

Abstract
BACKGROUND AND PURPOSE
Among several pharmacological properties, analgesia is the most common feature shared by either opioid or cannabinoid systems. Cannabinoids and opioids are distinct drug classes that have been historically used separately or in combination to treat different pain states. In the present study, we characterized the signal transduction pathways mediated by cannabinoid CB2 and µ-opioid receptors in quiescent and LPS-stimulated murine microglial cells.
EXPERIMENTAL APPROACH
We examined the effects of µ-opioid and CB2 receptor stimulation on phosphorylation of MAPKs and Akt and on IL-1β, TNF-α, IL-6 and NO production in primary mouse microglial cells.
KEY RESULTS
Morphine enhanced release of the proinflammatory cytokines, IL-1β, TNF-α, IL-6, and of NO via µ-opioid receptor in activated microglial cells. In contrast, CB2 receptor stimulation attenuated morphine-induced microglial proinflammatory mediator increases, interfering with morphine action by acting on the Akt-ERK1/2 signalling pathway.
CONCLUSIONS AND IMPLICATIONS
Because glial activation opposes opioid analgesia and enhances opioid tolerance and dependence, we suggest that CB2 receptors, by inhibiting microglial activity, may be potential targets to increase clinical efficacy of opioids.
Keywords: Akt, CB2 receptors, ERK1/2, microglia, morphine, nitric oxide, proinflammatory cytokines, signalling pathways
Introduction
Opioids produce their pharmacological effects by acting mainly through three types of receptors, namely µ, δ and κ (Satoh and Minami, 1995; receptor nomenclature follows Alexander et al. (2011). Opioids are the drugs of choice for treating severe pain, despite the development of tolerance, dependence and hyperalgesia (da Fonseca Pacheco et al., 2008). The cellular and molecular mechanisms involved in this phenomenon are complex and may involve receptor desensitization and endocytosis, intracellular signalling hyperactivity, secondary activation of excitatory amino acid receptors, and subsequent intracellular cascades, as well as glial activation and the release of proinflammatory mediators (Mayer et al., 1999; Watkins et al., 2005). Glial activation by opioids is an important phenomenon to understand, as it opposes opioid analgesia and enhances opioid tolerance, dependence and other negative side-effects, such as respiratory depression (Watkins et al., 2009). In particular, there is good evidence that glia and glia-derived proinflammatory mediators, such as IL-1β, TNF-α and IL-6, could be involved in tolerance to the anti-nociceptive properties of morphine. Glial cells are also considered to be crucial sources of nitric oxide (NO), responsible for morphine tolerance (Mayer et al., 1999; Chen and Sommer, 2009). Repeated morphine treatment can activate glia and hence up-regulate these various mediators (Raghavendra et al., 2002; 2004; Johnston et al., 2004; Watkins et al., 2005) through the MAPK, PKC and PI3K/Akt pathways, key players in the intracellular signalling cascade leading to the development of morphine tolerance (Mayer et al., 1999; Watkins et al., 2001; Raghavendra et al., 2002; 2004; Galeotti et al., 2006; Cunha et al., 2010).
In the nervous system, neurotransmission and neuroinflammation are mediated by the endocannabinoid signalling system (Fernández-Ruiz, 2009; Marrs et al., 2010). To date, two cannabinoid receptors have been identified by molecular cloning – CB1 and CB2 receptors. The CB1 receptors are expressed by the neurons and regulate the release of neurotransmitters, while CB2 receptors are expressed by the microglia, regulating their motility and immunomodulator production (Atwood and Mackie, 2010; Pertwee et al., 2010). In the nervous system, activation of CB1 and CB2 receptors is induced by the endocannabinoids, arachidonoylethanolamide (anandamide, AEA) and 2-arachidonoylglycerol, produced by the neurons and glia. However, ample evidence suggests that additional receptors may contribute to the behavioural, vascular and immunological actions of Δ9-tetrahydrocannabinol (THC), the major psychoactive constituent of marijuana, and endocannabinoids (Begg et al., 2005). In particular, it has been established that GPR55 is an additional novel cannabinoid receptor (Lauckner et al., 2008).
Microglia, a specialized population of macrophages found in the CNS, are quiescent in normal brain. However, after CNS injury or after interaction of LPS with toll-like receptor (TLR)-4 during bacterial infection (González-Scarano and Baltuch, 1999), these cells can be activated by cytokines produced by infiltrating immune effector cells. Thus, LPS stimulation of the microglia is a useful model for the study of mechanisms underlying neuronal injury by various proinflammatory and neurotoxic factors released by activated microglia (Jung et al., 2010).
CB receptor agonists produce pain relief in a variety of animal models (Richardson, 2000). Cannabinoids act on glia and neurons to inhibit the release of proinflammatory molecules, including IL-1β, TNF-α and NO (Molina-Holgado et al., 1997; 2002; Shohami et al., 1997; Puffenbarger et al., 2000; Cabral et al., 2001), and enhance the release of the anti-inflammatory cytokines IL-4, IL-10 (Klein et al., 2000) and IL-1 receptor antagonist (Molina-Holgado et al., 2003). It is known that the anti-inflammatory properties of CB receptor agonists are expressed through the activation of CB2 receptors (Klein and Newton, 2007; Romero-Sandoval et al., 2009; Correa et al., 2010; Hsieh et al., 2011). In particular, the activation of CB2 receptors expressed in brain microglia during neuroinflammation (Benito et al., 2008; Atwood and Mackie, 2010) reduced NO production and TNF-α in primary microglia (Ehrhart et al., 2005; Merighi et al., 2012). Furthermore, such activation protects against human microglial neurotoxicity by enhancing IL-10 production (Klegeris et al., 2003; Correa et al., 2005; 2010; Eljaschewitsch et al., 2006). The activation of CB2 receptors also reduces the release of proinflammatory factors in animal models of peritoneal hypoxia-ischemia and Huntington's disease (Benito et al., 2008). In the periphery, both CB1 and CB2 receptors participate in pain control (Malan et al., 2001).
Interestingly, receptors for opioids and cannabinoids are coupled to similar intracellular signalling mechanisms, leading to a decrease in cAMP production through the activation of Gi proteins (Satoh and Minami, 1995; Pertwee et al., 2010). Therefore, following the discovery that opioids and cannabinoids produce not only similar biochemical effects but also similar pharmacological effects, the interaction between these two classes of drugs has been extensively studied (Manzaneres et al., 1999). Cannabinoids can enhance the antinociceptive properties of opioids (Cichewicz, 2004; Wilson et al., 2008; Parolaro et al., 2010) and adolescent exposure to chronic THC blocks opiate dependence in maternally deprived rats (Morel et al., 2009). However, the molecular signalling underlying the participation of cannabinoids in the side effects induced by opioids is still unknown. Nevertheless, it is possible that cannabinoids may interfere with the tolerance and dependence effects induced by opioids because the administration of low-dose combinations of cannabinoids and opioids seems to be an alternative regimen that reduces the need to escalate opioid dose, while increasing opioid potency.
Therefore, the aim of the present study was to explore whether and how CB2 receptor stimulation affected opioid actions on activated microglia, with a view to improving pain control by increasing the clinical efficacy of opioids.
Methods
Animals
All animal care and experimental procedures conformed to the guidelines issued by the European Council (86/609/EEC) and were approved by the local Animal Care and Ethics Committee. The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). One-day-old Balb/c mice (50 in total) were obtained from Charles River (Calco, Italy).
Cell line, reagents and antibodies
CHO cells transfected with the human recombinant CB2 receptor cDNA (CHO-hCB2) were purchased from PerkinElmer (Milan, Italy). Tissue culture media and growth supplements were obtained from Cambrex, Bergamo, Italy. U0126 (MEK-1 and MEK-2 inhibitor, soluble in DMSO); human anti-ACTIVE®MAPK (phosphorylated Thr183/Tyr185) and human anti-ERK1/2 pAb were provided by Promega (Milan, Italy). Phosphorylated (Thr180/Tyr182) and total p38, phosphorylated (Thr183/Tyr185) and total JNK1/2 antibodies were from Cell Signalling Technology (Celbio, Milan, Italy). JWH-015 (1-propyl-2-methyl-3-(1-naphthoyl)indole) (soluble in ethanol), SH5 (inhibitor of Akt), blocking peptide for pAb to CB2 receptor (ALX-153-027) and anti-CB2 receptor rabbit polyclonal antibody (ALX-210-198) were from Enzo Life Sciences (Vinci-Biochem, Florence, Italy). The immunogen for the CB2 receptor antibody was a synthetic peptide corresponding to aa 20–33 of the human CB2 receptor N-terminal. Anti-MOR-1 (H80) (sc-15310), small interfering RNA (siRNA) for the CB2 receptor (sc-39913) and MOR-1 siRNA (sc-35958) were from Santa Cruz Biotechnology (DBA, Milan, Italy). AM 251 (N-(piperidin-1-yl)-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-1H-pyrazole-3-carboxamide) and AM 630 (6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone(6-iodopravadoline) (both soluble in DMSO) were from Tocris Bioscience (Bristol, UK). RNAiFect™ Transfection Kit was from Qiagen (Milan, Italy). Unless otherwise stated, all other chemicals were purchased from Sigma (Milan, Italy).
Primary microglial cell cultures
Primary glial cultures were prepared as described in a previous study (Molina-Holgado et al., 2002). Briefly, after anaesthesia (Zoletil 100, 30 mg kg-1, Virbac Laboratories, Carros, France) and decapitation, forebrains from newborn Balb/c mice were excised, meninges removed and tissue dissociated mechanically. Cells were re-suspended in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin, then plated on poly-D-lysine-coated (5 µg·mL−1) 75 cm2 flasks (Falcon; Celbio, Milan, Italy). After 15 days, the flasks were shaken vigorously to remove loosely adherent microglia. The supernatant was plated on multi-well culture plates for 2 h, and the medium was changed to remove non-adherent cells. Cells were grown in a humidified environment containing 5% CO2 at a constant temperature of 37°C. The purity of microglial cultures was assessed by examining cell morphology under phase-contrast microscopy, and was confirmed by flow cytometry with Mac-1 anti-CD11b antibody (BD Pharmingen, Milan, Italy).
