

Abstract
BACKGROUND AND PURPOSE
We recently reported that broad spectrum agonist-induced activation of presynaptic group III metabotropic glutamate (mGlu) receptors within the substantia nigra pars compacta using L-2-amino-4-phosphonobutyrate provided functional neuroprotection in the 6-hydroxydopamine lesion rat model of Parkinson's disease. The aim of this study was to establish whether selective activation of the mGlu4 receptor alone could afford similar functional neuroprotection.
EXPERIMENTAL APPROACH
The neuroprotective effects of 8 days of supranigral treatment with a positive allosteric modulator of mGlu4 receptors, (+/−)-cis-2-(3,5-dichlorphenylcarbamoyl)cyclohexanecarboxylic acid (VU0155041), were investigated in rats with unilateral 6-hydroxydopamine lesions. The effects of VU0155041 treatment on motor function were assessed using both habitual (cylinder test) and forced (adjusted stepping, amphetamine-induced rotations) behavioural tests. Nigrostriatal tract integrity was examined by analysis of tyrosine hydroxylase, dopa decarboxylase or dopamine levels in the striatum and tyrosine hydroxylase-positive cell counts in the substantia nigra pars compacta.
KEY RESULTS
VU0155041 provided around 40% histological protection against a unilateral 6-hydroxydopamine lesion as well as significant preservation of motor function. These effects were inhibited by pre-treatment with (RS)-α-cyclopropyl-4-phosphonophenylglycine, confirming a receptor-mediated response. Reduced levels of inflammatory markers were also evident in the brains of VU0155041-treated animals.
CONCLUSIONS AND IMPLICATIONS
Allosteric potentiation of mGlu4 receptors in the substantia nigra pars compacta provided neuroprotective effects in the 6-hydroxydopamine rat model A reduced inflammatory response may contribute, in part, to this action. In addition to the reported symptomatic effects, activation of mGlu4 receptors may also offer a novel approach for slowing the progressive degeneration observed in Parkinson's disease.
Keywords: group III mGlu receptor, 6-hydroxydopamine lesion, metabotropic glutamate receptor, mGlu4, neuroprotection, nigrostriatal tract, Parkinson's disease, substantia nigra
Introduction
Parkinson's disease is a debilitating neurodegenerative disorder that affects 1% of the population over 65, whereby degeneration of the dopaminergic neurones in the substantia nigra pars compacta (SNpc) results in a myriad of motor symptoms including tremor, postural instability and difficulty in initiating movement. Despite extensive research efforts, current treatments for Parkinson's disease rely on restoring striatal dopaminergic transmission through the use of the dopamine precursor, L-3,4-dihydroxyphenylalanine (L-DOPA) or dopamine agonists. Although extremely effective in addressing the motor symptoms of the disease, these drugs fail to address the ongoing progressive degeneration of dopaminergic neurones in the nigrostriatal tract. As a result, increasing doses of drug are required to stabilize the deterioration in symptoms often resulting in adverse effects, namely L-DOPA-induced dyskinesia and psychosis (Stocchi et al., 1997).
In Parkinson's disease, loss of striatal dopamine evokes numerous downstream changes within the basal ganglia, including increased firing of the subthalamic nucleus (STN) (Vila et al., 1999). The subsequent increased release of glutamate in the STN target areas such as the substantia nigra pars reticulata (SNpr) and internal globus pallidus, leads to inhibition of thalamocortical feedback, which contributes to the generation of motor deficits associated with Parkinson's disease. Furthermore, the STN also sends direct projections to the SNpc (Smith and Bolam, 1990; Iribe et al., 1999), which under Parkinsonian conditions may lead to a parallel increase in glutamate release and contribute to the progressive degeneration in the SNpc via an excitotoxic mechanism (Rodriguez et al., 1998). Accordingly, reducing STN firing via subthalamotomy or deep brain stimulation reduces symptoms in animal models and Parkinson's disease patients (Peppe et al., 2004; Alvarez et al., 2005; Windels et al., 2005), while chemical inactivation of the STN (through kainic acid lesioning) also reduces nigrostriatal tract damage induced by the dopaminergic toxin, 6-hydroxydopamine (6-OHDA) (Piallat et al., 1996). Given that these surgical procedures are invasive, expensive and available in few centres worldwide, alternative strategies for combating the effects of increased STN activity are desirable. One approach would be to use pharmacological agents that may reduce glutamate release from STN terminals in the SN.
Group III metabotropic glutamate receptors have the appropriate anatomical distribution and functional effects and are therefore considered possible targets for treatment of motor and degenerative processes in Parkinson's disease (see Duty, 2010). These Gi/Go-coupled receptors, which include mGlu4, mGlu7 and mGlu8 receptors (nomenclature follows Alexander et al., 2011), are found predominantly on presynaptic elements of both GABAergic and glutamatergic synapses where they serve a modulatory role (Conn and Pin, 1997). Accordingly, microdialysis studies from our group have demonstrated that activation of group III metabotropic glutamate (mGlu) receptors in the rodent globus pallidus using the broad spectrum agonists L-2-amino-4-phosphonobutyrate (L-AP4) or L-serine-O-phosphate L-SOP reduces GABA release (MacInnes and Duty, 2006). More importantly here, in agreement with in vitro electrophysiological studies showing that L-AP4 inhibits glutamate transmission across the subthalamonigral synapse (Wittmann et al., 2001), we recently demonstrated that activation of group III mGlu receptors in the SN in vivo leads to a reduction in glutamate release (Austin et al., 2010). We proposed that this mechanism at least partly explained the ability of L-AP4 to protect against 6-OHDA-induced nigrostriatal tract degeneration in rats (Vernon et al., 2005; 2007; Austin et al., 2010), an effect that was accompanied by preservation of motor behaviour (Austin et al., 2010).
The pharmacological identity of the group III receptor(s) mediating this protective effect against a 6-OHDA unilateral lesion is not yet known. However, we recently demonstrated a particularly high intensity of immunoreactivity for mGlu4 receptors in the SNpc and showed activation of nigral mGlu4 receptors, using the positive allosteric modulator N-phenyl-7-(hydroxylimino)cyclopropa[b]chromen-1a-carboxamide (PHCCC), reduced glutamate release from nigral slices in vitro and reversed akinesia in the reserpine-treated rat model of Parkinson's disease (Broadstock et al., 2011). Thus, the mGlu4 receptor looks a particularly promising target. In support of this suggestion, a previous report has demonstrated that a systemic injection of PHCCC, given 30 min prior to toxin injection, provided around 50% protection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced loss of striatal dopamine content and reduction in TH-positive cell numbers in the SNpc in mice (Battaglia et al., 2006). While these results with PHCCC are encouraging, the compound suffers from low potency (EC50 at mGlu4 receptors approximately 4 µM in vitro), poor aqueous solubility and demonstrates antagonism at mGlu1 receptors with a potency similar to that at mGlu4 receptors (Marino et al., 2003). Given that antagonism of mGlu1 receptors can provide protection in vivo against ischaemic or 6-OHDA-induced toxicity in its own right (Bruno et al., 1999; Vernon et al., 2005), this pharmacological profile of PHCCC is less than ideal for teasing out the protective efficacy of targeting mGlu4 receptors. To overcome these liabilities, the more recently described positive allosteric modulator (PAM) of mGlu4 receptors, VU0155041; (+/−)-cis-2-(3,5-dichlorphenylcarbamoyl)cyclohexanecarboxylic acid, was selected for all studies reported here. Discovered by high-throughput screening, this lead compound was shown to be approximately eightfold more potent than PHCCC at mGlu4 receptors, did not demonstrate any significant potentiator or antagonist activity at other mGlu receptor subtypes and is more readily soluble in aqueous solvents. In addition, this compound has demonstrated robust symptomatic activity in two rodent models of Parkinson's disease, dose-dependently decreasing haloperidol-induced catalepsy and reserpine-induced akinesia in rats (Niswender et al., 2008a). The model chosen for these studies was the 6-OHDA lesion rat model in which a battery of behavioural tests can be undertaken, allowing the efficacy of neuroprotective agents against both histological and motor deficits to be assessed.
In addition to showing enhanced glutamatergic signalling akin to Parkinson's disease (Hassani et al., 1996; Breit et al., 2006), the 6-OHDA model also displays a pronounced inflammatory response including microglial activation in the striatum and SNpc and elevated striatal levels of inflammatory markers such as TNF-α (Mogi et al., 2000; Cicchetti et al., 2002; Duty and Jenner, 2011). Given that mGlu4 receptors are expressed on glial cells (Taylor et al., 2003; Yao et al., 2005), it is possible that anti-inflammatory actions may contribute to any protective effects of mGlu4 receptor stimulation.
The aims of this study were therefore to investigate the contribution of mGlu4 receptors to mediating neuroprotective effects in 6-OHDA lesioned rats using the selective PAM, VU0155041, and to start to elucidate the potential mechanisms underlying this response. The findings of these studies demonstrate that activation of mGlu4 receptors may offer a novel approach to attenuating the ongoing degeneration of nigrostriatal neurones in Parkinson's disease and support involvement of an anti-inflammatory action.
