

Abstract
BACKGROUND AND PURPOSE
Oral salmon calcitonin (sCT), a dual-action amylin and calcitonin receptor agonist, improved glucose homeostasis in diet-induced obese rats. Here, we have evaluated the anti-diabetic efficacy of oral sCT using parameters of glycaemic control and beta-cell morphology in male Zucker diabetic fatty (ZDF) rats, a model of type 2 diabetes.
EXPERIMENTAL APPROACH
Male ZDF rats were treated with oral sCT (0.5, 1.0 or 2 mg·kg−1) or oral vehicle twice daily from age 8 to 18 weeks. Zucker lean rats served as control group. Fasting and non-fasted blood glucose, glycosylated haemoglobin (HbA1c) and levels of pancreas and incretin hormones were determined. Oral glucose tolerance test and i.p. glucose tolerance test were compared, and beta-cell area and function were evaluated.
KEY RESULTS
Oral sCT treatment dose-dependently attenuated fasting and non-fasted hyperglycaemia during the intervention period. At the end of the study period, oral sCT treatment by dose decreased diabetic hyperglycaemia by ∼9 mM and reduced HbA1c levels by 1.7%. Furthermore, a pronounced reduction in glucose excursions was dose-dependently observed for oral sCT treatment during oral glucose tolerance test. In addition, oral sCT treatment sustained hyperinsulinaemia and attenuated hyperglucagonaemia and hypersecretion of total glucagon-like peptide-1 predominantly in the basal state. Lastly, oral sCT treatment dose-dependently improved pancreatic beta-cell function and beta-cell area at study end.
CONCLUSIONS AND IMPLICATIONS
Oral sCT attenuated diabetic hyperglycaemia in male ZDF rats by improving postprandial glycaemic control, exerting an insulinotropic and glucagonostatic action in the basal state and by preserving pancreatic beta-cell function and beta-cell area.
Keywords: anti-diabetic agent, hyperglycaemia, salmon calcitonin, type 2 diabetes, beta cells
Introduction
Type 2 diabetes is characterized by hyperglycaemia resulting from islet dysfunction, manifested as impaired insulin secretion in the presence of insulin resistance (Kahn, 2003). Furthermore, augmented glucagon secretion resulting in excessive hepatic glucose production markedly contributes to the pathophysiology of type 2 diabetes (Dunning and Gerich, 2007). Thus, anti-diabetic therapy should optimally reverse these defects and target insulin action, pancreatic function and hyperglucagonaemia, thereby attenuating the development of fasting and postprandial hyperglycaemia in patients with type 2 diabetes (Wajchenberg, 2007).
Several oral anti-diabetic agents are available to improve a hyperglycaemic state by increasing insulin secretion (e.g. sulfonylureas), improving insulin sensitivity (e.g. thiazolidinediones) and decreasing endogenous glucose production (e.g. metformin); however, adequate glycaemic control of type 2 diabetic patients is difficult to achieve. Low and deteriorating efficacy and/or risk of hypoglycaemia, cardiac failure, weight gain and bone fractures are limiting factors associated with current treatments (Granberry et al., 2007; Amiel et al., 2008; Krentz et al., 2008). Novel promising hormone-based therapies resembling an endogenous mode of action, such as glucagon-like peptide (GLP)-1 and amylin analogues, are now emerging. However, it is still unclear whether these approaches possess intrinsic abilities to improve beta-cell health (Krentz et al., 2008).
Our group recently showed that an oral form of salmon calcitonin (sCT), a known amylin receptor agonist, possessed glucoregulatory abilities in diet-induced obese rats, a response that partially mimicked the effects of amylin, but in addition also provided direct effects on blood glucose and insulin levels, with an indication of an improvement in beta-cell function and insulin action (Feigh et al., 2011). While it was observed that oral sCT resembled well-described amylin actions as regards energy homeostasis by influencing food intake and energy expenditure thereby regulating body weight (Lutz et al., 2000), the direct effect on glucose homeostasis could not be solely explained by an amylin-like inhibitory effect on gastric emptying (Young et al., 1995) and glucagonostatic action (Gedulin et al., 1997).
One possible explanation for the glucoregulatory capacity of sCT is its dual agonist effect on both the amylin and the calcitonin receptor (Purdue et al., 2002), in contrast to amylin only binding to and activating the amylin receptor (Tilakaratne et al., 2000). Another important distinct feature is the novel oral delivery system that mimics the physiological pathway of endogenous secretion, where the highest concentrations are found in the splanchnic and portal circulation as seen for other hormones involved in the regulation of appetite and glucose homeostasis (Malkov et al., 2005; Steinert et al., 2009).
Based on the above findings, we hypothesized that oral sCT could attenuate the development of diabetic hyperglycaemia in an animal model of progressive type 2 diabetes, the male Zucker diabetic fatty (ZDF) rat. The hyperglycaemic, hyperinsulinaemic and hyperglucagonaemic phenotypes observed in male ZDF rats resemble those commonly observed in patients with type 2 diabetes (Bergman et al., 2002; Dunning and Gerich, 2007; Kahn et al., 2009). Furthermore, the progressive and irreversibly beta-cell deterioration and failure observed in male ZDF rats (Etgen and Oldham, 2000) makes this animal model highly suitable for the evaluation of therapeutic interventions as regards pancreatic preservation and/or degeneration in type 2 diabetes.
Oral sCT treatment in the ZDF rat model has not yet been examined; therefore, to test our hypothesis, we characterized the effect of multiple doses of oral sCT, compared to their vehicle (5-CNAC), with respect to glucose homeostasis in ZDF rats. To test the hypothesis that oral sCT could delay or attenuate progression of type 2 diabetes in ZDF rats, treatment was initiated during the ‘pre-diabetes’ phase and continued into the beta-cell deterioration stage. Further, to elucidate the mode of action of oral sCT, we explored postprandial glycaemia during glucose tolerance testing and investigated the involvement of pancreas and incretin glucoregulatory hormones in mediating glycaemic control. Finally, we examined pancreatic beta-cell morphology as regards chronic treatment.
Methods
sCT therapy
For oral delivery of sCT, the carrier agent 5-CNAC (N-(5-chlorosalicyloyl)-8-aminocaprylic acid) was obtained from Biomics Biotechnologies Co., Ltd. (Nantong, China) and prepared by mixing recombinant sCT at 0.5, 1 or 2 mg·kg−1 (Chinese Peptide Company, Zhejiang, China) with 150 mg·kg−1 of 5-CNAC. Vehicle-treated rats were given 5-CNAC only. All animals were dosed by oral gavage in a volume of 5 mL·kg−1 body weight twice daily.
Experimental animals
All studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010;McGrath et al., 2010). The experiment was approved by the Experimental Animal Committee of the Danish Ministry of Justice and was performed according to the European Standard for Good Clinical Practice (2008/561–1450). Male ZDF (fa/fa) rats and male Zucker (+/+; ZL) lean rats were obtained at the age of 7 weeks from Charles River Laboratories (Kisslegg, Deutschland). All animals were housed at the animal facility at Nordic Bioscience with a constant temperature (21–23°C) and relative humidity (55–65%) on a 12 h light/dark cycle with free access to Purina 5008 rat chow (Brogaarden, Lynge, Denmark) and tap water.
At the age of 8 weeks, the animals were randomized into five groups based upon fasting blood glucose and body weight (Table 1). The doses of oral sCT were based on previous experiments (Feigh et al., 2011). Animals were treated for a total of 10 weeks, until 18 weeks of age. Body weight and food consumption were measured weekly.
Table 1.
