

Abstract
The complexity of terpenoid natural products has drawn significant interest, particularly since their common (poly)isoprenyl origins were discovered. Notably, much of this complexity is derived from the highly variable cyclized and/or rearranged nature of the observed hydrocarbon skeletal structures. Indeed, at least in some cases it is difficult to immediately recognize their derivation from poly-isoprenyl precursors. Nevertheless, these diverse structures are formed by sequential elongation to acyclic precursors, most often with subsequent cyclization and/or rearrangement. Strikingly, the reactions used to assemble and diversify terpenoid backbones share a common carbocationic driven mechanism, although the means by which the initial carbocation is generated does vary. High-resolution crystal structures have been obtained for at least representative examples from each of the various types of enzymes involved in producing terpenoid hydrocarbon backbones. However, while this has certainly led to some insights into the enzymatic structure–function relationships underlying the elongation and simpler cyclization reactions, our understanding of the more complex cyclization and/or rearrangement reactions remains limited. Accordingly, selected examples are discussed here to demonstrate our current understanding, its limits, and potential ways forward.
1 Introduction
Terpenoids, named for their original isolation from conifer turpentine secretions, are derived from five-carbon isoprene units and, hence, sometimes termed isoprenoids.1 However, the resulting natural products are often cyclized and sometimes also rearranged, occasionally in such a complex manner as to confound recognition of their isoprenoid origin. Much of this diversity results from the manifold ways in which the constituent isoprene units can be linked together and then cyclized and/or rearranged. The ensuing complexity can perhaps best be appreciated by noting that over 55 000 such natural products are known.2
Terpenoids are most simply stratified by the number of constituent isoprene units (Fig. 1).1 Because the original terminology was derived from investigations of turpentine, where the first compounds to be isolated contained ten carbons (C10), which were defined at that time as monoterpenes, individual isoprene units are considered hemiterpenes, with monoterpenes actually containing two isoprene units. Terpenoids containing three isoprene units are termed sesquiterpenes, and those containing four isoprene units diterpenes. While quite unusual, there are five isoprene unit containing sesterterpenes, as well as seven isoprene unit containing sesquarterpenes. By contrast, six isoprene unit containing triterpenes are widely distributed, with the eight isoprene unit containing tetraterpenes also fairly common. Even longer chain isoprenoids are universally found as the prenyl chains on the quinone electron carriers (i.e., ubiquinone, as well as plastiquinone), along with the dolichols involved in glycoprotein production. Finally, natural products of mixed biosynthetic origin that includes prenylation can be termed meroterpenes.
Fig. 1.

A scheme depicting general terpenoid nomenclature and corresponding precursors (OPP, diphosphate; PPi, inorganic pyrophosphate).
The isoprene units that make up the terpenoids are derived from isopentenyl diphosphate (IPP), and its carbon–carbon double-bond isomer dimethylallyl diphosphate (DMAPP). DMAPP exhibits the allylic ester linked diphosphate that plays a key role as a facile leaving group in assembly and diversification of terpenoid natural products. Specifically, by heterolytic cleavage to generate not only a diphosphate anion but also an allylic (resonance-stabilized) carbocation, leading to carbon–carbon bond formation, typically by addition to a carbon–carbon double-bond. This type of reaction is used to both generate the acyclic precursors to the various chain length terpenoids, as catalyzed by the prenylelongases described in Section 2, as well as in many of the subsequent cyclization and/or rearrangement reactions, which are catalyzed by the terpene synthases described in Section 3. These enzymes together make up a broadly conserved superfamily, the underlying homology of which is only recognizable at the tertiary structure level, that has been termed the class I terpenoid synthases.3 In addition, while also catalyzing reactions that are carbocationic in nature, there is a separate enzyme superfamily that utilizes a protonation-initiated mechanism to drive cyclization (and sometimes subsequent rearrangement). It has been more recently suggested,4 and now demonstrated,5 that these enzymes exhibit a conserved protein fold, and they have been termed the class II terpenoid cyclases, and are described in Section 4. Here the impact of high-resolution structural characterization of these enzymes will be reviewed.
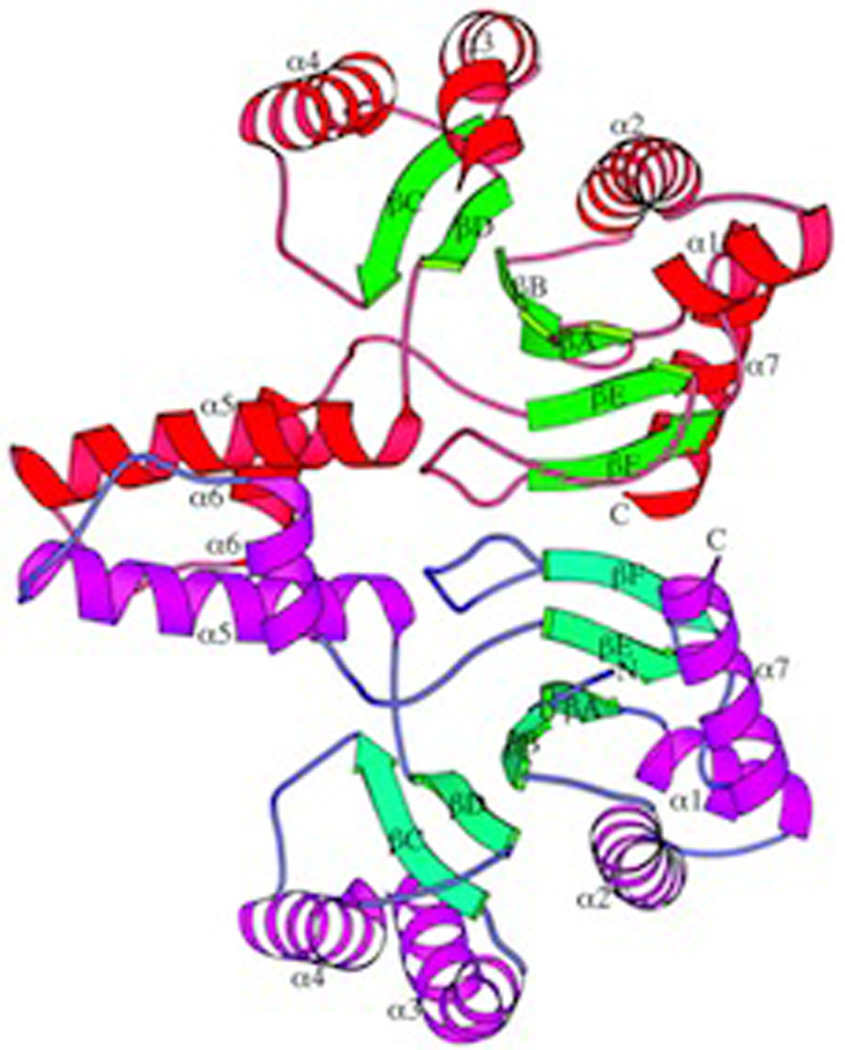
In order to provide context for the following descriptions of the various terpenoid synthase protein structures that have been determined to date (Table S1‡), two general findings are presented here, although more detailed discussions can be found later (Sections 5 and 6). First, as indicated above, the terpenoid synthase superfamilies reviewed here were defined on the basis of structural homology revealed by protein structure determination, with the class I enzymes defined by an α-helical bundle domain, and the class II enzymes by a pair of double α-barrel domains. Intriguingly, there was an ancient fusion of these two enzyme classes in plant diterpene synthases, leading to designation of the relevant domains as the class I associated α domain and class II associated β and γ domains, although these are actually found in a primary sequence order of γβα.4 Notably, these ancestral bifunctional diterpene synthases, presumably required for production of the gibberellin phytohormone intermediate ent-kaurene, seem to have given rise to all plantmono-, sesqui-, and di- terpene synthases. In particular, early gene duplication and sub-functionalization led to separate class II diterpene cyclases and subsequently acting class I diterpene synthases that, in turn, gave rise to the expanded families of class II diterpene cyclases and class I terpene synthases observed in plants. While all of the known class II diterpene cyclases retain the γβα tridomain structure, in the vast majority of plant class I terpene synthases the γ domain has been lost, leaving a βα didomain structure (Fig. 2). The second general finding noted here is that a critical role for orientation of the substrate(s) to bring the π bond of carbon–carbon double-bonds into the correct position for addition to the carbocation(s) formed in the relevant reaction has long been appreciated (e.g., Fig. 1),6 and has largely been born out by substrate/intermediate analog containing terpenoid synthase structures, where suitable such preorganization is often observed.7 Indeed, in those cases where conformation of the analog is inconsistent with that inferred from mechanistic considerations, the binding of that small molecule is considered not to be relevant to the catalyzed reaction.