Methods
Animals
All animal care and experimental procedures complied with the UK Animals (Scientific Procedures) Act 1986, and every effort was made to minimize animal numbers and suffering. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). Male Sprague-Dawley rats (B&K, Hull, UK or Harlan, Carshalton, UK), weighing 270–300 g (62 in total), were used in these studies. Food and water were provided ad libitum. Animals were housed in a temperature- and humidity-controlled environment with a 12-h light/dark cycle.
Protocol for supranigral cannulation to facilitate drug/toxin delivery
Under general anaesthesia (isoflurane, 4–5% induction and 2–3% maintenance in 95% O2/5% CO2), 12 mm, 23-gauge stainless steel guide cannulae were stereotaxically implanted, 2 mm above the SNpc [co-ordinates; antero-posterior (AP), −4.8 mm; medio-lateral, ± 2.0 mm; ventro-medial −6.3 mm] co-ordinates relative to the skull surface at Bregma (Paxinos and Watson, 1998). In all cases, bilateral cannulation was performed to maximize the chances of obtaining a patent cannula in each animal and thereby reduce numbers used. Cannulae were secured into position using dental cement and 30-gauge stainless steel stylets inserted to prevent blockage.
Protocol for inducing 6-OHDA lesion and delivering supranigral VU0155041 injections
A minimum of 4 days following cannulation above the SNpc, animals received a single, unilateral injection of 6-OHDA. Thirty minutes before 6-OHDA injection, the rats were pre-treated with desipramine (25 mg·kg−1 i.p.) and pargyline (5 mg·kg−1 i.p.). Animals were then anaesthetized briefly (as above), while 6-OHDA (12 µg in 2.5 µL of 0.2% ascorbic acid in 0.9% saline) was injected at a rate of 1.25 µL·min−1 into the SNpc (2 mm below the guide cannula). Animals were placed in cages on thermostatically heated mats until fully conscious.
For completion of the dose–response study, a total of 30 rats were used in groups of n= 7 or n= 8. However, a wrongly-placed cannula (identified through histological assessment) reduced final numbers in the highest dose group to n= 6. Animals were treated with VU0155041 (10–100 nmol in 4 µL of PBS) or vehicle (1× PBS) 1 h before the 6-OHDA lesion and daily for 7 days thereafter. In this case, unilateral supranigral infusions (2 µL·min−1) were made into conscious animals via the same cannula as the 6-OHDA, only with the needle resting 1 mm above the SNpc. The motor impairment of these animals was examined and then post mortem measures of nigral cell counts, striatal TH and dopa decarboxylase (DDC) expression and nigral inflammatory markers were taken. In a separate study where striata were taken for HPLC analysis of dopamine content, a total of 12 rats were treated as above with either vehicle (n= 6) or a single maximal dose of VU0155041 (100 nmol; n= 6). In order to examine receptor specificity, a total of 20 rats were used. These animals were treated as above, but (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG) (75 nmol in 4 µL 0.1 M NaOH in 1× PBS) or vehicle for CPPG (4 µL 0.1 M NaOH in 1× PBS) was infused unilaterally above the SNpc, 30 min before each injection of VU0155041 (100 nmol). A third group received vehicle injections alone. In this study, motor impairment of the animals was examined together with post mortem measures of nigral cell counts and striatal DA content only.
Protocol for assessing motor impairment
The severity of motor impairment following a 6-OHDA lesion was assessed using three behavioural tests. The cylinder test of forelimb akinesia was performed 1 day before and 5 days after the lesion. Animals were placed individually in a transparent, perspex cylinder (21 cm diameter × 34 cm height) and the number of upward reaches video-recorded in 5 min intervals. The use of the contralateral paw was then assessed as a percentage of total reaches made, both pre- and post-lesion, by observers unaware of the treatments. The adjusted steps test measure of contralateral forelimb use was performed 1 day prior to and 6 days post-lesion. The rat was held with the torso slightly raised and the hind limbs and ipsilateral forelimb gently restrained and then moved slowly sideways in backhand and forehand directions across a 90 cm distance with the contralateral forelimb-bearing weight. The number of steps made by the animal's contralateral paw was recorded in each direction and the average of three repeat runs each day was taken. The use of the contralateral paw after the lesion was expressed as a percentage of use before the lesion. Amphetamine-induced rotational behaviour was assessed on day 7 after the lesion. Animals were harnessed in jackets tethered to an automated rotometer and placed in 40 cm diameter hemispherical bowls and recorded for a 10 min baseline period before the injection of d-amphetamine (5 mg·kg−1 i.p.). Full 360° ipsiversive rotations were recorded in 5 min intervals for up to 60 min after the injection by observers unaware of the treatments. Data were quantified over the full 60 min by measuring the area under the time-course curve (AUC).
Histological and neurochemical protocols
On the final day of VU0155041 dosing (day 8), animals from the dose–response study were terminally anaesthetized using pentobarbital (100 mg·kg−1) and then transcardially perfused with 0.1 M PBS, followed by 4% paraformaldehyde (PFA) in 0.1 M PBS. The brains were removed and stored in PFA at 4°C until further processing for immunohistochemistry and cannula placement confirmation. Animals from the HPLC study and from the CPPG study were killed by CO2 asphyxiation, the brains removed and freshly dissected striata snap frozen on card-ice before storing at −80°C until further processing for HPLC measurement of dopamine and its metabolites. The nigral tissue blocks were post-fixed in 4% PFA before being analysed for TH-positive cell counts (CPPG study) or cannula placement confirmation (both studies).
For immunohistochemical assays, fixed rostral (striatum) and caudal (SN) segments of brain were dehydrated, defatted and then paraffin embedded. Coronal sections (6 µm) were taken through the striatum at +1.2, +0.2 and −0.26 mm AP from Bregma and through the SN at −4.8, −5.3 and −5.8 mm AP from Bregma (Paxinos and Watson, 1998). For TH and DDC immunohistochemistry, sections were de-paraffinized in xylene and 100% industrial methylated spirit (IMS) before a 10 min incubation in 3% hydrogen peroxide (H2O2) to block endogenous peroxidase activity. Following standard antigen retrieval with 1 mM citric acid and a 10 min incubation with blocking buffer (1% BSA in 0.1 M PBS and 10% sodium azide), sections were incubated with the primary antibody (rabbit polyclonal anti-α-tyrosine hydroxylase 1:1250; or rabbit polyclonal anti-DDC 1:500; both Chemicon, Millipore, Watford, UK), overnight at room temperature. For ionized calcium-binding adaptor molecule 1 (IBA-1) and glial fibrillary acidic protein (GFAP) immunocytochemistry, coronal sections were deparaffinized and rehydrated, and endogenous peroxidase was quenched with 0.3% H2O2 for 30 min. The slides were placed in pepsin (0.2 g Sigma-p-7000 pepsin, Sigma-Aldrich, St. Louis, MO, USA; in 50 mL 0.01 M HCl) for 30 min, washed and non-specific binding was blocked with 1.5% normal goat serum (Vectastain rabbit IgG ABC kit; Vector Laboratories, Peterborough, UK) diluted in PBS for 20 min. Sections were then incubated with primary rabbit polyclonal anti-IBA-1 (1:5000; Wako Chemicals, Richmond, VA, USA) or anti-GFAP (pre-diluted neat solution; BioGenex Laboratories Inc., San Ramon, CA, USA) for 18 h at room temperature. After washing with PBS (3 × 5 min), all sections were incubated with a biotinylated secondary antibody (Vectastain rabbit IgG ABC kit) for 30 min, followed by 3 × 5 min washes with PBS. The horseradish peroxidase conjugate (Vectastain rabbit IgG ABC kit) was then applied for 30 min, followed by 3 × 5 min PBS rinses. Antibody binding was visualized using the chromagen 3, 3V-diaminobenzidine (DAB substrate kit, Vector SK-4100). Sections were finally dehydrated in 100% IMS, cleared in xylene, cover-slipped with DPX mountant and allowed to dry before being analysed using light microscopy. Digital images of the striata were captured using an Epson Perfection V700 Photo scanner or Aperio ScanScope XT scanner and analysed using Scion Image Software (Scion Corporation, Frederick, MD, USA). Optical density was recorded in four quadrants of striatum and a background cortical region, the latter of which was subtracted from striatal readings. In order to control for variations in staining intensity between sections, the density of each lesioned striatum was expressed as a percentage of that in the respective contralateral (intact) striatum. For the nigra, staining results were viewed on a Zeiss apotome microscope and recorded using Axiovision LE software (Carl Zeiss Ltd., Hertfordshire, UK).
