

Abstract
BACKGROUND AND PURPOSE
Recently, the DNA damage response (DDR) has emerged as a promising target for anticancer drug development. In our previous study, we identified several DDR-inhibiting compounds via high-content screening of a small molecule library using γH2AX foci as a biomarker. Here, we studied the effects of the DNA damage response inhibitor DDRI-18 (3,3′-(1H,3′H-5,5′-bibenzo[d]imidazole-2,2′-diyl)dianiline) on DDR.
EXPERIMENTAL APPROACH
Osteosarcoma U2OS cells were treated with etoposide to induce DDR. The nuclear foci of γH2AX and other signalling molecules in DDR were visualized by immunofluorescence and quantified using an IN Cell Analyzer. The DNA repair capacity of cells was analysed using the comet assay and in vivo DNA end-joining assay. Cell survival after drug treatment was quantified using the MTT assay, and apoptotic cell death was analysed by Annexin V staining and flow cytometry.
KEY RESULTS
DDRI-18 inhibited the non-homologous end-joining (NHEJ) DNA repair process and delayed the resolution of DNA damage-related proteins (γH2AX, ATM and BRCA1) from DNA lesions at a later phase of DDR. Furthermore, DDRI-18 enhanced the cytotoxic effects of anticancer DNA-damaging drugs, including etoposide, camptothecin, doxorubicin and bleomycin. This synergistic effect on cell death was shown to be due to caspase-dependent apoptosis.
CONCLUSIONS AND IMPLICATIONS
We identified a chemical compound, DDRI-18, that has chemosensitization activity. Although the target molecule and mechanism of action of DDRI-18 remain unknown, DDRI-18 is an effective chemosensitizing agent and may improve the therapy with classical anticancer drugs.
Keywords: DNA damage response, small molecule, anticancer agents, γH2AX
Introduction
The DNA damage response (DDR) is a highly evolved complex signalling network that leads to damage repair while modulating numerous cellular processes. Upon induction of DNA damage, eukaryotic cells activate cell cycle checkpoint mechanisms and DNA repair machineries to fix damaged DNA. When DNA damage is too severe to repair, cells with damaged DNA are removed from the organism by apoptotic cell death. These cellular responses induced by DNA damage, including cell cycle checkpoint, DNA repair and apoptosis, are collectively called the DDR. DDR is essential for the maintenance of the genomic integrity of eukaryotic cells (Zhou and Elledge, 2000; Bassing and Alt, 2004).
Currently, the induction of DNA damage is a major therapeutic strategy for killing tumour cells. Most non-surgical cancer therapies, including radiotherapy and chemotherapeutic drugs, generate DNA damage to induce tumour cell death. Thus, DDR induced by DNA-damaging agents is an important biological process in cancer therapy. There are two aspects of DDR: cell death and cell protection. DDR induced by anticancer therapeutic agents primarily initiate apoptosis, resulting in tumour cell death. However, protective aspects of DDR, such as DNA repair and cell cycle arrest, which promote tumour cell survival, can also be activated by anticancer therapeutic agents. These protective responses may reduce the efficacy of DNA damage-based cancer therapies and lead to resistance (Longley and Johnston, 2005). Thus, specific inhibition of the DNA repair pathway or cell cycle arrest mechanism might increase the cytotoxic effects of DNA-damaging anticancer therapies (Ding et al., 2006; Lord et al., 2006; Tse et al., 2007; Helleday et al., 2008).
Post-translational modifications such as phosphorylation, sumolylation and acetylation regulate the DDR pathway. More than 700 proteins, including phosphoinositide-3-kinase-related protein kinase (PIKK) family members (ATM, ATR and DNA-PK) and cell cycle checkpoint kinases, are phosphorylated in response to DNA damage (Huen and Chen, 2008). Several specific inhibitors of protein kinases involved in DDR have been reported to enhance the efficacy of conventional DNA-damaging anticancer therapies. For example, NU7026, a specific inhibitor of DNA-dependent protein kinase (DNA-PK), which is a key molecule in non-homologous end joining (NHEJ) DNA repair, potentiated the cytotoxicity of ionizing radiation (IR) and topoisomerase II poison (Veuger et al., 2003; Willmore et al., 2004) and restored the sensitivity of resistant cells to DNA damage-induced apoptosis (Deriano et al., 2005). Another DNA-PK inhibitor, NU7441, also enhanced the cytotoxicity of IR and etoposide (Zhao et al., 2006). XL-844, an inhibitor of the checkpoint kinases CHK1 and CHK2, which are involved in cell cycle arrest upon DNA damage, increased the radiosensitivity of tumour cells (Riesterer et al., 2011). AZD7762, another inhibitor of CHK1 and CHK2, potentiated the cellular response to DNA-damaging agents in the p53-negative cancer cell line (Zabludoff et al., 2008). These examples illustrate that small molecular inhibitors that abolish DDR can be efficacious when combined with conventional chemotherapeutic drugs, and several such inhibitors are currently in clinical trials (Hickson et al., 2004; Lara et al., 2005; Zhao et al., 2006; Ratnam and Low, 2007; Lapenna and Giordano, 2009; Noriko Hosoya, 2009).
In addition to this combination therapeutic strategy, novel approaches targeting DDR have been proposed. In many cases, tumour cells are deficient in one or more DNA repair pathways and tend to rely on remaining, intact DNA repair pathways for survival. Thus, specific inhibition of these pathways may exclusively kill tumour cells without affecting normal cells that have a full repertoire of DNA repair pathways and can escape the harmful effects of specific inhibition of one DNA repair pathway (Bryant et al., 2005; Farmer et al., 2005; Kennedy et al., 2007). As an example of this strategy, the PARP inhibitor is known to be effective against tumours associated with the BRCA1 or BRCA2 mutations (Fong et al., 2009), demonstrating that targeting specific DDR mechanisms is a promising anticancer strategy.