Fig. 2.

The proposed evolutionary relationships among the terpenoid synthases.
2 Prenylelongases
The coupling of multiple isoprene units relies on ionization of an allylic diphosphate ester-containing precursor, followed by intermolecular addition of the resulting allylic carbocation to a carbon–carbon double-bond (i.e., a class I type reaction). Typically, this occurs from C1 of the allylic carbocation to C4′ of the terminal carbon–carbon double-bond of IPP, to generate the “head-to-tail” linkage most often found in multiple isoprenyl unit containing terpenoids. This is then followed by deprotonation at C2′ of the IPP unit, yielding either a cis or trans carbon–carbon double bond, and forming a labile allylic diphosphate ester bond in the elongated precursor (e.g., Fig. 1). Alternatively, it also is possible for the initially generated allylic carbocation to add to the carbon–carbon double-bond of allylic isoprenyl precursors, leading to formation of a distributed cyclopropyl carbocation and alternative couplings. For example, direct deprotonation to a cyclopropyl, or rearrangement to a branched or cyclobutyl, carbocation, with subsequent deprotonation to form an elongated product. In each of these cases, the resulting product no longer contain an allylic diphosphate ester bond, but at least in some cases further transformations lead to a compound that can undergo subsequent cyclization via protonation-initiated reactions (e.g., the triterpene precursor squalene – as described below in Section 4). While these enzymes can be generally described as prenyltransferases, they might be more accurately designated as prenylprenyltransferases, in part to distinguish them from the phylogenetically unrelated enzymes that transfer isoprenoid moieties onto other types of molecules, but here we use the simpler descriptor prenylelongase. Such separation of the prenylelongases from other prenyltransferases is consistent with the observed homology, including distinct structural folds exhibited by the other prenyltransferases, even those from other natural product biosynthesis – e.g. those that prenylate aromatic small molecules,8–10 whose structures differ from that of the prenylelongases described below.
2.1 Trans-isoprenyl diphosphate synthases
The isoprenyl diphosphate synthases (IDSs) that generate trans double-bonds form a conserved enzyme family. Of particular importance here, these form the prototypical precursors to mono-, sesqui-, and di- terpenes – i.e., the transoid acyclic head-to-tail joined geranyl, farnesyl, and geranylgeranyl diphosphates, respectively (Fig. 1). Specifically, by the sequential addition of IPP to the allylic precursor DMAPP, forming (E)-geranyl diphosphate (GPP), with subsequent addition of IPP to GPP to form (E,E)-farnesyl diphosphate (FPP), and further addition of IPP to (E,E)-FPP to form (E,E,E)-geranylgeranyl diphosphate (GGPP). While these enzymes have been termed short-chain trans-IDSs, they are related to those forming longer-chain trans-isoprenyl diphosphates, and given the somewhat surprising ease with which product chain length can be altered, we group them all together here. These trans-IDSs are in fact the prototypical class I terpenoid synthases, a structure for which was determined first (see below), and which seem likely to have been the ancestral enzymes in this superfamily given their requisite role in producing the trans-isoprenyl chains found in the ubiquitous quinone electron carriers.
2.1.1 (E,E)-Farnesyl diphosphate synthase
Given the nearly ubiquitous production of (E,E)-FPP (e.g., based on the use of these as allylic precursors/primers for the production of longer chain isoprenyls), it is perhaps not surprising that the first structure determined for an IDS, indeed for any terpenoid synthase, was that of such an FPP synthase, specifically that from the domestic chicken, Gallus gallus.11 While this initial structure did not contain any substrate, intermediate, or product analogs, it revealed the α-helical bundle fold that would come to be associated with class I terpenoid synthases (i.e., as the α domain), with putative active site nestled within, based in part on the presence therein of two conserved aspartate-rich motifs – these have been termed the first and second (FARM and SARM, respectively) – and generally appear as DDxxD (Fig. 3). In particular, the aspartates from these motifs had already been shown to be catalytically important,12 and their side-chains were found protruding into this cavity. Later reported structures containing substrate analogs definitively demonstrated that this internal cavity was the active site.13
Fig. 3.
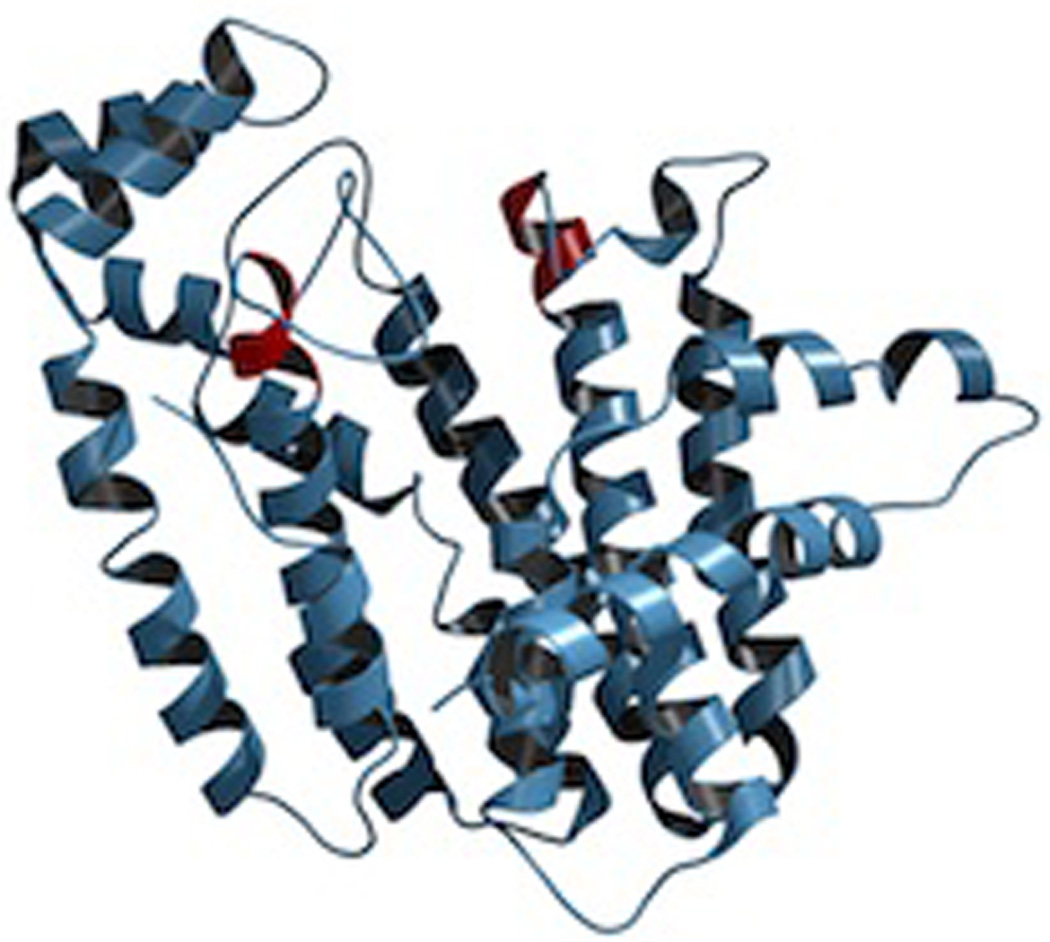
A depiction of avian farnesyl diphosphate synthase (ribbon diagram, with location of aspartate-rich motifs indicated by red coloring). Reprinted with permission from ref. 7. Copyright 2006 American Chemical Society.