Although manual rather than the more comprehensive stereological analysis was performed here, studies comparing these two modes of counting in this same 6-OHDA model have found no significant difference in the outcome between these measures, supporting the validity of our analysis (Iczkiewicz et al., 2010). TH-immunoreactive cells were thus counted in the contralateral and ipsilateral hemispheres of the brain at three regions of the SNpc (−4.8, −5.3 and −5.8 mm AP from Bregma) where the central region is clearly separated from the ventral tegmental area by the medial terminal nucleus (MT) of the accessory optic tract. Within the nigral sections, viable TH-positive cells were counted at ×50 magnification using image analysis software (Image J; National Institutes of Health, Bethesda, MD, USA). As TH staining can be achieved in non-viable cells, only intact round cells displaying a clear nucleus and cytoplasm were included in these analyses. The density of IBA-1-positive cells demonstrating amoeboid morphology typical of activated microglia was assessed in the contralateral and ipsilateral hemispheres of the SNpc (−5.3 mm AP from Bregma). GFAP-positive cells demonstrating the hallmark star-shaped characteristic of an activated astrocyte were also compared between adjacent hemispheres at a central region of the SNpc (−5.3 mm AP from Bregma). For each animal, three adjacent sections were counted at each level of the SNpc and then pooled to give a total mean cell count for the ipsilateral and contralateral hemispheres. An inter-class correlation (one-way random effects model; SPSS version 19, SPSS Inc., Chicago, IL, USA) was performed to assess the reliability of cell number and staining density measures for IBA-1 and GFAP both between triplicate measures and between animals in a given treatment group. Cronbach's α (coefficient of reliability) was greater than 0.9 in 15 out of 16 cases, with average correlations greater than 0.928 (P < 0.001). These results indicate that the data were highly correlated and support reliability in the quantification of neuronal cell number/density for IBA1 and GFAP within the intact and lesioned SNpc.
Striatal dopamine content was measured using HPLC. Frozen striata were homogenized in ice-cold buffer (0.1 M perchloric acid, 0.1 mM EDTA, 2.5 mg·L−1 ascorbate) and then were centrifuged at 20 000×g for 15 min at 4°C. The filtered supernatant was assayed for dopamine using a Triathlon autosampler, a Rheos 4000 pump, an Intro electrochemical detector (Antec, Zoeterwoude, the Netherlands; +0.75 V) and a Hypersil BDS analytical column (150 × 2.1 mm, 3 mm C18; Fisher Scientific, Loughborough, UK). The mobile phase consisted of 100 mM NaH2PO4 and 100 mM H3PO4, mixed until a pH of 2.6 was obtained, added to 2 mM 1-octane sulphonic acid (OSA), 1 mM EDTA and 13% methanol obtaining a final pH of 2.8. The flow rate was set at 200 µL·min−1 without recycling, and the limit of electrochemical detection by the Antec reference electrode (+0.75 V) was approximately 0.1 nM. Chromatographic analysis was carried out using Millennium 32 software (Waters, Elstree, UK). In addition to analysing dopamine content of each sample, dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) content was also analysed to obtain a turnover ratio and measure of dopamine metabolism to account for a potential intrinsic compensatory response following 6-OHDA lesion.
Cannula placement was confirmed by visual inspection of TH-stained sections in both the dose–response and the CPPG studies. Only one animal from the 100 nmol VU0155041 group was excluded for aberrantly placed cannulae, yielding a 98% success rate for correct placement.
Data handling and statistical analysis
All data are expressed as mean ± SEM, where n represents the number of animals in each experimental group. Statistical analyses were performed using Graph Pad Prism (version 5.0) and, in all cases, P < 0.05 was taken to indicate significance.
The use of the contralateral paw in the cylinder test, expressed as % of total reaches, was compared before and after the lesion in the different treatment groups using a two-way anova with Bonferroni's post hoc test. In the adjusted stepping test, the use of the contralateral paw after the lesion was expressed as % of use before the lesion, and differences in the treatment groups were compared using one-way anova with Dunnett's post hoc test. The number of amphetamine-induced rotations in 5 min intervals was compared between treatment groups using a two-way anova with Bonferroni's post hoc test.
For TH and DDC immunohistochemistry in the striatum, the average OD of three sections, corrected for background cortical staining, were obtained at each rostrocaudal level for each animal. In-depth analyses revealed there were no differences between any of the three rostrocaudal levels; therefore, a single average value representing OD of TH and DDC staining in the whole striatum was obtained for each animal for the final analysis shown here. The mean OD of the 6-OHDA lesioned side, expressed as % of the contralateral, intact hemisphere, was compared between treatment groups using a one-way anova with Dunnett's post hoc test. For the SN, the average number of TH-positive cells in three adjacent sections for each animal across the three rostrocaudal levels was obtained, and the mean of these values was taken per treatment group. Statistical comparison was made between the different treatment groups of mean TH-positive cell counts in the 6-OHDA lesioned side, expressed as % of the contralateral, intact hemisphere, using a one-way anova with Dunnett's post hoc test. For IBA-1 and GFAP staining in the SN, the average IBA-1 density or number of GFAP-positive cells between the contralateral and intact hemispheres in three adjacent sections at a central region of the SNpc was obtained, and the mean of these values was taken per treatment group. Statistical comparisons of mean IBA-1 density measures or GFAP-positive cell counts in the 6-OHDA lesioned hemisphere, expressed as % of the contralateral, intact hemisphere, were made between different treatment groups using a one-way anova with Dunnett's post hoc test.
HPLC
Dopamine, DOPAC and HVA content of striatal samples were converted from peak areas of the chromatogram using a calibration curve of pure reference standards and expressed as ng·g−1 protein. One-way anova with Dunnett's post hoc test was used to compare dopamine content of the 6-OHDA lesioned side, expressed as % of the contralateral, intact side between treatment groups. A two-way anova with Dunnett's post hoc test was used to compare dopamine turnover ratios in lesioned and non-lesioned striatal hemispheres between treatment groups.
Materials
VU0155041 was obtained from Ascent Scientific Ltd., Bristol, UK. CPPG was obtained from Tocris Cookson, Bristol, UK. All other reagents were obtained from Sigma-Aldrich, Gillingham, Dorset, UK.
Results
VU0155041 protects against 6-OHDA-induced degeneration of the nigrostriatal tract in rats
In the intact SNpc, a mean total of 306.3 ± 7.4 TH-positive cells were counted across the three rostrocaudal regions, that is, ∼102 cells per section. This correlates well with the 104 cells per section reported in our previous study (Austin et al., 2010) and the ∼100 cells per section in other studies (e.g. Iczkiewicz et al., 2010). In the 6-OHDA-lesioned side, the mean total number of TH-positive cells across three rostrocaudal levels declined to 26.5 ± 5.7 in the lesioned hemisphere (i.e. ∼12 cells per section) of vehicle-treated 6-OHDA lesioned animals (n= 8), demonstrating that less than 9% of cells remained. In contrast, supranigral treatment with VU0155041 protected against TH-positive cell loss, whereby significant protection was reached with a 100 nmol dose of VU0155041 (P < 0.001; one-way anova with a Dunnett's post hoc test; Figure 1). The number of TH-positive cells remaining across three rostrocaudal levels in the lesioned hemisphere following 100 nmol VU0155041 treatment was 131.3 ± 11.6, approaching 40% of the intact hemisphere where the mean total number of cells across the three levels was 321.1 ± 4.9 (n= 6). Although Nissl stained cell counts were not performed here to further confirm cell preservation with VU0155041, subsequent measurements taken at the striatal level support these nigral data.
Figure 1.
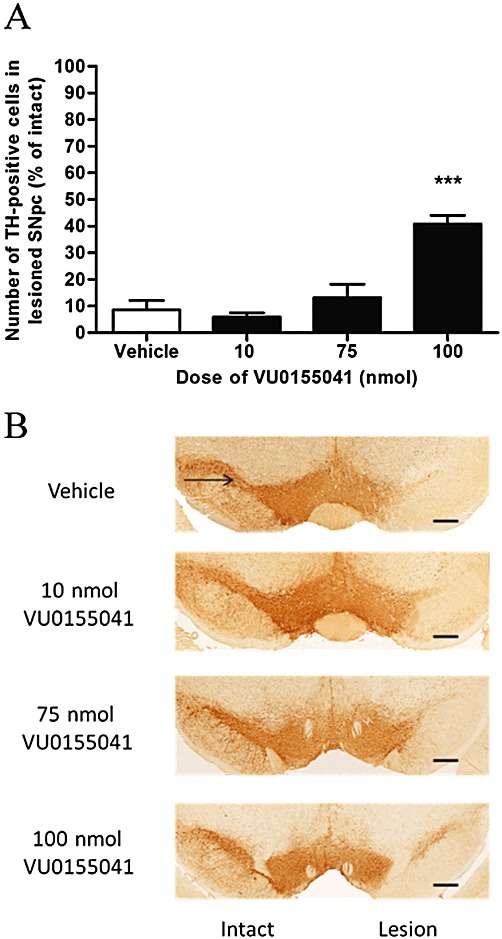
(A) Effect of 7 days of treatment with a range of doses of VU0155041 against 6-OHDA-induced loss of TH-positive cells in the SNpc. Data are mean ± SEM (n= 8 for vehicle; n= 7, 8 and 6 for increasing doses of VU0155041). ***P < 0.001, significantly different from vehicle; one-way anova, Dunnett's post hoc test. (B) Representative photomicrographs showing TH staining in the SNpc in the intact and lesion hemispheres of the different treatment groups. Scale bar: 500 µm.
TH levels were also significantly preserved in the striatum of animals treated with 100nmol VU0155041 (Figure 2; n= 6), in contrast to the low level remaining in the lesioned striatum of vehicle-treated animals (P < 0.001, n= 8; one-way anova, Dunnett's post hoc test). The levels of another marker of dopaminergic neurones, DDC, were also significantly preserved to a similar extent in the striatum of VU0155041-treated animals compared with vehicle-treated animals (n= 8; P < 0.05; one-way anova with Dunnett's post hoc test; Figure 3), confirming that the higher levels of TH were not simply a reflection of increased TH expression.