Cell cultures
Cells were maintained in DMEM (primary microglia) or Ham's (CHOh-CB2 cells) medium containing 10% fetal calf serum, penicillin (100 U·mL−1), streptomycin (100 µg·mL−1), and L-glutamine (2 mM) at 37°C in 5% CO2/95% air. Geneticin (G418, 0.4 mg·mL−1) was added to CHO-hCB2 cells. Cells were split two or three times weekly at a ratio between 1:5 and 1:10.
Flow cytometry of primary microglial cells
Aliquots of 0.5 × 106 cells were incubated for 40 min at 4°C with either specific phycoerythrin (PE)-labelled antibodies, or isotype-matched irrelevant IgG-PE (Beckman Coulter, Fullerton, CA, USA) as negative control. Cells were washed with PBS and characterized for CD11b and glial fibrillary acidic protein (GFAP) expression by flow cytometry with PE-labelled anti-CD11b MoAb (BD Pharmingen) and the fluorescein isothiocyanate (FITC)-labelled anti-GFAP MoAb (BD Pharmingen). In particular, GFAP immunophenotyping was performed in permeabilized cells, using IntraPrep™ fixing/permeabilization reagent (Beckman Coulter) (Gobbi et al., 2003). Analysis was performed on an Epics XL flow cytometer (Beckman Coulter) using Expo ADC software (Beckman Coulter).
Primary microglial cell exposure to cannabinoids, opioids and LPS treatment
LPS, a cell wall component of Gram-negative bacteria, is a potent activator of glia. Hence, microglial cells were treated with 1 µg·mL−1 LPS (from Escherichia coli, serotype 055:B5) (soluble in cell culture medium) before commencing incubation with CB and opioid receptor ligands. Unless otherwise stated, the concentration of morphine (Salars, Como, Italy), naloxone, Tyr-DAla-Gly-[NMePhe]-NH(CH2)2 (DAMGO), cyc[DPen2, DPen5]enkephalin (DPDE), 5α,7α,8β-(-)-N-methyl-N-(7-[1-pyrrolidinyl]-1-oxasipro(4,5)dec-8-yl)benzeneacetamide (U69593), D-Phe-cyc[Cys-Tyr-D-Trp-Arg-Thr-Pen]-Thr-NH2 (CTAP), JWH-015 (Enzo Life Sciences, Vinci-Biochem, Vinci, Florence, Italy), AM 630 and AM 251 was 100 nM, which is the ligand concentration able to occupy 99% of the receptors at equilibrium. Microglia were then maintained in DMEM containing cannabinoids, opioids or their vehicle, and harvested after treatment at the indicated times.
Nitrite assay for primary microglial cells
NO synthase activity was assessed indirectly by measuring nitrite (NO2-) accumulation in the cell culture media using a colorimetric kit (Calbiochem, Milan, Italy). At the end of the treatment period, the nitrite concentration in the conditioned media was determined according to a modified Griess method (Green et al., 1982). Briefly, the NADH-dependent enzyme nitrate reductase was used to convert the nitrate to nitrite prior to quantification of the absorbance, measured at 540 nm by a spectrophotometric microplate reader (Fluoroskan Ascent Labsystems, Stockholm, Sweden). Values were obtained by comparison with reference concentrations of sodium nitrite.
elisa
The levels of IL-1β, TNF-α and IL-6 protein secreted by the cells in the medium were determined by elisa kits (R&D Systems). In brief, subconfluent cells were changed into fresh medium in the presence of solvent or various concentrations of drugs. The medium was collected, and IL-1β, TNF-α and IL-6 protein concentrations were measured by elisa according to the manufacturer's instructions. The results were normalized to the number of cells per plate. The data are presented as mean ± SE from four independent experiments performed in triplicate.
Western blotting for primary microglial cells
Western blot assay was performed as previously described (Merighi et al., 2009). Aliquots of total protein sample (50 µg) were analysed using antibodies specific for phosphorylated or total p44/p42 MAPK (1:5000 dilution), phosphorylated or total p38 (1:1000 dilution), phosphorylated or total JNK1/2 (1:1000 dilution), phosphorylated or total Akt (1:1000 dilution), for CB2 and µ-opioid receptors. Specific reactions were revealed with enhanced chemiluminescence Western blotting detection reagent (Amersham Corp., Arlington Heights, IL, USA). The membranes were then stripped and re-probed with tubulin (1:250) to ensure equal protein loading.
Densitometry analysis
The intensity of each immunoblot assay band was quantified using a VersaDoc Imaging System (Bio-Rad, Milan, Italy). Mean densitometric data from independent experiments was normalized to the results obtained with control cell cultures. The ratio of phospho-protein to total protein was reported in a densitometric analysis.
Treatment of primary microglial cells with siRNA
Microglial cells were plated in six-well plates and grown to 50–70% confluence before transfection. Transfection of siRNACB2 or siRNAµ was performed at a concentration of 100 nM using RNAiFect™ Transfection Kit (Qiagen). Cells were cultured in complete media and total proteins were isolated at 24, 48 and 72 h for Western blot analysis of CB2 and µ-opioid receptor protein. A randomly chosen non-specific siRNA was used under identical conditions as control (Merighi et al., 2005; 2007).
Statistical analysis
All data are reported as mean ± SEM of independent experiments and are indicated in the figure legends. Each experiment was performed by using the microglial cells derived from one single mouse, and was performed in triplicate. The experiments were repeated at least four times as indicated from n values that represent the number of mice used.
Data sets were examined by anova for comparisons between multiple groups and Dunnett's test for comparing a control group to all other groups (when necessary). A P value < 0.05 was considered statistically significant.
Results
CB2 and µ-opioid receptor expression in primary mouse microglial cells
The expression of the myeloid cell surface antigen CD11b was analysed in primary microglial cells by flow cytometry. Cells were treated with specific MoAbs or isotype-matched irrelevant MoAbs. Microglia were negative for the astrocyte-specific protein GFAP but showed significant positive staining for CD11b, as compared to the isotype control, thereby indicating high expression levels of the microglial cell marker CD11b (Figure 1A).
Figure 1.
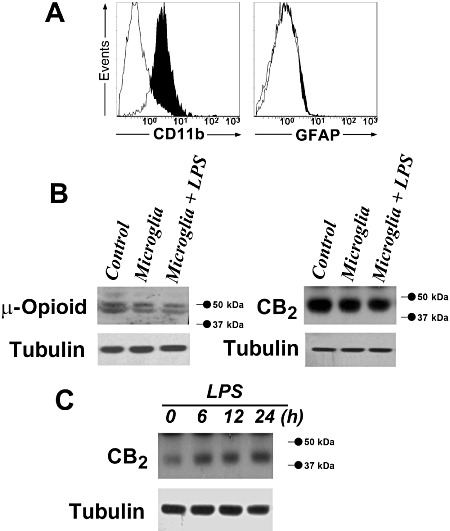
Detection of CB2 and µ-opioid receptors in primary microglial cells. (A), Cell surface expression of CD11b and intracellular expression of GFAP by flow cytometry analysis. Primary microglial cells were treated with specific monoclonal antibodies (black histograms) or with isotype-matched irrelevant monoclonal antibodies (empty histograms, controls). (B), CB2 and µ-opioid receptor detection by Western blot assay in quiescent and LPS-activated (1 µg·mL−1 for 30 min) primary microglial cells. Tubulin shows equal loading of protein. The expression of CB2 receptors in CHO-hCB2 cells and of µ-opioid receptors in mouse brain extracts, used as positive controls (Control), is shown. (C), CB2 receptor detection by Western blot assay in quiescent and LPS-activated (1 µg·mL−1 for 6, 12 and 24 h) primary microglial cells. Tubulin shows equal loading of protein.
The expression of CB2 receptors in CHO-hCB2 cells (used as positive control), in quiescent and LPS-activated primary microglial cells is shown in Figure 1B. The molecular weight of the protein detected in these cells was 50 kDa, comparable with the calculated molecular weight of CB2 receptors. To ascertain the specificity of the CB2 receptor antibody used in Western blots, antigen preabsorption experiments were carried out with the corresponding blocking peptide. Co-incubation with the immunizing peptide completely prevented the signal (data not shown). CB2 receptor protein expression was not modified by 30-min treatment with 1 µg·mL−1 LPS (Figure 1B). Similarly, the expression of µ-opioid receptors in mouse brain extracts (used as positive control) in quiescent and LPS-activated primary microglial cells is shown in Figure 1B. Therefore, CB2 and µ-opioid receptors were expressed in primary mouse microglial cells. To evaluate whether LPS induced changes in CB2 receptor expression, we assayed CB2 receptors over 24 h of LPS treatment. In agreement with published data (Carlisle et al., 2002), LPS 1 µg·mL−1 produced a time-dependent increase in CB2 receptor expression over the period from 6 to 24 h (Figure 1C).