Given the requirement for divalent magnesium ions (Mg2+), it had been supposed that the DDxxD motifs would be involved in binding these enzymatic co-factors. This was verified by the later reported substrate analog containing crystal structures of avian FPP synthase.13 However, the exact roles postulated for these residues required refinement upon determination of substrate analog co-crystal structures for the human FPP synthase,14 particularly that containing the unreactive allylic substrate analog dimethylallyl thiolodiphosphate along with IPP.15 In particular, it then became clear that the FARM and SARM chelate a trio of Mg2+ ions that are involved in binding and ionization of the diphosphate moiety of the allylic substrate (Fig. 4). Determination of substrate analog containing structures for the FPP synthase from Escherichia coli further indicated that the released diphosphate anion may act as the catalytic base,14 which seems to be recurring theme in these types of enzymes, as will become evident below. By contrast, the diphosphate of IPP, which is retained in the resulting product, is more simply bound to a number of conserved basic residues.
Fig. 4.
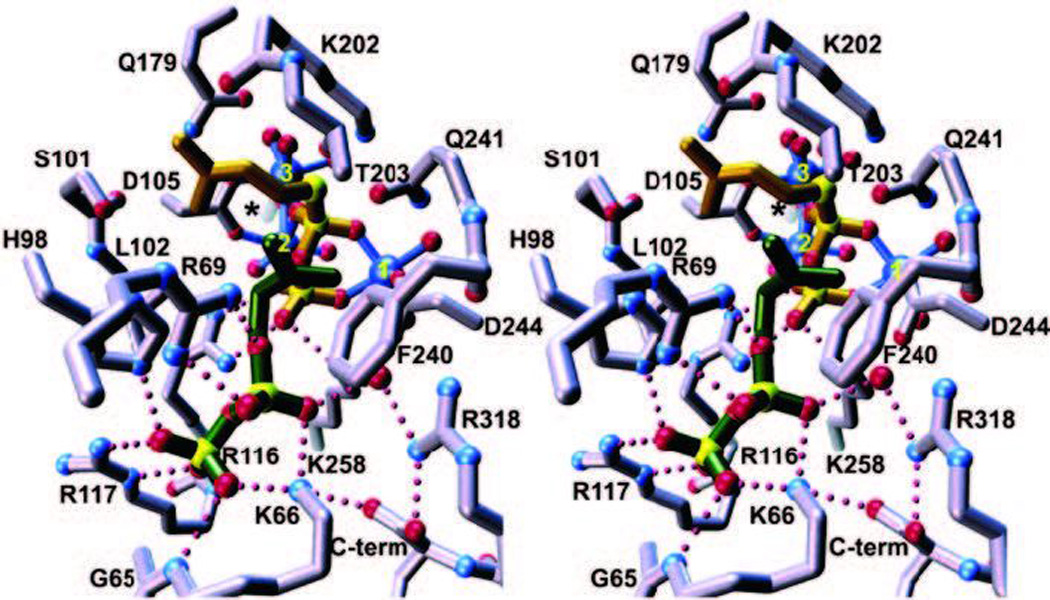
A depiction of the active site of the farnesyl diphosphate synthase from bacteria (E. coli), which is a homodimer (the subunit in the fore-ground is purple, that in the background, which does form part of the active site cavity, in red). The trio of Mg2+ ions are shown as blue spheres labeled 1–3, dimethylallyl-S-thiolodiphosphate in yellow, and isopentenyl diphosphate in green. Asp-111 from the first aspartate-rich motif is only indicated by an asterisk for clarity. Metal ion–ligand interactions are shown as solid blue lines, while hydrogen bond interactions are shown as dotted magenta lines. Reprinted with permission from ref. 14. Copyright 2004 The American Society for Biochemistry and Molecular Biology.
The early avian FPP synthase structures led to significant insights into the enzymatic determinants of product chain-length. In particular, careful examination of the enzyme crystal structures coupled to comparison of the amino acid sequences from FPP versus longer chain trans-IDSs suggested that the residues 4 and 5 positions upstream of the FARM would be important in this regard (Fig. 5). The presence of at least one aromatic residue at these positions, which made up the “bottom” of the active site cavity, was a conserved feature of FPP synthases, but smaller residues were found at these positions in longer chain producing homologs. Strikingly, substitution of smaller residues for these aromatic residues (which are both phenylalanines) in the avian FPP synthase led to the production of longer chain isoprenyl diphosphates, and similar studies also have been reported with FPP synthases from diverse other organisms.16 While crystal structures have since been reported for bacterial and the human FPP synthases as well,14,15,17 providing some details regarding the binding of bisphosphonate pharmaceuticals to this molecular target, the insights into chain length control generated from the original avian FPP synthase structures remains one of the most striking successes resulting from structural analysis of the enzymes involved in terpene biosynthesis. Indeed, similar types of investigations involving substitution of small for large (or large for small) have been reported with many other IDSs, as discussed below.
Fig. 5.
A depiction of avian farnesyl diphosphate synthase showing aromatic residues (pink) that help dictate product chain length (as defined by cavity size shown as meshwork enclosure) as well as apartate-rich motifs with a bound Mg2+ (green sphere). Reprinted with permission from ref. 13. Copyright 1996 National Academy of Sciences, USA
2.1.2 (E,E,E)-geranylgeranyl diphosphate synthase
Sequence alignments indicated that the chain length determinants described above for FPP synthases were not universal, with the homologous (E,E,E)-GGPP synthases divided into three classes based on the appearance of aromatic/bulky residues at the 4th and/or 5th position prior to the FARM, as well as the exact conservation of the FARM itself (DDxxD versus DDxxxxD). The first GGPP synthase crystal structure to be determined was that of the yeast (Saccharomyces cerevisiae) enzyme, which has the prototypical DDxxD FARM motif, but does not contain a bulky residue at either the 4th or 5th position upstream of this.18 Beside the expected conserved α domain fold, there were some additional parallels to the FPP synthase in that the “floor” of the active site was also constructed from bulky aromatic or large aliphatic residues (albeit these are found downstream rather than upstream of the FARM), with substitution of these by smaller residues enabling production of longer chain isoprenyl diphosphates. Furthermore, by substituting a tyrosine for the serine found at the 4th position upstream of the FARM, it was possible to convert this GGPP synthase to selectively produce FPP instead, emphasizing the importance of the previously obtained insight into chain length determinants.