Figure 2.
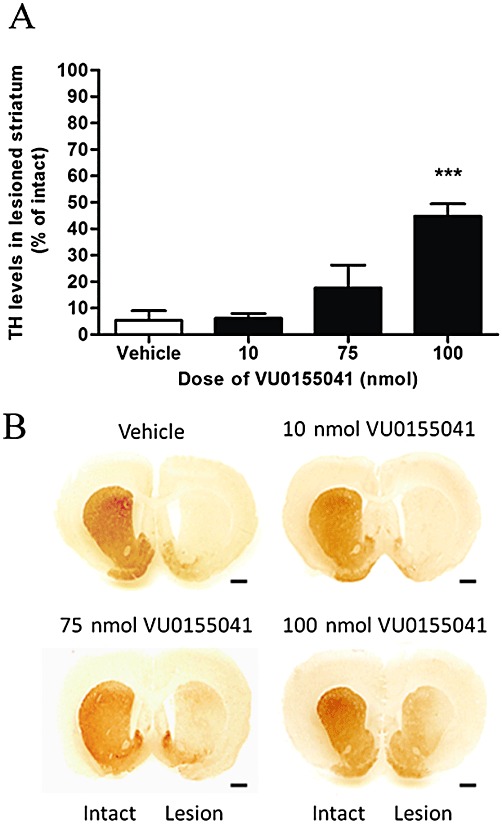
(A) Effect of 7 days' treatment with a range of doses of VU0155041 against the 6-OHDA-induced reduction in TH levels in the lesioned striatum. Data are mean ± SEM (n= 8 for vehicle; n= 7, 8 and 6 for increasing doses of VU0155041). ***P < 0.001, significantly different from vehicle; one-way anova, Dunnett's post hoc test. (B) Representative photomicrographs showing TH staining in the striatum in the intact and lesion hemispheres of the different treatment groups. Scale bar: 500 µm.
Figure 3.
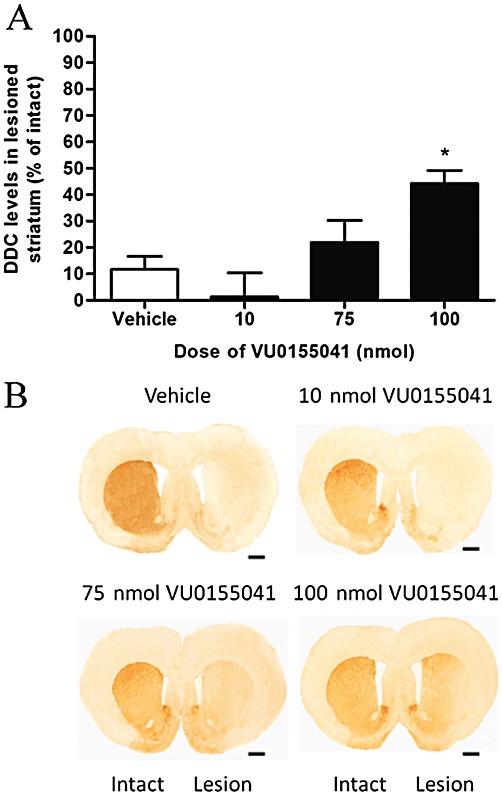
(A) Effect of 7 days of treatment with a range of doses of VU0155041 against the 6-OHDA-induced reduction in DDC levels in the lesioned striatum. Data are mean ± SEM (n= 8 for vehicle; n= 7, 8 and 6 for increasing doses of VU0155041). *P < 0.05, significantly different from vehicle; one-way anova, Dunnett's post hoc test. (B) Representative photomicrographs showing TH staining in the striatum in the intact and lesion hemispheres of the different treatment groups. Scale bar: 500 µm.
In the separate study where striata were taken for HPLC analysis, in vehicle-treated 6-OHDA lesioned animals, striatal dopamine content fell by ∼84% from 17 144 ± 756 ng·g−1 in the intact hemisphere to 2680 ± 1578 ng·g−1 in the lesioned hemisphere (n= 6). Treatment with 100 nmol VU0155041 preserved striatal dopamine content in the lesioned hemisphere, which remained at 13 214 ± 718 ng·g−1 compared with 17 409 ± 364 ng·g−1 in the intact hemisphere (n= 6), representing a fall of only 24% (P < 0.001 vs. vehicle; one-way anova, Dunnett's post hoc test). There was no significant change in dopamine turnover between groups which rose slightly in each case in the lesioned hemisphere; ratios rose from 0.14 ± 0.01 to 0.18 ± 0.02 and from 0.14 ± 0.01 to 0.16 ± 0.01 in the vehicle and 100 nmol VU0155041 treatment groups, respectively.
VU0155041 reduces the 6-OHDA-induced increase in inflammatory markers in the SNpc
Analysis of IBA-1, a marker of increased microglial activation, showed that a unilateral injection of 6-OHDA markedly increased microglial activation in the lesioned SNpc compared with the intact non-lesioned hemisphere (Figure 4A). Following a unilateral 6-OHDA lesion, the mean total IBA-1 density at a central region of the SNpc increased to almost three-fold that of the intact hemisphere in vehicle-treated animals (n= 8). However, following VU0155041 treatment (100 nmol, n= 6), this increase in IBA-1 density was significantly reduced compared with vehicle-treated animals (P < 0.05; one-way anova with Dunnett's post hoc test). Qualitative analysis (Figure 4B) revealed that IBA-1-positive cells in the intact hemisphere displayed morphology typical of resting or quiescent cells, while in the lesioned hemisphere, a marked increase in IBA-1-positive cells displaying morphology typical of activated cells was observed, demonstrating a switch from ramified to ameboid morphology in microglia.
Figure 4.
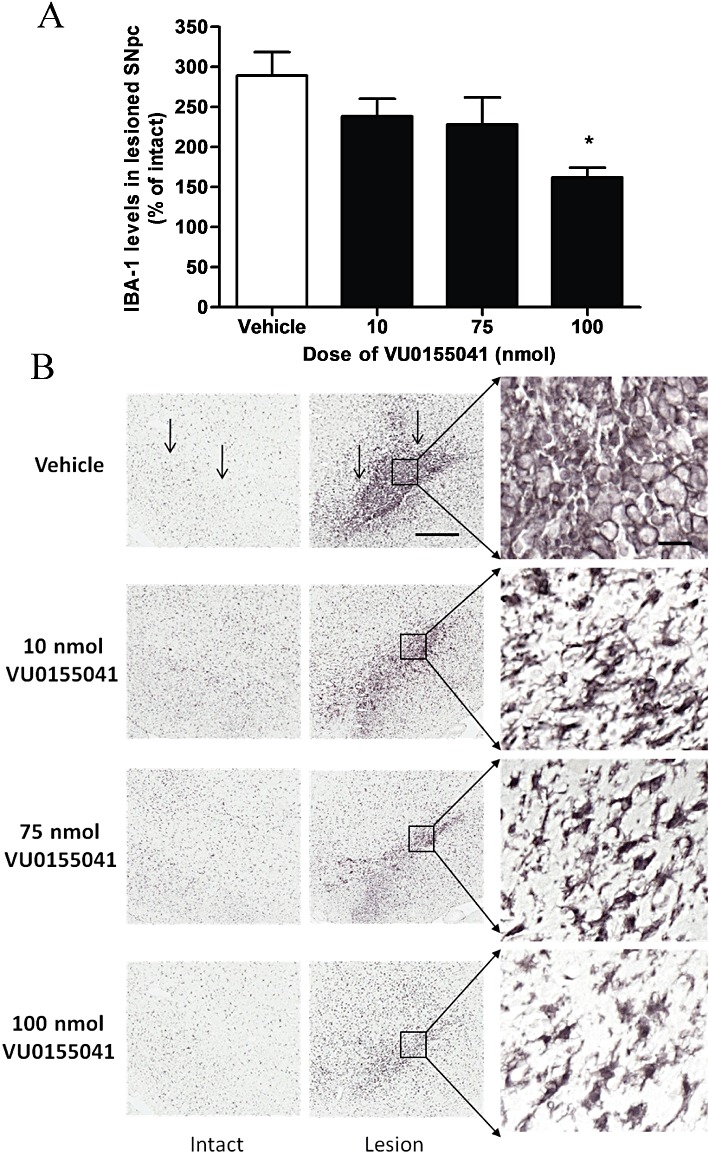
(A) Effect of 7 days of treatment with a range of doses of VU0155041 against the 6-OHDA-induced increase in IBA-1-positive cells in the SNpc. Data are mean ± SEM (n= 8 for vehicle; n= 7, 8 and 6 for increasing doses of VU0155041). *P < 0.05, significantly different from vehicle; one-way anova, Dunnett's post hoc test. (B) Representative photomicrographs showing IBA-1 staining in the SNpc in the intact and lesion hemispheres of the different treatment groups. Scale bars: 200 µm on main panel, 50 µm on higher magnification images. Vertical arrows indicate the dorsal border of the SNpc.