Effect of opioid receptor ligands on cytokine and nitrite production in primary microglial cells
To investigate the effect of opioid receptor ligands on microglial activation, the ability of morphine to induce inflammatory mediator production, such as IL-1β, TNF-α, IL-6 and nitrite, was examined in LPS-stimulated microglial cells. The TLR-4 agonist LPS was used to induce IL-1β, TNF-α, IL-6 and NO (as nitrite) release from primary microglial cells. Preliminary studies in our laboratory demonstrated that LPS induced release of these mediators from primary microglial cells in a concentration-dependent manner, with maximum release occurring at a concentration of 1 µg·mL−1 (data not shown). As shown in Figure 2, non-stimulated microglial cells showed a very low level of IL-1β, TNF-α, IL-6 and nitrite but LPS triggered a robust increase in the release of these mediators into the culture media. Then, we went on to investigate how the activation of opioid receptors interferes with the signalling pathways modulated by LPS by maintaining primary microglial cells in LPS-supplemented (1 µg·mL−1) DMEM in combination with the opioid receptor agonist morphine (100 nM) for 24 h. As shown in Figure 2, LPS-induced cytokine and nitrite release was significantly increased in the presence of morphine in primary microglial cells. The stimulatory response of morphine on cytokine and nitrite production was reversed in the presence of the broad-range opioid receptor-antagonist naloxone (Figure 2). Furthermore, DAMGO (100 nM), a µ-opioid receptor-selective agonist, mimicked the effects of morphine on microglial cytokine and nitrite induction (Figure 2). Conversely, DPDPE and U-69593, δ- and k-receptor-selective agonists, respectively, had no effect on LPS-induced microglial cytokine and nitrite production (Figure 2). Finally, pretreatment of microglial cells with CTAP (100 nM), a selective antagonist of µ-opioid receptors, before treatment with morphine (100 nM) or DAMGO (100 nM), abolished the effects of opioid receptor agonists on LPS-induced cytokine and nitrite production by microglia (Figure 2).
Figure 2.
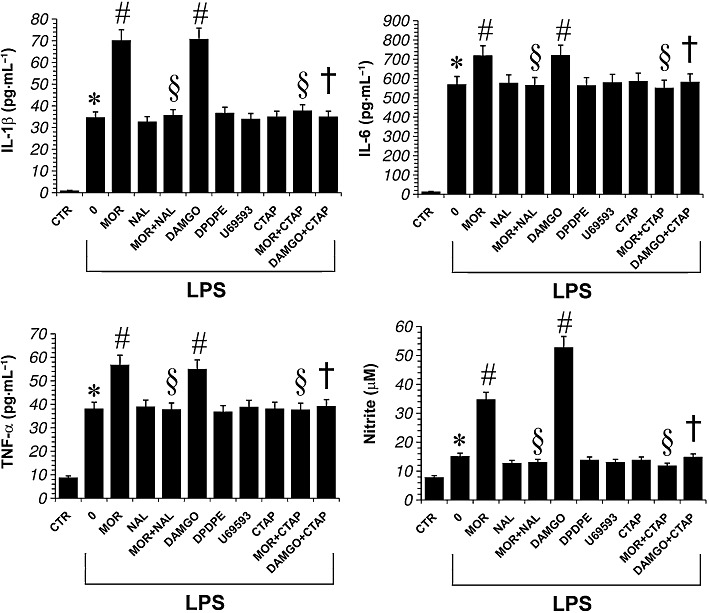
Effect of opioid receptor ligands on LPS-induced production of proinflammatory cytokines and nitrite in primary microglial cells. Microglial cells were treated with either LPS alone, LPS + morphine 100 nM (MOR), LPS + naloxone, a non-selective opioid receptor antagonist (Nal; 100 nM,), LPS + DAMGO, a selective µ-opioid receptor agonist (100 nM), LPS + DPDPE, a selective δ-opioid receptor agonist (100 nM), LPS + U69593, a selective κ-opioid receptor agonist (100 nM) or LPS + CTAP, a selective µ-opioid receptor antagonist (100 nM) for 24 h. The antagonists were added 30 min before morphine. Data shown are mean ± SEM values of four separate experiments performed in triplicate (n= 4). *P < 0.01 significantly different from control conditions (absence of drugs, CTR); #P < 0.01 significantly different from LPS conditions (0); §P < 0.01 significantly different from LPS + MOR; †P < 0.01 significantly different from LPS + DAMGO; analysis was by anova followed by Dunnett's test.
Influence of CB2 receptors on µ-opioid receptor-induced cytokine and nitrite production in primary microglial cells
The effect of CB2 receptor stimulation on µ-opioid receptor-induced cytokine and nitrite production in primary microglial cells was studied using JWH-015, a CB receptor agonist known to bind more readily to CB2 than CB1 receptors (Merighi et al., 2010). By this means we tested whether CB2 receptors can modulate cytokine and nitrite production in activated microglial cells. As shown in Figure 3, JWH-015 (100 nM) significantly decreased LPS-induced IL-1β, TNF-α, IL-6 and nitrite levels. Furthermore, co-administration of LPS (1 µg·mL−1), morphine (100 nM) and JWH-015 (100 nM) significantly attenuated the morphine-induced increases in cytokine and nitrite production (Figure 3). The anti-inflammatory response of JWH-015 in LPS-activated microglial cells treated with morphine was reversed in the presence of the CB2 receptor antagonist AM 630 (100 nM) (Figure 3). In contrast, the CB1 antagonist AM 251 (100 nM) did not affect the ability of JWH-015 to down-regulate the increase in IL-1β, TNF-α, IL-6 and nitrite induced by morphine in LPS-activated microglial cells (Figure 3).
Figure 3.
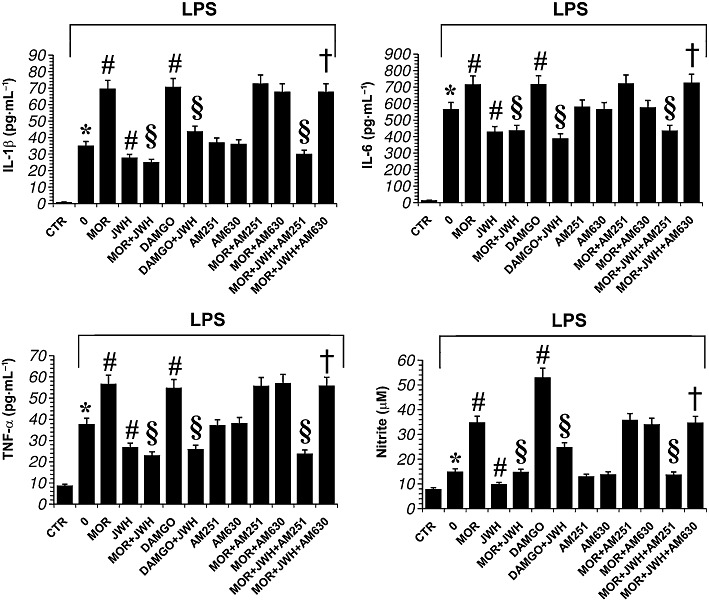
Effect of opioid and CB receptor ligands on LPS-induced production of proinflammatory cytokine and nitrite in primary microglial cells. Microglial cells were treated with either LPS alone, LPS + morphine (MOR; 100 nM), LPS + JWH-015 a CB2 receptor agonist (JWH; 100 nM)), LPS + DAMGO (100 nM), LPS + AM 251 a selective CB1 receptor antagonist (100 nM) or LPS + AM 630 a selective CB2 receptor antagonist (100 nM) for 24 h. The antagonists were added 30 min before morphine. Data shown are mean ± SE values of four separate experiments performed in triplicate (n= 4). *P < 0.01 significantly different from control conditions (absence of drugs, CTR); #P < 0.01 significantly different from LPS conditions (0); §P < 0.01 significantly different from LPS + MOR; †P < 0.01 significantly different from LPS + MOR + JWH; analysis was by anova followed by Dunnett's test.
Signalling induced by morphine on quiescent and activated microglial cells
We went on to investigate whether the stimulatory effect of morphine on cytokine and nitrite release induced by LPS is mediated via the MAPK and Akt pathways. We tested whether morphine could induce ERK1/2, p38, JNK1/2 and Akt phosphorylation in primary murine microglial cells, treated for 30 min at 37°C, in a concentration-dependent manner. As shown in Figure 4A, morphine (1–1000 nM) increased p-ERK1/2 and pAkt expression levels but did not modulate pp38 and pJNK1/2. In particular, morphine (100 nM) induced a rapid and sustained (up to 60 min) stimulation of ERK1/2 and pAkt (Figure 4B). Furthermore, we have investigated how the activation of microglial cells interferes with the signalling pathways modulated by morphine. Thus, microglial cells were maintained in DMEM containing LPS (1 µg·mL−1), and the ability of morphine (100 nM) to modulate ERK1/2 and Akt phosphorylation was evaluated at 15, 30 and 60 min. As shown in Figure 4B, LPS stimulation of microglial cells resulted in a rapid (15 min) increase in ERK1/2 and Akt phosphorylation, which was maximal at 30 min and declined towards basal levels within 60 min. In the presence of morphine (100 nM), ERK1/2 and Akt phosphorylation were significantly higher than LPS alone. To evaluate whether the morphine-induced changes in ERK1/2 and Akt phosphorylation levels were µ-opioid receptor-dependent, we used the selective antagonist of µ-opioid receptors, CTAP. Primary microglial cells were pretreated with CTAP, then exposed to morphine for 30 min. Morphine-induced p-ERK1/2 and p-Akt increased levels were reduced with CTAP pretreatment (Figure 4C), indicating that the effect of morphine was mediated via the µ-opioid receptor subtype.
Figure 4.