One of earliest events in the cellular response against DNA double strand breaks (DSB) is the phosphorylation of serine 139 of a histone H2 variant, H2AX, by ATM kinase at the DSB sites. Phosphorylated H2AX, called γH2AX, can be easily detected as nuclear foci at DSB sites after immunofluorescence staining. These γH2AX foci are useful as markers for DSBs or DDRs. (Rothkamm and Lobrich, 2003; Bonner et al., 2008). In our previous study, we confirmed the usefulness of γH2AX foci as a marker to determine the extent of DNA damage and developed a quantitative cell-based high-content screening method to measure DDR. This γH2AX foci quantification method can be used to detect compounds that inhibit the early stages of signalling events upon DNA damage as well as compounds that affect the repair process of DNA damage at later stages. We validated the usefulness of this method using two characterized DDR inhibitors. Treatment with the first inhibitor, KU55933 (an ATM-specific inhibitor), decreased the number of γH2AX foci during the early stages of etoposide-induced DNA damage response. However, a DNA-PK inhibitor, NU7026, delayed γH2AX foci resolution during the later phases of DNA repair. Using this method, we screened a chemical library and identified several chemicals that have DDR-inhibiting activity (Kim et al., 2011). In this report, we describe in detail the effects of a novel compound, DDRI-18 (3,3′-(1H,3′H-5,5′-bibenzo[d]imidazole-2,2′-diyl)dianiline), on DDR.
Methods
Cell culture and transfection
U2OS osteosarcoma cells were cultured in DMEM supplemented with 10% FBS (Hyclone, South Logan, UT, USA), 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin and 2 mM glutamine, at 37°C in a 5% CO2 atmosphere. Plasmid (1 µg) transfection was performed in 12-well plates using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions, and transfected cells were harvested 48 h later.
Materials and antibodies
DDRI-18 (C26H20N6) was kindly provided by the Korea Chemical Bank (Daejeon, Korea). Doxorubicin (DOX), cisplatin, 5-fluorouracil (5-FU), KU55933 (C21H17NO3S2) and NU7026 (C17H15NO3) were purchased from Calbiochem (San Diego, CA). Etoposide (ETO), camptothecin (CPT) and bleomycin were purchased from Sigma (St. Louis, MO, USA). Z-VAD-fmk was purchased from Tocris (Bristol, UK). Anti-γH2AX (Ser139) and anti-phospho BRCA1 (Ser1423) antibodies were purchased from Bethyl Laboratories (Montgomery, TX). Anti-phospho ATM (Ser1981) antibody was purchased from Millipore (Temecula, CA, USA). Antibodies against caspase 3 and PARP-1 were purchased from Cell Signaling (Danvers, MA) and BD Pharmingen (San Jose, CA, USA), respectively. All other chemicals and reagents were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA), unless specified otherwise.
Immunofluorescence
One day before treatment, cells were seeded at a concentration of 1 × 104 per well in black 96-well plates with clear flat bottoms (Costar, Corning, NY, USA). After treatment, the cells were rinsed with PBS, fixed with 3.7% formaldehyde in PBS for 20 min at room temperature (RT) and permeabilized with 0.02% Triton X-100 in PBS. Non-specific binding was blocked by incubating the cells with 1% BSA and 0.02% Triton X-100 in PBS for 30 min at RT. The cells were sequentially incubated at RT with primary antibodies (1:500) for 3 h, Alexa Fluor 488-conjugated anti-rabbit IgG antibody (1:500; Molecular Probe, Carlsbad, CA, USA) for 1 h and finally with Hoechst 33342 (10 µg·mL−1; Molecular Probe) for 10 min. The cells were washed three times with 0.02% Triton X-100 in PBS for 10 min after each incubation and were visualized using an IN Cell Analyzer 1000 (GE Healthcare, Buckinghamshire, UK).
Acquisition and analysis of images
The total area of γH2AX, phospho-ATM and phospho-BRCA1 foci was measured using an IN Cell Analyzer 1000. Images of stained cells were acquired from the automated fluorescence microscope platform of the IN Cell Analyzer using a 20× objective lens. Images from more than 5 fields per well were collected to obtain data from 200–400 cells. The filter sets used for detection of Hoechst 33342 and Alexa Fluor 488 signals were D360/40 (excitation)-HQ535/50 (emission) and D475/20 (excitation)-HQ 535/20 (emission), respectively. The acquired images were analysed using the Multi Target Analysis (MTA) module of the IN Cell Analyzer 1000 Workstation software (v3.4) according to the manufacturer's instructions.
Comet assay
The comet assay was performed under alkaline conditions based on the Trevigen kit procedure. Cells treated with chemical compounds, and DNA-damaging agents were harvested at the indicated time point. The mixture of 5 × 102 cells in 5 µL of PBS and 50 µL of comet low-melting agarose (LMA) was placed onto a slide, solidified at 4°C for 10 min, immersed in lysis solution at 4°C for 40 min and electophoresed at 21 V for 30 min in alkaline buffer (300 mM NaOH, 1 mM EDTA) in the dark. The slide was immersed in distilled water followed by 70% EtOH and then dried. DNA was stained with ethidium bromide and analysed using KOMET 5.5 software (Kinetic Imaging, Ltd, Nottingham, UK).
In vivo DNA end-joining assay
The plasmid-based DNA end-joining in vivo assay was performed as described by Shi et al. (2009). The pEGFP-N1 vector (Clontech, Mountain View, CA, USA) was linearized by BsrG1 restriction enzyme, which cut the plasmid within the GFP coding region, and transfected into U2OS cells using lipofectamin 2000 (Invitrogen). After 48 h, the fluorescence intensity of GFP was analysed using FACSCaliber (BD, San Jose, CA, USA).
Western blotting
Whole-cell lysates were prepared by suspending 2 × 106 cells in 100 µL of lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 2 mM PMSF, 1 mM Na3VO4 and 1X protease inhibitor; Roche, Mannheim, Germany). The cell lysates were kept on ice for 10 min, sonicated briefly and cleared by centrifugation at 15 000 ×g for 10 min at 4°C. Equal amounts of lysate protein were separated on a 4–12% gradient or 15% SDS-PAGE and transferred to enhanced chemiluminescence (ECL) nitrocellulose membranes (GE Healthcare). After being blocked with 5% skim milk in TBST (20 mM Tris, pH 7.5, 135 mM NaCl and 0.05% Tween 20), the membranes were incubated with the indicated antibodies. The membranes were washed and then incubated with horseradish peroxidase-conjugated anti-rabbit IgG (1:5000; Vector, Burlingame, CA, USA). After extensive washing, the proteins were visualized by chemiluminescence using ECL reagent (GE Healthcare).
