

Abstract
BACKGROUND AND PURPOSE
Sympathetic nervous system (SNS) hyperactivity is characteristic of chronic heart failure (HF) and significantly worsens prognosis. The success of β-adrenoceptor antagonist (β-blockers) therapy in HF is primarily attributed to protection of the heart from the noxious effects of augmented catecholamine levels. β-Blockers have been shown to reduce SNS hyperactivity in HF, but the underlying molecular mechanisms are not understood. The GPCR kinase-2 (GRK2)–α2adrenoceptor–catecholamine production axis is up-regulated in the adrenal medulla during HF causing α2-adrenoceptor dysfunction and elevated catecholamine levels. Here, we sought to investigate if β-blocker treatment in HF could lower SNS activation by directly altering adrenal GRK2 levels.
EXPERIMENTAL APPROACH
Four weeks after myocardial infarction-induced HF, adult rats were randomized to 10-week treatment with vehicle (HF/C) or bisoprolol (HF/B). Cardiac function and dimensions were measured. In heart and adrenal gland, GRK2 levels were assessed by RT-PCR and Western blotting and adrenoceptors studied with radioligand binding. Catecholamines and α2adrenoceptors in adrenal medulla chromaffin cell cultures were also measured.
KEY RESULTS
Bisoprolol treatment ameliorated HF-related adverse cardiac remodelling and reduced plasma catecholamine levels, compared with HF/C rats. Bisoprolol also attenuated adrenal GRK2 overexpression as observed in HF/C rats and increased α2adrenoceptor density. In cultures of adrenal medulla chromaffin cells from all study groups, bisoprolol reversed HF-related α2adrenoceptor dysfunction. This effect was reversed by GRK2 overexpression.
CONCLUSION AND IMPLICATIONS
Blockade of β-adrenoceptors normalized the adrenal α2adrenoceptor-catecholamine production axis by reducing GRK2 levels. This effect may contribute significantly to the decrease of HF-related sympathetic overdrive by β−blockers.
Keywords: heart failure, sympathetic overactivity, adrenal GRK2, catecholamines, β-blocker
Introduction
Heart failure (HF) is a major and growing public health problem in the Western world (Rengo et al., 2004; Hunt et al., 2009). Abnormalities in cardiac β-adrenoceptor signalling and function represent a salient characteristic of chronic HF pathophysiology (Feldman et al., 2005; Tilley and Rockman, 2006). Increased levels and activity of GPCR kinase-2 (GRK2) play a critical role in promoting β1-adrenoceptor down-regulation and G-protein uncoupling (desensitization) of both β1- and β2-adrenoceptor subtypes in the failing heart, resulting in a deterioration of overall cardiac function (Petrofski and Koch, 2003; Rengo et al., 2011; receptor nomenclature follows; receptor nomenclature follows Alexander et al., 2011). Moreover, in the past decade, our group has produced several lines of evidence demonstrating that inhibition of cardiac GRK2 decreases mortality and adverse ventricular remodelling while enhancing cardiac function in models of HF (Iaccarino et al., 2005; Raake et al., 2008; Rengo et al., 2009).
The failing human heart is adrenergically hyperactivated, which helps to maintain cardiac performance in the short term; but, with worsening HF, sympathetic nervous system (SNS) overdrive results in adverse biological effects on cardiomyocytes (Lymperopoulos et al., 2007a; 2009; 2011; Triposkiadis et al., 2009). Adrenergic hyperactivation is accompanied by a generalized neurohormonal activation, finally determining structural end-organ damages such as cardiac dilatation, hypertrophy and fibrosis (Lymperopoulos et al., 2007a; Triposkiadis et al., 2009). Treatment with β−adrenoceptor antagonists (β-blockers) exerts a range of beneficial effects on cardiac function and, importantly, reduces HF-related morbidity and mortality (Bristow, 2000b; Foody et al., 2002). β-blockers can directly prevent catecholamine-mediated toxicity on cardiomyocytes by preventing catecholamines from binding to β-adrenoceptors (Bristow, 2000b; Foody et al., 2002). Importantly, it has also been reported that long-term β-blocker treatment is able to reduce HF-related SNS over-activity in experimental models of HF (Pleger et al., 2007; Rengo et al., 2009). as well as in humans (Nemanich et al., 1990; Andersson et al., 1994; Yoshikawa et al., 1996). However, the underlying molecular mechanism(s) responsible for this effect remains unidentified.
Recently, our group established experimentally that GRK2 up-regulation in the catecholamine-producing adrenal medulla of HF animals leads to enhanced down-regulation and uncoupling of α2-adrenoceptors (Lymperopoulos et al., 2007a; 2008; 2010). These receptors are physiologically expressed on the chromaffin cell membranes of the adrenal gland and of the SNS nerve terminals where they normally exert negative feedback control on catecholamine production and secretion (Lymperopoulos et al., 2007b). It appears that with progression of HF, adrenal α2-adrenoceptors become severely dysfunctional due to the actions of GRK2, thus contributing to the chronically elevated catecholamine levels and SNS activity seen with this disease.
In the present study, we investigated the specific adrenal consequences of β-blocker treatment in a rat HF model. We sought to determine if chronic bisoprolol treatment could potentially rescue the dysfunctional α2-adrenoceptor-catecholamine axis in HF and contribute to lower SNS activity and thus to protect the failing heart. By concentrating on GRK2 levels in our study, we have attempted to find not only the nodal target for adrenal dysfunction in HF but also a potential therapeutic mechanism to produce physiological sympatholysis.
Methods
All animal care and experimental protocols were in accordance with The Guide for Care and Use of Laboratory Animals of the National Institutes of Health (NIH Publication no. 85–23, Revised 1996) and were approved by the Ethics Committee for the Use of Animals in Research of our Institution. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). An expanded Methods section appears in the online-only data supplement.
Experimental groups
Sprague–Dawley male rats (∼300 g, total = 105) (Charles River Laboratories, Lecco, Italy) entered the study and were randomly assigned to one of the three experimental groups. Sham-operated rats and rats with surgically induced myocardial infarction (MI) were treated with vehicle (drinking water) for 14 weeks (Sham, n= 20; HF/C, n= 40). At 4 weeks post MI, one group of HF rats was assigned to a 10 week bisoprolol treatment (HF/B, n= 45), using 80 mg L-1 in drinking water (approximate dose of 10 mg-1 kg-1 day-1) (Watanabe et al., 2001; Pleger et al., 2007; Rengo et al., 2009). Bisoprolol treatment was started at one-fourth of the full dose, and dosages were doubled every week until administration of the full dose was achieved.
Experimental procedures
MI was induced by surgical ligation of the proximal tract of the left anterior descending coronary artery, as previously described (Leosco et al., 2008). Serial M-mode echocardiographic evaluations were performed at 4 weeks after surgery and at the end of the study protocols in anaesthetized (1.5% isofluorane) rats, using the VisualSONICS VeVo 770 imaging system with a 716 scan head, as described (Pleger et al., 2007; Leosco et al., 2008; Rengo et al., 2009). At the end of the study period, animals were killed by cervical dislocation under deep anaesthesia (ventilated with a mixture of O2 and 5% isoflurane).