Baseline characteristics in ZDF rats and ZL rats
| ZL | ZDF | ZDF | ZDF | ZDF | |
|---|---|---|---|---|---|
| Control | Vehicle | sCT | sCT | sCT | |
| 0.5 mg·kg-1 | 1 mg·kg-1 | 2 mg·kg-1 | |||
| n | 10 | 12 | 12 | 12 | 12 |
| Body weight (g) | 210.2 ± 4.1 | 278.0 ± 6.2*** | 279.1 ± 5.3*** | 277.0 ± 3.9*** | 273.4 ± 6.8*** |
| FBG (mM) | 4.8 ± 0.4 | 6.7 ± 0.8* | 6.7 ± 0.6* | 6.6 ± 0.6* | 6.6 ± 0.5* |
| Non-fasted BG (mM) | 5.7 ± 0.1 | 19.3 ± 1.5*** | 18.2 ± 1.5*** | 19.0 ± 1.4*** | 18.0 ± 0.9*** |
| FPIns (ng mL−1) | 0.43 ± 0.02 | 4.57 ± 0.37*** | 4.49 ± 0.16*** | 4.77 ± 0.45*** | 4.76 ± 0.30*** |
| FPGlu (ng mL−1) | 0.31 ± 0.00 | 0.24 ± 0.01*** | 0.26 ± 0.01*** | 0.25 ± 0.01*** | 0.26 ± 0.01*** |
*P < 0.05, ***P < 0.001 versus ZL-Control.
Data are means ± SEM.
FBG, fasting blood glucose; FPIns, fasting plasma insulin; FPGlu, fasting plasma glucagon; Non-fasted BG, non-fasted blood glucose.
Fasting and non-fasted glucose
Blood was collected from the tail vein at baseline and before morning drug administration once weekly during the study period. Whole blood glucose levels were determined with an ACCU-CHEK® Avia blood glucose meter (Roche Diagnostics, Rotkreuz, Switzerland).
Fasting insulin, glucagon, GLP-1 and GIP
At baseline and after 4 and 9 weeks of treatment, plasma insulin and glucagon levels were analysed using the Mercodia® Rat Insulin ELISA kit (Mercodia AB, Uppsala, Sweden) and Rat Glucagon (1–29) EIA kit (Bachem, Weil am Rhein, Germany), respectively. After 3 and 8 weeks of treatment, plasma total GLP-1 and total GIP levels were analysed using GLP-1 Total elisa and GIP Total elisa kit (Millipore, Billerica, MA) respectively.
Glucose tolerance testing and beta-cell function
After 4 weeks of treatment, the four treatment groups were subjected to an OGTT, and after 5 weeks, an IPGTT was performed. After an overnight fast of approximately 16 h and 15 min after the drug administration, rats received glucose by p.o. gavage (1 g·kg−1) (OGTT) or by i.p. injection (1 g·kg−1) (IPGTT), respectively. Blood samples were collected from the tail vein before drug administration (-15 min) and glucose challenge (0 min) and 30, 60, 120 and 240 min post challenge.
A beta-cell function index was calculated as the ratio of fasting insulin/fasting glucose (Topp et al., 2007). This ratio was calculated from the fasting plasma insulin and blood glucose values measured at baseline and after 9 weeks of treatment.
Tissue collection
During the tenth week of treatment, rats were given their morning dose and fasted for 5 h, and subsequently, whole blood glucose were determined as described above. HbA1c levels were determined using the DCA Vantage™ Analyzer (Siemens Healthcare Diagnostics, Deerfield, IL). The rats were then killed by cervical dislocation, and the pancreas was removed en bloc and placed in 4% formaldehyde for immunohistochemical analysis.
Immunohistochemistry
After an overnight fixation, tissue samples were washed and subsequently dissected of surrounding tissue, dehydrated and embedded in paraffin. The paraffin sections (5 µm in thickness) were dried on slides overnight at 37°C and were deparaffinized and rehydrated at room temperature. For antigen retrieval, the sections were placed in 10 mM citrate buffer (pH 6) and heated for 2 × 5 min at 800 W in a microwave oven to stain for insulin, and the endogenous peroxidase was then blocked by 1% H2O2 in 99% ethanol followed by blocking in 10% rabbit normal serum. Pancreatic beta-cells were stained using polyclonal guinea-pig anti-insulin (Dako A0564, Dako Denmark A/S, Glostrup, Denmark) followed by HRP-coupled polyclonal rabbit anti-guinea pig (Dako P0141, Dako Denmark A/S) and developed in 3-amino-9-ethyl-carbazole (AEC) (A5754) (Sigma-Aldrich Denmark A/S, Broendby, Denmark). Finally, slides were counterstained with Mayer's haematoxylin and mounted with Kayser Mounting Media.
Morphometric analysis
For the determination of the fractional beta-cell area, the entire pancreatic section was imaged at ×4 magnification using a Nikon Eclipse 80i microscope (Nikon Instruments Inc., Melville, NY) equipped with a PixeLINK camera (PixeLINK, Ottawa, Canada). Insulin-stained sections, derived from three to five levels of the embedded pancreatic tissue, were chosen for evaluation; and a composite image of each individual field was generated in the new CAST system (Visiopharm, Hoersholm, Denmark) to control the stage and data collection. The pancreas section stained positive for insulin was quantified as fractional beta-cell area per total pancreatic section, and subsequently, the mean for each individual animal was calculated. All analyses were performed in a blinded fashion.
Statistical analysis
All data are presented as mean ± SEM. The statistical analysis of drug effects versus vehicle effects was done by one-way anova followed by the Dunnett's post hoc test. Student's t-test was used to compare lean control group and vehicle. All analyses were carried out using GRAPHPAD PRISM software (GraphPad Prism, San Diego, CA). A value of P < 0.05 was considered to be significant.
Results
Effects of oral sCT treatment on body weight and food consumption during the study period
At the start of the study, body weight was similar in the four randomized obese ZDF groups, although body weight was significantly increased compared with ZL-Control rats (Table 1).
As expected, cumulative food intake and body weight were significantly increased in ZDF groups compared with ZL-Control littermates throughout the study period (Table 2), confirming the hyperphagic and obese state of male ZDF rats (Tokuyama et al., 1995). Interestingly, body weight gain tended to be more pronounced in the oral sCT treated rats by dose, with the ZDF-sCT 2 mg·kg−1 group being significantly heavier (P < 0.05) than the ZDF-Vehicle rats at study end. In contrast, a trend towards reduction in cumulative food intake was observed in oral sCT treated rats, especially during the last part of the study period, when compared with the ZDF-Vehicle group (Table 2).
Table 2.
Effects of 10 weeks of oral sCT treatment on body weight and food intake in ZDF rats
| ZL | ZDF | ZDF | ZDF | ZDF | |
|---|---|---|---|---|---|
| Control | Vehicle | sCT | sCT | sCT | |
| 0.5 mg·kg-1 | 1 mg·kg-1 | 2 mg·kg-1 | |||
| n | 10 | 12 | 12 | 12 | 12 |
| Body weight (g) | |||||
| After 5 weeks | 326.3 ± 4.4 | 387.2 ± 7.7### | 393.7 ± 9.6 | 398.6 ± 14.3 | 413.3 ± 8.4 |
| Study end | 372.5 ± 5.0 | 411.4 ± 9.0## | 432.9 ± 12.9 | 442.7 ± 16.2 | 461.0 ± 12.4# |
| Cumulative food intake (g per animal) | |||||
| After 5 weeks | 566.9 ± 21.6 | 1079.0 ± 31.5### | 1032.0 ± 24.7 | 1004.0 ± 19.8 | 1026.0 ± 25.9 |
| Study end | 1099.8 ± 29.0 | 2057.2 ± 50.0### | 1884.4 ± 40.7 | 1838.7 ± 39.1 | 1864.2 ± 43.2 |
##P < 0.01, ###P < 0.001 versus ZL-Control; #P < 0.05 versus ZDF-Vehicle.
Data are means ± SEM.