3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) assay
Cells were seeded at 5 × 103 per well in a 96-well plate and treated with anticancer drugs and chemical compounds at the indicated concentrations. After incubation for the indicated time, the medium was removed followed by washing with PBS, and 100 µL of MTT (0.5 mg·mL−1) was added prior to incubation in a CO2 incubator at 37°C for 2 h. After incubation, insoluble crystals were completely dissolved in DMSO. The absorbance at 540 nm was measured using a Versamax microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Annexin V and propidium iodide staining
Cells treated with anticancer drugs and chemical compounds were washed with PBS, resuspended in 100 µL of binding buffer and incubated with 5 µL of Annexin V-FITC (BD Pharmingen) and 10 µL of propidium iodide (50 µg·mL−1) for 15 min at RT in the dark. After addition of 900 µL of binding buffer, the samples were analysed using FACSCaliber (BD Pharmingen).
Statistical analysis
Data are presented as mean values ± SEM. All statistical analyses were performed with GraphPad Prism version 5.03 (GraphPad Software, San Diego, CA, USA). Comparisons between two groups were carried out using Student's t-test for unpaired data. Differences between groups were considered statistically significant at P < 0.05. Combination Index (CI) was calculated using CalcuSyn software (Biosoft, Cambridge, UK) based on the multiple drug-effect equation of Chou-Talalay.
Results
DDRI-18 delays γH2AX foci disappearance in DDR
Previously, we validated the usefulness of γH2AX foci quantification for the identification of chemicals that inhibit DDR processes and screened a chemical library containing 6800 compounds to identify novel compounds that inhibit DDR. Since γH2AX foci disappear after DNA damage repair is complete (Rothkamm and Lobrich, 2003; Bonner et al., 2008), inhibition of the DNA damage repair mechanism may retard resolution of γH2AX foci. In an initial screening, eight compounds delayed γH2AX foci disappearance at a later phase of DDR, presumably by inhibiting DNA damage repair Among these compounds, DDRI-18 had the greatest effect in retaining γH2AX foci. In Figure 1A, we confirmed the inhibitory activity of DDRI-18 on γH2AX foci disappearance, which reflects progression of DNA damage repair. Without DDRI-18, immunofluorescence from the γH2AX foci reached a peak level 1 h after treatment with etoposide and declined after removal of etoposide. At 3 h after etoposide removal, 48.6% of the γH2AX foci remained relative to the peak level (immunofluorescence declined from 62.5 ± 2.1 to 30.4 ± 1.8). However, in the presence of DDRI-18, 76.6% of the γH2AX foci remained (immunofluorescence declined from 59.3 ± 0.6 to 45.4 ± 3.5). DDRI-18 increased the level of retained γH2AX foci induced by etoposide at a later phase of DDR in a dose-dependent manner (Figure 1B). The chemical structure of DDRI-18 is shown in Figure 1C. The compound is symmetric and contains a pair of benzimidazole and aniline rings.
Figure 1.
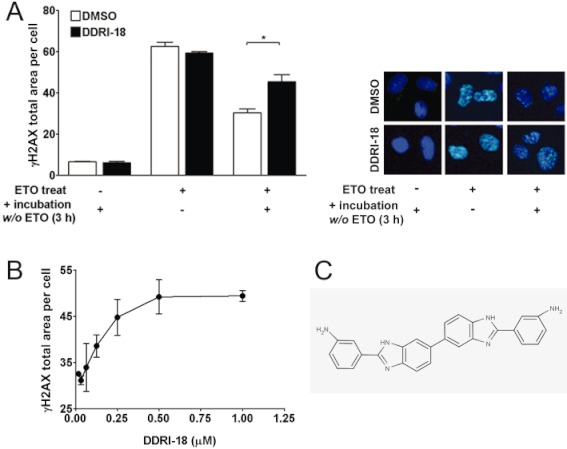
DDRI-18 delays γH2AX resolution at a later phase of DDR. (A) U2OS cells were pre-incubated with 2.5 µM DDRI-18 for 1 h and then exposed to 10 µM etoposide (ETO) for 1 h, followed by incubation in etoposide-free medium containing DDRI-18 for another 3 h. After incubation, the cells were fixed and processed for γH2AX immunofluorescence, and the γH2AX foci were analysed with an IN Cell Analyzer. Representative graphs and images from three independent experiments are shown. Values represent the means ± SEM (Student's t-test, P > 0.05). (B) U2OS cells were pre-incubated with DDRI-18 at the indicated concentrations for 1 h and then exposed to 10 µM etoposide for 1 h, followed by incubation in etoposide-free medium containing DDRI-18 for another 3 h. The γH2AX foci were visualized and analysed with an IN Cell Analyzer as described in (A). (C) Chemical structure of DDRI-18.
DDRI-18 inhibits DNA repair
Next, we examined whether retardation of γH2AX resolution by DDRI-18 was due to inhibition of DNA repair. We conducted an alkaline comet assay to determine the extent of unrepaired DNA damage after treatment with etoposide in the presence or absence of DDRI-18. In the absence of DDRI-18, the extent of DNA in the tail, which indicates damaged DNA, was maximal 1 h after treatment with etoposide and decreased to the basal level 3 h after removal of etoposide from the medium. In the presence of 2.5 µM DDRI-18, however, tail DNA did not decrease to the basal level 3 h after etoposide removal but remained at a higher level (Figure 2A). These comet assay data suggest that DDRI-18 inhibits the DNA damage repair process. In this experiment, DDRI-18 alone did not cause DNA damage after a 4 h treatment.
Figure 2.