RNA isolation and real-time RT-PCR
Cardiac total RNA isolations, reverse transcription to cDNA and quantitative real-time RT-PCR were carried out as previously described (Rengo et al., 2010; Zincarelli et al., 2010).
Western blotting
Western blots to assess protein levels of GRK2 (sc-562; Santa Cruz Biotechnology, Santa Cruz, CA) were performed using protein extracts from homogenized adrenal glands and hearts as described (Leosco et al., 2008; Rengo et al., 2010).
Preparation of tissue fractions
Tissue fractions were obtained as previously mentioned (Leosco et al., 2008).
Saturation ligand-binding
Plasma membranes from excised adrenal gland were prepared and saturation binding was performed using the α2-adrenoceptor radioligand [3H]RX821002, as reported (Lymperopoulos et al., 2007a).
B-adrenoceptor radioligand binding
Membrane fractions from the left ventricle (LV) were used for β-adrenoceptor radioligand binding studies using the non-selective β-adrenoceptor antagonist ligand [125I]cyanopindolol (125I-CYP) as previously described (Leosco et al., 2008). All assays were performed in triplicate, and receptor density (in fmol) was normalized to mg of membrane protein.
Measurement of infarct size
Infarct size was examined in all experimental groups at the end of the study period, as previously described (Rengo et al., 2009).
Adrenal chromaffin cell isolation and culture
To obtain chromaffin cells, excised adrenal glands from all three study groups were incubated in Locke's solution containing 1 mg·mL−1 collagenase (>200 U·mL−1; Biochrom KG, Berlin, Germany), as previously described (Lymperopoulos et al., 2007a; 2008).
In vitro catecholamine secretion measurements
Forty-eight hours post isolation, chromaffin cells were placed in a balanced salts buffer and stimulated with 20 µM nicotine for 30 min, following pretreatment with 10 µM UK14304 (Sigma Aldrich, Milan, Italy) or vehicle for 30 min. At the end of nicotine treatment, the supernatant was collected for determination of its catecholamine content. Adenoviral-transfected cells were infected 48 h post isolation, and cells were subjected to the various treatments 24 h after transfection (72 h post isolation).
Plasma and in vitro catecholamine secretion measurements
Plasma adrenaline and noradrenaline levels were determined by elisa, performed on rat plasma samples using the BI-CAT EIA kit from ALPCO Diagnostics (Windham, NH), as described previously (Lymperopoulos et al., 2007a; Rengo et al., 2010). In vitro adrenaline and noradrenaline secretion in the supernatant of cultured chromaffin cells were measured by using the same elisa kit, essentially as described previously (Lymperopoulos et al., 2007a; Rengo et al., 2010).
Adenoviruses
AdGRK2 or AdGFP recombinant adenoviruses were constructed as described and purified using two sequential rounds of CsCl density gradient ultracentrifugation (Lymperopoulos et al., 2008).
Statistics
Data were analysed by one-way anova, followed by a Bonferroni's post hoc analysis, or by unpaired t-testing, as appropriate. For all tests, P < 0.05 was considered statistically significant after Bonferroni corrections, if needed, and all data are reported as means ± SEM.
Materials
[3H]RX 821002 (specific activity 40–70 Ci mmol-1) was obtained from Perkin Elmer (Milan, Italy) and [125I]CYP (specific activity=2200 Ci mmol-1) from New England Nuclear Corp., Boston, MA, USA. Bisoprolol, and nicotine were from Sigma-Aldrich and isoflurane from Abbott (Latina, Italy).
Results
Effects of β-blocker therapy on cardiac function and dimensions
In order to evaluate the effects of β-blocker treatment in chronic HF, adult male Sprague–Dawley rats were first subjected to MI (coronary arterty ligation) or a sham operation. As shown in Figure 1, MI produced significant cardiac dysfunction and HF after 4 weeks. It was at this time that the rats with MI were randomly assigned to either bisoprolol or placebo treatment. As shown in Figure 1A and D, bisoprolol-treated (HF/B) or control rats (HF/C) had no discernable differences in cardiac function as measured by LV ejection fraction or dimensions (as measured by LV diastolic diameter) upon pretreatment echocardiography, as all rats had LV dysfunction 4 weeks post MI and before starting treatment, compared with Sham-operated control rats. After a 10 week treatment period, both HF groups still had significantly impaired cardiac function compared with sham controls. However, ejection fraction was significantly higher in rats receiving bisoprolol compared with HF controls, although, at the end of the study period, it did not exceed pretreatment values (Figure 1B and C; Table 1). Conversely, control treatment led to further deterioration of cardiac function after 10 weeks (Figure 1B and C; Table 1). Adverse LV remodelling, as measured by ventricular dilatation, also progressed further in control-treated HF rats and this was prevented by bisoprolol treatment (Figure 1E; Table 1). Moreover, β-blocker treatment resulted in significant reduction of heart rate (Figure 1F). Finally, there were no differences in infarct size between the two HF groups (Table 1), which was not surprising as bisoprolol treatment was started at 4 weeks post MI induction, a time by which the infarct scar is completely established. Taken together, these results strongly indicate that bisoprolol treatment is capable of preventing cardiac dysfunction and adverse remodelling in the post-MI failing heart.
Figure 1.
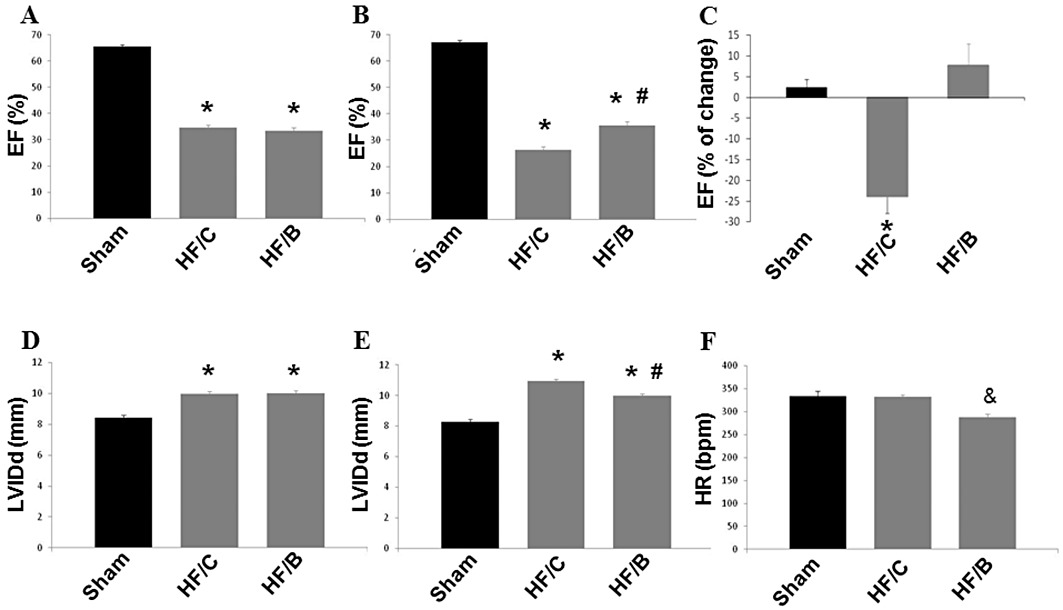
Ejection fraction (EF, as %) measured by echocardiography 4 weeks post MI (before treatments start) (A) and at the end of the study period, 10 weeks after bisoprolol or placebo treatments (B). (C) Percentage change in ejection fraction after the 10 weeks of treatment. LV internal diameter at diastole (LVIDd) measured by echocardiography before (D) and after treatment (E). (F) Heart rate (HR) at 14 weeks after MI. Sham, n= 12; HF/C, n= 18; HF/B, n= 20. Data are presented as mean ± SEM. *P < 0.05 versus sham; #P < 0.05 versus HF/C; & P < 0.05 versus sham and HF/C. anova analysis and Bonferroni test among all groups.
