

Abstract
Significance: The understanding of physiological and pathological processes involving protein oxidation, particularly under conditions of aging and oxidative stress, can be aided by proteomic identification of proteins that accumulate oxidative post-translational modifications only if these detected modifications are connected to functional consequences. The modification of tyrosine (Tyr) residues can elicit significant changes in protein structure and function, which, in some cases, may contribute to biological aging and age-related pathologies, such as atherosclerosis, neurodegeneration, and cataracts. Recent Advances: Studies characterizing proteins in which Tyr has been modified to 3-nitrotyrosine, 3,4-dihydroxyphenylalanine, 3,3′-dityrosine and other cross-links, or 3-chlorotyrosine are reviewed, with an emphasis on structural and functional consequences. Critical Issues: Distinguishing between inconsequential modifications and functionally significant ones requires careful biochemical and biophysical analysis of target proteins, as well as innovative methods for isolating the effects of the multiple modifications that often occur under oxidizing conditions. Future Directions: The labor-intensive task of isolating and characterizing individual modified proteins must continue, especially given the expanding list of known modifications. Emerging approaches, such as genetic and metabolic incorporation of unnatural amino acids, hold promise for additional focused studies of this kind. Antioxid. Redox Signal. 17, 1571–1579.
Introduction
Various oxidative post-translational modifications (PTMs) of proteins accumulate in cells during aging and disease, particularly age-related diseases such as atherosclerosis, cataracts, type 2 diabetes, and neurodegenerative diseases. Such modifications can alter protein structure and function by inducing conformational changes, partial or complete unfolding, cross-linking, and/or aggregation, or by modifying enzyme active sites, phosphorylation sites, or binding sites. These modifications can lead to either gain or loss of function and may serve physiological/regulatory purposes or contribute to (or at least accompany) pathogenesis (see Refs. 26, 33, 42 and references cited therein).
Modifications of tyrosine
Protein tyrosine (Tyr) residues are susceptible to an array of oxidative and nitrative modifications (Fig. 1), including the formation of 3-nitrotyrosine (3NY), which may be reduced to 3-aminotyrosine (1, 40); 3,4-dihydroxyphenylalanine (DOPA); 3,3′-dityrosine (DiY); 3-chlorotyrosine (3ClY); 3,5-dichlorotyrosine (DiClY); as well as brominated and iodinated forms (6, 33, 42). Further oxidation can convert DOPA to dopaquinone (DQ) or 3-aminotyrosine to the quinone imine, representing acceptors for additions. Subsequently, protein-bound conjugate DQ and quinone imine may propagate oxidative damage to DNA and lipids, as well as other proteins (28). Additionally, ortho-tyrosine (2-hydroxyphenylalanine, o-Tyr) and meta-tyrosine (3-hydroxyphenylalanine, m-Tyr) are products of reaction between hydroxyl radicals (or their metal-bound equivalents) and Phe (Refs. 26, 33, 42 and references cited therein). While mostly nonenzymatic, all of these modifications can preferentially accumulate on specific proteins, as well as specific residues within those proteins (1, 26, 33).
FIG. 1.

Endogenous post-translational modifications of protein-bound Tyr. EPO, eosinophil peroxidase; MPO, myeloperoxidase; TPO, thyroid peroxidase.
Consequences of modification
In many cases, oxidation targets proteins for degradation, perhaps due to the increased surface hydrophobicity that results from unfolding or misfolding (8, 11, 33, 42). However, after extensive oxidative modification and/or when cross-links and aggregates form, proteins can become resistant to and even inhibit proteolysis, leading to pathological accumulation and deposition (8, 11, 42). Proteolysis by both proteasomal and lysosomal pathways also tends to decline with aging and certain disease states, which may further contribute to cellular senescence and to pathologies characterized by protein deposition, such as atherosclerosis, cataract, macular degeneration, rheumatoid arthritis, and neurodegenerative diseases like Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS) (8, 11, 18, 33, 42).
The introduction of modified residues within proteins and any associated structural changes can create neo-epitopes for antibody recognition, and cross-reactivity with unmodified self-proteins can induce autoimmune disorders. This is particularly relevant for modified proteins that are absent during early development but accumulate with age (1, 26, 33).
In addition to these general consequences for proteins, oxidation can have specific effects on the function of a particular protein. For example, it can modify an active site, block a phosphorylation site, or disrupt a binding site for substrates, cofactors, or partner proteins. If a particular modification is reversible through repair or eliminated through protein turnover, it may serve a regulatory function and/or participate in cellular signaling pathways (Refs. 1, 26, 33, and references cited therein).
Importance of site specificity
While overall levels of a particular Tyr modification (e.g., protein-bound 3NY or DOPA) may or may not be measurably elevated in a particular tissue or cell sample, specific proteins may carry significant levels of such modifications (26, 33). When these proteins are of low abundance, the absolute quantity of the modification will not contribute significantly to the overall yield in the sample. Nevertheless, the modification may be physiologically important. For this reason, many proteomic studies have been undertaken to identify long lists of Tyr-modified proteins. However, the focus of the present review will be on progress made in characterizing the effects of specific protein modifications on structure, function, aggregation, and pathogenesis, with an emphasis on implications for aging. We will predominantly review studies published over the last 5 years.
