:127–133. doi: 10.1007/BF00315469. [DOI] [PubMed] [Google Scholar]
- Bexis S, Docherty JR. Role of α1-adrenoceptor subtypes in the effects of methylenedioxy methamphetamine (MDMA) on body temperature in the mouse. Br J Pharmacol. 2008;153:591–597. doi: 10.1038/sj.bjp.0707590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehrer JD, Moliterno DJ, Willard JE, Hillis LD, Lange RA. Influence of labetalol on cocaine-induced coronary vasoconstriction in humans. Am J Med. 1993;94:608–610. doi: 10.1016/0002-9343(93)90212-8. [DOI] [PubMed] [Google Scholar]
- Brody SL, Slovis CM, Wrenn KD. Cocaine-related medical problems: consecutive series of 233 patients. Am J Med. 1990;88:325–331. doi: 10.1016/0002-9343(90)90484-u. [DOI] [PubMed] [Google Scholar]
- Brogden RN, Sorkin EM. Ketanserin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in hypertension and peripheral vascular disease. Drugs. 1990;40:903–949. doi: 10.2165/00003495-199040060-00010. [DOI] [PubMed] [Google Scholar]
- Bruggisser M, Ceschi A, Bodmer M, Wilks MF, Kupferschmidt H, Liechti ME. Retrospective analysis of stimulant abuse cases reported to the Swiss Toxicological Information Centre during 1997–2009. Swiss Med Wkly. 2010;140:w13115. doi: 10.4414/smw.2010.13115. [DOI] [PubMed] [Google Scholar]
- Bunzow JR, Sonders MS, Arttamangkul S, Harrison LM, Zhang G, Quigley DI, et al. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol Pharmacol. 2001;60:1181–1188. doi: 10.1124/mol.60.6.1181. [DOI] [PubMed] [Google Scholar]
- Dafters RI. Hyperthermia following MDMA administration in rats: effects of ambient temperature, water consumption, and chronic dosing. Physiol Behav. 1995;58:877–882. doi: 10.1016/0031-9384(95)00136-7. [DOI] [PubMed] [Google Scholar]
- Derogatis LR, Rickels K, Rock AF. The SCL-90 and the MMPI: a step in the validation of a new self-report scale. Br J Psychiatry. 1976;128:280–289. doi: 10.1192/bjp.128.3.280. [DOI] [PubMed] [Google Scholar]
- Di Cara B, Maggio R, Aloisi G, Rivet JM, Lundius EG, Yoshitake T, et al. Genetic deletion of trace amine 1 receptors reveals their role in auto-inhibiting the actions of ecstasy (MDMA) J Neurosci. 2011;31:16928–16940. doi: 10.1523/JNEUROSCI.2502-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich A. The standardized psychometric assessment of altered states of consciousness (ASCs) in humans. Pharmacopsychiatry. 1998;31(Suppl. 2):80–84. doi: 10.1055/s-2007-979351. [DOI] [PubMed] [Google Scholar]
- Docherty JR, Green AR. The role of monoamines in the changes in body temperature induced by 3,4-methylenedioxymethamphetamine (MDMA, ecstasy) and its derivatives. Br J Pharmacol. 2010;160:1029–1044. doi: 10.1111/j.1476-5381.2010.00722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin C, Darracq L, Trovero F, Blanc G, Glowinski J, Cotecchia S, et al. α1b-adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J Neurosci. 2002;22:2873–2884. doi: 10.1523/JNEUROSCI.22-07-02873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont GJ, Verkes RJ. A review of acute effects of 3,4-methylenedioxymethamphetamine in healthy volunteers. J Psychopharmacol. 2006;20:176–187. doi: 10.1177/0269881106063271. [DOI] [PubMed] [Google Scholar]