Table 1.
Physical and echocardiographic data in sham-operated and HF rats at the end of the study period
| Sham-operated | HF/control | HF/Bisoprolol | |
|---|---|---|---|
| Physical data | |||
| BW (kg) | 0.463 ± 0.014 | 0.446 ± 0.010 | 0.470 ± 0.013 |
| HW (g) | 1.15 ± 0.04 | 1.40 ± 0.02* | 1.28 ± 0.04*† |
| HW/BW (g·kg−1) | 2.50 ± 0.08 | 3.14 ± 0.12* | 2.73 ± 0.09*† |
| Echocardiography | |||
| HR (bpm) | 333.9 ± 11.1 | 332.4 ± 4.66 | 287.5 ± 7.28& |
| LV EF (%) | 67.1 ± 0.9 | 26.3 ± 1.2* | 35.5 ± 1.7*† |
| LVIDd (mm) | 8.27 ± 0.17 | 10.95 ± 0.13* | 9.97 ± 0.15*† |
| LVIDs (mm) | 5.14 ± 0.16 | 9.51 ± 0.16* | 8.18 ± 0.20*† |
| LVAWDd (mm) | 1.72 ± 0.03 | 1.46 ± 0.05* | 1.45 ± 0.06* |
| LVPWDd (mm) | 1.68 ± 0.06 | 2.02 ± 0.06* | 2.17 ± 0.08*† |
| Infarct size (%) | – | 44.8 ± 3.7 | 45.2 ± 4.3 |
anova analysis and Bonferroni test were used among all three groups. Data are presented as mean ± SEM. *P < 0.05 versus Sham; †P < 0.05 versus HF/control, &P < 0.05 versus Sham and HF/control.
BW = body weight, HW = heart weight, HR = heart rate, LV EF = LV ejection fraction, LVIDd = LV internal diameter at diastole, LVIDs = LVID at systole, LVAWDd = LV anterior wall diameter at diastole, LVPWDd = LV posterior wall diameter at diastole.
Effects of β-blocker therapy on cardiac remodelling gene profile
Following 10 weeks of HF treatment (14 weeks post MI), we evaluated cardiac gene expression patterns related to ventricular remodelling in our experimental groups. As a marker of HF, we investigated expression of the mRNA for brain natriuretic peptide (BNP) in the LV and found this to be significantly increased in the HF/C group, while this was normalized in bisoprolol-treated animals (Figure 2A). We further examined, as markers of remodelling and fibrosis, cardiac mRNA levels of collagen type I (Col1) and TGFβ-1 (Figure 2B and C) and found mRNA levels of both of these markers significantly elevated in HF/C rats compared with sham controls; but both were markedly reduced in the bisoprolol-treated post-MI HF rats. These results demonstrate the positive effects of bisoprolol treatment on cardiac adverse remodelling also at the molecular level.
Figure 2.
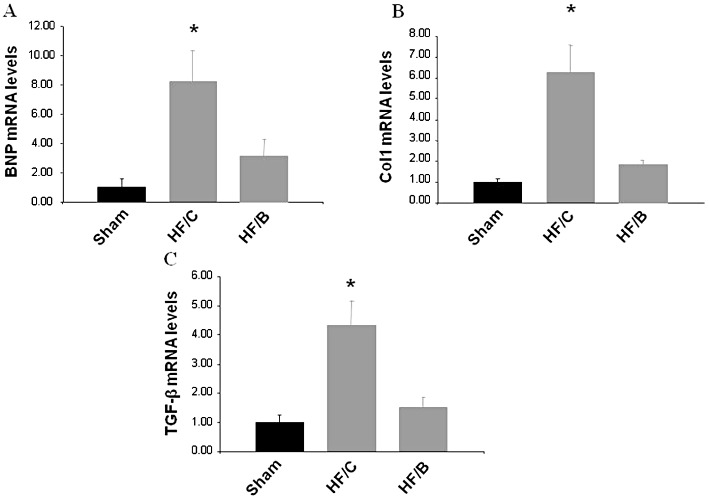
mRNA levels for (A) BNP, (B) collagen type I (Col1), (C) TGFβ1 in hearts from all experimental groups at the end of the study period (Sham, n= 10; HF/C, n= 12; HF/B, n= 12). All values were standardized to amplified 28S rRNA. *P < 0.05 versus Sham or HF/B groups. anova analysis and Bonferroni test among all groups. Data are presented as mean ± SEM and plotted as fold over sham values.
Effects of β-blocker therapy on plasma catecholamines in HF
We next investigated plasma catecholamine levels in our experimental groups at the end of the study period. Both adrenaline and noradrenaline were markedly elevated in HF rats compared with sham controls as expected (Figure 3). Ten weeks of bisoprolol treatment resulted in a significant reduction of circulating plasma levels of both noradrenaline and adrenaline compared with placebo group (Figure 3). However, our data show that bisoprolol treatment in HF did not induce a complete restoration of circulating catecholamine levels, as they remain significantly higher than those in sham control rats.
Figure 3.
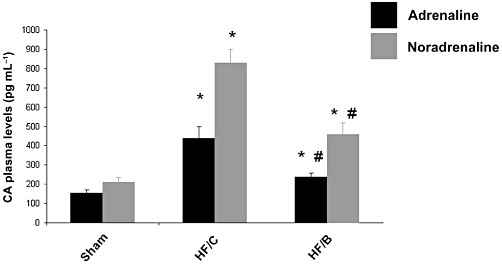
Plasma catecholamine (CA) levels in the three experimental groups at the end of the study. Data for noradrenaline and adrenaline are presented separately as means ± SEM. *P < 0.05 versus Sham or HF/B groups (n= 10 for each group); #P < 0.05 versus Sham or HF/C. anova analysis and Bonferroni test among all groups.
Adrenal GRK2 levels in HF after β-blocker therapy
Because we recently reported that GRK2, a critical regulator of adrenal catecholamine production, is up-regulated in the adrenal medulla of HF animals (Lymperopoulos et al., 2007a), the finding that bisoprolol administration significantly reduces circulating catecholamine levels in HF prompted us to evaluate how this treatment could affect GRK2 expression levels in the adrenal glands of HF animals. Consistent with our previous studies, adrenal GRK2 protein levels in the HF/C group were significantly elevated compared with sham control rats (Figure 4A). Interestingly, 10 weeks of bisoprolol treatment resulted in normal adrenal GRK2 levels indistinguishable from sham rats and in line with reduced circulating catecholamine levels (Figure 4A). Adrenal GRK2 mRNA levels were also increased in HF/C rats compared with sham; bisoprolol treatment was able to normalize GRK2 mRNA levels in the adrenals (Supplemental Figure S1). Of importance, we found similar increases of GRK2 levels in the myocardium of post-MI HF rats that was associated with significant β-adrenoceptor down-regulation compared with sham rats (Supplemental Figures S2 and S3). These results are consistent with our previous observations (Leosco et al., 2008; Rengo et al., 2009; 2010), and bisoprolol treatment resulted in a normalization of cardiac GRK2 protein levels and significantly increased β-adrenoceptor density in the heart compared with HF/C rats, and these levels were restored to normal (Sham) levels (Supplemental Figures S2 and S3).