3-Nitrotyrosine
Formation and significance
3-Nitrotyrosine (3NY) is the most commonly measured Tyr oxidation product, due to its relative chemical stability, the availability of anti-3NY antibodies, and its status as a marker of nitric oxide (NO)-dependent protein modifications. 3NY is generated by several pathways, including reactions of peroxynitrite (ONOO−) or nitrogen dioxide (•NO2), and catalysis by heme-containing proteins (26, 33). The nitration of Tyr can lead to conformational changes, unfolding, aggregation, immunogenicity, and/or partial or total inactivation of a target protein, although in some cases gain of function has been observed. These consequences may be attributed to changes in steric requirements, electrostatics, pKa of amino acid side chains, and/or surface hydrophobicity. Going beyond tables of proteomic identifications, there have been significant developments in the study of specific protein nitration sites and their effects on structure and function. The possibility of 3NY as a signaling moiety has also been proposed, and evidence has been presented for pathological roles of 3NY-containing proteins. For a comprehensive review, see Ref. 26 and references cited therein.
Inactivation of MnSOD by nitration of Tyr-34 in the active site
The activity of human manganese superoxide dismutase (MnSOD), which protects cells from damage by scavenging superoxide, is largely inhibited by nitration at Tyr-34 in the active site, and this modification has been observed in aging, ALS, AD, PD, and diabetes, among others (20, 26). Quint et al. (32) determined the crystal structures of both native and nitrated enzyme in order to investigate the mechanism of this inactivation (Fig. 2). Treatment with peroxynitrite gave 75% yield of 3NY-34 and reduced enzymatic activity to 20%±5% compared to that of the wild-type enzyme. Although mass spectrometry identified additional modification sites, 3NY-34 was the only modification present in the crystal structure. While no significant conformational changes were evident upon nitration, enzyme inhibition was attributed to steric interference with substrate binding and changes in the hydrogen bonding network. Electrostatic changes due to the lower phenolic pKa of 3NY relative to Tyr may also alter the finely-tuned redox potential in the active site and/or cause repulsion of the substrate, O2•−, although this hypothesis was not tested. The pKa of 3NY-34 in MnSOD is expected to be similar to that measured spectroscopically for 3NY-34 of the bacterial iron superoxide dismutase enzyme (FeSOD), which is 7.95, or approximately two units lower than that of Tyr (Ref. 32 and references cited therein). In this protein environment, this value is higher than the pKa ∼ 7.2 measured for 3NY in other peptides and proteins and in its free form (40, 46).
FIG. 2.

Active site of native (a) and nitrated (b) human MnSOD, shown with Mn and its ligands, His-26, Tyr-34, His-74, Asp-159, and His-163. Image derived from crystal structures stored in the Protein Data Bank (32); accession codes 2ADQ for native and 2ADP for nitrated. Figure generated using Protein Workshop (22).
In order to isolate 3NY-34 from all other modifications that can occur with oxidizing agents, Neumann et al. (25) used a genetic approach for site-specific incorporation of the unnatural amino acid 3NY at position 34 in recombinant rat mitochondrial MnSOD. Upon quantitative formation of this nitrated mutant, the enzyme showed 97% decrease in activity.
Nitration disrupts catalysis by GAPDH
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a crucial protein for cellular metabolism and energy, is a common entry on proteomic lists of nitrated proteins, including studies of aging (17) and AD (43), likely due to its role as a sensor of NO (27). In a mechanistic study, Palamalai et al. (27) recently determined that in vitro nitration of Tyr-311 and Tyr-317 in rabbit GAPDH by tetranitromethane resulted in loss of binding to NAD+, thereby destroying all catalytic activity of the enzyme. Computational analysis of the X-ray crystal structure predicted that these nitration sites are located very close to both the catalytic cysteine residue and the NAD+ binding site (Fig. 3). Furthermore, in silico nitration resulted in an even shorter predicted distance (1.4 Å) between 3NY-317 and NAD+, such that significant steric interference may occur. Alternatively, the movement of this residue at the base of an α-helix may disrupt this secondary structure. In fact, circular dichroism shows some loss of secondary structure following in vitro nitration.
FIG. 3.
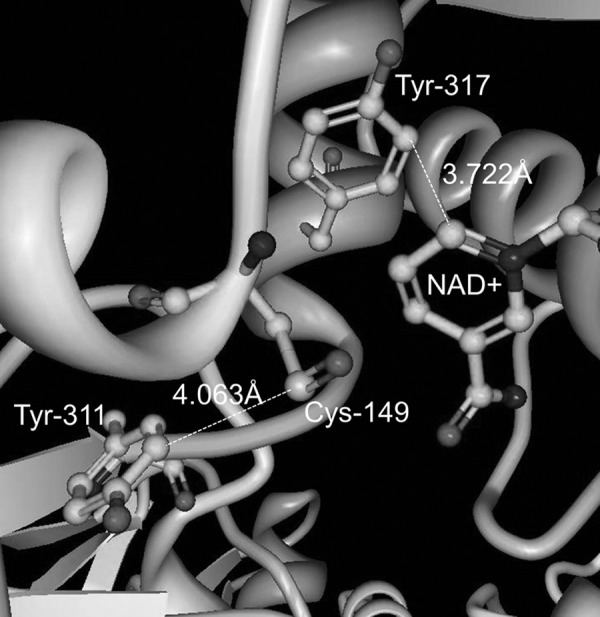
Nitration sites of rabbit muscle GAPDH, Tyr-311 and Tyr-317, and their proximity to Cys-149 and NAD+ in the active site. Image derived from the crystal structure stored in the Protein Data Bank (7); accession code 1J0X. Figure generated using Protein Workshop (22).