Following a unilateral 6-OHDA lesion, the mean total number of GFAP-positive cells was also markedly increased in vehicle-treated animals from 25.3 ± 2.9 in the intact hemisphere to 89.9 ± 7.6 in the lesion hemisphere, a rise of 442 ± 55% (n= 8). Treatment with VU0155401 reduced this increase in GFAP staining from 25.8 ± 2.7 in the intact hemisphere to 79.7 ± 2.4 cells in the lesion hemisphere, a rise of only 327 ± 39% (n= 6). However, this effect was not significant (P > 0.05; one-way anova, Dunnett's post hoc test; Supporting Figure S1).
VU0155041 preserves motor function in the 6-OHDA-lesioned rat
Supranigral injection of VU0155041 provided functional protection against 6-OHDA-induced loss of motor function. In the cylinder reaching test, pre-lesion use of the contralateral paw alone amounted to ∼30%, in line with our previous observations (Austin et al., 2010). At 5 days post-6-OHDA lesion, the use of the contralateral paw fell significantly with respect to pre-lesion scores in vehicle-treated animals (n= 8, P < 0.05; two-way anova, Bonferroni's post hoc test; Figure 5A). In contrast, animals treated with VU0155041 (100 nmol) showed no reduction in contralateral paw use post-lesion, although these animals did have a lower pre-lesion score against which this change was assessed (Figure 5A). In the adjusted stepping test performed 6 days post-6-OHDA lesion, the use of the contralateral paw in vehicle-treated animals fell to ∼50% of pre-lesion use, for both forehand and backhand responses. However, treatment with the maximal 100 nmol dose of VU0155041 significantly protected against loss of contralateral paw use compared with vehicle-treated animals, in both directions [Figure 5B; n= 6; P < 0.01 (forehand) and P < 0.001 (backhand); one-way anova, Dunnett's post hoc test]. Amphetamine-induced ipsiversive rotations, 7 days after the lesion, were also reduced in 6-OHDA lesioned animals treated with VU0155041 [100 nmol; AUC: 336 ± 61 (n= 6)] compared with vehicle [AUC: 443 ± 25 (n= 8)], although this effect failed to reach significance (one-way anova, Dunnett's post hoc test). However, the time course of ipsiversive rotations induced by amphetamine did reveal significant differences between vehicle- and 100 nmol VU0155041-treated animals at certain time points (Figure 5C; P < 0.01; two-way anova, Bonferroni's post hoc test).
Figure 5.
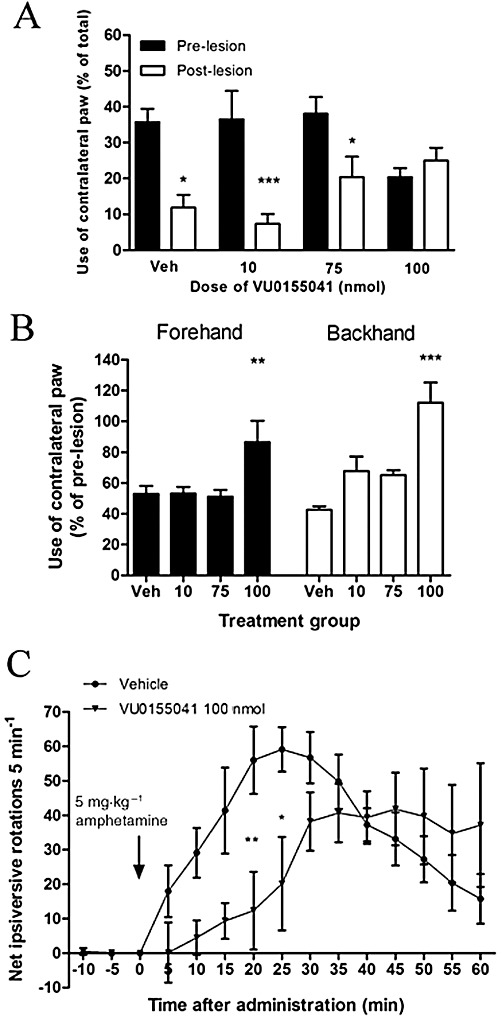
Effect of 7 days of treatment with a range of doses of VU0155041 against 6-OHDA-induced (A) reduced contralateral paw use in the cylinder test at 5 days post-lesion, (B) reduced contralateral paw use in forehand or backhand adjusted stepping test at 6 days post-lesion and (C) increased amphetamine-induced ipsiversive rotations over 60 min at 7 days post-lesion. Data are mean ± SEM [n= 8, vehicle (Veh); n= 6–8, VU0155041]. (A) *P < 0.05 and ***P < 0.001, significantly different from pre-lesion score two-way anova, Bonferroni's post hoc test. (B and C) *P < 0.05, **P < 0.01 and ***P < 0.001, significantly different from vehicle; one-way anova, Dunnett's post hoc test.
Protection with VU0155041 is inhibited by the group III mGlu receptor antagonist CPPG
In a separate study, the protective effects of 100 nmol VU0155041 in preserving TH-positive cells in the SNpc against a 6-OHDA lesion were found to be significantly inhibited by pre-treatment with 75 nmol CPPG 30 min before each dose of VU0155041 (Figure 6 A, B). In the lesioned hemisphere, preservation of TH-positive cells following VU0155041 treatment fell significantly following pre-treatment with CPPG (one-way anova, Dunnett's post hoc test). Preservation of striatal dopamine content following VU0155041 treatment was also lost by pre-treatment with 75 nmol CPPG (Figure 6C).
Figure 6.
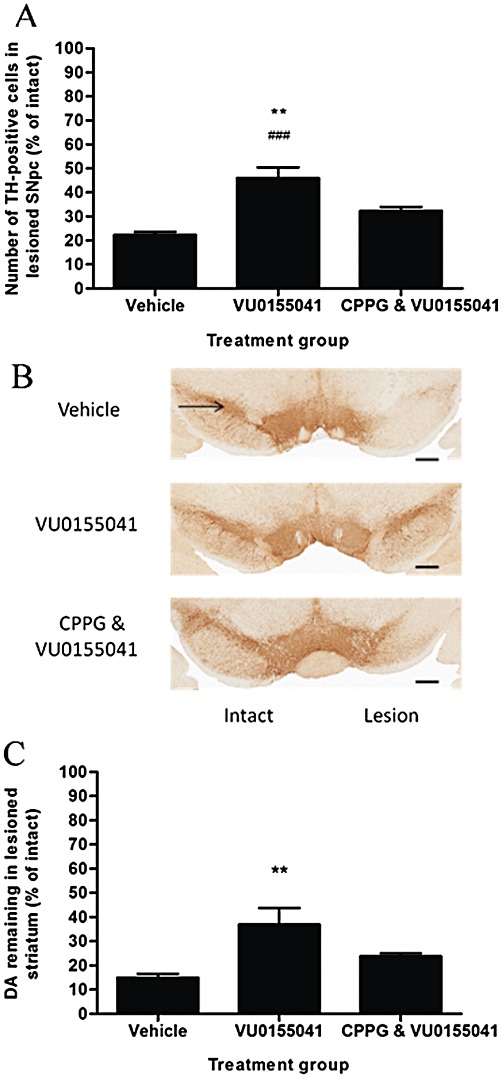
Effect of pre-treatment with 75 nmol CPPG on the protection afforded by 7 days of treatment with 100 nmol VU0155041 against (A) 6-OHDA-induced loss of TH-positive cells in the SNpc and (C) reduction in striatal dopamine (DA) content. Data are mean ± SEM (n= 6 for vehicle, n= 7 for VU0155041 treated alone and following CPPG). (A) **P < 0.01 and ###P < 0.001, significantly different from CPPG pre-treated group or vehicle-treated group, respectively; one-way anova, Dunnett's post hoc test. (C) **P < 0.01, significantly different from vehicle group. (B) Representative photomicrographs showing TH staining in the SNpc in the intact and lesion hemispheres of the different treatment groups. Scale bar: 500 µm.
The ability of VU0155041 to improve motor function was also compromised following pre-treatment with CPPG. As seen before, and this time from a similar pre-lesion starting point, VU0155041 prevented the significant decline in contralateral paw reaching seen in the vehicle-treated 6-OHDA lesioned animals (Figure 7A). In contrast, contralateral paw reaching in the cylinder test was not so well preserved at pre-lesion levels in the CPPG-treated animals, although this failed to reach significance (Figure 7A). In contrast, in the stepping test (Figure 7B), while use of the contralateral paw in VU0155041-treated animals was maintained in the forehand direction, this use was significantly reduced following CPPG pre-treatment (n= 7; P < 0.05, one-way anova, Bonferroni's post hoc test). Furthermore, the reduction in amphetamine-induced ipsiversive rotations following VU0155041 treatment with respect to vehicle-treated animals was inhibited at various time points following CPPG pre-treatment (Figure 7C; n= 7; P < 0.01, two-way anova with Bonferroni's post hoc test). Further analysis also revealed a tendency for the VU0155041-mediated reduction in amphetamine-induced rotational behaviour over the entire 60 min to be reversed by CPPG, although this failed to reach significance. Thus, AUC values of 520 ± 94 in vehicle-treated animals were reduced to 331 ± 85 following treatment with VU0155041 and vehicle (for CPPG), but to only 480 ± 84 in VU0155041- and CPPG-treated animals.