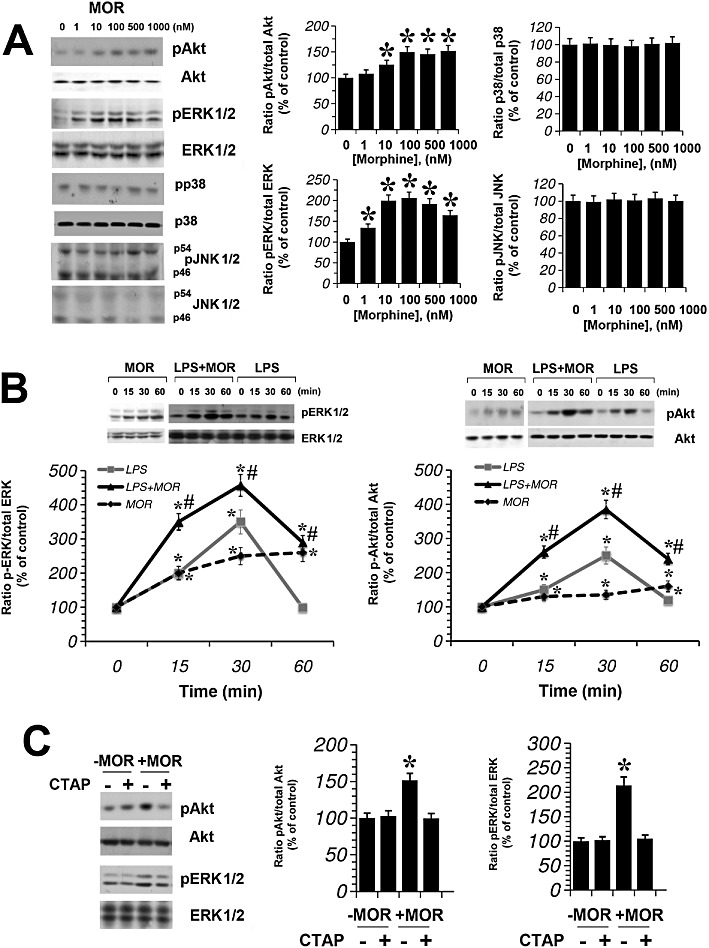
Morphine-enhanced Akt and ERK1/2 phosphorylation in a µ-opioid receptor-dependent manner. (A), Western blot analysis of Akt, ERK1/2, p38, and JNK1/2 phosphorylation in primary microglial cells incubated for 30 min with morphine (MOR) (1–1000 nM). The immunoblot signals were quantified using a VersaDoc Imaging System (Bio-Rad). The ratio of phospho-protein to total protein is used. The mean values of four independent experiments (one of which is shown) were normalized to the result obtained with morphine-untreated cell cultures (0). Densitometric analysis is shown. The unstimulated control (0, cells in the absence of morphine) was set to 100%. Data shown are mean ± SE values of four separate experiments performed in triplicate (n= 4). *P < 0.05 significantly different from unstimulated control; analysis was by anova followed by Dunnett's test. (B), Primary microglial cells were treated with morphine (100 nM) for 0, 15, 30 and 60 min, subjected to Western blot analysis and probed with anti-pERK1/2, anti-pAkt, then Akt and ERK1/2 antibody. The ratio of phospho-protein to total protein is used. Data shown are mean ± SE values of four separate experiments performed in triplicate (n= 4). *P < 0.01 significantly different from control conditions (0); #P < 0.01 significantly different from LPS conditions; analysis was by anova followed by Dunnett's test. (C), Image of Western blot membrane probed with anti-pERK1/2, anti-pAkt, then Akt and ERK1/2 antibody of primary microglial cells pre-treated for 30 min with 0 or 100 nM of the µ-opioid receptor antagonist CTAP, then treated for 30 min with 0 or 100 nM morphine. The ratio of phospho-protein to total protein is used. Data shown are mean ± SEM values of four separate experiments performed in triplicate (n= 4). *P < 0.01 significantly different from untreated cells; analysis was by anova followed by Dunnett's test.
Signalling induced by morphine and JWH-015 on LPS-activated microglial cells
Primary microglial cells were treated with either LPS alone, LPS + JWH-015 (100 nM) or LPS + morphine (100 nM) for 30 min. As shown in Figure 5, LPS significantly increased all kinases measured. Furthermore, while the CB2 receptor agonist JWH-015 failed to modulate the activity of LPS on p38 and JNK1/2, it significantly decreased ERK1/2 and Akt phosphorylation levels in LPS-activated cells (Figure 5). Similarly, the phosphorylation of ERK1/2 and Akt were significantly increased by morphine (100 nM) added LPS-activated cells, compared with treatment with LPS alone. In contrast, morphine did not modulate LPS-induced phosphorylation of p38 and JNK (Figure 5). Finally, when microglial cells were treated with LPS (1 µg·mL−1) plus morphine (100 nM) in combination with JWH-015 (100 nM), we found that JWH-015 decreased ERK1/2 and Akt activation induced by LPS plus morphine, without affecting pp38 and pJNK1/2 levels (Figure 5).
Figure 5.
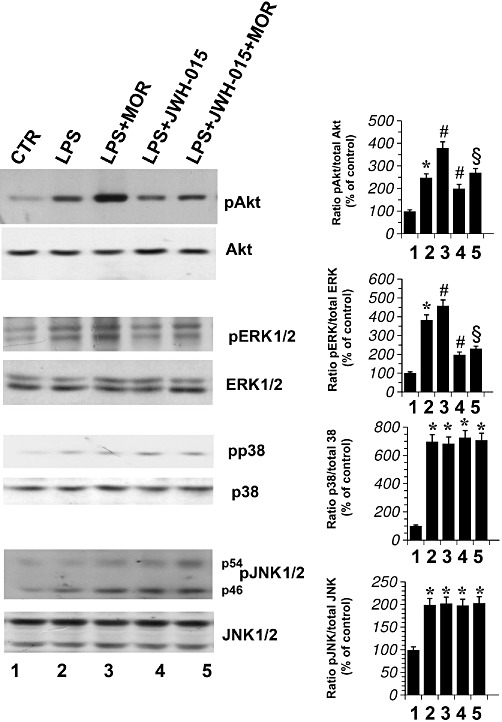
Effect of CB2 receptor stimulation in morphine-treated activated primary microglial cells. JWH-015 and morphine (MOR) effect on Akt, ERK1/2, p38 and JNK1/2 phosphorylation in primary microglial cells treated with LPS. Microglial cells were incubated with DMSO vehicle (CTR), with MOR (100 nM), or with JWH-015 (100 nM) alone and in combination in the presence of LPS 1 µg·mL−1 for 30 min. The mean values of four independent experiments (one of which is shown) were normalized to the result obtained in cells in the absence of LPS (CTR). Data shown are mean ± SEM values of four separate experiments performed in triplicate (n= 4). CTR was set to 100%. The immunoblot signals were quantified using a VersaDoc Imaging System (Bio-Rad). Densitometric analysis of kinase activation is shown. The ratio of phospho-protein to total protein is used. *P < 0.05 significantly different from CTR; #P < 0.05 significantly different from cells treated with LPS; §P < 0.05 significantly different from cells treated with LPS + morphine; analysis was by anova followed by Dunnett's test.
The effect of morphine and JWH-015 on ERK1/2 and Akt activation was reversed in the presence of the antagonists naloxone and AM 630 respectively (Figure 6). In contrast, the CB1 receptor antagonist AM 251 (100 nM) did not reverse the ability of JWH-015 to down-regulate the increase in p-ERK1/2 and p-Akt induced by morphine (Figure 6).
Figure 6.
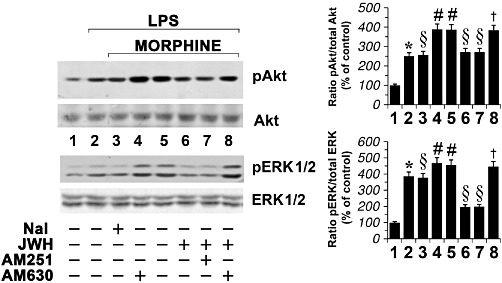
Effect of CB and opioid receptor blockade in morphine-treated activated primary microglial cells. Primary microglial cells were incubated with either DMSO vehicle (lane 1), morphine (MOR; 100 nM), naloxone a non-selective opioid receptor antagonist (Nal; 100 nM,), JWH-015 100 nM, AM 251 a selective CB1 receptor antagonist (100 nM) or AM 630 a selective CB2 receptor antagonist (100 nM) in the presence of LPS (1 µg·mL−1) for 30 min. The mean values of four independent experiments (one of which is shown) were normalized to the result obtained in cells in the absence of LPS (lane 1). Data shown are mean ± SEM values of four separate experiments performed in triplicate (n= 4). The unstimulated control (lane 1) was set to 100%. The immunoblot signals were quantified using a VersaDoc Imaging System (Bio-Rad). Densitometric analysis of kinase activation is shown. The ratio of phospho-protein to total protein is used. *P < 0.05 significantly different from untreated cells (lane 1); #P < 0.05 significantly different from cells treated with LPS (lane 2);§P < 0.05 significantly different from cells treated with LPS + morphine (lane 5); †P < 0.05 significantly different from LPS + morphine + JWH-015 (lane 6); analysis was by anova followed by Dunnett's test.
Akt and MEK-1/2 inhibition decreases LPS-induced inflammatory effects in microglial cells
To elucidate the mechanisms involved in the effects of morphine and JWH-015 on cutokine and nitrite production by activated microglial cells, we investigated the effects of inhibiting ERK1/2 or Akt signalling pathways,.with the MEK1/MEK2 inhibitor U0126 (1 µM) or the Akt inhibitor SH-5 (1 µM). To confirm the activity of these inhibitors, primary microglial cells were treated with U0126 (1 µM) and with SH-5 (1 µM) for 30 min and ERK-1/2 or Akt activation were measured. We observed that 30 min incubations of microglial cells with either U0126 or SH-5 resulted in significant decreases in active ERK-1/2 or Akt respectively, when compared with control cells incubated for 30 min with DMSO (U0126 = 32 ± 5% of control; n= 3; SH-5 = 28 ± 3% of control; n= 3). As shown in Figure 7, U0126 significantly reduced the production of TNF-α, IL-6 and nitrite induced by LPS-stimulated microglial cells. Similarly, the inhibition of the Akt pathway, by SH-5 (1 µM), resulted in a significant reduction of TNF-α, IL-6 and nitrite levels (Figure 7). However, while LPS-induced expression of IL-1β was significantly reduced by addition of SH-5, IL-1β expression was not affected by U0126 in LPS-treated microglia (Figure 7). These results suggest that ERK and Akt could mediate the enhancing effects of LPS on TNF-α, IL-6 and nitrite induction. In contrast, while the Akt pathway was involved in LPS-induced IL-1β production, the ERK pathway was not implicated.