DOPA, DQ, and Tyr-Mediated Cross-Links
DOPA and DQ
Hydroxyl radicals can convert Tyr to DOPA (14), and site-specificity for this reaction can be facilitated by redox-active transition metals bound to proteins or by localized sources of hydroxyl radicals, such as mitochondria (23, 48). Through reversible oxidation to dopaqinone (DQ), DOPA is part of a redox cycle capable of regenerating the reduced state of redox-active transition metals, particularly iron and copper, and of reducing metalloproteins (9, 12, 14). This phenomenon can perpetuate oxidative insult but also presents an opportunity for putative signaling functions, such as triggering antioxidant defenses (23). Protein-bound DOPA can also serve a protective role by scavenging radicals and chelating metals, although the latter function can alternatively direct oxidative damage to specific protein sites (23, 24, 41).
DOPA and DQ can also be formed enzymatically by tyrosinase or tyrosine hydroxylase (28). In addition, DOPA may be incorporated during protein synthesis when free DOPA is available to compete with Tyr, such as in levodopa-treated PD patients (10).
Conjugate addition
DQ and the related quinone imine (e.g., an oxidation product of 3-aminotyrosine) can serve as conjugate acceptors for nucleophilic groups on other amino acid side chains to generate protein cross-links (28). Protein aggregates may result that are resistant to proteolysis and can contribute to protein deposition/plaque formation in aging and various disease states (8, 11). Conversely, such cross-links can perform important functions. For example, the cofactor lysine tyrosylquinone is formed by the intramolecular addition of Lys-314 to the DQ moiety derived from Tyr-349 in lysyl oxidase (44).
3,3 ′-Dityrosine
Another route to Tyr-mediated protein cross-links involves the combination of two tyrosyl radicals to form 3,3′-dityrosine (DiY) (15, 42). Various pathways to the formation of tyrosyl radicals exist, mediated by hydroxyl radicals, peroxynitrite-derived species, peroxyl radicals, redox-active metals, γ-radiolysis, UV irradiation, heme, or hemeperoxidase enzymes (3, 15, 42). In the hydrophobic region of biological membranes, lipid-derived radicals can also play an intermediary role (3). Specificity may arise due to conformational requirements for the interaction of Tyr residues. DiY modification leads to selective proteolysis, and the released DiY is a stable and useful biomarker for oxidative stress, for example, as excreted in urine (15).
Although not as widely studied as 3NY, these other modifications have been linked to atherosclerosis (12), cataracts (13, 21), levodopa therapy for PD (10), and lysosomal dysfunction (10, 11). Some proteomic studies of DOPA, for example, have been published (19, 48), but here we will focus on recent mechanistic studies.
Age-related cataracts
The formation of age-related cataracts in humans is associated with up to 15-fold increases in protein-bound DOPA, as well as elevated levels of DiY and other protein-bound amino acid hydroxylation products, which can contribute to the protein cross-linking and browning that characterize cataractous lenses (13). DOPA and DiY formation may result from both Fenton-type, hydroxyl-radical chemistry (13), and reactions photosensitized by protein-bound tryptophan (Trp) metabolites (21). The free Trp metabolites kynurenine, 3-hydroxykynurenine, 3-hydroxykynurenine-O-β-D-glucoside, 4-(2-aminophenyl)-4-oxobutanoic acid, and 4-(2-amino-3-hydroxyphenyl)-4-oxobutanoic acid serve as filters for UVA light, as they absorb with high efficiency but are poor sensitizers for the formation of singlet oxygen or radical species. However, with aging, they become increasingly bound to lens proteins, either covalently or noncovalently (Fig. 4). Such protein-filter adducts accumulate in the lens nucleus, due at least in part to a diffusional barrier that develops at middle age. In this state, these compounds may lose their protective function and instead promote direct and indirect photooxidation of proteins, including generation of DOPA and DiY. At the same time, the pool of free, protective filter compounds is depleted, leaving the aging lens more susceptible to photochemical damage (21).
FIG. 4.

Trp metabolites can form covalent or noncovalent associations with proteins to become photosensitizers for protein oxidation. Figure based on information from Ref. 21.
When tested in vitro with soluble bovine lens proteins (21), all but one of these protein-Trp metabolite adducts led to increased peroxide formation (primarily hydrogen peroxide) in a process that is sensitive to D2O (increased yield) and azide (decreased yield), indicating the involvement of singlet oxygen. However, the levels of DOPA and DiY were metabolite dependent and insensitive to D2O. In addition, DOPA formation was independent of illumination and atmospheric oxygen. The authors postulated at least two reaction pathways to account for these differences: 1) the photosensitized formation of singlet oxygen, and 2) the auto-oxidation of the o-aminophenol moiety in 3-hydroxykynurenine or its metabolite, 4-(2-amino-3-hydroxyphenyl)-4-oxobutanoic acid, which generates superoxide. Furthermore, in the light-dependent pathway, the excited states of the filter compounds appear to have competing energy transfer pathways, either to oxygen or to protein-bound Tyr.
Regardless of the pathway, these phenomena generated covalent, nondisulfide protein cross-links and may also contribute to lens browning, two characteristic features of age-related nuclear cataracts (13, 21). The DOPA levels in the Trp metabolite study (21) were similar to those found in human type IV nigrescent cataract lenses, while DiY levels were much higher in the model system than in the cataract lenses. Cross-links observed by reducing SDS-PAGE may be due to DiY and/or reaction of nucleophilic side chains with the protein-bound oxidation products of DOPA (i.e., DQ) or the o-aminophenol moiety of 3-hydroxykynurenine or 4-(2-amino-3-hydroxyphenyl)-4-oxobutanoic acid (i.e., o-quinone imine).