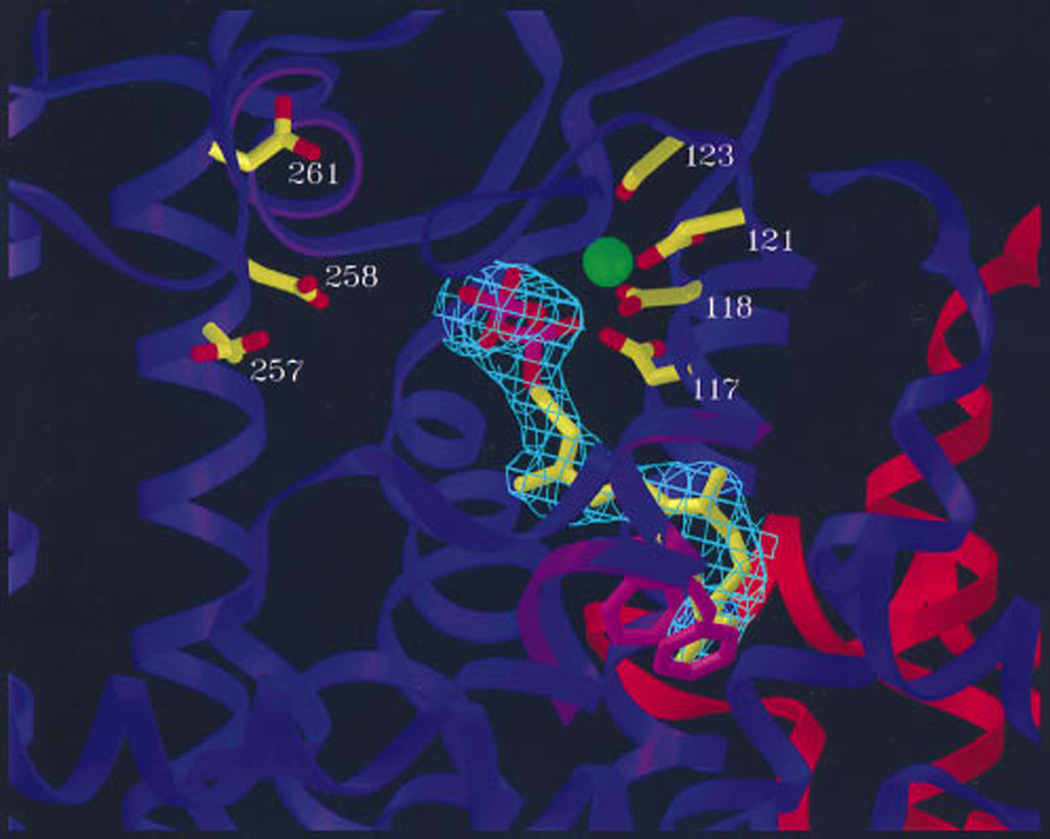
The structure of the human GGPP synthase also has been reported, and was found to consist of a hexameric assembly (a trimer of the previously observed homodimeric structures).19 Intriguingly, the diphosphate of the GGPP found in these structures was bound in the allylic substrate site (e.g., ligated to the Mg2+), rather than in the IPP site as would be expected for the initial enzymatic product, providing structural insight into the previously observed product inhibition effect (Fig. 6). A structure also has been reported for the mustard plant (Sinapis alba) GGPP synthase, wherein GGPP was bound with its diphosphate in the IPP site, but isoprenyl tail in the allylic precursor site, which presumably corresponds to the enzymatic product complex.20 Later structures of the yeast GGPP synthase with GGPP bound exhibited a similar configuration. These later reported structures of yeast GGPP synthase included some containing bisphosphates, demonstrating their ability to bind and inhibit not only FPP, but also GGPP synthases, including that from humans, as well as longer chain producing cis-IDSs from bacteria, as also mentioned below.21
Fig. 6.
A depiction of bound GGPP in the active site of the human GGPP synthase demonstrating binding of the diphosphate to Mg2+ ions in the allylic diphosphate site. Reprinted with permission from ref. 19. Copyright 2006 The American Society for Biochemistry and Molecular Biology.
2.1.3 (E)-geranyl diphosphate synthase
The only reported structure of a geranyl diphosphate synthase is representative of the heteromeric trans-IDSs. In particular, this crystal structure is of the geranyl diphosphate synthase from peppermint (Mentha piperita),22 which is composed of large and small subunits, with the large subunit being closely related to the homomeric trans-IDSs described above (particularly GGPP synthases) and assuming the expected α domain fold, while the small subunit is not related to these.23 Indeed, while the large subunit contains the requisite DDxxD motifs and presumably catalyzes elongation, the small subunit is not thought to directly participate in catalysis, although it has been shown to regulate chain length – e.g., dimerizing with other plant homomeric GGPP synthases with resulting selective production of GPP.24 However, the structure does not yet appear to have led to any insight into exactly how the small subunit regulates product chain length in the catalytic large subunit. Interestingly, while a structure has been reported for what was previously described as a GPP synthase from Arabidopsis thaliana, this enzyme also was reported to actually produce longer chain trans-isoprenyl diphosphates C25–C45 in length, consistent with the observed large active site cavity.25
2.1.4 Longer chain trans-isoprenyl diphosphate synthases
Structures for several trans-IDSs that produce longer chain lengths have been determined, and these exhibit the same α domain fold as other members of this enzymatic (class I) family and have enabled similar investigations of chain length determinants.25–29 In particular, it is possible to increase product length by substitution of bulky residues at the base of the active site cavity with smaller residues and visa versa (Fig. 7). For example, substitution of aromatic residues at the 4th and 5th position before the FARM converts an octaprenyl (C40) diphosphate synthase to selective production of GGPP instead.26 This further emphasizes the conserved nature of the trans-IDS family, with the observed diversification presumably enabled by such facile conversion to production of different chain lengths.
Fig. 7.