Figure 7.
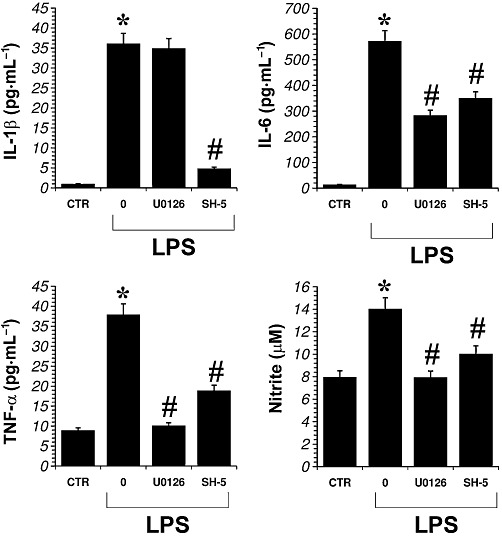
Effect of kinase inhibitors on LPS-induced production of proinflammatory cytokines and nitrite in primary microglial cells. Microglial cells were treated with LPS with either the MEK-1/2 inhibitor U0126 (1 µM) or the Akt inhibitor SH-5 (1 µM) for 24 h. The inhibitors were added 30 min before LPS. Data shown are mean ± SEM values of four separate experiments performed in triplicate (n= 4); *P < 0.01 significantly different from control conditions (CTR); #P < 0.01 significantly different from LPS conditions; analysis was by anova followed by Dunnett's test.
CB2 and µ-opioid receptor gene silencing in microglial cells
To confirm the apparent role of CB2 receptors and to investigate the involvement of µ-opioid receptors, we reduced CB2 and µ-opioid receptor expression in primary microglial cells by siRNA transfection, in order to cause transient knockdown of the CB2 and µ-opioid receptor genes. Primary microglial cells were transfected with non-specific random control ribonucleotides (siRNA scramble, siRNActr) or with small interfering RNAs that target CB2 or µ-opioid receptor mRNAs (siRNACB2 or siRNAµ, respectively) for degradation.
As shown in Figure 8, CB2 and µ-opioid receptor protein expression were strongly reduced after 48 and 72 h of treatment with siRNACB2 and siRNAµ respectively. Therefore, 48 h after siRNACB2 or siRNAµ transfection, primary microglial cells were treated with either LPS (1 µg·mL−1), JWH-015 (100 nM) or morphine (100 nM), alone and in combination, for 30 min, after which ERK1/2- and Akt-phosphorylated protein levels were measured. This revealed that inhibition of CB2 receptor expression was sufficient to block the JWH-015-induced inhibition of ERK1/2 and Akt phosphorylation levels, increased by morphine in LPS-treated microglia (Figure 9). Furthermore, inhibition of the expression of µ-opioid receptors blocked morphine-induced increases in pERK1/2 and pAkt in microglia (Figure 9). These results clearly show the connection between CB2 receptor stimulation, morphine, ERKs and Akt signalling in activated primary microglial cells.
Figure 8.
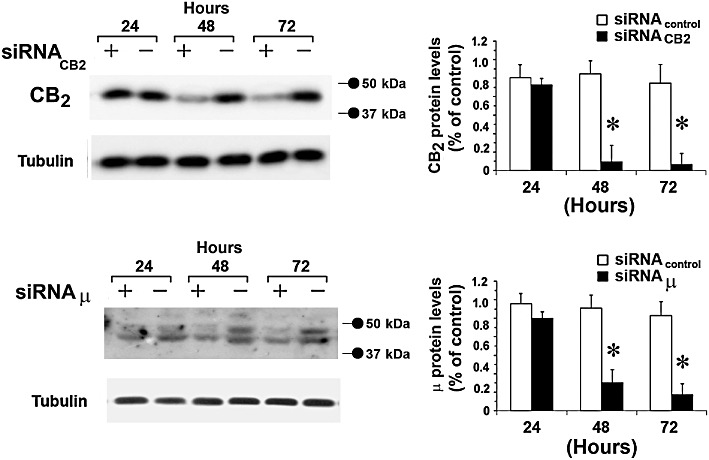
CB2 and µ-opioid receptor expression silencing. (A), Primary microglial cells were treated with either scrambled (-) (siRNAcontrol), with siRNACB2 (+) or with siRNAµ and cultured for 24, 48 and 72 h. Tubulin shows equal loading of protein. Densitometric quantification of CB2 and µ-opioid receptor by Western blot is shown; the immunoblot signals were quantified using a VersaDoc Imaging System (Bio-Rad). Plots are mean ± SEM values of four separate experiments performed in triplicate (n= 4); *P < 0.01 significantly different from control (scrambled siRNA transfected cells); analysis was by anova followed by Dunnett's test.
Figure 9.
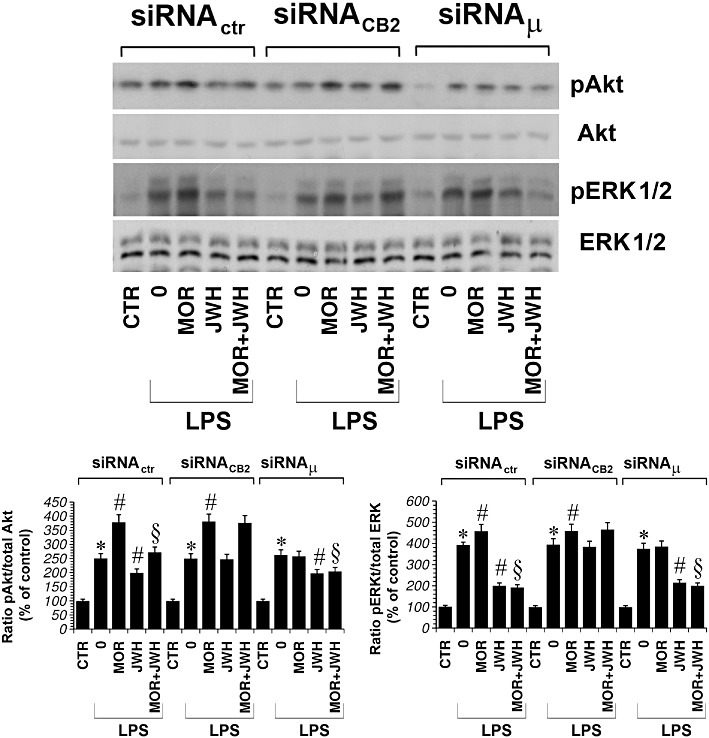
Primary microglial cells were treated with either siRNActr, siRNACB2 or siRNAµ for 48 h and cultured with LPS (1 µg·mL−1) alone or plus morphine (MOR; 100 nM), JWH-015 (JWH; 100 nM) alone and in combination for 30 min. Densitometric analysis of phosphorylated isoform is shown. The ratio of phospho-protein to total protein is used. The immunoblot signals were quantified using a VersaDoc Imaging System (Bio-Rad). The mean values of four independent experiments (one of which is shown) were normalized to the result obtained in cells in the absence of LPS. The unstimulated control was set to 100%. *P < 0.05 significantly different from unstimulated control (CTR); #P < 0.05 significantly different from cells treated with LPS; §P < 0.05 significantly different from LPS + MOR; analysis was by anova followed by Dunnett's test.
We also measured cytokine and nitrite levels in microglial cells in which CB2 or µ-opioid receptors were down-regulated. We found that, in microglial cells with µ-opioid receptors down-regulated, morphine did not significantly increase IL-1β, TNF-α, IL-6 and nitrite protein levels when compared to LPS. Similarly, the CB2 agonist JWH-015 did not significantly reduce cytokine and nitrite levels in microglial cells with down-regulated CB2 receptors, compared with those after LPS alone (Figure 10).
Figure 10.
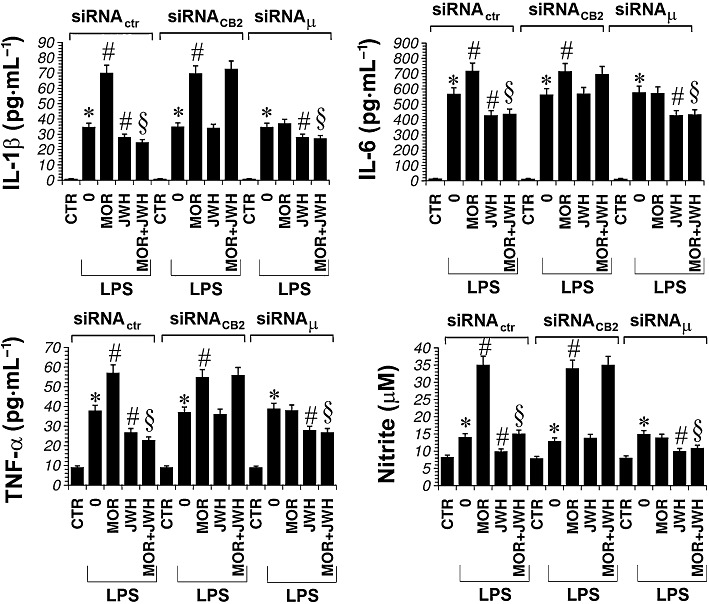
Primary microglial cells were treated with either siRNActr, siRNACB2 or with siRNAµ for 48 h and cultured with LPS (1 µg·mL−1) alone or plus morphine (MOR; 100 nM) or JWH-015 (JWH; 100 nM) alone and in combination for 24 h. The unstimulated control (CTR) was set to 100%. Data shown are mean ± SEM values of four separate experiments performed in triplicate (n= 4); *P < 0.05 significantly different from CTR; #P < 0.05 significantly different from LPS; §P < 0.05 significantly different from LPS + MOR; analysis was by anova followed by Dunnett's test.