Lysosomal accumulation
Using metabolic incorporation of DOPA into proteins, Dunlop et al. (11) showed that such proteins form autofluorescent, SDS-stable, proteolysis-resistant aggregates in J774 murine macrophages, similar to the “aging pigment” lipofuscin or its disease-related counterpart, ceroid. This aggregation is consistent with the ability of DOPA to oxidize to DQ and react with nucleophiles, such as cysteine residues, to form cross-links. The perinuclear punctate pattern of fluorescence and upregulation of the lysosomal proteases cathepsins B and L indicate accumulation of these aggregates in lysosomes. Lysosomes are rich in transition metals due to their role in autophagic degradation of metal-containing proteins, and DOPA redox cycling can continually regenerate these reactive species to fuel oxidative damage to the lysosomal membrane, setting off a lysosomal pathway to apoptosis, as illustrated in Figure 5 (see Ref. 18 for a review). Indeed, in a subsequent study (10), the authors repeated this result in THP1 human monocytes and used a variety of fluorescent staining/flow cytometry approaches to demonstrate lysosomal membrane permeabilization, depolarization of mitochondrial membranes, decreased mitochondrial cytochrome c levels, and increased apoptosis (measured as phosphatidylserine externalization by Annexin V-FITC, cellular DNA content by propidium iodide, and DNA fragmentation by TUNEL). Lysosomal membrane permeabilization occurred even in the presence of a pan-caspase inhibitor, demonstrating that it is not caused by an early release of caspases and thus indicating that it is the trigger for the apoptotic event. In the absence of the inhibitor, Western blots showed the cleavage of pro-caspase 3 to its active form, which is a key step that commits cells to apoptosis, and the resulting increase in activity was measured using a fluorigenic substrate. Lysosomal membrane permeabilization was also replicated in SH-SY5Y human neuroblastoma cells and MRC5 human lung fibroblasts.
FIG. 5.

Lysosomal pathway to apoptosis mediated by the accumulation of aggregated proteins following metabolic incorporation of DOPA. Figure based on information from Refs. 10 and 18.
Similar experiments with o-Tyr, m-Tyr, Tyr, and D-DOPA (a negative control for protein incorporation) showed that these effects are unique to protein-bound DOPA (10, 11). While o-Tyr did induce lower levels of apoptosis than DOPA, as well as lower levels of caspase 3 activation, it did not aggregate or accumulate, indicating a separate apoptotic pathway (10).
By specifically incorporating DOPA into cellular proteins in vitro, the study just described isolates this protein modification from other possible consequences of oxidative stress and defines the subsequent pathway to apoptosis. It also mimics long-term levodopa treatment of PD patients, for whom protein incorporation has been observed (10), and the apoptotic events observed may contribute to the suspected neurotoxicity of this treatment regimen. The lysosome-mediated apoptotic pathway may be particularly relevant for neurons and other post-mitotic cells that are more prone to accumulation of proteolysis-resistant proteins (10). Even without apoptotic events, the accumulation of nondegradable proteins or lipofuscin in lysosomes can lead to mitochondrial dysfunction, a phenomenon observed in long-lived post-mitotic senescent cells that may contribute to aging (18).
It should also be noted that sites of metabolic DOPA incorporation could very well be different from those sites altered by ROS in vivo. Perhaps this study is a better indication of events occurring in large-scale oxidative stress events [indeed, aggregation of DOPA-containing proteins occurs only when this oxidized amino acid is incorporated in larger amounts (11)], but not of localized effects that may result from highly specific redox events, such as the metal-catalyzed oxidation of Tyr in an enzyme's active site. Complementary proteomic studies could identify whether there are common sites for metabolic incorporation versus oxidative damage, and if any of these sites may be functionally significant, especially at lower levels of oxidative insult. Such proteomic studies and corresponding structural and functional studies for DOPA are currently lacking in the field.
3-Chlorotyrosine
Formation and significance
The enzyme myeloperoxidase (MPO) is secreted by neutrophils, monocytes, and some macrophages, and is a key component of inflammatory response (47). It uses hydrogen peroxide and chloride ions to generate hypochlorous acid (HOCl) as a defense against microorganisms, but it can also inflict oxidative damage on proteins and lipids (31). HOCl can react with protein-bound Tyr, and this is the only known source of 3-chlorotyrosine (3ClY) formation in humans, making this modification a good fingerprint for oxidative damage promoted by MPO (16, 31). HOCl can further chlorinate 3ClY to form DiClY (42). In addition, MPO can generate •NO2 from nitrite or ONOO−, which can generate 3NY, as well as tyrosyl radicals, leading to DiY and other Tyr-derived cross-links (26, 31).
3ClY has been found in many inflammatory disease states, such as in synovial fluid from rheumatoid arthritis patients (45). In addition, both MPO and its products, particularly 3ClY and 3NY, are known to be enriched in human atherosclerotic plaques and in plasma from patients with cardiovascular disease (5, 16, 47). Indeed, plasma MPO level has been shown to be an effective predictor of major adverse coronary events in patients presenting with chest pain to the emergency department (5).