Figure 7.
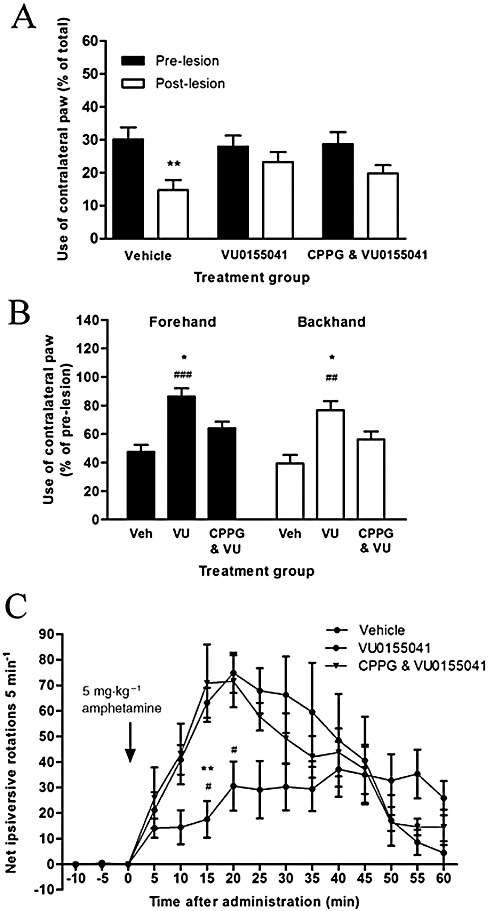
Effect of pre-treatment with 75 nmol CPPG on the protection afforded by 7 days of treatment with 100 nmol VU0155041 (VU) against 6-OHDA-induced (A) reduced contralateral paw use in the cylinder test at 5 days after the lesion, (B) reduced contralateral paw use in forehand or backhand adjusted stepping test at 6 days after the lesion and (C) increased amphetamine-induced ipsiversive rotations over 60 min at 7 days after the lesion. Data are mean ± SEM [n= 6 for vehicle (Veh), n= 7 for VU0155041 treated alone and following CPPG]. (A) **P < 0.01, significantly different from pre-lesion score; two-way anova, Bonferroni's post hoc test. (B) *P < 0.05, significantly different from CPPG; ##P < 0.01 and ###P < 0.001, significantly different from vehicle; one-way anova, Bonferroni's post hoc test. (C) **P < 0.01, significantly different from CPPG; #P < 0.05, significantly different from vehicle group; two-way anova, Bonferroni's post hoc test.
Discussion
We recently reported that activation of presynaptic group III mGlu receptors, using the broad spectrum agonist L-AP4 mediated functional neuroprotection in the 6-OHDA rat model of Parkinson's disease (Austin et al., 2010). The work presented here extends these findings, demonstrating for the first time that selective activation of mGlu4 receptors, using the PAM, VU0155041, is sufficient to provide beneficial effect. Furthermore, the effects were accompanied by a reduction in inflammatory markers, suggesting that a reduced inflammatory response may contribute, in part, to this protective response.
Neuroprotective effects of mGlu4 receptor activation in the SNpc
VU0155041 is a selective PAM for mGlu4 receptors, recently described by Niswender et al. (2008a) that allowed activation of the mGlu4 receptors in the SNpc to be probed without the drawbacks of mGlu1 receptor antagonism ascribed to the more commonly used mGlu4 receptor PAM, PHCCC (Marino et al., 2003). Administration of VU0155041 1 h before and for 7 days after the 6-OHDA lesion significantly protected the nigrostriatal tract. This was shown by the preservation of TH-positive cells in the SNpc and reduction in 6-OHDA-induced loss of striatal TH and DDC immunoreactivity. These changes were paralleled by elevated dopamine content in the striatum of animals treated with VU0155041, relative to that in vehicle-treated animals, which, in the absence of altered dopamine turnover, is likely to reflect functional preservation rather than simply intrinsic compensatory mechanisms.
Of important functional significance, VU0155041 also preserved motor function after the 6-OHDA lesion. Significant preservation of contralateral forelimb use was shown in both the cylinder and the adjusted stepping test, alongside a small reduction in amphetamine-induced ipsiversive rotations. Pre-treatment with the broad spectrum group III mGlu antagonist CPPG markedly attenuated both the behavioural end-points and the post mortem indices of preserved nigrostriatal tract function, confirming that VU0155041 produced these effects via a group III mGlu receptor-mediated process. As CPPG binds to the orthosteric site on mGlu4 receptors, distinct from that of the allosteric modulator, VU0155041, CPPG-mediated inhibition here most likely reflects block of endogenous glutamate binding to the orthosteric site on mGlu4 receptors, thereby indirectly reducing the efficacy of the PAM. At present, it remains to be determined whether higher doses of CPPG would have inhibited the residual responses of VU1055041. However, VU0155041 exhibits some intrinsic allosteric agonist activity in vitro, which is not blocked by the orthosteric antagonist, LY341495 (Niswender et al., 2008a). If similar actions occur in vivo, this may also explain the lack of complete reversal of VU0155041's actions by CPPG. The use of a selective mGlu4 receptor antagonist, when available, or examination of the protective effects of VU0155041 in mGlu4 receptor knockout mice, will help clarify the involvement of mGlu4 receptors in these actions.
The degree of protection seen with VU0155041 of around 40% for post mortem indices was less than the 60% achieved in our study using the broad spectrum agonist L-AP4 (Austin et al., 2010). Nevertheless, this suggests that a large proportion of the effects of L-AP4 were likely to be attributable to mGlu4 receptor activation. The maximal dose of VU0155041 tested here (100 nmol) was based on that used i.c.v. by Niswender et al. (2008a) against haloperidol-induced catalepsy and reserpine-induced akinesia. However, a recent study showing 500 nmol VU0155041 is tolerated intrathecally (Wang et al., 2011) paves the way for future examination of higher doses. In the meantime, a contribution from activation of additional group III mGlu receptors to the functional protection demonstrated previously with L-AP4 remains possible. The protective effects of VU0155041 are certainly consistent with the intense staining for mGlu4 receptors recently reported in the SNpc (Broadstock et al., 2011). Some of this immunoreactivity probably represents mGlu4 receptors located on excitatory (glutamatergic) terminals, as has been specifically demonstrated in the neighbouring SNpr (Kosinski et al., 1999; Corti et al., 2002). However, the precise anatomical location of mGlu4 receptors on either neurones or other cell types in the SNpc remains to be examined.
Potential mechanisms underlying neuroprotection mediated by mGlu4 receptor activation in the SNpc
Numerous mechanisms may underlie mGlu4 receptor-mediated neuroprotection (see Duty, 2010). Following the intranigral administration performed here, the two most likely mechanisms include inhibition of glutamate release in the SNpc and protection thereby from glutamate-mediated excitotoxicity, or reduced inflammatory actions. Previous data already provide some support for the glutamate hypothesis, while the present study has provided new evidence in support of an anti-inflammatory contribution.
Regarding the glutamate hypothesis, early demonstrations that L-AP4 depressed glutamate-mediated EPSP's in dopaminergic neurones of the SNpc in vitro supported the existence of presynaptic inhibitory group III mGlu receptors on descending glutamatergic inputs to this region (Wigmore and Lacey, 1998). mGlu4 receptors were subsequently identified as key subtypes involved as PHCCC similarly inhibited STN-evoked EPSC's in dopaminergic neurones of the SNpc (Valenti et al., 2005). In agreement with these electrophysiological findings, studies in this laboratory have shown that L-SOP and L-AP4 inhibit depolarization-evoked release of [3H]-D-aspartate from slices of rat SN in vitro, while local intranigral infusion of L-SOP reduces glutamate release in the SNpr in vivo (Austin et al., 2010). These effects of L-AP4 in vitro were potentiated by PHCCC, supporting involvement of mGlu4 receptors in restricting glutamate release in the SN in general (Broadstock et al., 2011). Further microdialysis studies will help establish whether inhibition of glutamate release in the SNpc contributes to the neuroprotective effects following mGlu4 receptor potentiation. In the MPTP study of Battaglia et al. (2006), protection against MPTP-induced degeneration was seen following injection of PHCCC directly into the globus pallidus, which lies upstream of the SNpc in the indirect basal ganglia circuit. Thus, mGlu4 receptor-mediated normalization of glutamatergic drive to the SNpc, either directly by actions in the SNpc itself or indirectly via correction of pallido-subthalamic firing and subsequent downstream reductions in STN glutamatergic drive to the SNpc, might underlie neuroprotection in these toxin-induced animal models of Parkinson's disease.