Discussion and conclusions
The presence of opioid receptors on glia and the ability of morphine to prime microglia for enhanced production of proinflammatory cytokines supports a possible direct interaction of morphine with glial cells (Chao et al., 1994). As microglial differentiation and immune function is regulated by activation of CB2 receptors (Stella, 2010), we set out to characterize the signalling pathways modulated by both µ-opioid and CB2 receptors expressed in microglial cells. CB2 and µ-opioid receptor ligands (agonist and antagonist) and CB2- and µ-opioid receptor-knockout microglial cells were used to determine the role of the CB and opioid system in primary microglial cells. We have described the mechanisms by which CB2 and opioid receptors modulate the MAPK signal response to LPS, an agent widely used experimentally to create inflammation in the brain (Lehnardt et al., 2002).
In this study, we demonstrated that (1) morphine enhanced the release of NO and the proinflammatory cytokines, IL-1β, IL-6, TNF-α, from activated microglial cells; (2) CB2 receptor stimulation attenuated morphine-induced microglial proinflammatory mediator increases; (3) morphine-induced microglial proinflammatory mediator increases were µ-opioid receptor dependent; and (4) CB2 receptor stimulation interfered with morphine action by acting on Akt-ERK1/2 signalling. Together, these results suggest that CB2 receptors are critical to the activity of microglia, opposing the morphine-induced release of NO and cytokines from microglial cells. Therefore, we suggest a novel interaction between µ-opioid- and CB2-receptor systems in microglia, which is likely to be mediated via the Akt/ERK1/2 pathways.
The influence of CB2 receptor stimulation on µ-opioid receptor-induced cytokine and nitrite production in primary microglial cells was studied using JWH-015, a CB receptor agonist known to bind more readily to CB2 than CB1 receptors (Merighi et al., 2010). However, JWH-015 also stimulates GPR55 (Lauckner et al., 2008). Similarly, different studies have demonstrated that the CB1 receptor antagonist AM251 induces GPR55 activity (Lauckner et al., 2008; Henstridge et al., 2010; Anavi-Goffer et al., 2012). Therefore, even if the activity of CB receptor ligands at GPR55 is influenced by the assay used to assess receptor-mediated downstream signalling (Henstridge et al., 2010), a role for GPR55 activation needs to be considered in further pharmacological studies of cannabinoid actions. Furthermore, a non-selective CB agonist enhanced morphine antinociception via the CB1 receptor, pointing to the involvement of CB1 receptors in cannabinoid antinociception produced by CB receptor ligands (Wilson et al., 2008).
The first report linking glia to morphine tolerance demonstrated that chronic systemic morphine increased glia activation in the spinal cord (Song and Zhao, 2001). Other authors have also shown that chronic morphine administration activated astroglia and microglia (Raghavendra et al., 2002; Cui et al., 2006). Activated microglial cells in the spinal cord may release proinflammatory cytokines and other substances thought to facilitate pain transmission (Watkins et al., 2001; 2003). Therefore, pharmacological attenuation of glial activation represents a novel approach for controlling neuropathic pain (Watkins et al., 2005). Neuropathic hyperalgesia could lead to lowered morphine efficacy and quicker development of morphine tolerance (Mayer et al., 1999), and some authors have suggested that uncontrolled activation of microglial cells after nerve injury can lead to altered activities of opioid systems or opioid-specific signalling (Watkins et al., 2005; 2007). It is already known that microglia release neuroexcitatory substances in response to morphine, thereby opposing its effects (Watkins et al., 2001; 2005; 2007). This raises an older hypothesis that suppression of glial activation and the resulting blockade of proinflammatory cytokine synthesis can improve morphine efficacy (Song and Zhao, 2001; Raghavendra et al., 2002; Watkins et al., 2007). The mechanism underlying the involvement of glial cells in morphine tolerance is unclear. It is possible that morphine can act directly on glial cells, triggering alterations in their morphology and functions (Raghavendra et al., 2002; 2004). Additionally, glial cells are also considered to be crucial sources of NO, cytokines and cyclooxygenase products that influence synaptic transmission in the CNS. Inhibition of these factors may delay morphine tolerance (Powell et al., 1999).
Many current studies aim to find substances inhibiting the biosynthesis of proinflammatory cytokines. Propentofylline, minocycline and ibudilast inhibit cytokines and decrease astroglia and microglia activation, thereby suppressing the development of neuropathic pain (Romero-Sandoval et al., 2008). The beneficial effects of minocycline are associated with a reduction of inducible NO synthase and cyclooxygenase-2 expression and a decrease in cytokine and prostaglandin release in microglia (Yrjanheikki et al., 1998; 1999). Further studies have shown that minocycline reduced microglial activation by inhibiting p38 MAPK in microglia and, in this way, delayed morphine tolerance (Romero-Sandoval et al., 2008). Ibudilast may counteract opioid tolerance by blocking the activation of glial cells in the spinal cord in rodents (Romero-Sandoval et al., 2008). In the present work, we found that in activated microglial cells, morphine increased Akt and ERK kinase phosphorylation. ERK1/2 kinases are known to regulate the production of proinflammatory mediators from glial cells (Watkins et al., 2001). Furthermore, p38 and ERK kinases have been implicated in the development of morphine-induced hyperalgesia and antinociceptive tolerance (Cui et al., 2006; Wang et al., 2009). Therefore, it is of interest that CB2 receptor stimulation was able to downregulate Akt and ERK1/2 activation induced by morphine in activated microglial cells. Our data indicate that the CB2 receptor did not mediate its effects through p38 and JNK1/2 kinases. However, β-caryophyllene, a CB2 receptor selective agonist, modulates JNK1/2 in LPS-stimulated monocytes (Gertsch et al., 2008). As microglia are considered as the resident macrophage-like cells in the brain, we suggest that this contrasting behaviour may be due to the different experimental conditions, for example, time of ligand incubation (30 min vs. 3 h for microglia and monocytes respectively) or LPS concentration (1 µg·mL−1 vs. 0.313 µg·mL−1 for microglia and monocytes respectively). As for ERK1/2 signalling, it has been previously observed that CB2 receptor stimulation leads to ERK-mediated cellular activation and anti-inflammatory effects in monocytes/macrophages and microglia (Gertsch et al., 2008; Correa et al., 2010). Similarly, a more recent study has demonstrated that CB2 receptor stimulation in microglial cells induced an anti-inflammatory phenotype and reduced migration via MKP-induced ERK dephosphorylation (Romero-Sandoval et al., 2009). Agonist at CB receptors inhibited the production of proinflammatory molecules, which were induced by LPS, in CNS glial cells (Molina-Holgado et al., 2002; Fachinetti et al., 2003; Ortega-Gutiérrez et al., 2005; Sheng et al., 2005; Correa et al., 2008; 2009). In particular, CB2 receptors influence the production of the potent inflammatory mediator NO, released from quiescent, and, to a greater extent, from activated microglia (Stella, 2010). In this work, we have described the activation of the ERK1/2 and Akt pathways by LPS leading to an increment in TNF-α, IL-6 and nitrite production. In contrast, while Akt is engaged as the signalling pathway generating IL-1β in LPS-activated microglia, the ERK pathway was not implicated. The involvement of the ERK-MAPK pathway in IL-1β production by LPS is controversial. Some reports show that the ERK cascade is important for LPS-stimulated production of IL-1β in macrophage cell lines and monocytes (Scherle et al., 1998; Caivano and Cohen, 2000). However, it has also been reported that the ERK pathway is not essential for IL-1β production in BV-2 microglia (Watters et al., 2002). Consistent with findings on BV-2 microglia cell lines, our study indicates that the regulation of LPS-stimulated IL-1β production is ERK-independent also in primary microglia.
According to previous studies showing that CB2 receptor mRNA and protein are modulated in vitro differentially in relation to cell activation state (Carlisle et al., 2002; Cabral et al., 2008), we have demonstrated that LPS increases CB2 receptor expression level in primary microglial cells. It is important to mention that CB2 receptors, identified in the healthy brain, mainly in glial elements, and, to a lesser extent, in certain subpopulations of neurons, are dramatically up-regulated in response to damaging stimuli, which supports the idea that the cannabinoid system behaves as an endogenous neuroprotective system. This CB2 receptor up-regulation has been found in many neurodegenerative disorders, which supports the beneficial effects found for CB2 receptor agonists in these pathologies (Fernández-Ruiz et al., 2011). Now, we have characterized, for the first time, the events occurring in LPS-activated microglia via CB2 receptor stimulation, which reduces not only ERK1/2- but also Akt-phosphorylation increases induced by LPS. Therefore, CB2 receptors expressed in microglia may participate in regulating neuroinflammation and provide neuroprotection by tempering morphine-induced cytokine and NO synthesis through ERK1/2 and Akt signalling in activated microglia. Interestingly, in microglia, we showed that the effects of morphine were mediated by the µ-opioid receptor subtype. This accords with previous observations describing the involvement of endocannabinoids in the peripheral antinociception induced by the µ-opioid receptor agonist morphine. In contrast, the release of endocannabinoids appears not to be involved in the peripheral antinociceptive effect induced by κ- and δ-opioid receptor agonists (da Fonseca Pacheco et al., 2008). At the same time, it would be possible that certain effects of CB2 receptor agonists in different models for inflammation and possibly their analgesic effects previously reported, actually reflect their interaction with endogenous opioids. In particular, it has been demonstrated that morphine is present in human gliomas (Olsen et al., 2005) and that it increases the proliferation of human glioblastoma cells (Lazarczyk et al., 2010). Because morphine is used to alleviate pain associated with cancers, this study suggests that a combination of CB2 receptor agonists to prevent morphine-induced proliferation may have clinically important implications.