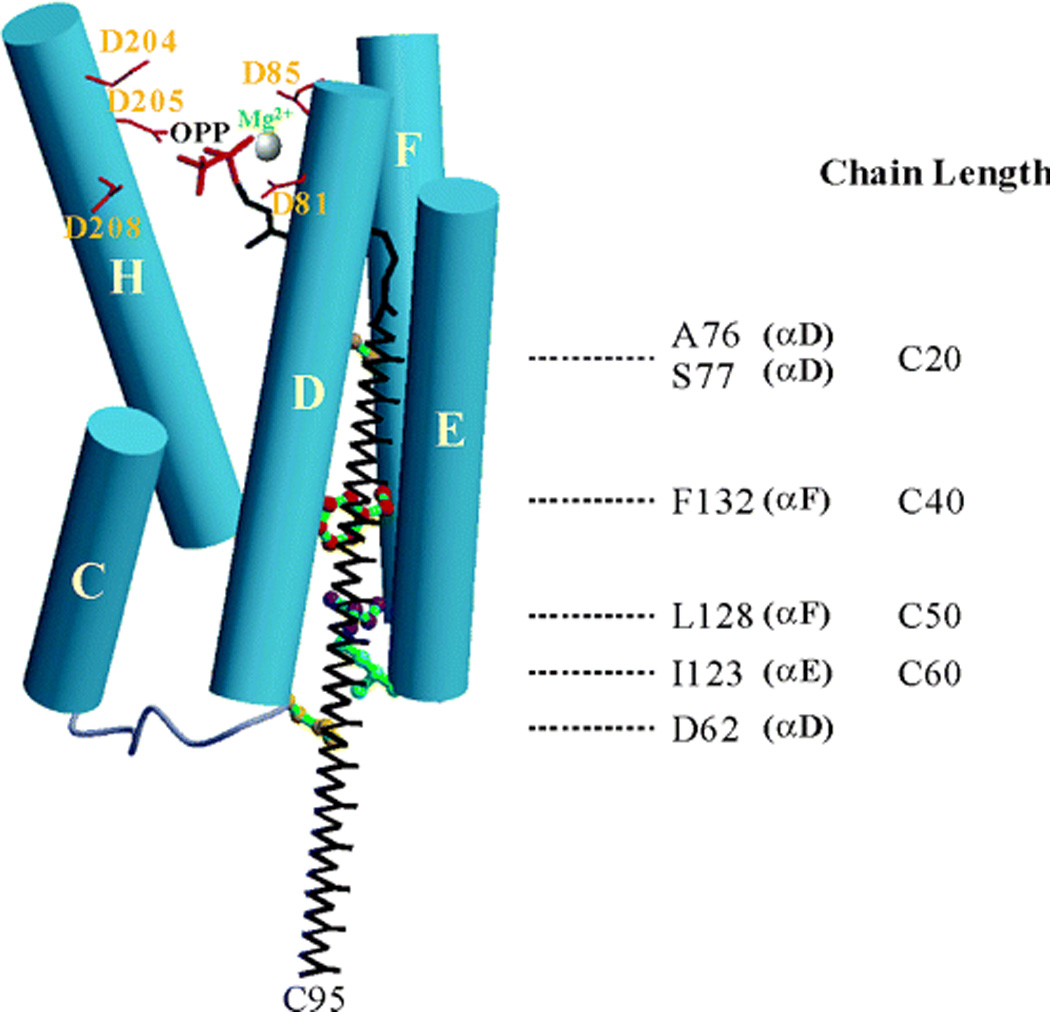
A depiction of the product chain length determinants for the octaprenyl diphosphate synthase from Thermotoga maritima. Shown are the aspartate-rich motifs as well as various residues that impact product chain length (as indicated on the side). Reprinted with permission from ref. 27. Copyright 2004 American Chemical Society.
2.2 Cis-isoprenyl diphosphate synthases
The production of head-to-tail joined cis-isoprenyl diphosphates, which requires alterations in the orientation of IPP relative to the allylic precursor, is catalyzed by a distinct family of prenylelongases. In particular, the determination of crystal structures for several of these cis-IDSs has been reported, which demonstrated that they exhibit a distinct tertiary fold (e.g., Fig. 8), along with providing some insight into the underlying enzymatic mechanism, as well as leading to studies on product chain length determinants.21,30–33 For example, a structure has been determined for a short chain cis-IDS from Mycobacterium tuberculosis.33 This enzyme adds IPP to GPP to produce (Z,E)-FPP, which provides the primer utilized by a subsequently acting decaprenyl diphosphate synthase, whose crystal structure also was reported.33 Comparison of these homologous structures then led to some insights into chain length determinants in at least the shorter chain producing enzyme, where substitution of an alanine for leucine led to some production of longer chain isoprenyl diphosphates.34
Fig. 8.
A depiction of the undecaprenyl diphosphate synthase from Escherichia coli (ribbon diagram) as an example of the distinct fold exhibited by cis-prenyl transferases. Reprinted with permission from ref. 31. Copyright 2001 The American Society for Biochemistry and Molecular Biology.
2.3 Cyclopropyl forming prenylelongases
While the addition of IPP enables the head-to-tail coupling reactions described above, it also is possible to couple two allylic precursors. It has long been recognized that squalene is produced by condensation of two molecules of FPP via the cyclopropyl containing intermediate presqualene diphosphate. In turn, squalene synthases are phylogenetically related to the enzymes that produce the carotenoid precursor phytoene by condensation of two molecules of GGPP. In both cases, the cyclopropyl group is cleaved, with loss of the diphosphate, to yield their eponymous acyclic olefin products (in the case of squalene, this also includes NADPH dependent reduction of the central double-bond). Somewhat surprisingly given the lack of any detectable sequence homology, determination of a crystal structure for the human squalene synthase revealed structural homology to the trans-IDSs – i.e., the same α domain fold, including the presence and arrangement of aspartate-rich motifs (Fig. 9).35 Notably, the resulting squalene, particularly the derived oxido-squalene, serves as a precursor for triterpenoid natural products, as described below. In addition, the production of certain unusual/irregular monoterpenes has been postulated to proceed via similar cyclopropyl forming addition reactions, albeit in this case from coupling of two molecules of DMAPP.36 More recently, it has been discovered that the relevant enzymes are closely related to trans-IDSs, as revealed by the paralogous relationship of chrysanthemyl diphosphate synthases to the (E,E)-FPP synthases found in the same plant.37,38 Furthermore, while no crystal structures are available for such an enzyme, chimeric analysis has revealed that these are capable of forming not only cyclopropyl, but branched or cyclobutanyl-containing monoterpenoid diphosphate products,39,40 which arise from rearrangement of an initially formed distributed cyclopropyl carbocation (Fig. 10).41 Thus, it seems likely that all cyclopropyl forming prenylelongases, regardless of final product outcome, will fall into the class I terpenoid synthase superfamily as well.
Fig. 9.
A depiction of the human squalene synthase, with comparison to FPP synthase, including location of asparate-rich motifs (as indicated). Reprinted with permission from ref. 35. Copyright 2000 The American Society for Biochemistry and Molecular Biology.
Fig. 10.
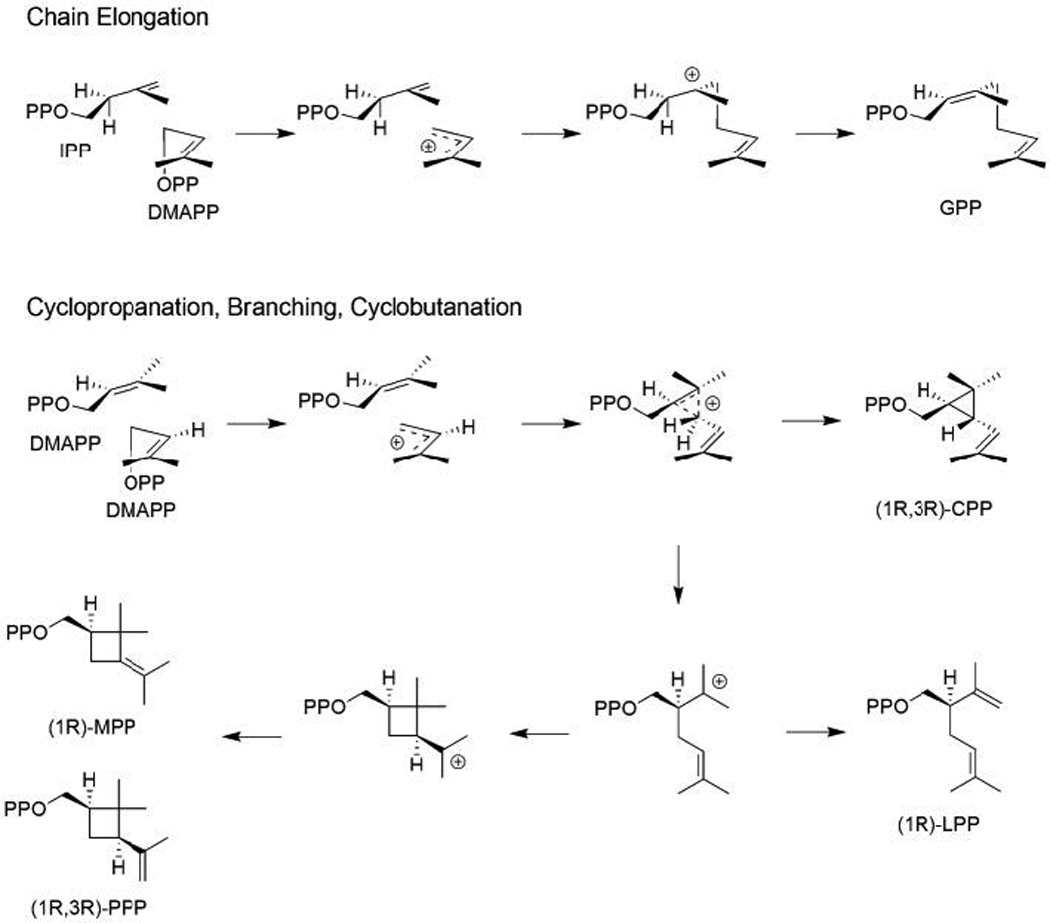
A mechanistic scheme for the biosynthesis of irregularly coupled isoprenoid diphosphates, with comparison to trans-prenylelongation. Reprinted with permission from Science, ref. 40. Copyright 2007 American Association for the Advancement of Science.