Chlorination of HDL may contribute to atherogenesis
Studies of specific 3ClY sites in proteins are scarce, but there has been interest in characterizing the role of MPO in the oxidative damage of high-density lipoprotein (HDL), particularly its major protein component, apolipoprotein A-I (apoAI). Figure 6 shows the locations of several important oxidation sites. While there is agreement in the literature that MPO selectively targets Tyr-192 (a highly conserved residue) in lipid-free apoAI for both chlorination and nitration, with lower levels observed at the other six Tyr residues (34, 50), the functional impact of this specific modification is controversial. There is agreement that MPO-mediated modification, and specifically the 3ClY and 3NY content of apoAI, correlates with loss of cholesterol efflux activity mediated by ATP-binding cassette transporter A1 (ABCA1) (34, 49), and that both chlorinated and nitrated apoAI are enriched in human plasma from patients with cardiovascular disease and even more so in atheromas isolated from such patients (49, 50). Shao et al. (34) further demonstrated that the specific modification of Tyr-192 to 3ClY shows a strong linear correlation with the loss of ABCA1-dependent cholesterol efflux activity. Additional, contradictory findings are briefly described below, and for recent related reviews from both groups actively researching this subject, see Refs. 35 and 39.
FIG. 6.
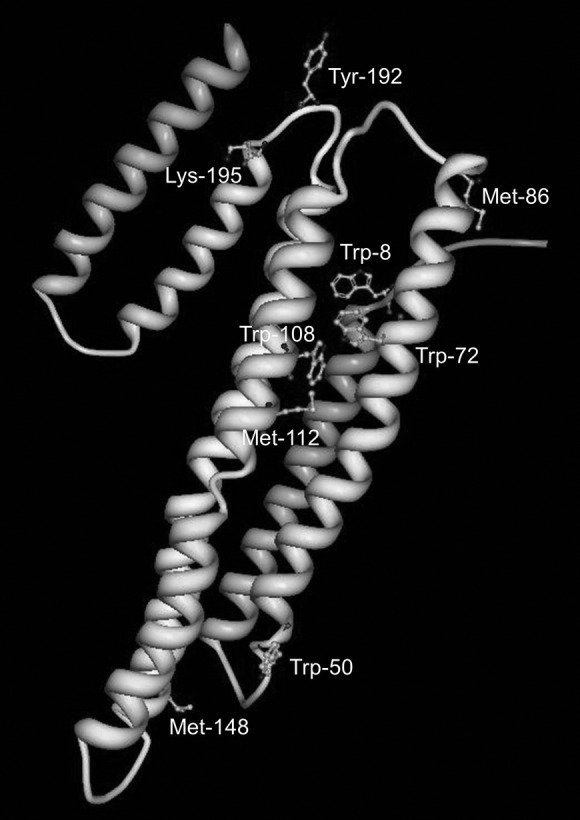
Oxidation sites of lipid-free human apoAI. Many of these sites are located near hinge regions that may be important for rearrangement upon lipid association and subsequent binding to ABCA1 for reverse cholesterol transport activity. Lys-195 facilitates chlorination of Tyr-192. Image derived from the crystal structure stored in the Protein Data Bank (2); accession code 2A01. Figure generated using Protein Workshop (22).
In a series of studies, the groups of Heinecke and Oram described roles for Tyr, Lys, and Met residues in the MPO-mediated inactivation of apoAI cholesterol efflux activity via ABCA1. While Tyr-192 was the major target for both chlorination and nitration by MPO (and also nitration by ONOO−), the latter modification did not significantly affect the biological activity (34). Nearby Lys residues on the same face of the amphipathic helix as Tyr, specifically in YXXK motifs, regiospecifically directed Tyr chlorination via chloramine intermediates formed on the ɛ-amino group of Lys (4, 36). Point mutations designed to add or remove Met residues near target Tyr residues showed their capacity to act as intramolecular antioxidants to inhibit the chlorination (36), as also demonstrated in a later study by Peng et al. (29).
Paradoxically, Shao et al. (36) presented evidence that Met oxidation, along with Tyr-192 chlorination, was required to impair cholesterol transport activity of apoAI. Mutation of Tyr-192 to the more MPO-oxidation-resistant Phe (Y192F) gave a functional apoAI that was protected slightly from chlorination-dependent loss of activity. Following quantitative oxidation of all three native Met residues to Met sulfoxide by the MPO system, their complete conversion back to Met by the methionine sulfoxide reductase enzyme PilB resulted in partial recovery of activity. While neither of these strategies led to full recovery on its own, the combination of the Y192F mutation and PilB treatment almost completely restored the activity to that of unmodified, wild-type apoAI, leading to the conclusion that both Tyr-192 chlorination and Met oxidation contribute to MPO-mediated functional impairment.
Toward an understanding of the mechanism underlying impaired reverse cholesterol transport, Shao et al. (37) showed that while both unmodified and nitrated apoAI were able to compete with 125I-labeled apoAI for binding to ABCA1, the MPO-treated protein could not. Furthermore, their data indicated that the lipid-binding ability of the protein was not compromised by MPO treatment. Since Tyr-192 and all three oxidized Met are in or near hinge regions of apoAI (Fig. 5), their modification may interfere with the rearrangement of its hairpin loops that is required for interaction with ABCA1. The resulting impairment of cholesterol removal from lipid-laden macrophages or foam cells would promote formation of atherosclerotic plaque (35).