Figure 4.
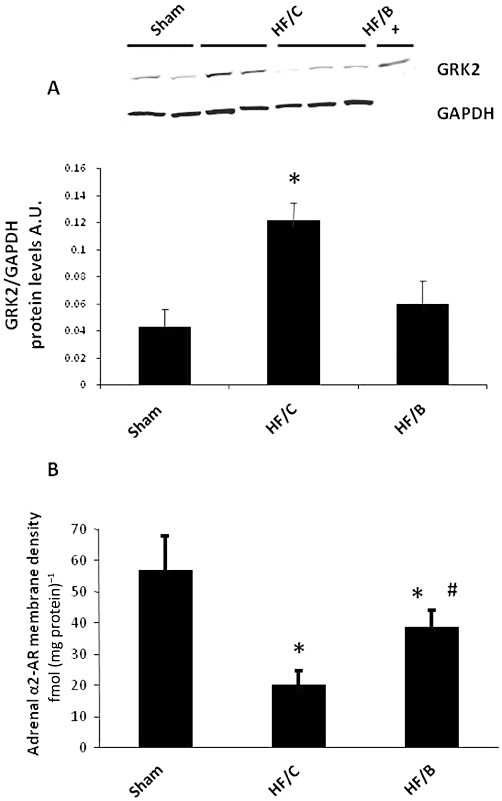
(A) GRK2 expression in adrenal homogenates purified from all three experimental groups at the end of the study period (n= 7 for each group). Representative Western blots (upper panel) and average densitometric quantitative analysis from blots showing the ratio of GRK2 to GAPDH (lower panel). += positive control. (B) Total α2-adrenoceptor (α2AR) density in plasma membranes purified from the adrenal glands of all three experimental groups at the end of the study (n= 6 for each group). *P < 0.05 versus Sham or HF/B; #P < 0.05 versus HF/C. Data are presented as mean ± SEM. anova analysis and Bonferroni test among all groups.
Adrenal α2-adrenoceptor density in HF after β-blocker therapy
Previous data from our group have demonstrated that during HF the increased expression and activity of GRK2 triggers a substantial α2-adrenoceptor dysregulation in the adrenal gland, resulting in loss of receptor inhibitory function and increased catecholamine secretion (Lymperopoulos et al., 2007a). This prompted us to investigate the effects of β-blocker therapy on adrenal α2-adrenoceptor density in our HF rats. As shown in Figure 4B, the HF/C group displayed marked adrenal α2-adrenoceptor down-regulation compared with healthy sham animals; however, bisoprolol treatment induced a significant improvement of adrenal α2-adrenoceptor density. These data strengthen the notion that β-blocker treatment reduces plasma catecholamine levels in HF by restoring the adrenal GRK2-α2-adrenoceptor-catecholamine secretion axis.
In vitro adrenaline release from isolated chromaffin cells
To better evaluate the effect of β-blocker treatment on α2-adrenoceptor dysfunction in HF, we examined catecholamine release from chromaffin cells isolated from the three experimental groups and treated in vitro with nicotine, a pharmacological stimulator of catecholamine release, and pretreated with or without the α2-adrenoceptor-specific agonist UK14304. In cultured chromaffin cells from all the three groups, nicotine induced similar levels of adrenaline release. In sham-derived cells, UK14304 blunted adrenaline release in response to the stimulus as expected; whereas in chromaffin cells derived from the HF/C group, the α2-adrenoceptor agonist was not able to inhibit adrenaline secretion in response to nicotine stimulation, indicating α2-adrenoceptor desensitization. Notably, when chromaffin cells from HF/B animals were stimulated with UK14304, nicotine-induced adrenaline release was significantly reduced, similar to that in cells from sham rats (Figure 5). Furthermore, in vivoβ-blocker treatment was also able to restore the function of α2-adrenoceptors towards inhibition of nicotine-induced noradrenaline secretion in chromaffin cells (Supplemental Figure S4). These data support the hypothesis that β-blocker treatment is able to reverse GRK2-mediated α2-adrenoceptor desensitization through reduction of GRK2 expression levels and thus can restore the inhibitory function of α2-adrenoceptors on catecholamine release from chromaffin cells during HF.
Figure 5.
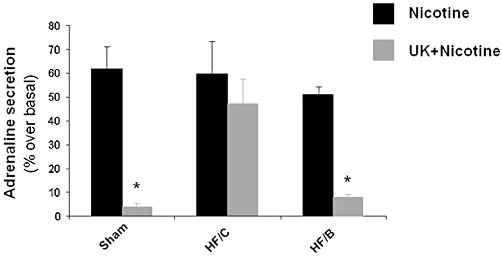
Secretion of adrenaline from cultures of chromaffin cells, isolated from the adrenals of sham, HF/C and HF/B rats, in response to nicotine (20 µmol·L−1), following pretreatment with vehicle (Nicotine) or with UK14304 (10 µmol·L−1; UK + Nicotine). *P < 0.05, versus HF/C-UK + Nicotine, n= 6. Data are presented as mean ± SEM. anova analysis and Bonferroni test among all groups.
Effects of GRK2 overexpression on adrenaline release from chromaffin cells isolated from HF bisoprolol at 14 weeks post MI
To test whether normalization of adrenal GRK2 protein levels is critical for the ability of bisoprolol to prevent HF-related α2-adrenoceptor down-regulation in vivo, we extracted the adrenal glands of bisoprolol-treated HF rats and cultured them for 24 h. Then the adrenal cells were infected either with a GFP adenovirus (Ad-GFP) as a control or Ad-GRK2. Induced expression of the transgenes (GRK2 and GFP) was confirmed by Western blotting in cellular protein extracts (Figure 6, upper panel), and 24 h after infection, catecholamine secretion from isolated chromaffin cells was tested using nicotine and the α2-adrenoceptor-specific agonist UK14304, as above. As shown in Figure 6 (lower panel), chromaffin cells extracted from bisoprolol-treated rats and infected in vitro with Ad-GFP were shown to have normalized negative feedback as UK14304 blocked nicotine-induced adrenaline release into the media. Importantly, when GRK2 was overexpressed in these cells, the inhibition of nicotine-induced catecholamine release by UK14304 was lost (Figure 6). Similar results have been obtained for noradrenaline secretion in the same setting of experiments (Supplemental Figure S5). These results strongly suggest that the effects of bisoprolol on restoring α2-adrenoceptor inhibitory function in HF are dependent on GRK2 levels/activity in the adrenal glands.
Figure 6.