3 Class I terpene synthases
The class I terpene synthases (TPSs) characteristically catalyze ionization of the allylic diphosphate ester bond in their isoprenyl substrates. This is mechanistically similar to the reactions catalyzed by the prenylelongases discussed above. Consistent with the derivation of trans-IDSs to alternative (cyclopropyl forming) coupling reactions, structural analysis revealed that TPSs also are related to the prenylelongases (i.e., are composed of structurally homologous α domains), although no sequence homology is readily evident. Thus, the TPSs fall into the class I terpenoid synthase superfamily. Interestingly, TPSs further generally react with transoid isoprenyl diphosphate substrates – i.e., the product of the ancestral trans-IDSs4 – although TPSs that react specifically with cisoid isoprenyl diphosphates are now known.42,43 In any case, following ionization, the initially formed allylic carbocation most often adds to an intramolecular carbon–carbon double-bond (albeit sometimes via formation of a rearranged tertiary diphosphate intermediate), resulting in cyclization, which can be followed by further cyclization and/or rearrangement (e.g., Fig. 11). However, this is not strictly necessary, as immediate deprotonation, with formation of an additional carbon–carbon double-bond instead, also is observed, leading to their more general designation as synthases – i.e., rather than cyclases (although this later nomenclature would apply to the majority of these enzymes). In any case, TPSs often catalyze very complex cyclization and/or rearrangement reactions. It also should be noted that these enzymes do not necessarily directly deprotonate the final carbocation, as capture of water also has been observed, with either direct deprotonation to form a hydroxyl group, or even subsequent (hetero)cyclization prior to deprotonation, forming a cyclic ether (as discussed below in Section 3.2.3). Indeed, it is even possible for readdition of the diphosphate anion to be used to quench the final carbocation after cyclization (as discussed below in Section 3.2.1), further emphasizing the ability of these enzymes to generate chemical complexity. Finally, as often noted, TPSs exhibit a wide range of catalytic promiscuity, while some are quite specific, others will yield a characteristic range of products from the same substrate.2,7,44
Fig. 11.

A scheme of representative mono- and sesqui- terpene cyclization reactions.
3.1 Sesquiterpene synthases
The first TPS structures to be determined were those of two FPP reactive (i.e., sesqui-)TPS.45,46 These structures revealed homology not only between TPSs from diverse organisms (bacteria and plants), but also of these to the trans-IDSs, leading to the initial definition of the (class I) terpenoid synthase superfamily.45 This structural homology includes not only the α-helical bundle tertiary fold (i.e. α domain), but also similar arrangement of divalent metal ion binding motifs therein. Although in the TPSs, while the first is similarly conserved as DDxx(D,E), the second has diverged to a (N,D)Dxx(S,T)xxxE consensus sequence, which is often referred to as the NSE/DTE motif.7 Just as in the trans-IDSs, these motifs in TPSs chelate a trio of divalent magnesium (or manganese) ions that are, in turn, used to bind the substrate diphosphate moiety, and are required for ionization of the allylic diphosphate ester bond to initiate catalysis.
3.1.1 Pentalenene synthase
One of these first TPSs crystal structures to be solved was that of the pentalene synthase from the bacteria Streptomyces UC5319,45 which revealed clear structural homology between the TPSs and trans-IDSs, later leading to the designation of this fold as the class I terpenoid synthase fold.3 However, this structure did not contain any ligands, or even Mg2+ co-factors, which resulted in some early confusion over the role of certain residues in the active site – e.g., those of the NSE/DTE motif (Fig. 12). Nevertheless, this was quickly clarified by comparison to other TPS structures, as well as follow-up mutational analysis.47 Notably, this included substitution of glutamates for the aspartates in the first motif, which was consistent with the observed conservation of this as DDxx(D,E) – i.e., there were deleterious effects on catalytic activity with such substitution for the first two, although not last, positions (all of which are aspartates in the wild type enzyme). Additional investigations of a potential catalytic base were unsuccessful, and led to the suggestion that this function may be fulfilled by the released diphosphate anion,48 which draws some parallels to the trans-IDSs, where at least the FPP synthase catalyzed reaction has been suggested to utilize such a co-product dependent mechanism as well (see Section 2.1.1 above).
Fig. 12.