Effects of oral sCT treatment on glycaemic control during the study period
Pre-intervention blood glucose levels were similar among the four pre-diabetic ZDF groups but were significantly elevated compared with their ZL-Control counterparts both in a fasting (P < 0.05) and non-fasted (P < 0.001) state (Table 1). As expected, glycaemic control was progressively deteriorated in the ZDF-Vehicle rats during the study period, and a pronounced diabetic hyperglycaemic state was observed from week five of intervention (at 13 weeks of age) (Figure 1A,B). In contrast, fasting and non-fasted blood glucose levels were almost unchanged in oral ZDF-sCT treated rats at 1 and 2 mg·kg−1 doses during the first 5 weeks of treatment and remained significantly attenuated compared with the ZDF-Vehicle group for the duration of the study (Figure 1A,B). At termination following a 5 h fast, the ZDF-Vehicle rats demonstrated severe hyperglycaemia (plasma glucose level >27 mmol·L−1); oral sCT dose-dependently and significantly attenuated hyperglycaemia, resulting in approximately 9 mmol·L−1 reductions in plasma glucose levels in rats treated with the 1 and 2 mg·kg−1 doses (P < 0.01) (Figure 1C). Furthermore, compared with ZDF-Vehicle rats, oral sCT treatment dose-dependently and significantly reduced glycosylated haemoglobin (HbA1c) levels after 5 weeks with a maximum effect of ∼1.7% observed in ZDF rats given 2 mg·kg−1 compared with ZDF-vehicle (P < 0.001) (data not shown). This reduction in HbA1C levels was sustained throughout the intervention period, with a maximum reducing effect of ∼1.7% at study end for ZDF rats given oral sCT at 1 and 2 mg·kg−1 (P < 0.01) (Figure 1D).
Figure 1.
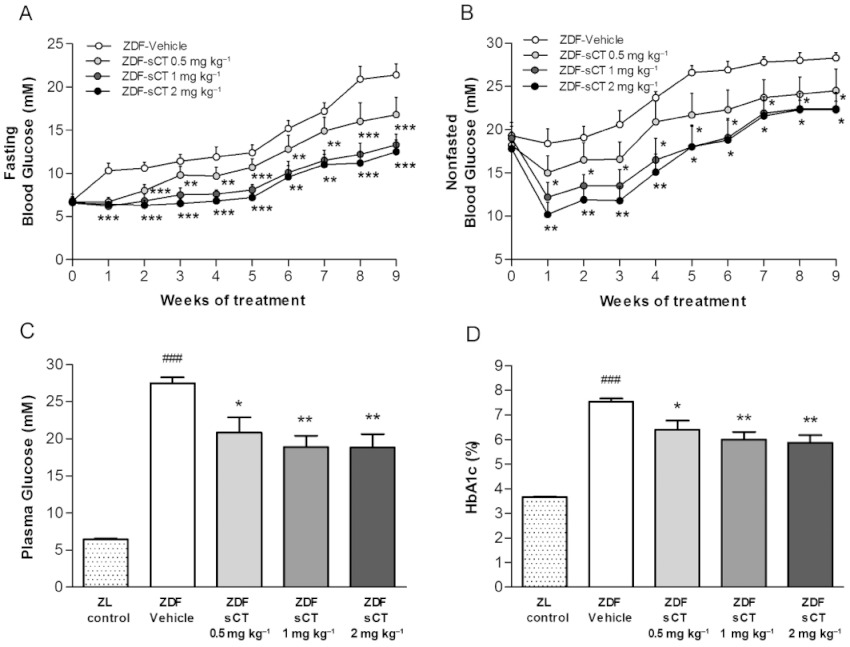
Effects of oral sCT treatment on glycaemic control in ZDF rats. (A) Fasting blood glucose levels during the study period. (B) Non-fasted blood glucose levels during the study period. (C) Plasma glucose levels at study end (5 h fasting). (D) HbA1c at study end. Data are presented as means ± SEM (n= 12 for ZDF rats, n= 10 for ZL-Control rats). ZL rats were used as normal control group. *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from ZDF-Vehicle group. ###P < 0.001 significantly different from ZL-Control group.
Effects of oral sCT treatment on pancreatic glucoregulatory hormones during the study period
At study start, fasting levels of plasma insulin and glucagon were similar between ZDF intervention groups. The pre-diabetic hyperinsulinaemic state in the ZDF rats was clearly demonstrated by significantly elevated fasting plasma insulin compared with ZL-Control rats (P < 0.001) (Table 1). In contrast, compared with the ZL-Control group, fasting plasma glucagon was significantly (P < 0.001) reduced in ZDF rats, possibly as a result of hyperinsulinaemic intra-islet paracrine suppression (Kawamori and Kulkarni, 2009). During the intervention period, a marked reduction in fasting plasma insulin from baseline (approximately 65%) was observed in ZDF-Vehicle rats, confirming a deterioration of the pancreatic beta-cell secretory function as these animals developed severe diabetes (Paulsen et al., 2010). In contrast, hyperinsulinaemia was dose-dependently sustained in the ZDF-sCT-treated groups with plasma insulin levels being significantly increased for sCT 1 mg·kg−1 (P < 0.01) and sCT 2 mg·kg−1 (P < 0.001) treated rats when compared with ZDF-Vehicle group at study end (Figure 2A). In contrast, the fasting plasma glucagon concentration was markedly increased (∼2.5-fold) in the ZDF-Vehicle group during the study period, indicating a defect in pancreatic alpha-cell function resulting in hyperglucagonaemia (Dunning and Gerich, 2007). The oral sCT treatment dose-dependently attenuated this hyperglucagonaemia throughout the study period, with plasma levels of glucagon being significantly reduced for sCT (1 and 2 mg·kg−1) treated groups compared with ZDF-Vehicle rats (P < 0.05) at study end (Figure 2B).
Figure 2.
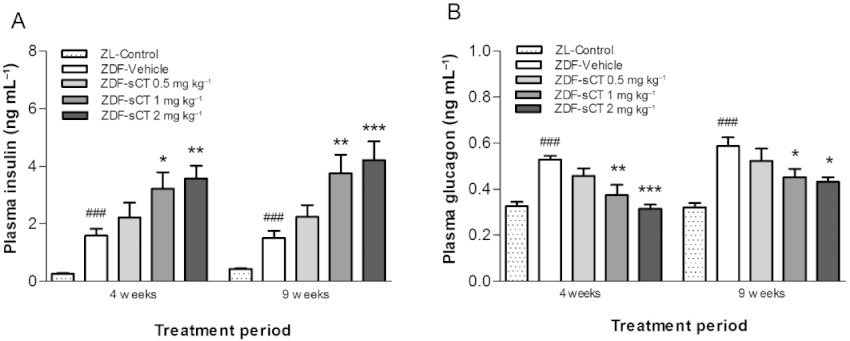
Effects of oral sCT treatment on pancreas glucoregulatory hormones in ZDF rats. (A) Fasting plasma insulin levels after 4 and 9 weeks of treatment. (B) Fasting plasma glucagon levels after 4 and 9 weeks of treatment. Data are presented as means ± SEM (n= 10–12 for ZDF rats, n= 8–10 for ZL-Control rats). ZL rats were used as normal control group. ###P < 0.001 significantly different from ZL-Control group. *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from ZDF-Vehicle group.
Effects of oral sCT treatment on incretin glucoregulatory hormones during the study period
After 3 and 8 weeks of intervention, ZDF-Vehicle rats exhibited a pronounced hypersecretion of incretin hormones with plasma total GLP-1 and GIP levels being significantly increased (P < 0.001) compared with their lean control littermates (Figure 3). Interestingly, plasma total GLP-1 concentration was dose-dependently decreased by oral sCT treatment during the intervention period with a significant reduction observed for sCT 1 mg·kg−1 (P < 0.05) and sCT 2 mg·kg−1 (P < 0.01) treated rats compared with ZDF-Vehicle group at study end (Figure 3A). In contrast, compared with ZDF-Vehicle, oral sCT treatment did not influence plasma levels of total GIP (Figure 3B).