Other researchers have also independently identified Tyr-192 in apoAI as a major target for chlorination by MPO, but their data has not supported a significant functional impact for this particular modification in murine macrophages. Rather, their mutation of all seven Tyr residues to Phe conferred no protective effect for cholesterol efflux activity following MPO treatment of apoAI (30). On the other hand, mutation of all four Trp residues to Phe did make apoAI resistant to MPO-mediated oxidation, leading to the conclusion that mono- and di-hydroxylated Trp is responsible for loss of apoAI's ABCA1-dependent cholesterol efflux activity (29).
Acknowledging these discrepancies, Shao et al. (36) suggest that different cell culture models may behave differently, specifically proposing that since their ABCA1-transfected baby hamster kidney cells express higher levels of ABCA1 than murine macrophages, the modest protective effect of replacing Tyr-192 with Phe is more easily detected. Furthermore, Shao and Heinecke (35) point to structural changes in the Trp-free mutant that may explain its resistance to MPO-mediated inactivation, rather than the absence of oxidizable Trp. They specifically cite the increase in α-helical content from 56% for the wild-type to 71% in the Trp-free mutant (29, 35).
There are likely many factors that contribute to HDL dysfunction in atherosclerosis (see Ref. 39 for a useful discussion), of which modification of Tyr and other amino acids is only one. Even so, specific characterization of the residues involved in oxidation-mediated protein damage may open new diagnostic and therapeutic avenues, both through the use of modified residues as biomarkers and the development of oxidation-resistant forms of apoAI as more effective anti-atherogenic agents for possible therapeutic use (35, 36, 38). In particular, the specificity of 3ClY as a marker for MPO activity and the link between MPO and disease progression may make it an important diagnostic and/or therapeutic tool.
Conclusions
Tyr modification has been demonstrated to have important consequences for protein function and structure in many cases. In others, there is still an opportunity to learn what impact these PTMs may have on particular proteins and what role they may play. As the work of identifying, isolating, and characterizing modified proteins under conditions relevant to biological aging and age-related diseases continues, novel insights and deeper understanding of physiological and pathological processes can ultimately lead to improved diagnostic methods and therapeutic interventions for patients.
Abbreviations Used
- 3ClY
3-chlorotyrosine
- 3NY
3-nitrotyrosine
- ABCA1
ATP-binding cassette transporter A1
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- apoAI
apolipoprotein A-I
- DiClY
3,5-dichlorotyrosine
- DiY
3,3′-dityrosine
- DOPA
3,4-dihydroxyphenylalanine
- DQ
dopaquinone
- EPO
eosinophil peroxidase
- FeSOD
iron superoxide dismutase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HDL
high-density lipoprotein
- HOBr
hypobromous acid
- HOCl
hypochlorous acid
- HPLC
high-performance liquid chromatography
- HPLC-MS/MS
high-performance liquid chromatography-tandem mass spectrometry
- MnSOD
manganese superoxide dismutase
- MPO
myeloperoxidase
- m-Tyr
meta-tyrosine, 3-hydroxyphenylalanine
- NO
nitric oxide
- •NO2
nitrogen dioxide
- o-Tyr
ortho-tyrosine, 2-hydroxyphenylalanine
- ONOO−
peroxynitrite
- PD
Parkinson's disease
- PTM
post-translational modification
- ROS
reactive oxygen species
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TPO
thyroid peroxidase
Acknowledgments
We gratefully acknowledge support by the NIH (AG12993, AG23551, AG25350), the Madison and Lila Self Graduate Fellowship program at The University of Kansas, Amgen, and the Department of Pharmaceutical Chemistry at The University of Kansas.
Disclosure Statement
No competing financial interests exist.
References
- 1.Abello N. Kerstjens HAM. Postma DS. Bischoff R. Protein tyrosine nitration: Selectivity, physicochemical and biological consequences, denitration, and proteomics methods for the identification of tyrosine-nitrated proteins. J Proteome Res. 2009;8:3222–3238. doi: 10.1021/pr900039c. [DOI] [PubMed] [Google Scholar]
- 2.Ajees AA. Anantharamaiah GM. Mishra VK. Hussain MM. Murthy HMK. Crystal structure of human apolipoprotein A-I: Insights into its protective effect against cardiovascular diseases. Proc Natl Acad Sci USA. 2006;103:2126–2131. doi: 10.1073/pnas.0506877103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Bartesaghi S. Wenzel J. Trujillo M. López M. Joseph J. Kalyanaraman B. Radi R. Lipid peroxyl radicals mediate tyrosine dimerization and nitration in membranes. Chem Res Toxicol. 2010;23:821–835. doi: 10.1021/tx900446r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergt C. Fu X. Huq NP. Kao J. Heinecke JW. Lysine residues direct the chlorination of tyrosines in YXXK motifs of apolipoprotein A-I when hypochlorous acid oxidizes high density lipoprotein. J Biol Chem. 2004;279:7856–7866. doi: 10.1074/jbc.M309046200. [DOI] [PubMed] [Google Scholar]