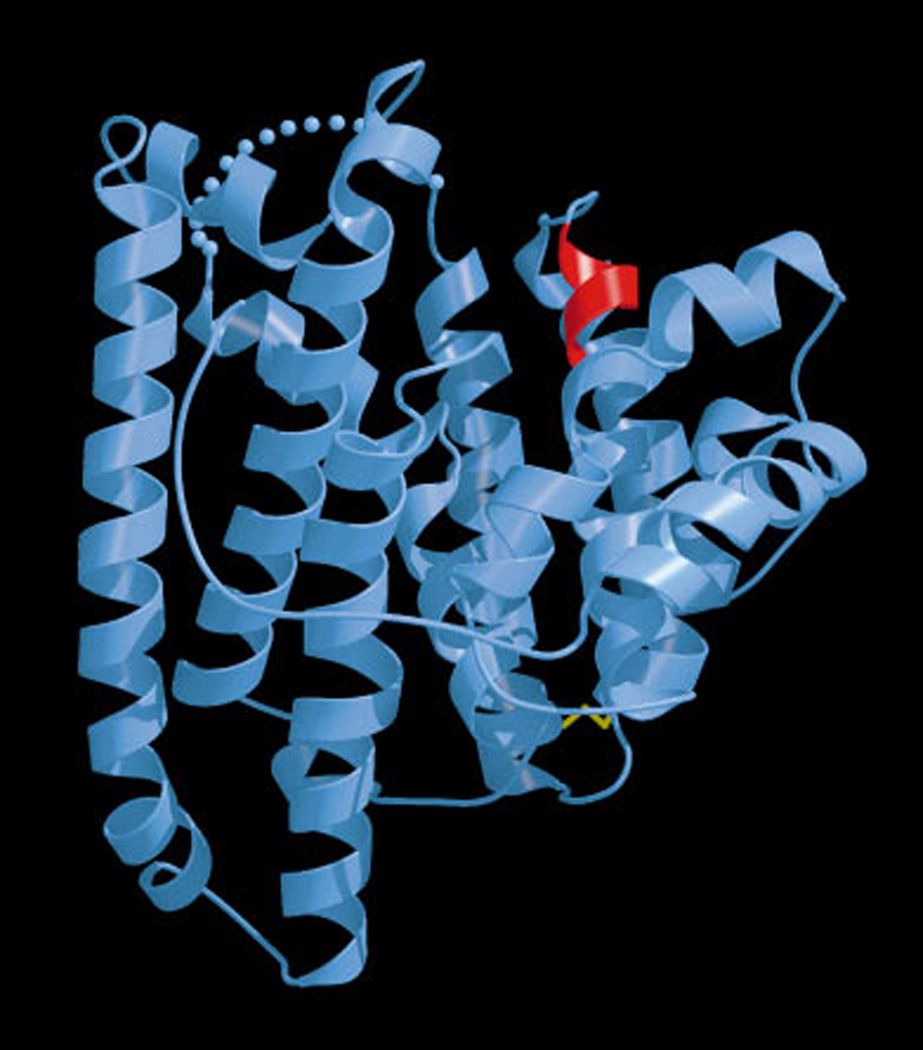
A depiction of pentalenene synthase as shown in the original report (reprinted with permission from Science, ref. 45), with only the DDxxD motif corresponding to the first aspartate-rich motif of the trans-isoprenyl diphosphate synthases highlighted in red. Copyright 1997 American Association for the Advancement of Science.
3.1.2 Epi-aristolochene synthase
Reported at the same time as that of pentalenene synthase were crystal structures of 5-epi-aristolochene synthase from tobacco (Nicotiana tabacum) – either alone or in the presence of one of two substrate analogs, farnesyl hydroxyphosphonate or trifluorofarnesyl diphosphate, both of which further contained a trio of Mg2+ ions bound to the DDxxD and NSE/DTE motifs.46 However, in addition to a C-terminal domain with the α-helical bundle class I terpenoid synthase fold, the tobacco epi-aristolochene synthase also contained an N-terminal α-helical domain (Fig. 13). The corresponding sequence is conserved among all the plant TPSs, although it is not involved in catalysis and appears to be relictual (i.e., this is the β domain mentioned in the Introduction – see also Fig. 2). Nevertheless, despite this βα didomain structural organization, it should be noted that the N-terminus of plant TPSs folds back to form part of the C-terminal α domain when substrate analogs are bound, helping shield the active site from bulk water. Interestingly, comparison of the tobacco epi-aristolochene synthase structure with the closely related premnaspirodiene synthase from Hyoscyamus muticus demonstrated that the residues lining the active site cavity were conserved between these functionally distinct enzymes, implying that subtle changes in active site geometry/contour imposed by differences in residues behind those that directly contact the substrate are sufficient to change product outcome. Indeed, guided by previous chimeric analysis,49 follow-up mutagenesis identified a set of residues that could be exchanged to interconvert product specificity of the parent enzyme.50 In addition, the reactions catalyzed by both epi-aristolochene and premnaspirodiene synthases proceed via a germacrene A intermediate, which requires reprotonation to lead to final product outcome (Fig. 14), and substitution of a tyrosine identified as a potential proton donor to phenylalanine in epi-aristolochene synthase resulted in an enzyme that specifically produced germacrene A, providing some support for this hypothesis.51 More recently, the ability of this enzyme to react with (Z,E)- as well as (E,E)-FPP also has been structurally examined.52
Fig. 13.

A depiction of the complex of epi-aristolochene synthase with farnesyl hydroxyphosphonate (FHP), as well as trio of Mg2+. The C-terminal class I terpenoid synthase domain is shown in orange, while the N-terminal domain commonly found in plant class I terpene synthases is shown in blue. Reprinted with permission from Science, ref. 46. Copyright 1997 American Association for the Advancement of Science.
Fig. 14.

The proposed cyclization mechanism for epi-aristolochene synthase.
3.1.3 Aristolochene synthases
Crystal structures for two distinct aristolochene synthases have been reported, one from the blue cheese mold Penicillium roqueforti,53 and the other, more recently determined, from the filamentous fungi Aspergillus terreus.54,55 These two fungal sesqui-TPSs both exhibit the expected α domain fold, but their sequences are relatively divergent, sharing only ~60% identity, with the enzyme from A. terreus exhibiting tight specificity for production of aristolochene, while that from P. roqueforti is less specific, and produces small amounts of the known (S)-(−)-germacrene A intermediate. Strikingly, the structure of the A. terreus enzyme in complex with inorganic pyrophosphate and Mg2+ demonstrates that its active site cavity is highly complementary to aristolochene, suggesting that such tight steric constraints contribute to the observed specificity (Fig. 15).54 Later reported structures of this aristolochene synthase in complex with various substrate analogs were used to further suggest a binding order for the trio of divalent metal ions, occurring in conjunction with binding of the substrate diphosphate moiety.55
Fig. 15.
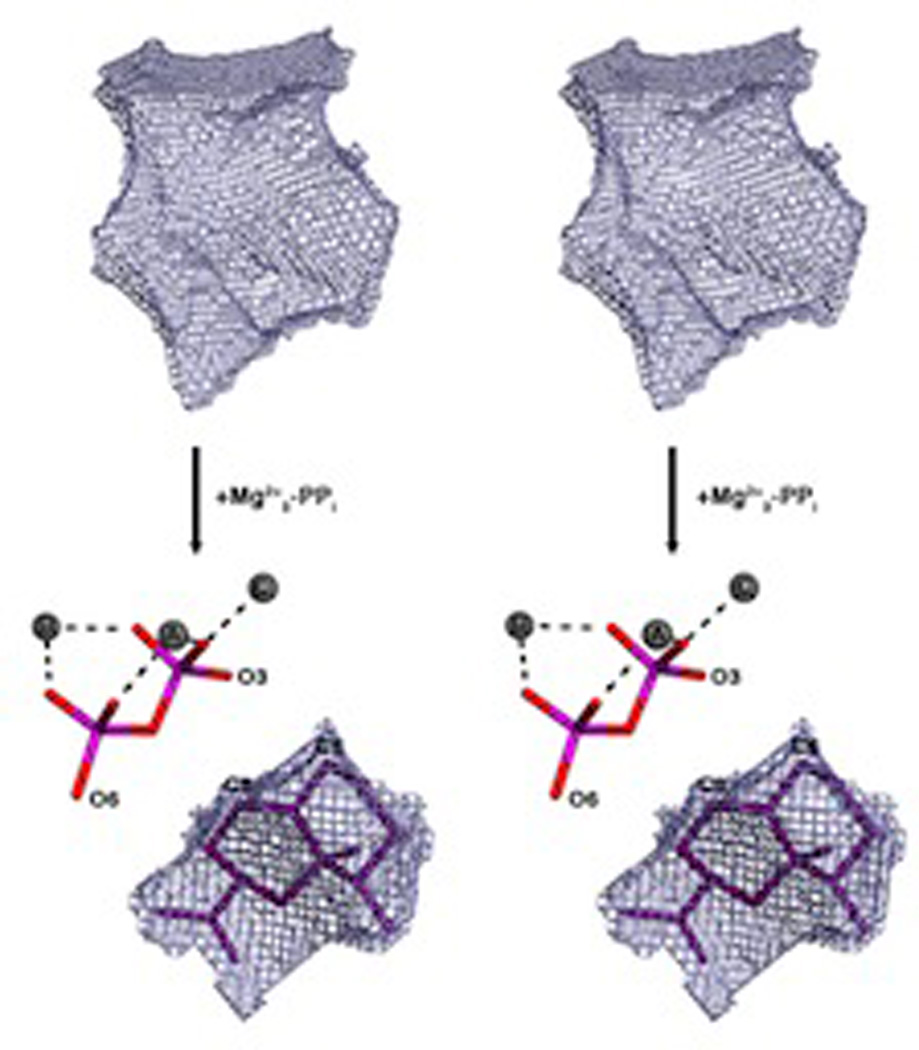
A stereoview of the active site cavity (meshwork enclosure) from aristolochene synthase in the absence and presence of ligands (Mg2+3-PPi only), demonstrating the highly complementary nature of this to the product, which is modeled into the remaining space (with Mg2+3-PPi also shown for clarity). Reprinted with permission from ref. 54. Copyright 2007 American Chemical Society.