Figure 3.
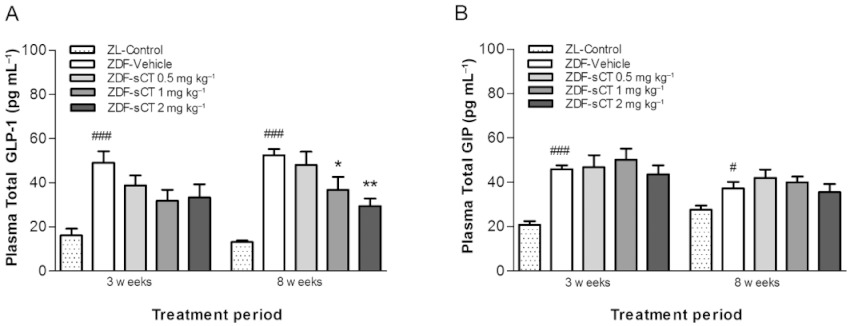
Effects of oral sCT treatment on incretin glucoregulatory hormones in ZDF rats. (A) Fasting plasma total GLP-1 level after 3 and 8 weeks of treatment. (B) Fasting plasma total GIP level after 3 and 8 weeks of treatment. Data are presented as means ± SEM (n= 8 for ZDF rats, n= 6 for ZL-Control rats). ZL rats were used as normal control group. #P < 0.05, ###P < 0.001 significantly different from ZL-Control group. *P < 0.05, **P < 0.01 significantly different from ZDF-Vehicle group.
Effects of oral sCT treatment on acute glycaemic control during glucose tolerance tests
As expected, ZDF-Vehicle rats showed overt glucose intolerance during both OGTT (Figure 4A) and IPGTT (Figure 4B). Thirty minutes after glucose administration, glycaemia was ∼26 mM in ZDF-Vehicle rats, which, in contrast, was dose-dependently and significantly reduced by 83% (P < 0.001) and 26% (P < 0.01) in oral sCT 2 mg·kg−1 treated rats during OGTT and IPGTT, respectively. Furthermore, throughout the observation period glycaemia during oral and i.p. glucose tolerance testing was dose-dependently and significantly reduced by oral sCT (Figure 4A,B). Calculation of total AUCs (0–240 min) demonstrated a dose-dependent reduction by 50% (P < 0.001) (Figure 4C) and 30% (P < 0.01) (Figure 4D) during OGTT and IPGTT, respectively, for the highest doses of oral sCT given compared with the ZDF-Vehicle group. However, when blood glucose excursion was determined over the first 30 min after glucose loading as AUC of change from baseline, oral sCT significantly and dose-dependently reduced this plasma glucose excursion, by approximately 70% in oral sCT 2 mg·kg−1 treated rats compared with ZDF-Vehicle (P < 0.001) during the OGTT (Figure 4E), albeit no change was observed during IPGTT (Figure 4F).
Figure 4.
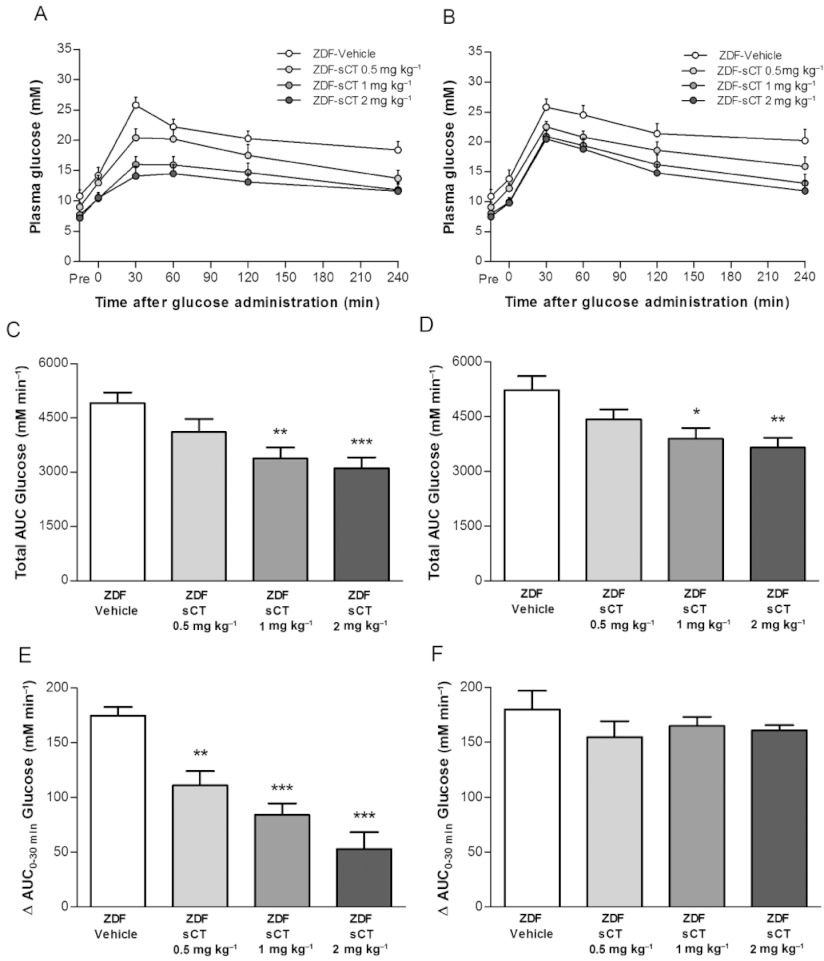
Effects of oral sCT treatment on acute glycaemic control after OGTT and IPGTT in ZDF rats. (A) Plasma glucose levels during an OGTT after 4 weeks of treatment. (B) Plasma glucose levels during an IPGTT after 5 weeks of treatment. (C) Total AUCs (0–240 min) calculated from the OGTT data for each group of rats. (D) Total AUCs (0–240 min) calculated from the IPGTT data for each group of rats. (E) AUC of change for plasma glucose excursions calculated during the first 30 min of the OGTT. (F) AUC of change for plasma glucose excursions calculated during the first 30 min of the IPGTT. Data are presented as means ± SEM (n= 10–12 per ZDF group). *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from ZDF-Vehicle group.
Effects of oral sCT treatment on pancreatic and incretin glucoregulatory hormones during OGTT
Interestingly, acute pre-glucose administration of oral sCT dose-dependently increased the plasma insulin level compared with ZDF-Vehicle group (P < 0.05 for ZDF-sCT 1 and 2 mg·kg−1) (Figure 5A). However, no further glucose-stimulated insulin secretion was observed during the 4 h observation period in oral sCT-treated rats; and, in contrast, an acute decline in plasma insulin was observed during the initial 30 min post-glucose dosing when compared with ZDF-Vehicle. In contrast, acute pre-glucose dosing with oral sCT dose-dependently decreased plasma glucagon levels compared with ZDF-Vehicle (P < 0.001 for ZDF-sCT 1 and 2 mg·kg−1) (Figure 5B). However, although absolute plasma insulin and glucagon concentrations were increased and suppressed, respectively, by oral sCT during the OGTT when compared with ZDF-Vehicle (Figure 5A,B), no significant difference in AUC of change was observed between the intervention groups (data not shown). To further explore the basal insulin secretion and glucagon suppression in response to acute oral sCT administration, pre-glucose dosing levels of incretin hormones GLP-1 and GIP were analysed on the OGTT day. Interestingly, plasma total GLP-1 was significantly decreased in ZDF-sCT 2 mg·kg−1 treated rats compared with ZDF-Vehicle (P < 0.01) (Figure 5C), but no pre-glucose dosing effect of oral sCT was observed on plasma total GIP (Figure 5D).