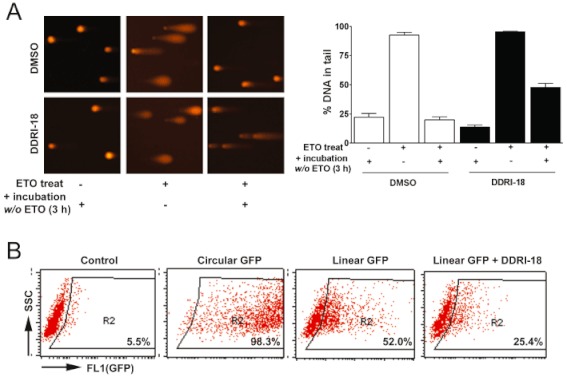
DDRI-18 inhibits DNA repair. (A) U2OS cells were pretreated with 2.5 µM DDRI-18 for 1 h and then exposed to 10 µM etoposide (ETO) for 1 h, followed by incubation in etoposide-free medium in the presence of DMSO or 2.5 µM DDRI-18 for 3 h. A comet assay was performed to detect tail DNA. The y-axis in the graphs indicates the percentage of DNA in the tail. (B) U2OS cells were transfected with linearized (by BsrG1) pEGFP-N1 and incubated in medium containing DMSO or 2.5 µM DDRI-18 for 48 h. GFP expression was analysed by FACSCaliber. Cells transfected with circular (uncut) plasmid served as a positive control, whereas mock-transfected cells (no pEGFP-N1) served as a negative control.
Next, we tested whether DDRI-18 inhibits the NHEJ mechanism, which is one of the principal pathways of repair of DSB. U2OS cells were transfected with a pEGFP-N1 plasmid linearized with the BsrG1 enzyme, which cleaves at the EGFP coding region. With competent host cell NHEJ machinery, linearized plasmid DNA can be repaired, and EGFP is expressed. In the absence of DDRI-18, EGFP was expressed in 52.0% of U2OS cells 48 h after transfection with linearized EGFP plasmid. In the presence of 2.5 µM DDRI-18, however, the percentage of EGFP-expressing cells was decreased to 25.4%, indicating that DDRI-18 inhibits NHEJ (Figure 2B). We also examined whether DDRI-18 affects cell cycle arrest induced by DNA-damaging agents; DDRI-18 did not affect G2M arrest induced by etoposide (Supplementary Figure S1). We suggest that DDRI-18 reduces DNA repair (not cell cycle arrest) activated by DNA-damaging agents such as etoposide by specifically inhibiting NHEJ, thereby increasing the retention of γH2AX at a later phase of DDR.
DDRI-18 attenuates resolution of DNA repair proteins at the DSB site after DNA damage
One of the earliest events in the cellular response to DSBs, the most severe form of DNA damage, is generation of a phosphorylated form of H2AX (called γH2AX) by activated ATM kinase at the site of the DSB. Several DNA repair-related proteins, such as BRCA1, are recruited to the DNA lesion. Regarding the inhibitory effect of DDRI-18 on DNA repair, we examined whether DDRI-18 could affect the recruitment of DNA repair-related proteins to the site of DNA damage. In this experiment, we detected the foci of γH2AX, p-ATM and p-BRCA1 through immunofluorescence staining. After treatment of U2OS cells with etoposide in the presence and absence of DDRI-18, we observed the level of γH2AX, ATM and BRCA1 foci in the damaged regions of DNA at the indicated time points. In the absence of any inhibitory chemicals, the levels of γH2AX, ATM and BRCA1 foci were maximal 1 h after treatment with 10 µM etoposide and gradually declined thereafter (Figure 3A). However, in the presence of DDRI-18, the accumulation of γH2AX, ATM and BRCA1 at DNA damage sites was sustained at later time points. Treatment of cells with NU7026, which is a specific inhibitor of DNA-PK and may inhibit the NHEJ mechanism, resulted in a similar pattern of sustained accumulation of the factors as seen after treatment with DDRI-18. However, KU55933, a specific inhibitor of ATM kinase (which phosphorylates H2AX), abolished foci formation of DNA repair proteins after DNA damage. Western blot analysis also confirmed that the levels of phosphorylated forms of DNA repair proteins 4 h after etoposide treatment were higher in cells treated with DDRI-18 than in vehicle-treated cells (Figure 3B). Collectively, these data suggest that DDRI-18 delays the resolution of DNA repair proteins accumulated at DNA damage regions, possibly by inhibiting the DNA damage repair machinery.
Figure 3.
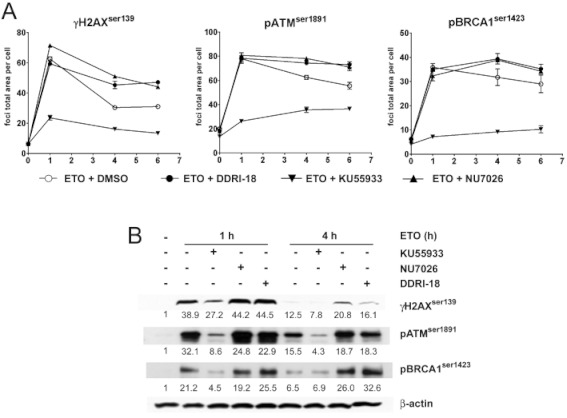
DDRI-18 attenuates resolution of DNA repair proteins after DNA damage. (A) U2OS cells were pre-incubated with inhibitors, including DMSO, 2.5 µM DDRI-18, 10 µM NU7026 or 10 µM KU55933, for 1 h and then exposed to 10 µM etoposide (ETO) for 1 h, followed by incubation in etoposide-free medium containing inhibitors for the indicated times. After incubation, the cells were fixed and processed for γH2AX, phospho-ATM, and phospho-BRCA1 immunofluorescence staining. The foci area per cell was analysed with an IN Cell Analyzer. Graphs from one representative experiment from three independent experiments are shown. Values represent the means ± SEM from triplicate experiments. (B) Protein extracts from U2OS cells treated with 10 µM etoposide alone, etoposide + 10 µM KU55933, 10 µM NU7026 or 2.5 µM DDRI-18 for the indicated time were analysed by Western blotting using antibodies against phospho-H2AX, -ATM and -BRCA1 and β-actin. Values shown represent the relative ratio of blotted protein to β-actin densitometry values (normalized to 1 in media only).