- Dumont GJ, Kramers C, Sweep FC, Touw DJ, van Hasselt JG, de Kam M, et al. Cannabis coadministration potentiates the effects of ‘ecstasy’ on heart rate and temperature in humans. Clin Pharmacol Ther. 2009;86:160–166. doi: 10.1038/clpt.2009.62. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Rundquist B, Aneman A, Friberg P, Dakak N, Kopin IJ, et al. Regional release and removal of catecholamines and extraneuronal metabolism to metanephrines. J Clin Endocrinol Metab. 1995;80:3009–3017. doi: 10.1210/jcem.80.10.7559889. [DOI] [PubMed] [Google Scholar]
- Elsinga PH, Hendrikse NH, Bart J, van Waarde A, Vaalburg W. Positron emission tomography studies on binding of central nervous system drugs and P-glycoprotein function in the rodent brain. Mol Imaging Biol. 2005;7:37–44. doi: 10.1007/s11307-005-0951-x. [DOI] [PubMed] [Google Scholar]
- Esler M, Jennings G, Lambert G, Meredith I, Horne M, Eisenhofer G. Overflow of catecholamine neurotransmitters to the circulation: source, fate, and functions. Physiol Rev. 1990;70:963–985. doi: 10.1152/physrev.1990.70.4.963. [DOI] [PubMed] [Google Scholar]
- Fantegrossi WE, Kiessel CL, Leach PT, Van Martin C, Karabenick RL, Chen X, et al. Nantenine: an antagonist of the behavioral and physiological effects of MDMA in mice. Psychopharmacology (Berl) 2004;173:270–277. doi: 10.1007/s00213-003-1741-2. [DOI] [PubMed] [Google Scholar]
- Farre M, Abanades S, Roset PN, Peiro AM, Torrens M, O'Mathuna B, et al. Pharmacological interaction between 3,4-methylenedioxymethamphetamine (ecstasy) and paroxetine: pharmacological effects and pharmacokinetics. J Pharmacol Exp Ther. 2007;323:954–962. doi: 10.1124/jpet.107.129056. [DOI] [PubMed] [Google Scholar]
- Forget B, Wertheim C, Mascia P, Pushparaj A, Goldberg SR, Le Foll B. Noradrenergic alpha1 receptors as a novel target for the treatment of nicotine addiction. Neuropsychopharmacology. 2011;35:1751–1760. doi: 10.1038/npp.2010.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman RR, Johanson CE, Tancer ME. Thermoregulatory effects of 3,4-methylenedioxymethamphetamine (MDMA) in humans. Psychopharmacology (Berl) 2005;183:248–256. doi: 10.1007/s00213-005-0149-6. [DOI] [PubMed] [Google Scholar]
- Graff DW, Williamson KM, Pieper JA, Carson SW, Adams KF, Jr, Cascio WE, et al. Effect of fluoxetine on carvedilol pharmacokinetics, CYP2D6 activity, and autonomic balance in heart failure patients. J Clin Pharmacol. 2001;41:97–106. doi: 10.1177/00912700122009746. [DOI] [PubMed] [Google Scholar]
- Green AR, Cross AJ, Goodwin GM. Review of the pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA or ‘Ecstasy’) Psychopharmacology. 1995;119:247–260. doi: 10.1007/BF02246288. [DOI] [PubMed] [Google Scholar]
- Grunau BE, Wiens MO, Brubacher JR. Dantrolene in the treatment of MDMA-related hyperpyrexia: a systematic review. CJEM. 2010;12:435–442. doi: 10.1017/s1481803500012598. [DOI] [PubMed] [Google Scholar]
- Hall AP, Henry JA. Acute toxic effects of ‘Ecstasy’ (MDMA) and related compounds: overview of pathophysiology and clinical management. Br J Anaesth. 2006;96:678–685. doi: 10.1093/bja/ael078. [DOI] [PubMed] [Google Scholar]