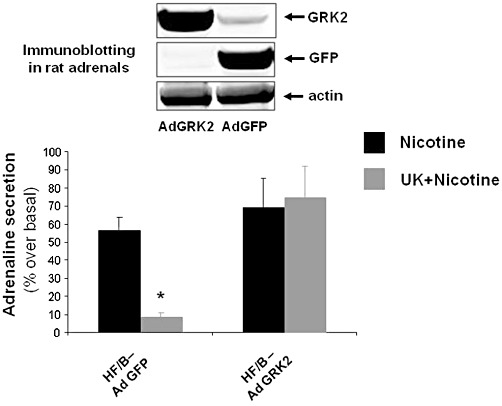
Representative Western blot showing expression of transgenes in cultures of chromaffin cells (upper panel). Secretion of adrenaline from chromaffin cells isolated from the adrenals of HF/B rats infected in vivo with AdGFP or AdGRK2 in response to nicotine treatment with or without pretreatment with UK14304 (UK + Nicotine) (lower panel). *P < 0.05,versus AdGRK2-UK + Nicotine, n= 6. Data are presented as mean ± SEM. anova analysis and Bonferroni test among all groups.
Discussion
In this study, we report a novel molecular mechanism that explains, at least in part, the ability of long-term β-blocker treatment in chronic HF to reduce the enhanced circulating catecholamine levels that significantly worsen the prognosis of patients affected by this condition (Lymperopoulos et al., 2007a; Triposkiadis et al., 2009). Ten weeks of bisoprolol treatment in rats with post-MI HF lowered adrenal GRK2 to normal levels and consequently markedly reduced adrenal catecholamine production via reversal of the dysfunction of adrenal α2-adrenoceptors, which are desensitized and down-regulated in HF. This finding is of particular physiological and clinical relevance since several experimental studies have reported that β-blocker treatment counteracts the catecholaminergic hyperactivation of chronic HF in animal models (Pleger et al., 2007; Rengo et al., 2009), not only by preventing adrenoceptor activation by agonist but also, chronically, by reducing circulating catecholamine levels. However, no evidence had been provided regarding the molecular mechanisms underlying this beneficial effect of β-blockade in HF.
Circulating catecholamines are derived from two main sources: the peripheral SNS nerve terminals, which release noradrenaline and the adrenal medulla, which produces mainly adrenaline and to a lesser extent, noradrenaline. In the adrenal gland, catecholamine production is tightly regulated by the adrenoceptors, in particular α2-adrenoceptors expressed in the chromaffin cells of the medullary region, which exerts tonic inhibition of catecholamine release, and also β2-adrenoceptors, which can facilitate catecholamine secretion (Lymperopoulos et al., 2007b). Like most other GPCRs, the α2-adrenoceptors can be desensitized by phosphorylation via GRKs, and chronic action by these kinases such as GRK2 can promote receptor down-regulation (i.e. receptor degradation). During HF, GRK2 is up-regulated in the adrenal medulla, causing α2-adrenoceptor dysfunction and ultimately leading to catecholamine hypersecretion from the chromaffin cells (Lymperopoulos et al., 2007a). By decreasing GRK2 levels in the adrenal gland, β-blocker treatment appears to restore adrenal α2-adrenoceptor density and signalling at the plasma membrane and catecholamine feedback inhibition, and this represents a mechanism whereby drugs such as bisoprolol (used in this study) can reduce SNS overdrive in chronic HF.
Overall, the success of β-blockers in HF treatment is attributed to their ability to block the constantly increased sympathetic overdrive present in the failing heart, thus reducing mortality and morbidity in HF patients (Bristow, 2000a; Foody et al., 2002). Moreover, in experimental models and in human HF, β-adrenoceptor blockade exerts several additional therapeutic effects, including improved cardiac reverse remodelling, blunted apoptosis, inhibited β-adrenoceptor internalization, reduced oxygen consumption and reduced risk of arrhythmias (Foody et al., 2002). In addition to these functional and clinical effects, β-blockade also appears to reduce the autonomic derangement and neurohumoral excitation in HF (Nemanich et al., 1990; Andersson et al., 1994; Yoshikawa et al., 1996). In particular, we have recently demonstrated that long-term metoprolol treatment in HF rats induces a significant reduction of both circulating noradrenaline and adrenaline levels (Rengo et al., 2009). This latter property of β-blocker treatment is particularly important for, at least, two main reasons. First, catecholamines exert negative effects on the heart and peripheral vasculature also through α-adrenoceptors, which are not antagonized by β-blockers (Lamba and Abraham, 2000). Second, SNS outflow can enhance and perpetuate the hyperactivation of other neurohormonal axes, such as the renin–angiotensin and endothelin systems (Goldsmith, 2004; Lee and Tkacs, 2008). Thus, β-blocker treatment, by reducing HF-related SNS overdrive, is also able to prevent the noxious effects of catecholamines on α2-adrenoceptors and to diminish activity of other cardiotoxic neurohormonal systems.
However, it is important to note that there are also studies showing no effects of β-blocker on circulating catecholamine levels (Santostasi et al., 1998; The RESOLVD Investigators, 2000; Blanchet et al., 2003; Foucart et al., 1991; Gilbert et al., 1996; Al-Hesayen et al., 2005; Lowes et al., 2002) and thus, the ability of β-blocker to reduce SNS hyperactivity in HF patients is still a matter of debate. Such discrepancies among studies might be attributed to several factors: (1) the particular β-blocker agent used (e.g. non-subtype-selective vs. β1-adrenoceptor-selective drugs); (2) duration of treatment (long-term vs. short-term); (3) HF severity (NYHA class and ejection fraction); (4) catecholamine levels before starting treatment (high vs. low levels). In our model of severe HF, due to the induction of a large MI (infarct size ∼45%), documented by the dramatically impaired ejection fraction, altered LV remodelling and high circulating catecholamine levels, we found almost a normalization of circulating catecholamine levels in HF/B compared with HF/C group.
As mentioned above, in the SNS nerve endings and in the adrenal glands, catecholamine secretion is tightly regulated by adrenoceptors acting as ‘presynaptic autoreceptors’ in this regard (Lymperopoulos et al., 2007b). Thus, the possibility of reducing catecholamine levels using β-blockers has already been suggested and is mechanistically linked to the blockade of the facilitatory β2-adrenoceptors presynaptic autoreceptor subtype (Foucart et al., 1988; 1991). In our study we chose bisoprolol, a β1-adrenoceptor-selective blocker, in order to preclude any involvement of the β2-adrenoceptors subtype, which is purported to be a facilitatory presynaptic autoreceptor in the adrenals and SNS nerve terminals (Foucart et al., 1988; 1991; Lymperopoulos et al., 2007a). Of note, the bisoprolol-blocked β1-adrenoceptors, contrary to the β2-adrenoceptors, appear not involved at all in neuronal or adrenal catecholamine release and hence in the modulation of catecholamine levels (Foucart et al., 1988; 1991; Lymperopoulos et al., 2007a). Moreover, several lines of evidence have shown that the β2-adrenoceptor signals and functions in a substantially different manner from the β1-adrenoceptor in cardiomyocytes. Importantly, the detrimental effects of sympathetic overdrive in the failing heart seem to be conferred mainly by cardiac β1- rather than β2-adrenoceptors (Zhu et al., 2005).