The presence of mGlu4 receptors on both astrocytes and microglia in culture (Taylor et al., 2003; Yao et al., 2005) raises the possibility that a further astroglial component may contribute to the overall protective actions of the modulators of mGlu4 receptors. Certainly, inflammation is believed to play a key role in the pathogenesis of Parkinson's disease (Mogi et al., 1994; Gerhard et al., 2006; Whitton, 2007; Tansey and Goldberg, 2010), and microglial activation and elevated inflammatory markers are similarly seen in the striatum and SNpc of rats following a 6-OHDA lesion (Mogi et al., 2000; Cicchetti et al., 2002). It is thought that following a pathological stimulus of this kind, substantial increases in pro-inflammatory cytokines and cell adhesion molecules occur, whereupon the activated microglia cluster around dopaminergic neurones and become phagocytic (Bronstein et al., 1995; Banati et al., 1998). The progressive degeneration is worsened by the release of chemo-attractants from the dying neurones, which leads to further activated microglia infiltrating the region. L-AP4 treatment of astrocytes has been shown to reduce the production of such pro-inflammatory chemokines (Besong et al., 2002), an effect abolished in astrocyte cultures from mGlu4 receptor knock out mice, implicating astroglial mGlu4 receptors in mediating potential anti-inflammatory actions. The decrease in GFAP-positive cells observed here following VU0155041 treatment supports a role for mGlu4 receptor activation in restricting 6-OHDA-induced astroglial-driven neurotoxicity in vivo. Treatment with the PAM for mGlu4 receptors also led to a significant reduction in activation of microglia in the SNpc of 6-OHDA rats, as shown by the reduced levels of IBA-1. Therefore, although these mechanisms have only just begun to be explored, these data support an anti-inflammatory component contributing to the overall protective potential of targeting mGlu4 receptors in Parkinson's disease. Such an anti-inflammatory role has been linked with mGlu4 receptor activation in the experimental autoimmune encephalomyelitis mouse model of multiple sclerosis, where PHCCC restrained the neuroinflammatory response (Fallarino et al., 2010). More interestingly, our preliminary immunofluorescent studies (Supporting Figure S2) suggested little co-localization of mGlu4 receptors and GFAP in the SNpc, suggesting that any anti-inflammatory actions of VU0155041 in this 6-OHDA model are unlikely to be astrocyte-mediated. However, whether these actions are mediated via neuronal- or microglial-residing mGlu4 receptors remains to be established and future co-localization studies will help elucidate this.
By initiating treatment before the lesion, the present study provided in vivo proof of concept but did not reflect the timing of drug intervention required in a clinical setting. Therefore, future studies should seek to investigate the potential of targeting mGlu4 receptors in a slower retrograde lesion model whereby treatment can be delayed until after the lesion. Achieving good brain penetration with mGlu4 receptor PAMs, in the absence of a compromised blood brain barrier, has proven difficult. Other researchers have checked the systemic availability of VU0155041 and found that, like many other mGlu4 receptor PAMs, it has a very low brain : plasma ratio of 0.01 (Doller et al., 2010). Here, using site-directed injections that circumvent these barriers, we have been able to carefully dissect the protective role of the mGlu4 receptors in the SNpc itself, but better tools are needed to facilitate the next stage of systemic studies towards translation of these data.
To conclude, these studies demonstrate that selective activation of mGlu4 receptors using the PAM V0155041 provides functional neuroprotection in the 6-OHDA rat model of Parkinson's disease. The notion that multiple mechanisms, including restricting glutamate release and provision of an anti-inflammatory component, may contribute to this protection is particularly appealing given the likelihood that multiple pathogenic events lead to Parkinson's disease and the lack of translation from animal models into a clinically proven neuroprotective strategy often seen when targeting one mechanism alone (see Duty and Jenner, 2011). Taken together with recent reports that mGlu4 receptor activation provides symptomatic relief in the reserpine and haloperidol rodent models of Parkinson's disease (Niswender et al., 2008a; East et al., 2010; Broadstock et al., 2011), the present study demonstrates targeting mGlu4 receptors may ultimately offer a unifying means to treat symptoms and ongoing degeneration in Parkinson's disease. With the current emphasis on developing systemically active PAMs for mGlu4 receptors (Niswender et al., 2008b; Williams et al., 2009; East et al., 2010; Engers et al., 2010), it is hoped that subsequent studies exploring the efficacy of chronic, systemically delivered agents will soon be possible.
Acknowledgments
Matthew J. Betts is funded by a KCL Case studentship with Eli Lilly & Co Ltd. We would like to thank Mark A. Ward, Carl Hobbs and Claire Cella for their assistance with the aspects of the histology; Jane Cooper for helping with neurochemical analysis; and Dr Domenico Spina for expert statistical advice.
Glossary
- 6-OHDA
6-hydroxydopamine
- CPPG
(RS)-α-cyclopropyl-4-phosphonophenylglycine
- DDC
dopa decarboxylase
- GFAP
glial fibrillary acidic protein
- IBA-1
ionized calcium-binding adaptor molecule 1
- L-AP4
L-2-amino-4-phosphonobutyrate
- mGlu
metabotropic glutamate
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PAM
positive allosteric modulator
- PHCCC
N-phenyl-7-(hydroxylimino)cyclopropa[b]chromen-1a-carboxamide
- SNpc
substantia nigra pars compacta
- STN
subthalamic nucleus
- VU0155041
(+/−)-cis-2-(3,5-dichlorphenylcarbamoyl)cyclohexanecarboxylic acid
Conflicts of interest
The authors state no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effect of sub-chronic VU0155041 treatment against 6-OHDA-induced activation of GFAP-positive cells in the SNpc (A), with representative photomicrographs demonstrating effect of each dose of VU0155041 tested on lesioned hemisphere (B).Data are mean ± SEM (n = 8 for vehicle; n = 7, 8 and 6 for increasing doses of VU0155041). Scale bars: 200 μm on main panel, 50 μm on higher magnification images. Vertical arrows indicate the dorsal border of the SNpc. Methodological details are described within the mainmanuscript.
Figure S2 Photomicrographs of immunofluorescence staining showing minimal co-localization of GFAPand mGlu4 receptors in the SNpc from naïve rat brain section (6 μm). Images show (A) mGlu4 receptors [red], (B) nuclei (Hoechst) [blue], (C) GFAP-positive cells [green] and (D) merged image of all three markers. In (D), white arrow indicates co-localization of mGlu4 receptor with GFAP-positive cell. Scale bar = 50 μm. White arrow indicates only region where co-localization may be seen. Briefly, 6 μm sections from the central region of the SNpc (-5.3 mm AP from Bregma) were incubated with the primary antibody rabbit polyclonal anti-mGlu4 primary antibody (ab53088 Abcam, 1:50) at room temperature overnight then for 1 hour with a goat anti-rat biotinylated secondary antibody (Sigma) and 1 hour at room temperature with a fluorescent AlexaFluor 594 streptavidin complex (Invitrogen, USA). Sections were then washed and subsequently incubated with a mouse monoclonal anti-GFAP (Sigma, G3893, 1:1000), primary antibody for 1 hour at room temperature, followed by goat anti-mouse-488 fluorescent antibody (Alexa Fluor, Invitrogen, 1:1000) solution containing Hoechst (1:5000). Fluorescent images of sections werecaptured using a Zeiss Apotome fluorescent microscope and analysed for co-localization using Axiovision image analysis software.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez L, Macias R, Lopez G, Alvarez E, Pavon N, Rodriguez-Oroz MC, et al. Bilateral subthalamotomy in Parkinson's disease: initial and long-term response. Brain. 2005;128:570–583. doi: 10.1093/brain/awh397. [DOI] [PubMed] [Google Scholar]
- Austin PJ, Betts MJ, Broadstock M, O'Neill MJ, Mitchell SN, Duty S. Symptomatic and neuroprotective effects following activation of nigral group III metabotropic glutamate receptors in rodent models of Parkinson's disease. Br J Pharmacol. 2010;160:1741–1753. doi: 10.1111/j.1476-5381.2010.00820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson's disease. Mov Disord. 1998;13:221–227. doi: 10.1002/mds.870130205. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Busceti CL, Molinaro G, Biagioni F, Traficante A, Nicoletti F, et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurosci. 2006;26:7222–7229. doi: 10.1523/JNEUROSCI.1595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besong G, Battaglia G, D'Onofrio M, Di Marco R, Ngomba RT, Storto M, et al. Activation of group III metabotropic glutamate receptors inhibits the production of RANTES in glial cell cultures. J Neurosci. 2002;22:5403–5411. doi: 10.1523/JNEUROSCI.22-13-05403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breit S, Lessmann L, Unterbrink D, Popa RC, Gasser T, Schulz JB. Lesion of the pedunculopontine nucleus reverses hyperactivity of the subthalamic nucleus and substantia nigra pars reticulata in a 6-hydroxydopamine rat model. Eur J Neurosci. 2006;24:2275–2282. doi: 10.1111/j.1460-9568.2006.05106.x. [DOI] [PubMed] [Google Scholar]
- Broadstock M, Austin P, Betts M, Duty S. Antiparkinsonian potential of targeting group III metabotropic glutamate receptor subtypes in the rodent substantia nigra pars reticulata. Br J Pharmacol. 2011;165:1034–1045. doi: 10.1111/j.1476-5381.2011.01515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein DM, Perez-Otano I, Sun V, Mullis Sawin SB, Chan J, Wu GC, et al. Glia-dependent neurotoxicity and neuroprotection in mesencephalic cultures. Brain Res. 1995;704:112–116. doi: 10.1016/0006-8993(95)01189-7. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Kingston A, O'Neill MJ, Catania MV, Di Grezia R, et al. Neuroprotective activity of the potent and selective mGlu1a metabotropic glutamate receptor antagonist, (+)-2-methyl-4-carboxyphenylglycine (LY367385): comparison with LY357366, a broader spectrum antagonist with equal affinity for mGlu1a and mGlu5 receptors. Neuropharmacology. 1999;38:199–207. doi: 10.1016/s0028-3908(98)00159-2. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci. 2002;15:991–998. doi: 10.1046/j.1460-9568.2002.01938.x. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Corti C, Aldegheri L, Somogyi P, Ferraguti F. Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience. 2002;110:403–420. doi: 10.1016/s0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- Doller D, Hong S-P, Uberti MA, Nerio MT, Brodbeck R, Breysse N, et al. 2010. Lu AF21934, a brain penetrant mGlu4 receptor positive allosteric modulator tool compound. Society for Neuroscience, Poster 655.28.