In conclusion, the novel finding of this study is the existence of a receptor–receptor interaction when the receptors are co-expressed in the same cells leading to the interaction of their intracellular pathways. In particular, CB2 receptor stimulation counteracts the ability of morphine to upregulate Akt and ERK1/2 activation induced by LPS, thus reducing NO and proinflammatory cytokine release, a process which is ERK- and Akt-dependent. The studies presented here are the first to assess the signalling mechanisms through which CB2 receptor stimulation modulates morphine effects on microglia and MAPK activation. The ability to modulate microglia and MAPK is very interesting because their activation in the central and peripheral nervous system contributes to morphine tolerance and dependence (Mayer et al., 1999; Watkins et al., 2001; Raghavendra et al., 2002; 2004; Galeotti et al., 2006; Cunha et al., 2010). Our results indicate a regulatory role for CB2 receptors in preventing excessive microglial cell response to injury in activated microglia. Based on the findings obtained in the present study, we will advance our research to reinforce the idea that the cannabinoid system exerts an important control on the tolerance and dependence effects induced by opioids. In particular, it will be interesting to evaluate the impact of low-dose combinations of cannabinoids and opioids to effectively treat acute and chronic pain, especially pain that may be resistant to opioids alone. It is well known that the use of cannabinoids, like that of the opioids, has liability for abuse potential. The use of marijuana as a therapeutic pain management tool has generated a great deal of publicity and controversy. However, it should be noted that CB2 receptor agonists, in comparison with CB1 agonists, lack the undesirable CNS side effects, like sedation and psychotomimetic effects (Fernández-Ruiz et al., 2011). Therefore, the development of selective CB2 receptor agonists might open new avenues of therapeutic intervention to reduce the release of proinflammatory mediators especially during morphine therapy. Accordingly, CB2 receptors may be potential targets for reducing morphine tolerance and dependence.
Acknowledgments
This work was supported by ‘Fondazione Cassa di Risparmio’ of Ferrara.
Glossary
- AM 251
N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- AM 630
6-iodopravadoline
- CHO-hCB2
CHO cells transfected with human CB2 receptor
- CTAP
D-Phe-cyc[Cys-Tyr-D-Trp-Arg-Thr-Pen]-Thr-NH2
- DAMGO
Tyr-DAla-Gly-[NMePhe]-NH(CH2)2
- DPDPE
cyc[DPen2, DPen5]enkephalin
- FBS
fetal bovine serum
- JWH-015
(2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone
- PE
phycoerythrin
- siRNA
small interfering RNA
- THC
Δ9tetrahydrocannabinol
- U69593
5α,7α,8β-(-)-N-methyl-N-(7-[1-pyrrolidinyl]-1-oxasipro(4,5)dec-8-yl)benzeneacetamide
Conflict of interest
On behalf of all the authors, I declare that we have no conflicts of interest.
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anavi-Goffer S, Baillie G, Irving AJ, Gertsch J, Greig IR, Pertwee RG, et al. Modulation of L-α-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. J Biol Chem. 2012;287:91–104. doi: 10.1074/jbc.M111.296020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol. 2010;160:467–479. doi: 10.1111/j.1476-5381.2010.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg M, Pacher P, Bátkai S, Osei-Hyiaman D, Offertáler L, Mo FM, et al. Evidence for novel cannabinoid receptors. Pharmacol Ther. 2005;106:133–145. doi: 10.1016/j.pharmthera.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Benito C, Tolón RM, Pazos MR, Núñez E, Castillo AI, Romero J. Cannabinoid CB2 receptors in human brain inflammation. Br J Pharmacol. 2008;153:277–285. doi: 10.1038/sj.bjp.0707505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral GA, Harmon KN, Carlisle SJ. Cannabinoid-mediated inhibition of inducible nitric oxide production by rat microglial cells: evidence for CB1 receptor participation. Adv Exp Med Biol. 2001;493:207–214. doi: 10.1007/0-306-47611-8_24. [DOI] [PubMed] [Google Scholar]
- Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F. CB2 receptors in the brain: role in central immune function. Br J Pharmacol. 2008;153:240–251. doi: 10.1038/sj.bjp.0707584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caivano M, Cohen P. Role of mitogen-activated protein kinase cascades in mediating lipopolysaccharide-stimulated induction of cyclooxygenase-2 and IL-1 beta in RAW264 macrophages. J Immunol. 2000;164:3018–3025. doi: 10.4049/jimmunol.164.6.3018. [DOI] [PubMed] [Google Scholar]
- Carlisle SJ, Marciano-Cabral F, Staab A, Ludwick C, Cabral GA. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int Immunopharmacol. 2002;2:69–82. doi: 10.1016/s1567-5769(01)00147-3. [DOI] [PubMed] [Google Scholar]
- Chao CC, Gekker G, Sheng WS, Hu S, Tsang M, Peterson PK. Priming effect of morphine on the production of tumor necrosis factor-a by microglia: implications in respiratory burst activity and human immunodeficiency virus-1 expression. J Pharmacol Exp Ther. 1994;269:198–203. [PubMed] [Google Scholar]
- Chen Y, Sommer C. The role of mitogen-activated protein kinase (MAPK) in morphine tolerance and dependence. Mol Neurobiol. 2009;40:101–107. doi: 10.1007/s12035-009-8074-z. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL. Synergistic interactions between cannabinoid and opioid analgesics. Life Sci. 2004;74:1317–1324. doi: 10.1016/j.lfs.2003.09.038. [DOI] [PubMed] [Google Scholar]
- Correa F, Mestre L, Docagne F, Guaza C. Activation of cannabinoid CB2 receptor negatively regulates IL-12p40 production in murine macrophages: role of IL-10 and ERK1/2 kinase signaling. Br J Pharmacol. 2005;145:441–448. doi: 10.1038/sj.bjp.0706215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa F, Docagne F, Clemente D, Mestre L, Becker C, Guaza C. Anandamide inhibits IL-12p40 production by acting on the promoter repressor element GA-12: possible involvement of the COX-2 metabolite prostamide E(2) Biochem J. 2008;409:761–770. doi: 10.1042/BJ20071329. [DOI] [PubMed] [Google Scholar]
- Correa F, Docagne F, Mestre L, Clemente D, Hernangómez M, Loría F, et al. A role for CB2 receptors in anandamide signalling pathways involved in the regulation of IL-12 and IL-23 in microglial cells. Biochem Pharmacol. 2009;77:86–100. doi: 10.1016/j.bcp.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Correa F, Hernangómez M, Mestre L, Loría F, Spagnolo A, Docagne F, et al. Anandamide enhances IL-10 production in activated microglia by targeting CB2 receptors: roles of ERK1/2, JNK, and NF-kappaB. Glia. 2010;58:135–147. doi: 10.1002/glia.20907. [DOI] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Cunha TM, Roman-Campos D, Lotufo CM, Duarte HL, Souza GR, Verri WA, et al. Morphine peripheral analgesia depends on activation of the PI3Kgamma/AKT/nNOS/NO/KATP signalling pathway. Proc Natl Acad Sci U S A. 2010;107:4442–4447. doi: 10.1073/pnas.0914733107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, et al. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation. 2005;2:29. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron. 2006;49:67–79. doi: 10.1016/j.neuron.2005.11.027. [DOI] [PubMed] [Google Scholar]
- Fachinetti F, Del Giudice G, Furegato S, Passarotto M, Leon M. Cannabinoids ablate release of TNFα in rat microglial cells stimulated with lypopolysaccharide. Glia. 2003;41:161–168. doi: 10.1002/glia.10177. [DOI] [PubMed] [Google Scholar]
- Fernández-Ruiz J. The endocannabinoid system as a target for the treatment of motor dysfunction. Br J Pharmacol. 2009;156:1029–1040. doi: 10.1111/j.1476-5381.2008.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Moreno-Martet M, Rodríguez-Cueto C, Palomo-Garo C, Gómez-Cañas M, Valdeolivas S, et al. Prospects for cannabinoid therapies in basal ganglia disorders. Br J Pharmacol. 2011;163:1365–1378. doi: 10.1111/j.1476-5381.2011.01365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Fonseca Pacheco D, Klein A, de Castro Perez A, da Fonseca Pacheco CM, de Francischi JN, Duarte ID. The mu-opioid receptor agonist morphine, but not agonists at delta- or kappa-opioid receptors, induces peripheral antinociception mediated by cannabinoid receptors. Br J Pharmacol. 2008;154:1143–1149. doi: 10.1038/bjp.2008.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeotti N, Stefano GB, Guarna M, Bianchi E, Ghelardini C. Signalling pathway of morphine induced acute thermal hyperalgesia in mice. Pain. 2006;123:294–305. doi: 10.1016/j.pain.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Gertsch J, Leonti M, Raduner S, Racz I, Chen JZ, Xie XQ, et al. Beta-caryophyllene is a dietary cannabinoid. Proc Natl Acad Sci U S A. 2008;105:9099–9104. doi: 10.1073/pnas.0803601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbi G, Mirandola P, Tazzari PL, Ricci F, Caimi L, Cacchioli A, et al. Flow cytometry detection of serotonin content and release in resting and activated platelets. Br J Haematol. 2003;121:892–896. doi: 10.1046/j.1365-2141.2003.04369.x. [DOI] [PubMed] [Google Scholar]