It should be mentioned here that whether the observed effects of bisoprolol on the adrenal GRK2–α2-adrenoceptor-catecholamine axis secretion axis are receptor-dependent still remains an open question that we plan to address in future experiments. For example, it is not clear from our present results that GRK2 lowering in the adrenal gland (and myocardium) is specifically due to β1-adrenoceptor blockade per se or whether there could be a receptor-independent event such as a more general beneficial actions of the drug on the heart, which indirectly feeds back to the adrenal medulla to suppress catecholamine secretion. However, we also investigated the effects of bisoprolol treatment on adrenal GRK2–α2-adrenoceptor-catecholamine production axis at an earlier time point: 2 weeks after initiation of bisoprolol treatment (6 weeks post MI). At this time point and, although there was no difference between the ejection fraction and left ventricular end-diastolic diameter between bisoprolol-treated and HF control groups, we still observed a significant reduction in the mRNA levels of HF-related remodelling/fibrosis genes in the bisoprolol-treated group (Supplemental Figures S6 and S7). However, we did not find any differences in GRK2 protein levels and α2-adrenoceptor function between bisoprolol and HF control groups (Supplemental Figures S8 and S9). Taken together, these latter results, although not conclusive, suggest that bisoprolol-dependent reverse remodelling preceded GRK2 and normalization of α2-adrenoceptors. Another important question that warrants further investigation in the future is whether these beneficial adrenal effects of bisoprolol are shared by other agents in this drug class (β-blockers) or at least by the other β1-adrenoceptor-selective blockers that have been shown to be beneficial in HF, such as metoprolol.
An interesting therapeutic issue may be explained by our data on adrenal GRK2- α2-adrenoceptor signalling following chronic β1-adrenoceptor blockade. The poor sympatho-inhibitory efficacy of the α2-adrenoceptor agonist moxonidine in HF patients might be ascribable to GRK2-dependent α2-adrenoceptor dysfunction. In fact, the MOXSE and MOXCON trials (Swedberg et al., 2000; 2002), two recent clinical trials of moxonidine for the treatment of HF, were discontinued because of excess mortality in the treatment group. A possible reason for the failure of these trials could have been the dysfunction of peripheral α2-adrenoceptors. Thus, cardiac sympathetic nerve terminals and the adrenal glands might not adequately respond to α2-adrenoceptor agonists because of the impaired receptor function. Importantly, in both studies, treatment with β-blockers. within 2 months before recruitment was an exclusion criterion. The present study demonstrates that β-blocker treatment was able to restore α2-adrenoceptor function in HF, and this might result not only in the observed reduction of circulating catecholamine levels but also, perhaps, in potentially increased efficacy of moxonidine in HF treatment. Therefore, β-blocker treatment coupled with α2-adrenoceptor agonists should be reconsidered.
Of note, these favourable molecular/physiological alterations in the adrenal gland post MI are associated with beneficial effects on the heart in terms of LV contractility and remodelling. In particular, bisoprolol treatment seems to prevent further progression of cardiac dysfunction. In fact, in HF/C rats we observed a progressive decrease in ejection fraction and increase in LV diameters by comparing the echocardiograpic results obtained at 4 and 10 weeks post MI. On the other hand, bisoprolol halted the progression of HF, as shown by the similar values in ejection fraction and LV diameters obtained by echocardiography at 4 and 10 weeks post MI. These findings are in complete accordance with previous reports indicating that β-blocker treatment affects ejection fraction and reduces LV diameters in the failing heart (Bristow, 2000b; Sabbah, 2004; Tevaearai and Koch, 2004). Moreover, the positive effects of bisoprolol treatment in the heart are evident also at the molecular level. GRK2 is normally up-regulated in the failing heart and significantly impairs cardiac β-adrenoceptor signalling and function (Petrofski and Koch, 2003; Rengo et al., 2011). Indeed, we found a marked up-regulation of GRK2 in hearts of untreated post-MI rats, whereas bisoprolol-treated rats showed a restoration of cardiac GRK2 protein levels. Normalization of cardiac GRK2 levels represents further additional molecular evidence for cardiac improvement by β-blockers in HF (Petrofski and Koch, 2003; Rengo et al., 2009). Of course, we cannot discriminate between the beneficial effect of bisoprolol obtained through direct β-adrenoceptor blockade in the heart and the indirect effect related to the reduction of the circulating catecholamine levels. These findings are in line with previous observations by our group in the failing myocardium (Pleger et al., 2007; Rengo et al., 2009) and in the aged heart (Leosco et al., 2007) and by others in hypertensive animals (Eckhart et al., 2002). Finally, markers of adverse cardiac remodelling were normalized after bisoprolol treatment such as BNP, TGF-β1, and Col-1 in HF rats.
In conclusion, the present study suggests a novel molecular mechanism for the beneficial effects of β-blocker treatment by curbing the autonomic derangements that confound and aggravate chronic HF. In addition, this study supports our previous findings that adrenal GRK2 up-regulation is an important pathogenic mechanism for the sympathetic overdrive in HF, and its lowering or inhibition represents a novel potential sympatholytic strategy for HF therapy.
Acknowledgments
This work was supported in part by the Italian ministry of University and Scientific Research, P.R.I.N. (Progetto di Ricerca di Interesse Nazionale) to Drs Nicola Ferrara, Dario Leosco, Giuseppe Rengo and Carmela Zincarelli and by postdoctoral fellowships to Drs Giuseppe Rengo and Anastasios Lymperopoulos from the American Heart Association (Great Rivers Affiliate). A. Lymperopoulos is supported by a Scientist Development Grant (SDG) from the American Heart Association.
Glossary
- Ad
adenovirus
- BNP
brain natriuretic peptide
- Col1
collagen type I
- GFP
green fluorescent protein
- GRK2
GPCR protein-coupled receptor kinase-2
- HF
heart failure
- HR
heart rate
- HW
heart weight
- 125I-CYP
[125I]Cyanopindolol
- LV
left ventricle
- LVIDd
LV internal diameter at diastole
- LVPWDd
LV posterior wall diameter at diastole
- MI
myocardial infarction
- SNS
sympathetic nervous system
Conflict of interest
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Adrenal GRK2 mRNA levels in all experimental groups at the end of the study period (Sham, n= 7; HF/C, n = 8; HF/B, n = 8). All values were standardized to amplified 28S rRNA. *P < 0.05 versus Sham or HF/B groups. ANOVA analysis and Bonferroni test among all groups. Data are presented as mean ± SEM and plotted as fold over sham values.
Figure S2 GRK2 expression in heart homogenates purified from all three experimental groups at the end of the studyperiod (n = 7 for each group). Representative Western blots (upper panel) and average densitometric quantitative analysis from blots showing the ratio of GRK2 to GAPDH (lower panel). *P < 0.05 versus Sham or HF/B. Data are presented as mean ± SEM. ANOVA analysis and Bonferroni test among all groups.
Figure S3 Total β-adrenoceptor density in plasma membranes purified from the hearts of all three experimentalgroups at the end of the study (n = 6 for each group). *P < 0.05 versus Sham or HF/B. Data are presented as mean ± SEM. ANOVA analysis and Bonferroni test among all groups.