DDRI-18 potentiates the cytotoxicity of anticancer drugs
Because several reports have indicated that anticancer drug-induced cytotoxicity is enhanced by co-treatment with a DNA repair inhibitor, we examined whether DDRI-18 treatment could sensitize tumour cells to anticancer drugs. MTT assays were carried out where U2OS cells were exposed to a range of etoposide doses in the presence of 2 or 8 µM DDRI-18 or DMSO for 24 and 48 h, respectively. The results show that DDRI-18 significantly enhanced the cytotoxic effect of etoposide (Figure 4A). After the 24 h incubation, 2 and 8 µM DDRI-18 increased the cytotoxicity of etoposide 2.9- and 6.5-fold, respectively (LD50 values were 120.60, 42.69 and 18.50 µM in the presence of DMSO alone and 2 and 8 µM DDRI-18, respectively). After the 48 h incubation, 2 µM DDRI-18 increased the cytotoxicity of etoposide 4.6-fold (LD50 values were 14.35 and 3.13 µM in the absence and presence of DDRI-18, respectively). DDRI-18 alone had no effect on cell viability after 24 h of incubation, whereas cell viability was slightly decreased after 48 h of incubation with DDRI-18 alone. Enhancement of etoposide-induced cytotoxicity by DDRI-18 was also observed in other human tumour cell lines, including HeLa, 2774, A549, SK-BR3, FRO and SK-Hep1. However, DDRI-18 did not potentiate the cytotoxicity of etoposide in human dermal fibroblasts (HDF) (Supplementary Figure S2).
Figure 4.
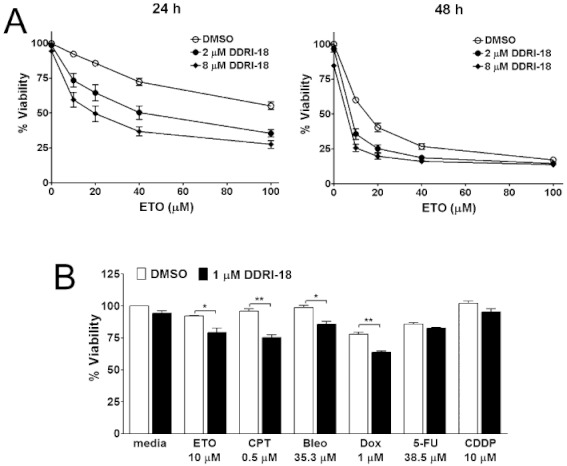
DDRI-18 potentiates the cytotoxicity of anticancer drugs. (A) U2OS cells were pretreated with 2 or 8 µM DDRI-18 for 1 h and then added with 10 µM etoposide, followed by a 24 h or 48 h incubation. After incubation, cell viability was measured by the MTT assay. In the presence of DDRI-18, cell death induced by etoposide was increased at 24 h and 48 h. Graphs and values represent the means ± SEM from three independent experiments. (B) U2OS cells were treated as described in (A) with the indicated concentrations of DDRI-18 and anticancer drugs, followed by a 24 h incubation, and then cell viability was evaluated using the MTT assay. Values represent the means ± SEM from three independent experiments (Students t-test): ***P < 0.0005; **P < 0.005; *P < 0.05.
Next, we tested chemosensitization activity of DDRI-18 when used in combination with other anticancer drugs. We treated U2OS cells with anticancer drugs such as camptothecin, bleomycin, doxorubicin, 5-FU and cisplatin in the presence and absence of 1 µM DDRI-18. The MTT assay showed that DDRI-18 significantly enhanced the cytotoxicity of the anticancer drugs etoposide, camptothecin, doxorubicin and bleomycin (Student's t-test, P-value 0.0228, 0.0017, 0.0109 and 0.0013, respectively). The cytotoxicity of 5-FU and cisplatin, by contrast, was only marginally increased by DDRI-18 and Student's t-test indicated that the increase was not statistically significant (Figure 4B). Taken together, these data suggest that DDRI-18 can potentiate the cytotoxic effects of diverse anticancer drugs in various tumour cell lines.
DDR-18 combined with etoposide triggers typical apoptosis
We investigated whether cell death induced by DDRI-18 plus etoposide was due to apoptosis. Firstly, we tested for phosphatidyl serine (PS) exposure and nuclear condensation, which are hallmarks of apoptosis. To detect PS exposure, we used Annexin V staining; at 24 h after treatment with etoposide plus DDRI-18, the percentage of cells stained with Annexin V, which represents PS exposure, reached 78.0%, whereas it was only 7.7%, 8.9% and 18.2% in cells treated with DMSO, DDRI-18 alone and etoposide alone, respectively (Figure 5A). To detect nuclear condensation, we used Hoechest 33342 staining. We observed increased nuclear condensation in cells treated with etoposide plus DDR-18 compared with untreated cells and cells treated with etoposide alone or DDRI-18 alone (Figure 5B).
Figure 5.