Figure 5.
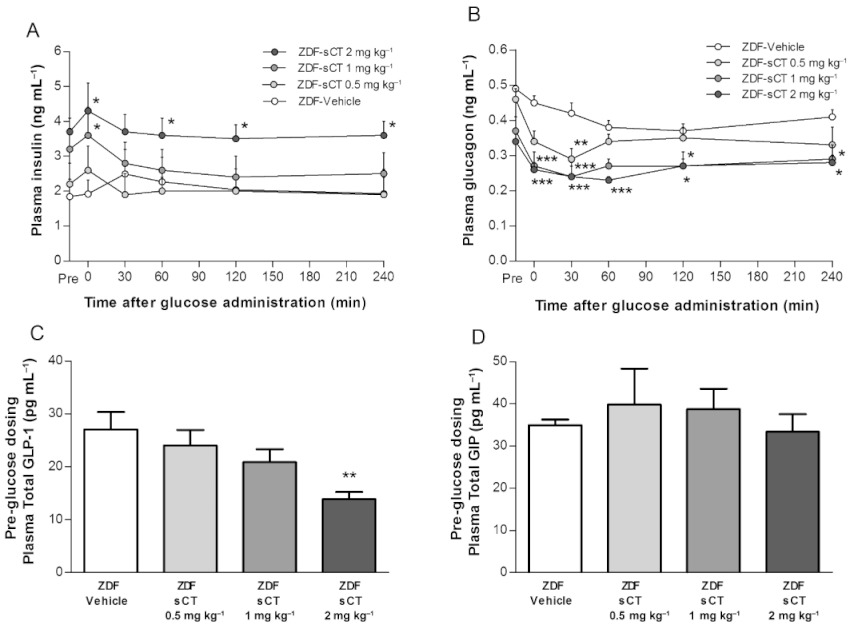
Effects of oral sCT treatment on pancreas and incretin glucoregulatory hormones after OGTT in ZDF rats. (A) Plasma insulin levels during OGTT administered after 4 weeks of treatment. (B) Plasma glucagon levels during OGTT administered after 4 weeks of treatment. (C) Pre-glucose dosing (time point 0) level of plasma total GLP-1 on OGTT day. (D) Pre-glucose dosing (time point 0) level of plasma total GIP on OGTT day. Data are presented as mean ± SEM (n= 8–10 per ZDF group). *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from ZDF-Vehicle group.
Effects of oral sCT treatment on pancreatic beta-cell area and function at study end
At 18 weeks of age, pancreatic islet structure in ZDF-Vehicle rats was highly irregular in shape and size compared with ZL-Control (Figure 6A,B), thus confirming the diabetic phenotype of the ZDF rat with deterioration of normal islet morphology (Paulsen et al., 2010). In contrast, rats treated with oral sCT 2 mg·kg−1 resulted in a more regular islet shape, although islet size was markedly increased compared with ZL-Control (Figure 6C). Immunohistochemistry for insulin demonstrated severe degranulation of beta cells in ZDF-Vehicle rats compared with ZL-Control rats (Figure 6A,B). Insulin-positive staining area, which reflects beta-cell area, was also significantly reduced by approximately 65% (P < 0.001) in the ZDF-Vehicle group versus ZL-Control group (Figure 6D). In contrast, oral sCT treatment dose-dependently demonstrated extensive insulin staining of beta cells (Figure 6C), with beta-cell area in the sCT 2 mg·kg−1 treated rats being comparable with that observed in ZL-Control rats (NS by one-way anova between intervention-groups, P < 0.05 by unpaired t-test for ZDF-sCT 2 mg·kg−1 compared with ZDF-Vehicle) (Figure 6D). A beta-cell function index was calculated as the ratio of fasting insulin/fasting glucose. Compared with baseline values, this index was markedly reduced (∼13-fold) at study end in ZDF-Vehicle rats (P < 0.05 vs. ZL-Control). In contrast, although beta-cell function index was reduced from baseline in oral sCT-treated rats, sCT dose-dependently and significantly increased this index (P < 0.05 for ZDF-sCT 1 mg·kg−1 and P < 0.01 for ZDF-sCT 2 mg·kg−1) at study end when compared with ZDF-Vehicle (Figure 6E).
Figure 6.
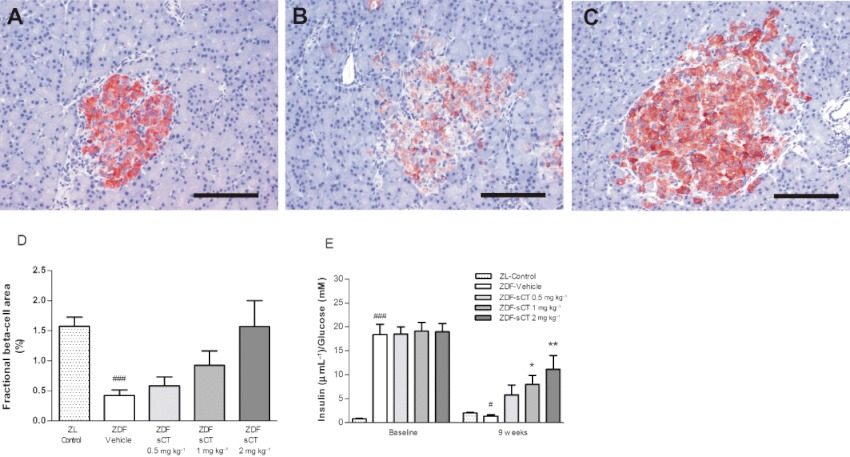
Effects of oral sCT treatment on pancreatic beta-cell area and function in ZDF rats. Representative insulin-stained images for (A) ZL-Control, (B) ZDF-Vehicle and (C) ZDF-sCT 2 mg·kg−1 treated rats are shown. (D) Fractional beta-cell area as % of total pancreatic area. (E) Beta-cell function index (fasting insulin/fasting glucose) at baseline and at study end. Data are presented as means ± SEM (n= 10–12 for ZDF rats, n= 10 for ZL-Control rats). #P < 0.05, ###P < 0.001 significantly different from ZL-Control group. *P < 0.05, **P < 0.01 significantly different from ZDF-Vehicle group. Scale bar = 200 µm.
Discussion
In the present study, male ZDF rats, an animal model of type 2 diabetes, were used to evaluate the effects of oral sCT treatment on glycaemic control, pancreatic and incretin hormonal profiles and pancreatic beta-cell morphology; we extended previous studies on the beneficial influence of oral sCT on glucose homeostasis in lean healthy and diet-induced obese rats (Feigh et al., 2011; 2012). We demonstrated that oral sCT dose-dependently attenuates the development of hyperglycaemia in severely diabetic ZDF rats as observed by reductions in fasting and non-fasted blood glucose and HbA1C. In addition, oral sCT, independently of incretin hormones, induced an insulinotropic and glucagonostatic action in the basal state. Furthermore, oral sCT treatment markedly improved postprandial glycaemic control during acute glucose tolerance testing. Lastly, oral sCT treatment preserved pancreatic beta-cell area and function relative to vehicle control.
Establishing an oral delivery system for gut-derived peptide drugs involved in the regulation of appetite and glucose homeostasis is of great importance to mimic the physiological pathway of endogenous secretion, where the highest concentrations are found in the splanchnic and portal circulation (Malkov et al., 2005; Steinert et al., 2010). The present novel findings introduce an approach for oral delivery of peptides resembling the action of pancreatic-derived hormones in type 2 diabetes, with focus on the amylin-like effects of sCT.
Several clinical studies in type 2 diabetic patients have demonstrated that the amylin-analogue pramlintide reduces the postprandial glucose excursion by delaying an exaggerated gastric emptying rate and reducing hyperglucagonaemia. However, pramlintide improves glucose homeostasis and HbA1C only when used as an adjunct to insulin therapy (Fineman et al., 2002; Hollander et al., 2003a; 2003b; Maggs et al., 2004).