Figure S4 In vitro noradrenaline secretion from chromaffin cells isolated from the adrenals of sham, HF/C and HF/B rats in response to 20μmol·L−1 nicotine treatment, following pretreatment with vehicle (Nicotine) or with 10μmol·L−1 UK14304 (UK +Nicotine). *P < 0.05, compared with HF/C-UK+ Nicotine, n = 6. Data are presented as mean± SEM. ANOVA analysis and Bonferroni test among all groups.
Figure S5 In vitro noradrenaline secretion from chromaffin cells isolated from the adrenals of HF/Brats infected in vivo with AdGFP or AdGRK2 in response to nicotine treatment with or without pretreatment with UK14304 (UK+ Nicotine). *P < 0.05, compared with AdGRK2-UK+Nicotine, n = 6. Data are presented as mean ± SEM. ANOVA analysis and Bonferroni test among all groups.
Figure S6 Ejection fraction (EF) as measured by echocardiography 6 weeks post MI (2 weeks after treatments start; upper panel); LV internal diameter at diastole (LVIDd) measured by echocardiography 2 weeks after treatments start (lower panel). HF control, n = 6; HF Bisoprolol, n = 6. Data are presented as mean ± SEM. ANOVA analysis and Bonferroni test among all groups.
Figure S7 Heart mRNA levels of TGFβ1 (upper panel) collagen type I (Col1) (lower panel); in all experimental groups at 2 weeks after treatment start (Sham, n = 6; HF control, n = 6; HF bisoprolol, n =6). All values were standardized to amplified 28S rRNA. *P < 0.05 versus Sham; #P < 0.05 versus HF control. ANOVA analysis and Bonferroni test among all groups. Data are presented as mean ± SEM and plotted as fold over sham values.
Figure S8 GRK2 expression in adrenal homogenates purified from all three experimental groups at 2 weeks post-treatment start (n = 5 for each group). Average densitometric quantitative analysis from blots showing the ratio of GRK2 to GAPDH. *P < 0.05 versus Sham. Data are presented as mean ± SEM. ANOVA analysis and Bonferroni test among all groups.
Figure S9 In vitro adrenaline secretion from chromaffin cells isolated from the adrenals of HF control and HF bisoprolol rats (2 weeks after treatment start) in response to 20 μmol·L−1 nicotine treatment, following pretreatment with vehicle (Nicotine) or with 10μmol·L−1 UK14304 (UK +Nicotine); n = 5 for each group. Data are presented as mean± SEM. ANOVA analysis and Bonferroni test among all groups.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hesayen A, Azevedo ER, Floras JS, Hollingshead S, Lopaschuk GD, Parker JD. Selective versus nonselective beta-adrenergic receptor blockade in chronic heart failure: differential effects on myocardial energy substrate utilization. Eur J Heart Fail. 2005;7:618–623. doi: 10.1016/j.ejheart.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Andersson B, Hamm C, Persson S, Wikström G, Sinagra G, Hjalmarson A, et al. Improved exercise hemodynamic status in dilated cardiomyopathy after beta-adrenergic blockade treatment. J Am Coll Cardiol. 1994;23:1397–1404. doi: 10.1016/0735-1097(94)90383-2. [DOI] [PubMed] [Google Scholar]
- Blanchet M, Ducharme A, Racine N, Rouleau JL, Tardif JC, Juneau M, et al. Effects of cold exposure on submaximal exercise performance and adrenergic activation in patients with congestive heart failure and the effects of beta-adrenergic blockade (carvedilol or metoprolol) Am J Cardiol. 2003;92:548–553. doi: 10.1016/s0002-9149(03)00723-9. [DOI] [PubMed] [Google Scholar]
- Bristow MR. Mechanistic and clinical rationales for using beta-blockers in heart failure. J Card Fail. 2000a;6:8–14. [PubMed] [Google Scholar]
- Bristow MR. β-Adrenergic receptor blockade in chronic heart failure. Circulation. 2000b;101:558–569. doi: 10.1161/01.cir.101.5.558. [DOI] [PubMed] [Google Scholar]
- Eckhart AD, Ozaki T, Tevaearai H, Rockman HA, Koch WJ. Vascular-targeted overexpression of G protein-coupled receptor kinase-2 in transgenic mice attenuates beta-adrenergic receptor signaling and increases resting blood pressure. Mol Pharmacol. 2002;61:749–758. doi: 10.1124/mol.61.4.749. [DOI] [PubMed] [Google Scholar]
- Feldman DS, Carnes CA, Abraham WT, Bristow MR. Mechanisms of disease: β-adrenergic receptors in signal transduction and pharmacogenomics in heart failure. Nat Clin Pract Cardiovasc Med. 2005;2:475–483. doi: 10.1038/ncpcardio0309. [DOI] [PubMed] [Google Scholar]
- Foody JM, Farrell MH, Krumholz HM. beta-Blocker therapy in heart failure: scientific review. JAMA. 2002;287:883–889. doi: 10.1001/jama.287.7.883. [DOI] [PubMed] [Google Scholar]
- Foucart S, Nadeau R, de Champlain J. Local modulation of adrenal catecholamines release by beta-2 adrenoceptors in the anaesthetized dog. Naunyn Schmiedebergs Arch Pharmacol. 1988;337:29–34. doi: 10.1007/BF00169473. [DOI] [PubMed] [Google Scholar]
- Foucart S, de Champlain J, Nadeau R. Modulation by beta-adrenoceptors and angiotensin II receptors of splanchnic nerve evoked catecholamine release from the adrenal medulla. Can J Physiol Pharmacol. 1991;69:1–7. doi: 10.1139/y91-001. [DOI] [PubMed] [Google Scholar]
- Gilbert EM, Abraham WT, Olsen S, Hattler B, White M, Mealy P, et al. Comparative hemodynamic, left ventricular functional, and antiadrenergic effects of chronic treatment with metoprolol versus carvedilol in the failing heart. Circulation. 1996;94:2817–2825. doi: 10.1161/01.cir.94.11.2817. [DOI] [PubMed] [Google Scholar]
- Goldsmith SR. Interactions between the sympathetic nervous system and the RAAS in heart failure. Curr Heart Fail Rep. 2004;1:45–50. doi: 10.1007/s11897-004-0024-5. [DOI] [PubMed] [Google Scholar]
- Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. 2009 Focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and management of heart failure in adults a Report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines developed in collaboration with the international society for heart and lung transplantation. J Am Coll Cardiol. 2009;53:e1–e90. doi: 10.1016/j.jacc.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Iaccarino G, Barbato E, Cipolletta E, De Amicis V, Margulies KB, Leosco D, et al. Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. Eur Heart J. 2005;26:1752–1758. doi: 10.1093/eurheartj/ehi429. [DOI] [PubMed] [Google Scholar]
- Lamba S, Abraham WT. Alterations in adrenergic receptor signaling in heart failure. Heart Fail Rev. 2000;5:7–16. doi: 10.1023/A:1009885822076. [DOI] [PubMed] [Google Scholar]
- Lee CS, Tkacs NC. Current concepts of neurohormonal activation in heart failure: mediators and mechanisms. AACN Adv Crit Care. 2008;19:364–385. doi: 10.1097/01.AACN.0000340718.93742.c4. [DOI] [PubMed] [Google Scholar]
- Leosco D, Rengo G, Iaccarino G, Filippelli A, Lymperopoulos A, Zincarelli C, et al. Exercise training and beta-blocker treatment ameliorate age-dependent impairment of beta-adrenergic receptor signaling and enhance cardiac responsiveness to adrenergic stimulation. Am J Physiol Heart Circ Physiol. 2007;293:H1596–H1603. doi: 10.1152/ajpheart.00308.2007. [DOI] [PubMed] [Google Scholar]