- Halpern P, Moskovich J, Avrahami B, Bentur Y, Soffer D, Peleg K. Morbidity associated with MDMA (ecstasy) abuse: a survey of emergency department admissions. Hum Exp Toxicol. 2011;30:259–266. doi: 10.1177/0960327110370984. [DOI] [PubMed] [Google Scholar]
- Harris GC, Hedaya MA, Pan WJ, Kalivas P. β-adrenergic antagonism alters the behavioral and neurochemical responses to cocaine. Neuropsychopharmacology. 1996;14:195–204. doi: 10.1016/0893-133X(95)00089-V. [DOI] [PubMed] [Google Scholar]
- Henry JA, Jeffreys KJ, Dawling S. Toxicity and deaths from 3,4-methylenedioxymethamphetamine (‘ecstasy’) Lancet. 1992;340:384–387. doi: 10.1016/0140-6736(92)91469-o. [DOI] [PubMed] [Google Scholar]
- Hoffman RS. Cocaine and beta-blockers: should the controversy continue? Ann Emerg Med. 2008;51:127–129. doi: 10.1016/j.annemergmed.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Hysek CM, Vollenweider FX, Liechti ME. Effects of a β-blocker on the cardiovascular response to MDMA (ecstasy) Emerg Med J. 2010;27:586–589. doi: 10.1136/emj.2009.079905. [DOI] [PubMed] [Google Scholar]
- Hysek CM, Simmler LD, Ineichen M, Grouzmann E, Hoener MC, Brenneisen R, et al. The norepinephrine transporter inhibitor reboxetine reduces stimulant effects of MDMA (‘ecstasy’) in humans. Clin Pharmacol Ther. 2011;90:246–255. doi: 10.1038/clpt.2011.78. [DOI] [PubMed] [Google Scholar]
- Hysek CM, Brugger R, Simmler LD, Bruggisser M, Doncelli M, Grouzmann E, et al. Effects of the α2-adrenergic agonist clonidine on the pharmacodynamics and pharmacokinetics of methylenedioxymethamphetamine in healthy volunteers. J Pharmacol Exp Ther. 2012;340:286–294. doi: 10.1124/jpet.111.188425. [DOI] [PubMed] [Google Scholar]
- Kolbrich EA, Goodwin RS, Gorelick DA, Hayes RJ, Stein EA, Huestis MA. Physiological and subjective responses to controlled oral 3,4-methylenedioxymethamphetamine administration. J Clin Psychopharmacol. 2008;28:432–440. doi: 10.1097/JCP.0b013e31817ef470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange RA, Cigarroa RG, Flores ED, McBride W, Kim AS, Wells PJ, et al. Potentiation of cocaine-induced coronary vasoconstriction by β-adrenergic blockade. Ann Intern Med. 1990;112:897–903. doi: 10.7326/0003-4819-112-12-897. [DOI] [PubMed] [Google Scholar]
- Liechti ME, Vollenweider FX. Acute psychological and physiological effects of MDMA (‘Ecstasy’) after haloperidol pretreatment in healthy humans. Eur Neuropsychopharmacol. 2000;10:289–295. doi: 10.1016/s0924-977x(00)00086-9. [DOI] [PubMed] [Google Scholar]
- Liechti ME, Vollenweider FX. Which neuroreceptors mediate the subjective effects of MDMA in humans? A summary of mechanistic studies. Hum Psychopharmacol. 2001;16:589–598. doi: 10.1002/hup.348. [DOI] [PubMed] [Google Scholar]
- Liechti ME, Saur MR, Gamma A, Hell D, Vollenweider FX. Psychological and physiological effects of MDMA (‘Ecstasy’) after pretreatment with the 5-HT2 antagonist ketanserin in healthy humans. Neuropsychopharmacology. 2000;23:396–404. doi: 10.1016/S0893-133X(00)00126-3. [DOI] [PubMed] [Google Scholar]
- Liechti ME, Gamma A, Vollenweider FX. Gender differences in the subjective effects of MDMA. Psychopharmacology (Berl) 2001;154:161–168. doi: 10.1007/s002130000648. [DOI] [PubMed] [Google Scholar]