In the present study, oral sCT treatment exerted amylin-like effects by inducing a pronounced reduction in postprandial glycaemia during OGTT in ZDF rats, as also recently demonstrated in lean healthy and diet-induced obese rats (Feigh et al., 2011; 2012). Although the effects of oral sCT upon gastric emptying were not measured directly, the pronounced reduction in glucose excursion during the initial phase following glucose administration during OGTT, but not IPGTT, do suggest a mode of action involving glucose absorption from the GI tract. In support of this, we and others observed a delaying effect of sCT on gastric emptying in lean healthy rats (Reidelberger et al., 2002; Feigh et al., 2011). Furthermore, in line with previous observations in diet-induced obese rats (Feigh et al., 2011), a marked suppression of hyperglucagonaemia was observed in the present study, which would thereby reduce hepatic glucose production and postprandial hyperglycaemia (Dunning and Gerich, 2007). Thus, as for amylin agonism (Mack et al., 2011), oral sCT treatment improved postprandial glycaemic control by delaying gastric emptying and exerting a glucagonostatic action.
The primary target of action of amylin agonism appears to be mediated by a central mechanism involving the area postrema with amylin binding sites present on the vagal nuclei in the brain (Lutz et al., 1998; Potes et al., 2010). Alternatively, the amylin-like effect of oral sCT upon gastric emptying and hyperglucagonaemia could be mediated through the incretin hormone GLP-1 (Holst et al., 2008). However, we observed that oral sCT treatment reduced plasma levels of total GLP-1 throughout the study period. Thus, as regards the amylin-like effects of oral sCT, the likely site of action is through centrally located amylin receptors.
Importantly, in contrast to amylin, sCT is known to elicit a dual agonist effect, as it binds to and activates both the calcitonin receptor (CT receptor) and the amylin receptor (AMY receptor), and, interestingly, with stronger binding affinity than amylin itself. Thus, by binding to and activating both the CT receptor and the AMY receptor, sCT elicits responses in multiple organs (Hay et al., 2004; 2005), thus providing one potential explanation for the potent ability of sCT to improve glucose metabolism.
A novel finding in the present study is the insulinotropic action exerted by oral sCT in the basal state, thereby markedly improving glucose homeostasis. As mentioned above, the present data do not indicate an incretin effect mediating this sustained hyperinsulinaemia in ZDF rats (Kiec-Klimczak et al., 2011), as oral sCT treatment did not enhance levels of GLP-1 and GIP. In contrast, oral sCT, by following the endogenous physiological pathway and thereby resulting in higher drug concentrations in the splanchnic circulation could cause pronounced exposure to receptors and nerves located in the pancreatic and portal area. In support of this hypothesis, stimulation of hepatic vagal afferents resulting in hypothalamic mediated insulin secretion has been demonstrated to improve hyperglycaemia in ZDF rats (Chen et al., 2010). Additionally, in preliminary experiments, the CT receptor was localized by immunohistochemistry as being dispersed throughout the pancreas islet including beta cells (unpublished data). However, sCT exposure in isolated islets from lean healthy rats, although inducing a cAMP response, did not potentiate glucose-stimulated insulin secretion (unpublished data). Thus, we speculate, that oral sCT could function as a direct and/or indirect insulinotropic peptide and thereby improve glucose homeostasis in severely diabetic animals, although this awaits further investigations with a more mechanistic approach.
The hyperphagia of ZDF rats is a result of dysfunctional leptin signalling as well as caloric wasting because of marked glucosuria (Etgen and Oldham, 2000). Surprisingly, in the present study, we did not observe a reduction in body weight, despite a trend towards a reduction in food intake, which contrasts with our previous findings in diet-induced obese rats (Feigh et al., 2011) and with the well-described anorectic action typically observed for sCT and amylin (Roth et al., 2006; 2008; Chelikani et al., 2007). Interestingly, the effects of amylin and sCT as regards energy balance are known to be attenuated in leptin deficient systems, such as ob/ob mice, ZDF and fa/fa rats (Eiden et al., 2002; Trevaskis et al., 2010). In contrast, in diet-induced obesity-related leptin resistance, an amylin-mediated restoration of leptin responsiveness has been extensively demonstrated in rodent models (Trevaskis et al., 2008; 2010; Turek et al., 2010). From a therapeutic perspective, only very few humans worldwide present with genetically based leptin deficiency, whereas the majority of obese people presents with leptin resistance; thus, combined leptin/amylin agonism has now gone through clinical trials as a potential approach to obesity drug development (Chan et al., 2009). A thorough study in ob/ob and db/db mice, representing leptin deficiency versus leptin receptor defect, respectively, would be required to fully interpret the lack of effect on body weight during oral sCT treatment. Additionally, the positive effect on energy balance despite a decrease in food intake could be due to a reduction in glucosuria because of marked improvement in glycaemic control, as also previously observed in severely diabetic ZDF rats (Larsen et al., 2008).
Of note, although oral sCT treatment markedly improved glycaemic control, irrespective of inducing weight loss, the impressive glucoregulatory effect lessened towards the end of the study period. To our knowledge, only very few studies have investigated anti-diabetic efficacy of monotherapeutic interventions in severely diabetic ZDF rats. At 18 weeks of age, the vehicle-treated rats demonstrated a pronounced diabetic hyperglycaemic state with pancreatic islet destruction. Thus, in analogy to human type 2 diabetes (Kruger et al., 2011), we speculate that combination therapy could be a prerequisite for sustained glycaemic control in late-stage diabetic ZDF rats. In support of this theory, the GLP-1 analogue liraglutide improved glycaemic control only in combination therapy with the insulin sensitizer pioglitazone in severely diabetic ZDF rats, although the glucoregulatory efficacy, in line with the present study, lessened towards the end of the study period (∼17 weeks of age) (Larsen et al., 2008). Thus, an investigation into the anti-diabetic efficacy of oral sCT in combination therapy is warranted. Furthermore, we speculate that the lack of effect of oral sCT treatment on weight loss in the ZDF rat, possibly due to leptin deficient signalling, could influence the chronic anti-hyperglycaemic efficacy. It is well known that a reduction in caloric consumption in type 2 diabetic patients by pharmacological intervention is accompanied by improved glycaemic control (Rowe et al., 2005; Scheen et al., 2006), and supporting this, oral sCT treatment induced a pronounced vehicle-corrected weight loss (∼15%) and markedly improved obesity-related insulin resistance and sustained glycaemic control in leptin-resistant DIO rats (Feigh et al., 2011).
To our knowledge this is the first study investigating the effect of amylin agonists on pancreatic beta-cell function and area in a severely diabetic animal model, and these data strongly support the notion that oral sCT possesses a potent ability to prevent beta-cell degradation and preserve secretory function. Interestingly, amylin treatment does not protect against beta-cell loss (Edelman et al., 2008; Trevaskis et al., 2010), which could indicate that the effect of sCT is primarily mediated through the calcitonin receptor, rather than the amylin receptor. Alternatively, the oral route of administration allows the sCT to have an effect on the pancreas; however, this requires further studies.
In conclusion, these data for the first time show that in severely diabetic ZDF rats, (i) oral sCT attenuates the development of diabetic hyperglycaemia, (ii) oral sCT improves postprandial glycaemic control through delaying gastric emptying, (iii) oral sCT exerts a basal state insulinotropic and glucagonostatic action and (iv) oral sCT induces a pronounced improvement in beta-cell health and function. The oral formulation of sCT thus appears to possess insulin-like effects and our results highlight the importance of following the physiological pathway of endogenous secretion when attempting to mimic the action of gut hormones.
Acknowledgments
This study received funding from the Danish Research Foundation (Den Danske Forskningsfond) and the Danish Ministry of Science, Technology and Innovation.