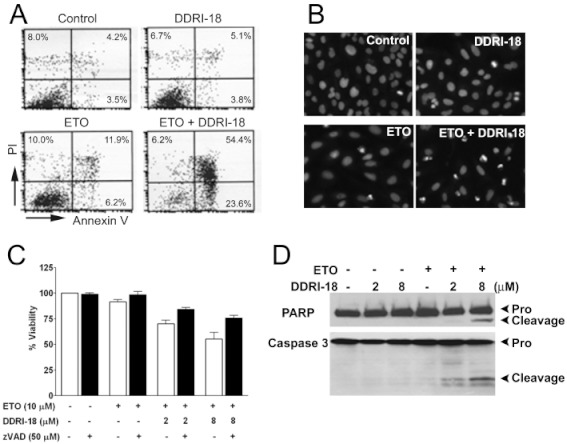
Cell death induced by etoposide in combination with DDRI-18 is caspase-dependent apoptosis. (A, B) U2OS cells were incubated in the presence of 5 µM DDRI-18 and 10 µM etoposide (ETO) for 24 h. Apoptotic cells were detected by flow cytometry after Annexin V–FITC + PI staining (A), or by nuclear condensation monitored by Hoechst 33342 staining (B). (C) U2OS cells were pretreated with 50 µM z-VAD-fmk for 1 h before treatment with the indicated concentrations of DDRI-18 and etoposide. After 24 h, cell viability was evaluated by the MTT assay. Graphs and values represent the means ± SEM from three independent experiments. (D) Protein extracts from U2OS cells treated with the indicated concentrations of etoposide and DDRI-18 for 24 h were analysed by Western blotting using antibodies against PARP-1 and caspase 3. The proform of PARP (116 kDa), cleaved PARP (85 kDa), proform of caspase 3 (35 kDa) and cleaved caspase 3 (17 kDa) are indicated.
To elucidate the involvement of caspases in cell death induced by etoposide plus DDRI-18, the cells were pretreated with the pan-caspase inhibitor, z-VAD-fmk. In the presence of z-VAD-fmk, cell death induced by etoposide plus DDRI-18 was inhibited (Figure 5C). Furthermore, cleaved caspase 3 and cleaved PARP, which are markers of caspase activation, were detected only in cells treated with etoposide plus DDRI-18 (Figure 5D). Collectively, these results suggest that cell death induced by etoposide plus DDRI-18 is caspase-dependent apoptosis.
Discussion
The cellular response to DNA damage is emerging as a promising target for cancer therapy. Specific inhibitors of DDR, including cell cycle arrest and DNA repair, are expected to enhance the efficacy of anticancer drugs and to decrease side effects. Previously, we reported on the usefulness of γH2AX foci quantification to identify chemicals that inhibit early signal transduction pathways and later DNA damage repair processes that are activated in response to DNA damage. Using this method, we screened a chemical library containing 6800 compounds to identify candidate chemicals that inhibit DDR. In this study, we analysed the effects of one candidate molecule, DDRI-18, on DNA damage repair, especially on NHEJ. Because γH2AX foci disappear after completion of DNA damage repair (Rothkamm and Lobrich, 2003; Nakada et al., 2008), inhibition of DNA damage repair is reflected in retardation of the resolution of γH2AX foci. It has been reported that specific inhibitors of DNA repair, such as NU7026 and NU7441, delay the resolution of γH2AX foci at DSB sites induced by IR (Riballo et al., 2004) or etoposide (Cowell et al., 2005; Zhao et al., 2006), respectively. In this report, we characterized DDRI-18 as an inhibitor of DNA damage repair. The comet assay (Figure 2A) and in vivo DNA end joining assay (Figure 2B) revealed that DDRI-18 inhibited the DNA repair process. However, DDRI-18 did not augment DNA damage induction because the percentage of tail DNA (damaged DNA), which was maximal after 1 h of treatment with etoposide, was the same with or without DDRI-18. Also, DDRI-18 alone had no effect on tail DNA induction after 4 h of incubation (Figure 2A) or on γH2AX foci formation (a DSB marker) after 6 h of incubation (data not shown). Collectively, these data indicate that DDRI-18 itself does not induce DNA damage. We observed that DDRI-18 did not affect cell cycle arrest induced by DNA-damaging agents (DDRI-18 did not affect G2M arrest induced by etoposide) (Supplementary Figure S1). Taken together, these data suggest that DDRI-18 inhibits the DNA repair process but does not affect cell cycle arrest after DNA damage.
Upon DNA damage, several proteins involved in DDR as sensors, transducers and effectors are activated and recruited to the DNA damage sites to perform DNA repair and co-localize with γH2AX foci, which are biomarkers of DSBs (Harper and Elledge, 2007). Protein interactions and modifications such as phosphorylation, ubiquitination and sumolylation at the DNA lesion can regulate the whole DDR process. Accumulated DNA repair proteins at the DNA damage sites resolve with DNA damage repair. Accordingly, small molecules that inhibit DNA repair retard the resolution of γH2AX foci at the DNA damage sites (Riballo et al., 2004; Cowell et al., 2005; Zhao et al., 2006; Kim et al., 2011). We measured the accumulation and resolution of γH2AX and DNA repair proteins at DSB sites after treatment with DNA-damaging agents in the presence and absence of DDRI-18 (Figure 3A). We observed that DDRI-18 delayed resolution of DNA repair-related proteins, as did NU7026.
Next, we evaluated whether DDRI-18 inhibited the DNA-PK dependent NHEJ DNA repair pathway, similar to NU7026 (DNA-PK inhibitor), since the DDRI-18 inhibitory patterns were similar to NU7026 (Figure 3A). In the presence of DDRI-18 and NU7026, the level of γH2AX foci at the later phases after etoposide treatment was higher than those in the presence of DDRI-18 or NU7026 alone (Supplementary Figure S3). We calculated the combination index (CI) using CalcuSyn software (Biosoft, Cambridge, UK) based on the multiple drug-effect equation of Chou-Talalay. In this analysis, a CI value < 0.9 implies synergism, CI = 0.9–1.1 implies additive and CI > 1.1 indicates antagonism. CI values at the ED50, ED75 and ED90 doses were 0.8854, 0.33208 and 0.18683, respectively, suggesting that DDRI-18 and NU7026 have nearly additive to synergistic effects at this concentration range. Taken together, these results suggest that DDRI-18 may have a different mode of action from NU7026 and may inhibit the DNA-PK independent NHEJ DNA repair, which has been studied in several previous reports (Yano et al., 2008; Hendrickson et al., 2010). However, the specific target of the DDRI-18 inhibitory mechanism remains unknown.