- González-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- Henstridge CM, Balenga NA, Schröder R, Kargl JK, Platzer W, Martini L, et al. GPR55 ligands promote receptor coupling to multiple signalling pathways. Br J Pharmacol. 2010;160:604–614. doi: 10.1111/j.1476-5381.2009.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh GC, Pai M, Chandran P, Hooker BA, Zhu CZ, Salyers AK, et al. Central and peripheral sites of action for CB2 receptor mediated analgesic activity in chronic inflammatory and neuropathic pain models in rats. Br J Pharmacol. 2011;162:428–440. doi: 10.1111/j.1476-5381.2010.01046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, et al. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–7365. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung WK, Lee DY, Park C, Choi YH, Choi I, Park SG, et al. Cilostazol is anti-inflammatory in BV2 microglial cells by inactivating nuclear factor-kappaB and inhibiting mitogen-activated protein kinases. Br J Pharmacol. 2010;159:1274–1285. doi: 10.1111/j.1476-5381.2009.00615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klegeris A, Bissonnette CJ, McGeer PL. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br J Pharmacol. 2003;139:775–786. doi: 10.1038/sj.bjp.0705304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein TW, Newton CA. Therapeutic potential of cannabinoid-based drugs. Adv Exp Med Biol. 2007;601:395–413. doi: 10.1007/978-0-387-72005-0_43. [DOI] [PubMed] [Google Scholar]
- Klein TW, Lane B, Newton CA, Friedman H. The cannabinoid system and cytokine network. Proc Soc Exp Biol Med. 2000;225:1–8. doi: 10.1177/153537020022500101. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A. 2008;105:2699–2704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarczyk M, Matyja E, Lipkowski AW. A comparative study of morphine stimulation and biphalin inhibition of human glioblastoma T98G cell proliferation in vitro. Peptides. 2010;31:1606–1612. doi: 10.1016/j.peptides.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, et al. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malan TP, Ibrahim MM, Deng H, Liu Q, Mata HP, Vanderah T, et al. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- Manzaneres J, Corchero J, Romero JJ, Fernandez-Ruiz JA, Ramos JÁ FJA. Pharmacological and biochemical interactions between opioids and cannabinoids. Trends Pharmacol Sci. 1999;20:287–294. doi: 10.1016/s0165-6147(99)01339-5. [DOI] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci USA. 1999;96:7731–7736. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, et al. A3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/Akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in A375 human melanoma cells. J Biol Chem. 2005;280:19516–19526. doi: 10.1074/jbc.M413772200. [DOI] [PubMed] [Google Scholar]
- Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Simioni C, et al. Caffeine inhibits adenosine-induced accumulation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and interleukin-8 expression in hypoxic human colon cancer cells. Mol Pharmacol. 2007;72:395–406. doi: 10.1124/mol.106.032920. [DOI] [PubMed] [Google Scholar]
- Merighi S, Simioni C, Gessi S, Varani K, Mirandola P, Tabrizi MA, et al. A2B and A3 adenosine receptors modulate vascular endothelial growth factor and interleukin-8 expression in human melanoma cells treated with etoposide and doxorubicin. Neoplasia. 2009;11:1064–1073. doi: 10.1593/neo.09768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merighi S, Simioni C, Gessi S, Varani K, Borea PA. Binding thermodynamics at the human cannabinoid CB1 and CB2 receptors. Biochem Pharmacol. 2010;79:471–477. doi: 10.1016/j.bcp.2009.09.009. [DOI] [PubMed] [Google Scholar]
- Merighi S, Gessi S, Varani K, Simioni C, Fazzi D, Mirandola P, et al. Cannabinoid CB2 receptor modulates microglial cells stimulated with lypopolysaccharide: role of ERK-1/2 kinase signalling in nitric oxide release. Br J Pharmacol. 2012;165:1773–1788. doi: 10.1111/j.1476-5381.2011.01673.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Molina-Holgado F, Lledo A, Guaza C. Anandamide suppresses nitric oxide and TNF-alpha responses to Theiler's virus or endotoxin in astrocytes. Neuroreport. 1997;8:1929–1933. doi: 10.1097/00001756-199705260-00027. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Molina-Holgado E, Guaza C, Rothwell NJ. Role of CB1 and CB2 receptors in the inhibitory effects of cannabinoids on lipopolysaccharide-induced nitric oxide release in astrocyte cultures. J Neurosci Res. 2002;67:829–836. doi: 10.1002/jnr.10165. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Pinteaux E, Moore JD, Molina-Holgado E, Guaza C, Gibson RM. Endogenous interleukin-1 receptor antagonist mediates anti-inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J Neurosci. 2003;23:6470–6474. doi: 10.1523/JNEUROSCI.23-16-06470.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel LJ, Giros B, Daugé V. Adolescent exposure to chronic delta-9-tetrahydrocannabinol blocks opiate dependence in maternally deprived rats. Neuropsychopharmacology. 2009;34:2469–2476. doi: 10.1038/npp.2009.70. [DOI] [PubMed] [Google Scholar]
- Olsen P, Rasmussen M, Zhu W, Tonnesen E, Stefano GB. Human gliomas contain morphine. Med Sci Monit. 2005;11:MS18–MS21. [PubMed] [Google Scholar]
- Ortega-Gutiérrez S, Molina-Holgado E, Guaza C. Effect of anandamide uptake inhibition in the production of nitric oxide and in the release of cytokines in astrocyte cultures. Glia. 2005;52:163–168. doi: 10.1002/glia.20229. [DOI] [PubMed] [Google Scholar]
- Parolaro D, Rubino T, Viganò D, Massi P, Guidali C, Realini N. Cellular mechanisms underlying the interaction between cannabinoid and opioid system. Curr Drug Targets. 2010;11:393–405. doi: 10.2174/138945010790980367. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: beyond CB1 and CB2. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell KJ, Hosokawa A, Bell A, Sutak M, Milne B, Quirion R, et al. Comparative effects of cyclo-oxygenase and nitric oxide synthase inhibition on the development and reversal of spinal opioid tolerance. Br J Pharmacol. 1999;127:631–644. doi: 10.1038/sj.bjp.0702587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffenbarger RA, Boothe AC, Cabral GA. Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia. 2000;29:58–69. [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980–9989. doi: 10.1523/JNEUROSCI.22-22-09980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology. 2004;29:327–334. doi: 10.1038/sj.npp.1300315. [DOI] [PubMed] [Google Scholar]
- Richardson JD. Cannabinoids modulate pain by multiple mechanisms of action. J Pain. 2000;1:2–14. [Google Scholar]
- Romero-Sandoval EA, Horvath RJ, DeLeo JA. Neuroimmune interactions and pain: focus on glial-modulating targets. Curr Opin Investig Drugs. 2008;9:726–734. [PMC free article] [PubMed] [Google Scholar]
- Romero-Sandoval EA, Horvath R, Landry RP, DeLeo JA. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol Pain. 2009;5:25. doi: 10.1186/1744-8069-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Minami M. Molecular pharmacology of the opioid receptors. Pharmacol Ther. 1995;68:343–364. doi: 10.1016/0163-7258(95)02011-x. [DOI] [PubMed] [Google Scholar]
- Scherle PA, Jones EA, Favata MF, Daulerio AJ, Covington MB, Nurnberg SA, et al. Inhibition of MAP kinase kinase prevents cytokine and prostaglandin E2 production in lipopolysaccharide-stimulated monocytes. J Immunol. 1998;161:5681–5686. [PubMed] [Google Scholar]
- Sheng WS, Hu S, Min X, Cabral GA, Lokensgard JR, Peterson PK. Synthetic cannabinoid WIN55,212-2 inhibits generation of inflammatory mediators by IL-1beta-stimulated human astrocytes. Glia. 2005;49:211–219. doi: 10.1002/glia.20108. [DOI] [PubMed] [Google Scholar]
- Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72:169–177. doi: 10.1016/s0165-5728(96)00181-6. [DOI] [PubMed] [Google Scholar]
- Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–286. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- Stella N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia. 2010;58:1017–1030. doi: 10.1002/glia.20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ma W, Chabot JG, Quirion R. Cell-type specific activation of p38 and ERK mediates calcitonin gene-related peptide involvement in tolerance to morphine-induced analgesia. FASEB J. 2009;23:2576–2586. doi: 10.1096/fj.08-128348. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Spinal cord glia: new players in pain. Pain. 2001;93:201–205. doi: 10.1016/S0304-3959(01)00359-1. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol. 2003;521:1–21. [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends Neurosci. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Ledeboer A, Wieseler-Frank J, Milligan ED, Maier SF. Norman Cousins Lecture. Glia as the ‘bad guys’: implications for improving clinical pain control and the clinical utility of opioids. Brain Behav Immun. 2007;21:131–146. doi: 10.1016/j.bbi.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF. The ‘toll’ of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci. 2009;30:581–591. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters JJ, Sommer JA, Pfeiffer ZA, Prabhu U, Guerra AN, Bertics PJ. A differential role for the mitogen-activated protein kinases in lipopolysaccharide signaling: the MEK/ERK pathway is not essential for nitric oxide and interleukin 1beta production. J Biol Chem. 2002;277:9077–9087. doi: 10.1074/jbc.M104385200. [DOI] [PubMed] [Google Scholar]
- Wilson AR, Maher L, Morgan MM. Repeated cannabinoid injections into the rat periaqueductal gray enhance subsequent morphine antinociception. Neuropharmacology. 2008;55:1219–1225. doi: 10.1016/j.neuropharm.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]