- Liechti ME, Kunz I, Kupferschmidt H. Acute medical problems due to ecstasy use. Case-series of emergency department visits. Swiss Med Wkly. 2005;135:652–657. doi: 10.4414/smw.2005.11231. [DOI] [PubMed] [Google Scholar]
- Martin TL, Chiasson DA, Kish SJ. Does hyperthyroidism increase risk of death due to the ingestion of ecstasy? J Forensic Sci. 2007;52:951–953. doi: 10.1111/j.1556-4029.2007.00463.x. [DOI] [PubMed] [Google Scholar]
- Mazzeo RS, Carroll JD, Butterfield GE, Braun B, Rock PB, Wolfel EE, et al. Catecholamine responses to α-adrenergic blockade during exercise in women acutely exposed to altitude. J Appl Physiol. 2001;90:121–126. doi: 10.1152/jappl.2001.90.1.121. [DOI] [PubMed] [Google Scholar]
- Mills EM, Banks ML, Sprague JE, Finkel T. Pharmacology: uncoupling the agony from ecstasy. Nature. 2003;426:403–404. doi: 10.1038/426403a. [DOI] [PubMed] [Google Scholar]
- Mills EM, Rusyniak DE, Sprague JE. The role of the sympathetic nervous system and uncoupling proteins in the thermogenesis induced by 3,4-methylenedioxymethamphetamine. J Mol Med. 2004;82:787–799. doi: 10.1007/s00109-004-0591-7. [DOI] [PubMed] [Google Scholar]
- Mo W, Arruda JA, Dunea G, Singh AK. Cocaine-induced hypertension: role of the peripheral sympathetic system. Pharmacol Res. 1999;40:139–145. doi: 10.1006/phrs.1999.0503. [DOI] [PubMed] [Google Scholar]
- Morgan T. Clinical pharmacokinetics and pharmacodynamics of carvedilol. Clin Pharmacokinet. 1994;26:335–346. doi: 10.2165/00003088-199426050-00002. [DOI] [PubMed] [Google Scholar]
- Newton TF. A perhaps unexpected role of norepinephrine in actions of MDMA. Clin Pharmacol Ther. 2011;90:215–216. doi: 10.1038/clpt.2011.125. [DOI] [PubMed] [Google Scholar]
- O'Mathuna B, Farre M, Rostami-Hodjegan A, Yang J, Cuyas E, et al. The consequences of 3,4-methylenedioxymethamphetamine induced CYP2D6 inhibition in humans. J Clin Psychopharmacol. 2008;28:523–529. doi: 10.1097/JCP.0b013e318184ff6e. [DOI] [PubMed] [Google Scholar]
- Omvik P, Lund-Johansen P, Myking O. Effects of carvedilol on atrial natriuretic peptide, catecholamines, and hemodynamics in hypertension at rest and during exercise. J Cardiovasc Pharmacol. 1992;19(Suppl. 1):S90–S96. doi: 10.1097/00005344-199219001-00018. [DOI] [PubMed] [Google Scholar]
- Pan WH, Sung JC, Fuh SM. Locally application of amphetamine into the ventral tegmental area enhances dopamine release in the nucleus accumbens and the medial prefrontal cortex through noradrenergic neurotransmission. J Pharmacol Exp Ther. 1996;278:725–731. [PubMed] [Google Scholar]
- Parrott AC. MDMA and temperature: a review of the thermal effects of ‘ecstasy’ in humans. Drug Alcohol Depend. 2012;121:1–9. doi: 10.1016/j.drugalcdep.2011.08.012. [DOI] [PubMed] [Google Scholar]
- Parrott AC, Lock J, Conner AC, Kissling C, Thome J. Dance clubbing on MDMA and during abstinence from Ecstasy/MDMA: prospective neuroendocrine and psychobiological changes. Neuropsychobiology. 2008;57:165–180. doi: 10.1159/000147470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen NP, Blessing WW. Cutaneous vasoconstriction contributes to hyperthermia induced by 3,4-methylenedioxymethamphetamine (ecstasy) in conscious rabbits. J Neurosci. 2001;21:8648–8654. doi: 10.1523/JNEUROSCI.21-21-08648.