It has been reported that specific inhibitors of DDR, including DNA repair and cell cycle arrest, potentiate the cytotoxicity of DNA-damaging agents. KU55933, a specific ATM inhibitor, enhances the cytotoxicity of IR and topoisomerase poisons (Hickson et al., 2004). XL-844 and AZD7762, inhibitors of cell cycle kinases, also potentiate the cytotoxic effects of DNA-damaging agents (Zabludoff et al., 2008; Lapenna and Giordano, 2009; Riesterer et al., 2011). Additionally, SU11752, NU7026 and NU7441, specific inhibitors of DNA-PK involved in NHEJ DNA repair, potentiate the cytotoxicity of DNA-damaging agents (Ismail et al., 2004; Willmore et al., 2004; Zhao et al., 2006). Because small molecules that inhibit the DNA damage response can render tumour cells more sensitive to DNA-damaging agents, including IR, etoposide, doxorubicin and camptothecin, we expected that DDRI-18 would also potentiate the cytotoxic effects of DNA-damaging agents. As expected, DDRI-18 significantly enhanced the cytotoxicity of etoposide, camptothecin, doxorubicin and bleomycin. The cytotoxic effects of 5-FU and cisplatin were also enhanced marginally by DDRI-18, although the effect was not statistically significant. In our previous report, 5-FU (anti-metabolite) and cisplatin (alkylator), which are not direct DSB inducers, did not induce γH2AX foci formation, in contrast to etoposide and doxorubicin (topoisomerase II inhibitor), camptothecin (topoisomerase I inhibitor) and bleomycin (radiomimetic) (Kim et al., 2011). These data suggest that DDRI-18 is a good candidate as an anticancer sensitizer when used in combination with anticancer agents that induce the formation of DSBs and γH2AX foci. It is interesting that DDRI-18 could inhibit NHEJ pathway in that NHEJ is one of major mechanisms of DSB repair. Regarding the prospect of DDRI-18 as an anticancer sensitizing agent, it is quite promising that DDRI-18 did not increase the cytotoxicity of etoposide in non-transformed cells such as HDF, demonstrating some degree of cancer specificity (Supplementary Figure S2). Furthermore, neither γH2AX foci formation nor cell cycle arrest was induced by DDRI-18 alone in HDF cells (data not shown).
Most DNA-damaging agents induce tumour cell death through the apoptotic pathway. In the case of DDRI-18 in combination with etoposide, cell death was due to apoptosis because it was accompanied by increased PS exposure, as determined by positive staining with Annexin V, and by increased nuclear condensation and cleavage by PARP, which are hallmarks of apoptosis. Inhibition of cell death by the pancaspase inhibitor z-VAD-fmk suggested that the apoptosis induced by etoposide plus DDRI-18 is caspase-dependent. Apoptosis is an attractive approach for cancer treatment because it does not cause inflammation, in contrast to necrosis. Therefore, DDRI-18 may prove to be a promising sensitizer for anticancer drugs.
We did not clarify the target or exact mechanism of action of DDRI-18. The molecular structure of DDRI-18 includes two benzimidazole and aniline (phenylamine group) moieties. These structural features are also found in DNA-intercalating Hoechst dye, which binds to the minor grooves of DNA (Albert et al., 1999). Because of this structural similarity, DDRI-18 might bind to the minor grooves of DNA and increase DNA rigidity, possibly resulting in inhibition of the DNA repair process. To evaluate this hypothesis, we conducted a gel mobility shift and fluorescence assay to determine whether DDRI-18 could intercalate with DNA. DDRI-18 did not change DNA mobility. However, it is possible that DDRI-18 is a weak DNA intercalator since DDRI-18 showed dim but significant fluorescence after an extended exposure (data not shown). This weak DNA-intercalating activity may play a role in the DNA damage repair pathway, especially for the DNA lesion induced by topoisomerase inhibition in that the chemosensitization effects of DDRI-18 are greatest when combined with topoisomerase inhibitors. However, this hypothesis needs to be substantiated by further study.
Next, we tested whether Hoechst 33342 could also affect DDR. However, the effects of Hoechst 33342 on γH2AX foci formation and disappearance were unlike those of DDRI-18 in that Hoechst 33342 abolished formation of γH2AX foci right after the induction of DNA damage (at the 1 h time point). DAPI, another DNA-staining dye that contains indole (an analogue of benzimidazole) and phenyl groups, did not affect γH2AX foci formation (data not shown). So even though DDRI-18, Hoechst33342 and DAPI share similar structural moieties (benzimidazole or indole with phenyl group), they exhibited totally different effects on the kinetics of γH2AX foci formation and disappearance, suggesting that the inhibition of DDR by DDRI-18 does not result from its direct binding to DNA.
In conclusion, we identified a chemical compound, DDRI-18, that dramatically potentiates the cytotoxicity of various DNA-damaging agents, including etoposide, camptothecin and bleomycin. Its chemosensitization activity appears to be due to inhibition of DNA damage repair and is accompanied by retardation of γH2AX foci resolution and loss of the DDR signalling pathway induced by DNA-damaging agents. However, the mechanism of action of DDRI-18, including its target of action, has yet to be elucidated.
Acknowledgments
We thank Ms Mi-ae Kim for technical support and Korea Chemical Bank for providing the chemical library. This work was supported by the National Cancer Center Grants 1110031 and 1110032.
Glossary
- CDDP
cisplatin
- CPT
camptothecin
- DDR
DNA damage response
- DDRI-18
DNA damage response inhibitor 18 (3,3′-(1H,3′H-5,5′-bibenzo[d]imidazole-2,2′-diyl)dianiline))
- DNA-PK
DNA-dependent protein kinase
- DSB
DNA double strand breaks
- DOX
doxorubicin
- ETO
etoposide
- 5-FU
5-fluorouracil
- MTT
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide
- NHEJ
non-homologous end-joining
- PS
phosphatidylserine
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 DDRI-18 did not affect etoposide-induced G2/M arrest. U2OS cells were pretreated with 2.5µM DDRI-18 for 1 h and then treated with 1 µM etoposide, followed by a 24 h incubation. After incubation, the cells were fixed and processed for propidium iodide staining, and DNA contents were analysed using FACSCaliber.
Figure S2 The effects of DDRI-18 on cytotoxicity of etoposide in various cell lines. Various cells were pretreated with 2 µM DDRI-18 for 1 h and then treated with etoposide at the indicated concentrations, followed by a 24 h incubation. After incubation, cell viability was measured using the MTT assay. Representative results from three independent experiments performed in triplicate are shown.