Author contributions: MF researched data, contributed to discussion and wrote the manuscript.
KVA, AVNW, STP, ACB-J and CH researched data. JEH and HB-N contributed to data interpretation and discussion. KH, CC and MAK contributed to discussion and reviewed and edited the manuscript.
Glossary
- AMY-R
amylin receptor
- AUC
area under the curve
- CTR
calcitonin receptor
- GIP
glucose-dependent insulinotropic polypeptide
- GLP-1
glucagon-like peptide-1
- HbA1c
glycosylated haemoglobin
- IPGTT
i,p. glucose tolerance test
- OGTT
oral glucose tolerance test
- sCT
salmon calcitonin
- ZDF
Zucker diabetic fatty
- ZL
Zucker lean
Conflict of interest
The authors declare that there is no duality of interest associated with this manuscript.
References
- Amiel SA, Dixon T, Mann R, Jameson K. Hypoglycaemia in Type 2 diabetes. Diabet Med. 2008;25:245–254. doi: 10.1111/j.1464-5491.2007.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman RN, Finegood DT, Kahn SE. The evolution of beta-cell dysfunction and insulin resistance in type 2 diabetes. Eur J Clin Invest. 2002;32(Suppl 3):35–45. doi: 10.1046/j.1365-2362.32.s3.5.x. [DOI] [PubMed] [Google Scholar]
- Chan JL, Roth JD, Weyer C. It takes two to tango: combined amylin/leptin agonism as a potential approach to obesity drug development. J Investig Med. 2009;57:777–783. doi: 10.2310/JIM.0b013e3181b91911. [DOI] [PubMed] [Google Scholar]
- Chelikani PK, Haver AC, Reidelberger RD. Effects of intermittent intraperitoneal infusion of salmon calcitonin on food intake and adiposity in obese rats. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1798–R1808. doi: 10.1152/ajpregu.00386.2007. [DOI] [PubMed] [Google Scholar]
- Chen J, Pasricha PJ, Yin J, Lin L, Chen JD. Hepatic electrical stimulation reduces blood glucose in diabetic rats. Neurogastroenterol Motil. 2010;22:1109–e286. doi: 10.1111/j.1365-2982.2010.01556.x. [DOI] [PubMed] [Google Scholar]
- Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev. 2007;28:253–283. doi: 10.1210/er.2006-0026. [DOI] [PubMed] [Google Scholar]
- Edelman S, Maier H, Wilhelm K. Pramlintide in the treatment of diabetes mellitus. BioDrugs. 2008;22:375–386. doi: 10.2165/0063030-200822060-00004. [DOI] [PubMed] [Google Scholar]
- Eiden S, Daniel C, Steinbrueck A, Schmidt I, Simon E. Salmon calcitonin – a potent inhibitor of food intake in states of impaired leptin signalling in laboratory rodents. J Physiol. 2002;541:1041–1048. doi: 10.1113/jphysiol.2002.018671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etgen GJ, Oldham BA. Profiling of Zucker diabetic fatty rats in their progression to the overt diabetic state. Metabolism. 2000;49:684–688. doi: 10.1016/s0026-0495(00)80049-9. [DOI] [PubMed] [Google Scholar]
- Feigh M, Henriksen K, Andreassen KV, Hansen C, Henriksen JE, Beck-Nielsen H, et al. A novel oral form of salmon calcitonin improves glucose homeostasis and reduces body weight in diet-induced obese rats. Diabetes Obes Metab. 2011;13:911–920. doi: 10.1111/j.1463-1326.2011.01425.x. [DOI] [PubMed] [Google Scholar]
- Feigh M, Nielsen RH, Hansen C, Henriksen K, Christiansen C, Karsdal MA. Oral salmon calcitonin improves fasting and postprandial glycemic control in lean healthy rats. Horm Metab Res. 2012;44:130–134. doi: 10.1055/s-0031-1298027. [DOI] [PubMed] [Google Scholar]
- Fineman M, Weyer C, Maggs DG, Strobel S, Kolterman OG. The human amylin analog, pramlintide, reduces postprandial hyperglucagonemia in patients with type 2 diabetes mellitus. Horm Metab Res. 2002;34:504–508. doi: 10.1055/s-2002-34790. [DOI] [PubMed] [Google Scholar]
- Gedulin BR, Rink TJ, Young AA. Dose-response for glucagonostatic effect of amylin in rats. Metabolism. 1997;46:67–70. doi: 10.1016/s0026-0495(97)90170-0. [DOI] [PubMed] [Google Scholar]
- Granberry MC, Hawkins JB, Franks AM. Thiazolidinediones in patients with type 2 diabetes mellitus and heart failure. Am J Health Syst Pharm. 2007;64:931–936. doi: 10.2146/ajhp060446. [DOI] [PubMed] [Google Scholar]
- Hay DL, Christopoulos G, Christopoulos A, Sexton PM. Amylin receptors: molecular composition and pharmacology. Biochem Soc Trans. 2004;32:865–867. doi: 10.1042/BST0320865. [DOI] [PubMed] [Google Scholar]
- Hay DL, Christopoulos G, Christopoulos A, Poyner DR, Sexton PM. Pharmacological discrimination of calcitonin receptor: receptor activity-modifying protein complexes. Mol Pharmacol. 2005;67:1655–1665. doi: 10.1124/mol.104.008615. [DOI] [PubMed] [Google Scholar]
- Hollander P, Ratner R, Fineman M, Strobel S, Shen L, Maggs D, et al. Addition of pramlintide to insulin therapy lowers HbA1c in conjunction with weight loss in patients with type 2 diabetes approaching glycaemic targets. Diabetes Obes Metab. 2003a;5:408–414. doi: 10.1046/j.1463-1326.2003.00295.x. [DOI] [PubMed] [Google Scholar]
- Hollander PA, Levy P, Fineman MS, Maggs DG, Shen LZ, Strobel SA, et al. Pramlintide as an adjunct to insulin therapy improves long-term glycemic and weight control in patients with type 2 diabetes: a 1-year randomized controlled trial. Diabetes Care. 2003b;26:784–790. doi: 10.2337/diacare.26.3.784. [DOI] [PubMed] [Google Scholar]
- Holst JJ, Deacon CF, Vilsboll T, Krarup T, Madsbad S. Glucagon-like peptide-1, glucose homeostasis and diabetes. Trends Mol Med. 2008;14:161–168. doi: 10.1016/j.molmed.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Zraika S, Utzschneider KM, Hull RL. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia. 2009;52:1003–1012. doi: 10.1007/s00125-009-1321-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamori D, Kulkarni RN. Insulin modulation of glucagon secretion: the role of insulin and other factors in the regulation of glucagon secretion. Islets. 2009;1:276–279. doi: 10.4161/isl.1.3.9967. [DOI] [PubMed] [Google Scholar]
- Kiec-Klimczak ME, Pach DM, Pogwizd ME, Hubalewska-Dydejczyk AB. Incretins yesterday, pleiotropic gastrointestinal hormones today:glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) Recent Pat Endocr Metab Immune Drug Discov. 2011;5:176–182. doi: 10.2174/187221411797265863. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krentz AJ, Patel MB, Bailey CJ. New drugs for type 2 diabetes mellitus: what is their place in therapy? Drugs. 2008;68:2131–2162. doi: 10.2165/00003495-200868150-00005. [DOI] [PubMed] [Google Scholar]
- Kruger DF, Boucher JL, Banerji MA. Utilizing current diagnostic criteria and treatment algorithms for managing type 2 diabetes mellitus. Postgrad Med. 2011;123:54–62. doi: 10.3810/pgm.2011.07.2304. [DOI] [PubMed] [Google Scholar]