- 5.Brennan M-L. Penn MS. Van Lente F. Nambi V. Shishehbor MH. Aviles RJ. Goormastic M. Pepoy ML. McErlean ES. Topol EJ. Nissen SE. Hazen SL. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 6.Coval ML. Taurog A. Purification and iodinating activity of hog thyroid peroxidase. J Biol Chem. 1967;242:5510–5523. [PubMed] [Google Scholar]
- 7.Cowan-Jacob SW. Kaufmann M. Anselmo AN. Stark W. Grütter MG. Structure of rabbit-muscle glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr D Biol Crystallogr. 2003;59:2218–2227. doi: 10.1107/s0907444903020493. [DOI] [PubMed] [Google Scholar]
- 8.Davies KJA. Shringarpure R. Preferential degradation of oxidized proteins by the 20S proteasome may be inhibited in aging and in inflammatory neuromuscular diseases. Neurology. 2006;66:S93–S96. doi: 10.1212/01.wnl.0000192308.43151.63. [DOI] [PubMed] [Google Scholar]
- 9.Dean RT. Fu S. Stocker R. Davies MJ. Biochemistry and pathology of radical-mediated protein oxidation. Biochem J. 1997;324:1–18. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunlop RA. Brunk UT. Rodgers KJ. Proteins containing oxidized amino acids induce apoptosis in human monocytes. Biochem J. 2011;435:207–216. doi: 10.1042/BJ20100682. [DOI] [PubMed] [Google Scholar]
- 11.Dunlop RA. Dean RT. Rodgers KJ. The impact of specific oxidized amino acids on protein turnover in J774 cells. Biochem J. 2008;410:131–140. doi: 10.1042/BJ20070161. [DOI] [PubMed] [Google Scholar]
- 12.Fu S. Davies MJ. Stocker R. Dean RT. Evidence for roles of radicals in protein oxidation in advanced human atherosclerotic plaque. Biochem J. 1998;333:519–525. doi: 10.1042/bj3330519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu S. Dean R. Southan M. Truscott R. The hydroxyl radical in lens nuclear cataractogenesis. J Biol Chem. 1998;273:28603–28609. doi: 10.1074/jbc.273.44.28603. [DOI] [PubMed] [Google Scholar]
- 14.Gieseg SP. Simpson JA. Charlton TS. Duncan MW. Dean RT. Protein-bound 3,4-dihydroxyphenylalanine is a major reductant formed during hydroxyl radical damage to proteins. Biochemistry. 1993;32:4780–4786. doi: 10.1021/bi00069a012. [DOI] [PubMed] [Google Scholar]
- 15.Giulivi C. Traaseth NJ. Davies KJA. Tyrosine oxidation products: Analysis and biological relevance. Amino Acids. 2003;25:227–232. doi: 10.1007/s00726-003-0013-0. [DOI] [PubMed] [Google Scholar]
- 16.Hazen SL. Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99:2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanski J. Hong SJ. Schöneich C. Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J Biol Chem. 2005;280:24261–24266. doi: 10.1074/jbc.M501773200. [DOI] [PubMed] [Google Scholar]
- 18.Kurz T. Terman A. Gustafsson B. Brunk U. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol. 2008;129:389–406. doi: 10.1007/s00418-008-0394-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S. Chen Y. Luo H. Wu AA. Wilde M. Schumacker PT. Zhao Y. The first global screening of protein substrates bearing protein-bound 3,4-dihydroxyphenylalanine in Escherichia coli and human mitochondria. J Proteome Res. 2010;9:5705–5714. doi: 10.1021/pr1005179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacMillan-Crow LA. Crow JP. Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998;37:1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]
- 21.Mizdrak J. Hains PG. Truscott RJW. Jamie JF. Davies MJ. Tryptophan-derived ultraviolet filter compounds covalently bound to lens proteins are photosensitizers of oxidative damage. Free Radic Biol Med. 2008;44:1108–1119. doi: 10.1016/j.freeradbiomed.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Moreland J. Gramada A. Buzko O. Zhang Q. Bourne P. The Molecular Biology Toolkit (MBT): A modular platform for developing molecular visualization applications. BMC Bioinformatics. 2005;6:21. doi: 10.1186/1471-2105-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson M. Foxwell AR. Tyrer P. Dean RT. Protein-bound 3,4-dihydroxy-phenylanine (DOPA), a redox-active product of protein oxidation, as a trigger for antioxidant defences. Int J Biochem Cell Biol. 2007;39:879–889. doi: 10.1016/j.biocel.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Nelson M. Foxwell AR. Tyrer P. Dean RT. Radical sequestration by protein-bound 3,4-dihydroxyphenylalanine. Int J Biochem Cell Biol. 2010;42:755–761. doi: 10.1016/j.biocel.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 25.Neumann H. Hazen JL. Weinstein J. Mehl RA. Chin JW. Genetically encoding protein oxidative damage. J Am Chem Soc. 2008;130:4028–4033. doi: 10.1021/ja710100d. [DOI] [PubMed] [Google Scholar]
- 26.Pacher P. Beckman JS. Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palamalai V. Miyagi M. Mechanism of glyceraldehyde-3-phosphate dehydrogenase inactivation by tyrosine nitration. Protein Sci. 2010;19:255–262. doi: 10.1002/pro.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pattison DI. Dean RT. Davies MJ. Oxidation of DNA, proteins and lipids by DOPA, protein-bound DOPA, and related catechol(amine)s. Toxicology. 2002;177:23–37. doi: 10.1016/s0300-483x(02)00193-2. [DOI] [PubMed] [Google Scholar]