- Leosco D, Rengo G, Iaccarino G, Golino L, Marchese M, Fortunato F, et al. Exercise promotes angiogenesis and improves beta-adrenergic receptor signaling in the post-ischaemic failing rat heart. Cardiovasc Res. 2008;78:385–394. doi: 10.1093/cvr/cvm109. [DOI] [PubMed] [Google Scholar]
- Lowes BD, Gilbert EM, Abraham WT, Minobe WA, Larrabee P, Ferguson D, et al. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med. 2002;346:1357–1365. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Funakoshi H, Eckhart AD, Koch WJ. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007a;13:315–323. doi: 10.1038/nm1553. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Koch WJ. Adrenal adrenoceptors in heart failure: fine-tuning cardiac stimulation. Trends Mol Med. 2007b;13:503–511. doi: 10.1016/j.molmed.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Zincarelli C, Soltys S, Koch WJ. Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol Ther. 2008;16:302–307. doi: 10.1038/sj.mt.6300371. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Soltys S, Koch WJ. An adrenal β-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc Natl Acad Sci USA. 2009;106:5825–5830. doi: 10.1073/pnas.0811706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Gao E, Ebert SN, Dorn GW, 2nd, Koch WJ. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J Biol Chem. 2010;285:16378–16386. doi: 10.1074/jbc.M109.077859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Koch WJ. Adrenal β-arrestin-1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J Am Coll Cardiol. 2011;57:356–365. doi: 10.1016/j.jacc.2010.08.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemanich JW, Veith RC, Abrass IB, Stratton JR. Effects of metoprolol on rest and exercise cardiac function and plasma catecholamines in chronic congestive heart failure secondary to ischemic or idiopathic cardiomyopathy. Am J Cardiol. 1990;66:843–848. doi: 10.1016/0002-9149(90)90362-5. [DOI] [PubMed] [Google Scholar]
- Petrofski JP, Koch WJ. The β-adrenergic receptor kinase (βARK1) in heart failure. J Mol Cell Cardiol. 2003;35:1167–1174. doi: 10.1016/s0022-2828(03)00243-8. [DOI] [PubMed] [Google Scholar]
- Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115:2506–2515. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, et al. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008;103:413–422. doi: 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengo F, Leosco D, Iacovoni A, Rengo G, Golino L, Borgia F, et al. Epidemiology and risks factor for heart failure in the elderly. Ital Heart J. 2004;5(Suppl. 9):3S–10S. [PubMed] [Google Scholar]
- Rengo G, Lymperopoulos A, Zincarelli C, Donniacuo M, Soltys S, Rabinowitz JE, et al. Myocardial adeno-associated virus serotype 6-betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009;119:89–98. doi: 10.1161/CIRCULATIONAHA.108.803999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengo G, Leosco D, Zincarelli C, Marchese M, Corbi G, Liccardo D, et al. Adrenal GRK2 lowering is an underlying mechanism for the beneficial sympathetic effects of exercise training in heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H2032–H2038. doi: 10.1152/ajpheart.00702.2009. [DOI] [PubMed] [Google Scholar]
- Rengo G, Lymperopoulos A, Leosco D, Koch WJ. GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol. 2011;50:785–792. doi: 10.1016/j.yjmcc.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbah HN. Biologic rationale for the use of beta-blockers in the treatment of heart failure. Heart Fail Rev. 2004;9:91–97. doi: 10.1023/B:HREV.0000046363.59374.23. [DOI] [PubMed] [Google Scholar]
- Santostasi G, Fraccarollo D, Dorigo P, Egloff C, Miraglia G, Marinato PG, et al. Early reduction in plasma norepinephrine during beta-blocking therapy with metoprolol in chronic heart failure. J Card Fail. 1998;4:177–184. doi: 10.1016/s1071-9164(98)80004-3. [DOI] [PubMed] [Google Scholar]
- Swedberg K, Bergh CH, Dickstein K, McNay J, Steinberg M. The effects of moxonidine, a novel imidazoline, on plasma norepinephrine in patients with congestive heart failure. Moxonidine Investigators. J Am Coll Cardiol. 2000;35:398–404. doi: 10.1016/s0735-1097(99)00565-3. [DOI] [PubMed] [Google Scholar]
- Swedberg K, Bristow MR, Cohn JN, Dargie H, Straub M, Wiltse C, et al. Moxonidine Safety and Efficacy (MOXSE) Investigators. Effects of sustained-release moxonidine, an imidazoline agonist, on plasma norepinephrine in patients with chronic heart failure. Circulation. 2002;105:1797–1803. doi: 10.1161/01.cir.0000014212.04920.62. [DOI] [PubMed] [Google Scholar]
- Tevaearai HT, Koch WJ. Molecular restoration of beta-adrenergic receptor signaling improves contractile function of failing hearts. Trends Cardiovasc Med. 2004;14:252–256. doi: 10.1016/j.tcm.2004.07.002. [DOI] [PubMed] [Google Scholar]
- The RESOLVD Investigators. Effects of metoprolol CR in patients with ischemic and dilated cardiomyopathy. The Randomized Evaluation of Strategies for Left Ventricular Dysfunction Pilot Study. Circulation. 2000;101:378–384. doi: 10.1161/01.cir.101.4.378. [DOI] [PubMed] [Google Scholar]
- Tilley DG, Rockman HA. Role of β-adrenergic receptor signaling and desensitization in heart failure: new concepts and prospects for treatment. Exp Rev Cardiovasc Ther. 2006;4:417–432. doi: 10.1586/14779072.4.3.417. [DOI] [PubMed] [Google Scholar]
- Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54:1747–1762. doi: 10.1016/j.jacc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Ohta Y, Inoue M, Ma M, Wahed MI, Nakazawa M, et al. Bisoprolol improves survival in rats with heart failure. J Cardiovasc Pharmacol. 2001;38:S55–S58. doi: 10.1097/00005344-200110001-00012. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Handa S, Anzai T, Nishimura H, Baba A, Akaishi M, et al. Early reduction of neurohumoral factors plays a key role in mediating the efficacy of beta-blocker therapy for congestive heart failure. Am Heart J. 1996;131:329–336. doi: 10.1016/s0002-8703(96)90362-2. [DOI] [PubMed] [Google Scholar]
- Zhu W, Zeng X Zheng M, Xiao RP. The Enigma of β2Adrenergic Receptor Gi Signaling in the Heart. The Good, the Bad, and the Ugly. Circ Res. 2005;97:507–509. doi: 10.1161/01.RES.0000184615.56822.bd. [DOI] [PubMed] [Google Scholar]
- Zincarelli C, Soltys S, Rengo G, Koch WJ, Rabinowitz JE. Comparative cardiac gene delivery of adeno-associated virus serotypes 1-9 reveals that AAV6 mediates the most efficient transduction in mouse heart. Clin Transl Sci. 2010;3:81–89. doi: 10.1111/j.1752-8062.2010.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.