- Larsen PJ, Wulff EM, Gotfredsen CF, Brand CL, Sturis J, Vrang N, et al. Combination of the insulin sensitizer, pioglitazone, and the long-acting GLP-1 human analog, liraglutide, exerts potent synergistic glucose-lowering efficacy in severely diabetic ZDF rats. Diabetes Obes Metab. 2008;10:301–311. doi: 10.1111/j.1463-1326.2008.00865.x. [DOI] [PubMed] [Google Scholar]
- Lutz TA, Senn M, Althaus J, Del PE, Ehrensperger F, Scharrer E. Lesion of the area postrema/nucleus of the solitary tract (AP/NTS) attenuates the anorectic effects of amylin and calcitonin gene-related peptide (CGRP) in rats. Peptides. 1998;19:309–317. doi: 10.1016/s0196-9781(97)00292-1. [DOI] [PubMed] [Google Scholar]
- Lutz TA, Tschudy S, Rushing PA, Scharrer E. Amylin receptors mediate the anorectic action of salmon calcitonin (sCT) Peptides. 2000;21:233–238. doi: 10.1016/s0196-9781(99)00208-9. [DOI] [PubMed] [Google Scholar]
- Mack CM, Smith PA, Athanacio JR, Xu K, Wilson JK, Reynolds JM, et al. Glucoregulatory effects and prolonged duration of action of davalintide: a novel amylinomimetic peptide. Diabetes Obes Metab. 2011;13:1105–1113. doi: 10.1111/j.1463-1326.2011.01465.x. [DOI] [PubMed] [Google Scholar]
- Maggs DG, Fineman M, Kornstein J, Burrell T, Schwartz S, Wang Y, et al. Pramlintide reduces postprandial glucose excursions when added to insulin lispro in subjects with type 2 diabetes: a dose-timing study. Diabetes Metab Res Rev. 2004;20:55–60. doi: 10.1002/dmrr.419. [DOI] [PubMed] [Google Scholar]
- Malkov D, Angelo R, Wang HZ, Flanders E, Tang H, Gomez-Orellana I. Oral delivery of insulin with the eligen technology: mechanistic studies. Curr Drug Deliv. 2005;2:191–197. doi: 10.2174/1567201053586001. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen SJ, Vrang N, Larsen LK, Larsen PJ, Jelsing J. Stereological assessment of pancreatic beta-cell mass development in male Zucker Diabetic Fatty (ZDF) rats: correlation with pancreatic beta-cell function. J Anat. 2010;217:624–630. doi: 10.1111/j.1469-7580.2010.01285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potes CS, Turek VF, Cole RL, Vu C, Roland BL, Roth JD, et al. Noradrenergic neurons of the area postrema mediate amylin's hypophagic action. Am J Physiol Regul Integr Comp Physiol. 2010;299:R623–R631. doi: 10.1152/ajpregu.00791.2009. [DOI] [PubMed] [Google Scholar]
- Purdue BW, Tilakaratne N, Sexton PM. Molecular pharmacology of the calcitonin receptor. Receptors Channels. 2002;8:243–255. [PubMed] [Google Scholar]
- Reidelberger RD, Kelsey L, Heimann D. Effects of amylin-related peptides on food intake, meal patterns, and gastric emptying in rats. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1395–R1404. doi: 10.1152/ajpregu.00597.2001. [DOI] [PubMed] [Google Scholar]
- Roth JD, Hughes H, Kendall E, Baron AD, Anderson CM. Antiobesity effects of the beta-cell hormone amylin in diet-induced obese rats: effects on food intake, body weight, composition, energy expenditure, and gene expression. Endocrinology. 2006;147:5855–5864. doi: 10.1210/en.2006-0393. [DOI] [PubMed] [Google Scholar]
- Roth JD, Trevaskis JL, Wilson J, Lei C, Athanacio J, Mack C, et al. Antiobesity effects of the beta-cell hormone amylin in combination with phentermine or sibutramine in diet-induced obese rats. Int J Obes (Lond) 2008;32:1201–1210. doi: 10.1038/ijo.2008.91. [DOI] [PubMed] [Google Scholar]
- Rowe R, Cowx M, Poole C, Mcewan P, Morgan C, Walker M. The effects of orlistat in patients with diabetes: improvement in glycaemic control and weight loss. Curr Med Res Opin. 2005;21:1885–1890. doi: 10.1185/030079905X74943. [DOI] [PubMed] [Google Scholar]
- Scheen AJ, Finer N, Hollander P, Jensen MD, Van Gaal LF. Efficacy and tolerability of rimonabant in overweight or obese patients with type 2 diabetes: a randomised controlled study. Lancet. 2006;368:1660–1672. doi: 10.1016/S0140-6736(06)69571-8. [DOI] [PubMed] [Google Scholar]
- Steinert RE, Poller B, Castelli MC, Friedman K, Huber AR, Drewe J, et al. Orally administered glucagon-like peptide-1 affects glucose homeostasis following an oral glucose tolerance test in healthy male subjects. Clin Pharmacol Ther. 2009;86:644–650. doi: 10.1038/clpt.2009.159. [DOI] [PubMed] [Google Scholar]
- Steinert RE, Poller B, Castelli MC, Drewe J, Beglinger C. Oral administration of glucagon-like peptide 1 or peptide YY 3-36 affects food intake in healthy male subjects. Am J Clin Nutr. 2010;92:810–817. doi: 10.3945/ajcn.2010.29663. [DOI] [PubMed] [Google Scholar]
- Tilakaratne N, Christopoulos G, Zumpe ET, Foord SM, Sexton PM. Amylin receptor phenotypes derived from human calcitonin receptor/RAMP coexpression exhibit pharmacological differences dependent on receptor isoform and host cell environment. J Pharmacol Exp Ther. 2000;294:61–72. [PubMed] [Google Scholar]
- Tokuyama Y, Sturis J, Depaoli AM, Takeda J, Stoffel M, Tang J, et al. Evolution of beta-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes. 1995;44:1447–1457. doi: 10.2337/diab.44.12.1447. [DOI] [PubMed] [Google Scholar]
- Topp BG, Atkinson LL, Finegood DT. Dynamics of insulin sensitivity, -cell function, and -cell mass during the development of diabetes in fa/fa rats. Am J Physiol Endocrinol Metab. 2007;293:E1730–E1735. doi: 10.1152/ajpendo.00572.2007. [DOI] [PubMed] [Google Scholar]
- Trevaskis JL, Coffey T, Cole R, Lei C, Wittmer C, Walsh B, et al. Amylin-mediated restoration of leptin responsiveness in diet-induced obesity: magnitude and mechanisms. Endocrinology. 2008;149:5679–5687. doi: 10.1210/en.2008-0770. [DOI] [PubMed] [Google Scholar]
- Trevaskis JL, Turek VF, Wittmer C, Griffin PS, Wilson JK, Reynolds JM, et al. Enhanced amylin-mediated body weight loss in estradiol-deficient diet-induced obese rats. Endocrinology. 2010;151:5657–5668. doi: 10.1210/en.2010-0590. [DOI] [PubMed] [Google Scholar]
- Turek VF, Trevaskis JL, Levin BE, Dunn-Meynell AA, Irani B, Gu G, et al. Mechanisms of amylin/leptin synergy in rodent models. Endocrinology. 2010;151:143–152. doi: 10.1210/en.2009-0546. [DOI] [PubMed] [Google Scholar]
- Wajchenberg BL. beta-cell failure in diabetes and preservation by clinical treatment. Endocr Rev. 2007;28:187–218. doi: 10.1210/10.1210/er.2006-0038. [DOI] [PubMed] [Google Scholar]
- Young AA, Gedulin B, Vine W, Percy A, Rink TJ. Gastric emptying is accelerated in diabetic BB rats and is slowed by subcutaneous injections of amylin. Diabetologia. 1995;38:642–648. doi: 10.1007/BF00401833. [DOI] [PubMed] [Google Scholar]