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramoska E, Sacchetti AD. Propranolol-induced hypertension in treatment of cocaine intoxication. Ann Emerg Med. 1985;14:1112–1113. doi: 10.1016/s0196-0644(85)80934-3. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, et al. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Rudnick G, Wall SC. The molecular mechanism of ‘ecstasy’[3,4-methylenedioxy-methamphetamine (MDMA)]: serotonin transporters are targets for MDMA-induced serotonin release. Proc Natl Acad Sci USA. 1992;89:1817–1821. doi: 10.1073/pnas.89.5.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyniak DE, Banks ML, Mills EM, Sprague JE. Dantrolene use in 3,4-methylenedioxymethamphetamine (ecstasy)-mediated hyperthermia. Anesthesiology. 2004;101:263. doi: 10.1097/00000542-200407000-00053. [DOI] [PubMed] [Google Scholar]
- Rusyniak DE, Tandy SL, Hekmatyar SK, Mills E, Smith DJ, Bansal N, et al. The role of mitochondrial uncoupling in 3,4-methylenedioxymethamphetamine-mediated skeletal muscle hyperthermia and rhabdomyolysis. J Pharmacol Exp Ther. 2005;313:629–639. doi: 10.1124/jpet.104.079236. [DOI] [PubMed] [Google Scholar]
- Schmitz N, Hartkamp N, Kiuse J, Franke GH, Reister G, et al. The Symptom Check-List-90-R (SCL-90-R): a German validation study. Qual Life Res. 2000;9:185–193. doi: 10.1023/a:1008931926181. [DOI] [PubMed] [Google Scholar]
- Selken J, Nichols DE. Alpha1-adrenergic receptors mediate the locomotor response to systemic administration of (±)-3,4-methylenedioxymethamphetamine (MDMA) in rats. Pharmacol Biochem Behav. 2007;86:622–630. doi: 10.1016/j.pbb.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda K, Nisijima K, Yoshino T, Kuboshima K, Iwamura T, Yui K, et al. Risperidone attenuates and reverses hyperthermia induced by 3,4-methylenedioxymethamphetamine (MDMA) in rats. Neurotoxicology. 2008;29:1030–1036. doi: 10.1016/j.neuro.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Sofuoglu M, Sewell RA. Norepinephrine and stimulant addiction. Addict Biol. 2009;14:119–129. doi: 10.1111/j.1369-1600.2008.00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofuoglu M, Brown S, Babb DA, Pentel PR, Hatsukami DK. Carvedilol affects the physiological and behavioral response to smoked cocaine in humans. Drug Alcohol Depend. 2000a;60:69–76. doi: 10.1016/s0376-8716(99)00143-x. [DOI] [PubMed] [Google Scholar]
- Sofuoglu M, Brown S, Babb DA, Pentel PR, Hatsukami DK. Effects of labetalol treatment on the physiological and subjective response to smoked cocaine. Pharmacol Biochem Behav. 2000b;65:255–259. doi: 10.1016/s0091-3057(99)00201-4. [DOI] [PubMed] [Google Scholar]
- Sofuoglu M, Poling J, Hill K, Kosten T. Atomoxetine attenuates dextroamphetamine effects in humans. Am J Drug Alcohol Abuse. 2009;35:412–416. doi: 10.3109/00952990903383961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn-Langguth H, Schloos J. Evidence of significant contribution of carvedilol metabolites to β-receptor antagonism: p.o. and i.v. data. Naunyn Schmiedebergs Arch Pharmacol. 1996;353:S158. [Google Scholar]