Figure S3 The effects of DDRI-18 and NU7026 are nearly additive to synergistic. U2OS cells were incubated for 1 h in medium containing inhibitors at the serial concentrations. Cells were then exposed to 10 µM etoposide for 1 h, followed by incubation in etoposide-free medium containing inhibitors for another 3 h. After incubation, the cells were fixed and processed for γH2AX immunofluorescence, and the γH2AX foci were analysed using an IN Cell Analyzer. The CI value for combinations of DDRI-18 and NU7026 was calculated using CalcuSyn software. (CI =0.9–1.1, additivity; CI < 0.9, synergy; CI > 1.1, antagonism. ED50, ED75 and ED90 means effective dose that results in 50%, 75% and 90% of inhibitory activity respectively).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Albert FG, Eckdahl TT, Fitzgerald DJ, Anderson JN. Heterogeneity in the actions of drugs that bind in the DNA minor groove. Biochemistry. 1999;38:10135–10146. doi: 10.1021/bi990382p. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst) 2004;3:781–796. doi: 10.1016/j.dnarep.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Cowell IG, Durkacz BW, Tilby MJ. Sensitization of breast carcinoma cells to ionizing radiation by small molecule inhibitors of DNA-dependent protein kinase and ataxia telangiectsia mutated. Biochem Pharmacol. 2005;71:13–20. doi: 10.1016/j.bcp.2005.09.029. [DOI] [PubMed] [Google Scholar]
- Deriano L, Guipaud O, Merle-Beral H, Binet JL, Ricoul M, Potocki-Veronese G, et al. Human chronic lymphocytic leukemia B cells can escape DNA damage-induced apoptosis through the nonhomologous end-joining DNA repair pathway. Blood. 2005;105:4776–4783. doi: 10.1182/blood-2004-07-2888. [DOI] [PubMed] [Google Scholar]
- Ding J, Miao ZH, Meng LH, Geng MY. Emerging cancer therapeutic opportunities target DNA-repair systems. Trends Pharmacol Sci. 2006;27:338–344. doi: 10.1016/j.tips.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- Hendrickson CL, Purkayastha S, Pastwa E, Neumann RD, Winters TA. Coincident in vitro analysis of DNA-PK-dependent and -independent nonhomologous end joining. J Nucleic Acids. 2010;2010:823917. doi: 10.4061/2010/823917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- Huen MS, Chen J. The DNA damage response pathways: at the crossroad of protein modifications. Cell Res. 2008;18:8–16. doi: 10.1038/cr.2007.109. [DOI] [PubMed] [Google Scholar]
- Ismail IH, Martensson S, Moshinsky D, Rice A, Tang C, Howlett A, et al. SU11752 inhibits the DNA-dependent protein kinase and DNA double-strand break repair resulting in ionizing radiation sensitization. Oncogene. 2004;23:873–882. doi: 10.1038/sj.onc.1207303. [DOI] [PubMed] [Google Scholar]
- Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117:1440–1449. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Jun DH, Kim HJ, Jeong KC, Lee CH. Development of a high-content screening method for chemicals modulating DNA damage response. J Biomol Screen. 2011;16:259–265. doi: 10.1177/1087057110392993. [DOI] [PubMed] [Google Scholar]
- Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev. 2009;8:547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- Lara PN, Jr, Mack PC, Synold T, Frankel P, Longmate J, Gumerlock PH, et al. The cyclin-dependent kinase inhibitor UCN-01 plus cisplatin in advanced solid tumors: a California cancer consortium phase I pharmacokinetic and molecular correlative trial. Clin Cancer Res. 2005;11:4444–4450. doi: 10.1158/1078-0432.CCR-04-2602. [DOI] [PubMed] [Google Scholar]
- Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- Lord CJ, Garrett MD, Ashworth A. Targeting the double-strand DNA break repair pathway as a therapeutic strategy. Clin Cancer Res. 2006;12:4463–4468. doi: 10.1158/1078-0432.CCR-06-1269. [DOI] [PubMed] [Google Scholar]
- Nakada S, Chen GI, Gingras AC, Durocher D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008;9:1019–1026. doi: 10.1038/embor.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noriko Hosoya KM. Clinical importance of DNA repair inhibitors in cancer therapy. Mag Eur Med Oncol. 2009;2:9–14. [Google Scholar]
- Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res. 2007;13:1383–1388. doi: 10.1158/1078-0432.CCR-06-2260. [DOI] [PubMed] [Google Scholar]
- Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Riesterer O, Matsumoto F, Wang L, Pickett J, Molkentine D, Giri U, et al. A novel Chk inhibitor, XL-844, increases human cancer cell radiosensitivity through promotion of mitotic catastrophe. Invest New Drugs. 2011;29:514–522. doi: 10.1007/s10637-009-9361-2. [DOI] [PubMed] [Google Scholar]
- Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA. 2003;100:5057–5062. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Vivian CJ, Lee KJ, Ge C, Morotomi-Yano K, Manzl C, et al. DNA-PKcs-PIDDosome: a nuclear caspase-2-activating complex with role in G2/M checkpoint maintenance. Cell. 2009;136:508–520. doi: 10.1016/j.cell.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tse AN, Carvajal R, Schwartz GK. Targeting checkpoint kinase 1 in cancer therapeutics. Clin Cancer Res. 2007;13:1955–1960. doi: 10.1158/1078-0432.CCR-06-2793. [DOI] [PubMed] [Google Scholar]
- Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 2003;63:6008–6015. [PubMed] [Google Scholar]
- Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA, et al. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood. 2004;103:4659–4665. doi: 10.1182/blood-2003-07-2527. [DOI] [PubMed] [Google Scholar]
- Yano K, Morotomi-Yano K, Wang SY, Uematsu N, Lee KJ, Asaithamby A, et al. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 2008;9:91–96. doi: 10.1038/sj.embor.7401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7:2955–2966. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, et al. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66:5354–5362. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.