- Duty S. Therapeutic potential of targeting group III metabotropic glutamate receptors in the treatment of Parkinson's disease. Br J Pharmacol. 2010;161:271–287. doi: 10.1111/j.1476-5381.2010.00882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duty S, Jenner P. Animal models of Parkinson's disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol. 2011;164:1357–1391. doi: 10.1111/j.1476-5381.2011.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East SP, Bamford S, Dietz MG, Eickmeier C, Flegg A, Ferger B, et al. An orally bioavailable positive allosteric modulator of the mGlu4 receptor with efficacy in an animal model of motor dysfunction. Bioorg Med Chem Lett. 2010;20:4901–4905. doi: 10.1016/j.bmcl.2010.06.078. [DOI] [PubMed] [Google Scholar]
- Engers DW, Gentry PR, Williams R, Bolinger JD, Weaver CD, Menon UN, et al. Synthesis and SAR of novel, 4-(phenylsulfamoyl)phenylacetamide mGlu4 positive allosteric modulators (PAMs) identified by functional high-throughput screening (HTS) Bioorg Med Chem Lett. 2010;20:5175–5178. doi: 10.1016/j.bmcl.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallarino F, Volpi C, Fazio F, Notartomaso S, Vacca C, Busceti C, et al. Metabotropic glutamate receptor-4 modulates adaptive immunity and restrains neuroinflammation. Nat Med. 2010;6:897–902. doi: 10.1038/nm.2183. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Hassani OK, Mouroux M, Feger J. Increased subthalamic neuronal activity after nigral dopaminergic lesion independent of disinhibition via the globus pallidus. Neuroscience. 1996;72:105–115. doi: 10.1016/0306-4522(95)00535-8. [DOI] [PubMed] [Google Scholar]
- Iczkiewicz J, Broom L, Cooper JD, Wong AM, Rose S, Jenner P. The RGD-containing peptide fragment of osteopontin protects tyrosine hydroxylase positive cells against toxic insult in primary ventral mesencephalic cultures and in the rat substantia nigra. J Neurochem. 2010;114:1792–1804. doi: 10.1111/j.1471-4159.2010.06896.x. [DOI] [PubMed] [Google Scholar]
- Iribe Y, Moore K, Pang KC, Tepper JM. Subthalamic stimulation-induced synaptic responses in substantia nigra pars compacta dopaminergic neurons in vitro. J Neurophysiol. 1999;82:925–933. doi: 10.1152/jn.1999.82.2.925. [DOI] [PubMed] [Google Scholar]
- Kosinski CM, Risso BS, Conn PJ, Levey AI, Landwehrmeyer GB, Penney JB, et al. Localization of metabotropic glutamate receptor 7 mRNA and mGluR7a protein in the rat basal ganglia. J Comp Neurol. 1999;415:266–284. [PubMed] [Google Scholar]
- MacInnes N, Duty S. Group III metabotropic glutamate receptors act as hetero-receptor modulating evoked GABA release in the globus pallidus in vivo. Eur J Pharmacol. 2006;580:95–99. doi: 10.1016/j.ejphar.2007.10.030. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Valenti O, Conn PJ. Glutamate receptors and Parkinson's disease: opportunities for intervention. Drugs Aging. 2003;20:377–397. doi: 10.2165/00002512-200320050-00006. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from Parkinsonian patients. Neurosci Lett. 1994;165:208–210. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- Mogi M, Togari A, Tanaka K, Ogawa N, Ichinose H, Nagatsu T. Increase in level of tumor necrosis factor-α in 6-hydroxydopamine-lesioned striatum in rats is suppressed by immunosuppressant FK506. Neurosci Lett. 2000;289:165–168. doi: 10.1016/s0304-3940(00)01275-1. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, et al. Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol. 2008a;74:1345–1358. doi: 10.1124/mol.108.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Lebois EP, Luo Q, Kim K, Muchalski H, Yin H, et al. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4): part I. Discovery of pyrazolo[3,4-d]pyrimidines as novel mGluR4 positive allosteric modulators. Bioorg Med Chem Lett. 2008b;18:5626–5630. doi: 10.1016/j.bmcl.2008.08.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1998. [Google Scholar]
- Peppe A, Pierantozzi M, Bassi A, Altibrandi MG, Brusa L, Stefani A, et al. Stimulation of the subthalamic nucleus compared with the globus pallidus internus in patients with Parkinson disease. J Neurosurg. 2004;101:195–200. doi: 10.3171/jns.2004.101.2.0195. [DOI] [PubMed] [Google Scholar]
- Piallat B, Benazzouz A, Benabid AL. Subthalamic nucleus lesion in rats prevents dopaminergic nigral neuron degeneration after striatal 6-OHDA injection: behavioural and immunohistochemical studies. Eur J Neurosci. 1996;8:1408–1414. doi: 10.1111/j.1460-9568.1996.tb01603.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxicity in Parkinson's disease: a target for neuroprotection. Ann Neurol. 1998;44:S175–S188. doi: 10.1002/ana.410440726. [DOI] [PubMed] [Google Scholar]
- Smith AD, Bolam JP. The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci. 1990;13:259–265. doi: 10.1016/0166-2236(90)90106-k. [DOI] [PubMed] [Google Scholar]
- Stocchi F, Nordera G, Marsden CD. Strategies for treating patients with advanced Parkinson's disease with disastrous fluctuations and dyskinesias. Clin Neuropharmacol. 1997;20:95–115. doi: 10.1097/00002826-199704000-00001. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Pocock JM. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J Neurosci. 2003;23:2150–2160. doi: 10.1523/JNEUROSCI.23-06-02150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ. Group III metabotropic glutamate-receptor-mediated modulation of excitatory transmission in rodent substantia nigra pars compacta dopamine neurons. J Pharmacol Exp Ther. 2005;313:1296–1304. doi: 10.1124/jpet.104.080481. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Palmer S, Datla KP, Zbarsky V, Croucher MJ, Dexter DT. Neuroprotective effects of metabotropic glutamate receptor ligands in a 6-hydroxydopamine rodent model of Parkinson's disease. Eur J Neurosci. 2005;22:1799–1806. doi: 10.1111/j.1460-9568.2005.04362.x. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Zbarsky V, Datla KP, Dexter DT, Croucher MJ. Selective activation of group III metabotropic glutamate receptors by L-(+)-2-amino-4-phosphonobutryic acid protects the nigrostriatal system against 6-hydroxydopamine toxicity in vivo. J Pharmacol Exp Ther. 2007;320:397–409. doi: 10.1124/jpet.106.108159. [DOI] [PubMed] [Google Scholar]
- Vila M, Marin C, Ruberg M, Jimenez A, Raisman-Vozari R, Agid Y, et al. Systemic administration of NMDA and AMPA receptor antagonists reverses the neurochemical changes induced by nigrostriatal denervation in basal ganglia. J Neurochem. 1999;73:344–352. doi: 10.1046/j.1471-4159.1999.0730344.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Jiang W, Yang R, Li Y. Spinal metabotropic glutamate receptor 4 is involved in neuropathic pain. Neuroreport. 2011;22:244–248. doi: 10.1097/WNR.0b013e3283453843. [DOI] [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigmore MA, Lacey MG. Metabotropic glutamate receptors depress glutamate-mediated synaptic input to rat midbrain dopamine neurones in vitro. Br J Pharmacol. 1998;123:667–674. doi: 10.1038/sj.bjp.0701662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R, Niswender CM, Luo Q, Le U, Conn PJ, Lindsley CW. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4). Part II: challenges in hit-to-lead. Bioorg Med Chem Lett. 2009;19:962–966. doi: 10.1016/j.bmcl.2008.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windels F, Carcenac C, Poupard A, Savasta M. Pallidal origin of GABA release within the substantia nigra pars reticulata during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2005;25:5079–5086. doi: 10.1523/JNEUROSCI.0360-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann M, Marino MJ, Bradley SR, Conn PJ. Activation of group III mGluRs inhibits GABAergic and glutamatergic transmission in the substantia nigra pars reticulata. J Neurophysiol. 2001;85:1960–1968. doi: 10.1152/jn.2001.85.5.1960. [DOI] [PubMed] [Google Scholar]
- Yao HH, Ding JH, Zhou F, Wang F, Hu LF, Sun T, et al. Enhancement of glutamate uptake mediates the neuroprotection exerted by activating group II or III metabotropic glutamate receptors on astrocytes. J Neurochem. 2005;92:948–961. doi: 10.1111/j.1471-4159.2004.02937.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.