- Sprague JE, Brutcher RE, Mills EM, Caden D, Rusyniak DE. Attenuation of 3,4-methylenedioxymethamphetamine (MDMA, Ecstasy)-induced rhabdomyolysis with α1- plus β3-adrenoreceptor antagonists. Br J Pharmacol. 2004a;142:667–670. doi: 10.1038/sj.bjp.0705823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague JE, Mallett NM, Rusyniak DE, Mills E. UCP3 and thyroid hormone involvement in methamphetamine-induced hyperthermia. Biochem Pharmacol. 2004b;68:1339–1343. doi: 10.1016/j.bcp.2004.03.049. [DOI] [PubMed] [Google Scholar]
- Sprague JE, Moze P, Caden D, Rusyniak DE, Holmes C, Goldstein DS, et al. Carvedilol reverses hyperthermia and attenuates rhabdomyolysis induced by 3,4-methylenedioxymethamphetamine (MDMA, Ecstasy) in an animal model. Crit Care Med. 2005;33:1311–1316. doi: 10.1097/01.ccm.0000165969.29002.70. [DOI] [PubMed] [Google Scholar]
- Sprague JE, Yang X, Sommers J, Gilman TL, Mills EM. Roles of norepinephrine, free fatty acids, thyroid status, and skeletal muscle uncoupling protein 3 expression in sympathomimetic-induced thermogenesis. J Pharmacol Exp Ther. 2007;320:274–280. doi: 10.1124/jpet.106.107755. [DOI] [PubMed] [Google Scholar]
- Studerus E, Gamma A, Vollenweider FX. Psychometric evaluation of the altered states of consciousness rating scale (OAV) PLoS ONE. 2010;5:e12412. doi: 10.1371/journal.pone.0012412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham TC, Guy S, McDermott BJ, Shanks RG, Riddell JG. The dose dependency of the α- and β-adrenoceptor antagonist activity of carvedilol in man. Br J Clin Pharmacol. 1995;40:19–23. doi: 10.1111/j.1365-2125.1995.tb04529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren LJ, Beemster P, Schoffelmeer AN. On the role of noradrenaline in psychostimulant-induced psychomotor activity and sensitization. Psychopharmacology. 2003;169:176–185. doi: 10.1007/s00213-003-1509-8. [DOI] [PubMed] [Google Scholar]
- Verrico CD, Miller GM, Madras BK. MDMA (ecstasy) and human dopamine, norepinephrine, and serotonin transporters: implications for MDMA-induced neurotoxicity and treatment. Psychopharmacology. 2007;189:489–503. doi: 10.1007/s00213-005-0174-5. [DOI] [PubMed] [Google Scholar]
- Wee S, Mandyam CD, Lekic DM, Koob GF. α1-Noradrenergic system role in increased motivation for cocaine intake in rats with prolonged access. Eur Neuropsychopharmacol. 2008;18:303–311. doi: 10.1016/j.euroneuro.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman P, Ho D, Cepeda-Benito A, Bellinger L, Nation J. Cocaine-induced hypophagia and hyperlocomotion in rats are attenuated by prazosin. Eur J Pharmacol. 2002;455:117–126. doi: 10.1016/s0014-2999(02)02616-x. [DOI] [PubMed] [Google Scholar]
- White TL, Justice AJ, de Wit H. Differential subjective effects of d-amphetamine by gender, hormone levels and menstrual cycle phase. Pharmacol Biochem Behav. 2002;73:729–741. doi: 10.1016/s0091-3057(02)00818-3. [DOI] [PubMed] [Google Scholar]
- Wittchen HU, Wunderlich U, Gruschwitz S, Zaudig M. SKID-I: Strukturiertes Klinisches Interview für DSM-IV. Göttingen: Hogrefe-Verlag; 1997. [Google Scholar]
- Woolverton WL. Evaluation of the role of norepinephrine in the reinforcing effects of psychomotor stimulants in rhesus monkeys. Pharmacol Biochem Behav. 1987;26:835–839. doi: 10.1016/0091-3057(87)90618-6. [DOI] [PubMed] [Google Scholar]
- Zerssen DV. Die Beschwerden-Liste. Münchener Informationssystem. München: Psychis; 1976. [Google Scholar]