- 29.Peng D-Q. Brubaker G. Wu Z. Zheng L. Willard B. Kinter M. Hazen SL. Smith JD. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler Thromb Vasc Biol. 2008;28:2063–2070. doi: 10.1161/ATVBAHA.108.173815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng D-Q. Wu Z. Brubaker G. Zheng L. Settle M. Gross E. Kinter M. Hazen SL. Smith JD. Tyrosine modification is not required for myeloperoxidase-induced loss of apolipoprotein A-I functional activities. J Biol Chem. 2005;280:33775–33784. doi: 10.1074/jbc.M504092200. [DOI] [PubMed] [Google Scholar]
- 31.Podrez EA. Abu-Soud HM. Hazen SL. Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic Biol Med. 2000;28:1717–1725. doi: 10.1016/s0891-5849(00)00229-x. [DOI] [PubMed] [Google Scholar]
- 32.Quint P. Reutzel R. Mikulski R. McKenna R. Silverman DN. Crystal structure of nitrated human manganese superoxide dismutase: Mechanism of inactivation. Free Radic Biol Med. 2006;40:453–458. doi: 10.1016/j.freeradbiomed.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 33.Scaloni A. Mass spectrometry approaches for the molecular characterization of oxidatively/nitrosatively modified proteins. In: Dalle-Donne I, editor; Scaloni A, editor; Butterfield DA, editor; Hoboken NJ, editor. Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases. Wiley-Interscience; 2006. pp. 59–99. [Google Scholar]
- 34.Shao B. Bergt C. Fu X. Green P. Voss JC. Oda MN. Oram JF. Heinecke JW. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. J Biol Chem. 2005;280:5983–5993. doi: 10.1074/jbc.M411484200. [DOI] [PubMed] [Google Scholar]
- 35.Shao B. Heinecke JW. Impact of HDL oxidation by the myeloperoxidase system on sterol efflux by the ABCA1 pathway. J Proteomics. 2011;74:2289–2299. doi: 10.1016/j.jprot.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shao B. Oda MN. Bergt C. Fu X. Green PS. Brot N. Oram JF. Heinecke JW. Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J Biol Chem. 2006;281:9001–9004. doi: 10.1074/jbc.C600011200. [DOI] [PubMed] [Google Scholar]
- 37.Shao B. Tang C. Heinecke JW. Oram JF. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J Lipid Res. 2010;51:1849–1858. doi: 10.1194/jlr.M004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith JD. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler Thromb Vasc Biol. 2010;30:151–155. doi: 10.1161/ATVBAHA.108.179226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith JD. Myeloperoxidase, inflammation, and dysfunctional high-density lipoprotein. J Clin Lipidol. 2010;4:382–388. doi: 10.1016/j.jacl.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sokolovsky M. Riordan JF. Vallee BL. Conversion of 3-nitrotyrosine to 3-aminotyrosine in peptides and proteins. Biochem Biophys Res Commun. 1967;27:20–25. doi: 10.1016/s0006-291x(67)80033-0. [DOI] [PubMed] [Google Scholar]
- 41.Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 42.Stadtman ER. Levine RL. Chemical modification of proteins by reactive oxygen species. In: Dalle-Donne I, editor; Scaloni A, editor; Butterfield DA, editor. Redox Proteomics: From Protein Modifications to Cellular Dysfunction and Diseases. Hoboken, NJ: Wiley-Interscience; 2006. pp. 3–23. [Google Scholar]
- 43.Sultana R. Poon HF. Cai J. Pierce WM. Merchant M. Klein JB. Markesbery WR. Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Wang SX. Mure M. Medzihradszky KF. Burlingame AL. Brown DE. Dooley DM. Smith AJ. Kagan HM. Klinman JP. A crosslinked cofactor in lysyl oxidase: Redox function for amino acid side chains. Science. 1996;273:1078–1084. doi: 10.1126/science.273.5278.1078. [DOI] [PubMed] [Google Scholar]
- 45.Whiteman M. Chu SH. Siau JL. Rose P. Sabapathy K. Schantz J-T. Cheung NS. Spencer JPE. Armstrong JS. The pro-inflammatory oxidant hypochlorous acid induces Bax-dependent mitochondrial permeabilisation and cell death through AIF-/EndoG-dependent pathways. Cell Signal. 2007;19:705–714. doi: 10.1016/j.cellsig.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 46.Yee CS. Seyedsayamdost MR. Chang MCY. Nocera DG. Stubbe J. Generation of the R2 subunit of ribonucleotide reductase by intein chemistry: Insertion of 3-nitrotyrosine at residue 356 as a probe of the radical initiation process. Biochemistry. 2003;42:14541–14552. doi: 10.1021/bi0352365. [DOI] [PubMed] [Google Scholar]
- 47.Zhang R. Brennan M-L. Fu X. Aviles RJ. Pearce GL. Penn MS. Topol EJ. Sprecher DL. Hazen SL. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X. Monroe ME. Chen B. Chin MH. Heibeck TH. Schepmoes AA. Yang F. Petritis BO. Camp DG. Pounds JG. Jacobs JM. Smith DJ. Bigelow DJ. Smith RD. Qian W-J. Endogenous 3,4-dihydroxyphenylalanine and dopaquinone modifications on protein tyrosine: Links to mitochondrially derived oxidative stress via hydroxyl radical. Mol Cell Proteomics. 2010;9:1199–1208. doi: 10.1074/mcp.M900321-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng L. Nukuna B. Brennan M-L. Sun M. Goormastic M. Settle M. Schmitt D. Fu X. Thomson L. Fox PL. Ischiropoulos H. Smith JD. Kinter M. Hazen SL. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng L. Settle M. Brubaker G. Schmitt D. Hazen SL. Smith JD. Kinter M. Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. J Biol Chem. 2005;280:38–47. doi: 10.1074/jbc.M407019200. [DOI] [PubMed] [Google Scholar]