Notably, labeling studies have demonstrated that the final aristolochene product incorporates a proton derived from water, strongly supporting a mechanism incorporating deprotonation to the stable germacrene A intermediate followed by protonation (i.e., rather than intermolecular proton transfer). Nevertheless, this is a tightly bound reaction intermediate, and the enzyme is unable to convert exogenous germacrene A to aristolochene. Intriguingly, while the P. roqueforti aristolochene synthase was suggested to use a tyrosine as the general acid to protonate the germacrene A intermediate, similar to the situation described for the tobacco epi-aristolochene synthase above, the analogous Tyr → Phe mutant still produced largely aristolochene.56 Substitution of smaller residues (e.g., alanine) for this Tyr led to production of largely acyclic farnesene, which was interpreted as indicating a role for this residue in folding the isoprenyl tail to enable cyclization to occur (i.e., by enforcing proximity of the C10–C11 double-bond to the allylic carbocation formed by initial ionization).57 On the other hand, substitution of leucine for a tryptophan suggested to stabilize the eudesmanyl carbocation formed following protonation of germacrene A (via aromatic π interactions), results in a mutant enzyme that produces only germacrene A, albeit with greatly reduced catalytic efficiency,58 and it should be noted that similar effects also are observed with conservative substitutions for the NSE/DTE, although not DDxxD, motif.56 In any case, the identity of the general acid in the aristolochene synthase remains unknown, and it is unclear how this is selectively accomplished only following ionization-initiated production of germacrene A from FPP.
3.1.4 Trichodiene synthase
Perhaps the structurally best-defined TPS is the trichodiene synthase from the fungus Fusarium sporotrichioides, for which multiple structures of not only the wild-type,59 but also several mutants,60–63 have been determined. The originally reported structures, of the wild-type enzyme, compared unliganded and pyrophosphate + Mg2+3 states, demonstrating that binding of the pyrophosphate co-product not only leads to binding of the trio of divalent metal ions, but conformational changes that “close” the active site.59 The ability of this pyrophosphate-Mg2+3 complex to trigger active site closure has since been observed in other TPSs as well.54,64 Some of the mutant structures contained an aza analog of the bisabolyl carbocation intermediate bound in the presence of pyrophosphate and Mg2+ to mimic this step in the reaction mechanism. However, the aza-bisabolene was observed in multiple or disordered conformations, much of which appears to be driven by orientation of the aza analog to allow ion-pairing interactions with the tightly bound pyrophosphate-Mg2+3 complex, indicating that the catalyzed reaction is under kinetic rather than thermodynamic control.63 Moreover, the various investigated mutants exhibit reduced product specificity/fidelity, and their structures reveal larger active site cavities. This contrasts with the high fidelity imposed by the tight steric constraints of the A. terreus aristolochene synthase discussed above, and together these results indicate that the promiscuous production of varied products is at least in part a reflection of the greater steric freedom afforded by large active site volume – i.e., both in terms of what intermediates can be formed, as well as their susceptibility to terminating deprotonation (which also has been proposed to be catalyzed by the pyrophosphate anion co-product).60–63
3.1.5 Delta-cadinene synthase
The structure of (+)-δ-cadinene synthase from tree cotton (Gossypium arboreum) has been reported, in both apo form as well as in complex with a fluorinated 2F-FPP substrate analog.65 This enzyme exhibits the βα didomain structure expected for plant TPSs. Interestingly, in place of the usual NSE/DTE motif this domain contains a DDxxE sequence that serves the same divalent metal binding purpose instead, which draws some further parallels to the analogous SARM motif found in the trans-IDSs.
3.1.6 Epi-isozizaene synthase
The structure of epi-isozizaene from the bacteria Streptomyces coelicolor A3(2) has been recently reported, including some follow-up mutagenesis studies.66 Perhaps most interesting were the effects from substitution of a number of aromatic residues suggested to stabilize carbocation intermediates, which produced a mixture of “simpler” sesquiterpenes – i.e., resulting from less complex cyclization and rearrangement than that required for production of epi-isozizaene. Determination of the structure of one such mutant demonstrated an increased active site cavity, consistent with the observed decreased fidelity in product outcome. However, this also confounds interpretation, leaving in question the relevance of aromatic π stabilization of specific carbocation intermediates versus simple steric increases in accessible reaction space.
3.1.7 Alpha-bisabolene synthase
The structure of α-bisabolene synthase from grand fir (Abies grandis) revealed not only the usual βα domain pair expected for plant TPSs, but an additional domain as well.67 This is the γ domain mentioned in the Introduction – see also Fig. 2, which appears to be relictual, and will be described in more detailed below as it was first observed and is most relevant in diterpene cyclases. It should be noted that this report included apo structures as well as those with various substrate analogs or inhibitors bound,67 with observation of similar loop closure over the relevant active site as described for previously determined terpene synthase structures.
3.2 Monoterpene synthases
The only GPP reactive (i.e., mono-) TPSs whose structures have been determined are all from plants, all three of which then exhibit the βα didomain structure associated with plant TPSs.7 Interestingly, these three structurally defined mono-TPSs provide examples of the alternative chemistries that can be utilized by TPSs. In particular, while limonene synthase quenches the final cyclized carbocation intermediate by deprotonation to form an olefin, cineole synthase provides an example of the incorporation of water to form a cyclic ether, and bornyl diphosphate synthase a unique example of readdition of the diphosphate anion to the cyclized final carbocation.
3.2.1 Limonene synthase
The structure of limonene synthase serves as an admirable introduction to monoterpene cyclization reactions.68 The C2–C3 trans double-bond of GPP prevents attack of the C1 allylic carbocation on the C6–C7 double-bond. Thus, in order for cyclization to occur, GPP is isomerized to linalyl diphosphate (LPP), enabling reposition of C1 (now part of the C1–C2 vinyl group) for addition to the C6–C7 double-bond upon reionization, forming a key cyclized α-terpinyl cation (Fig. 11). Previous mutational analysis indicated that a tandem pair of arginines at (or near) the N-terminus of the mature enzyme (plant mono- and di- terpene synthase are imported into plastids, with subsequent removal of the N-terminal targeting peptide), are specifically required for this initial isomerization.69 This not only indicated that that the N-terminal sequence folded back to form part of the class I active site despite the intervening β domain found in plant TPSs, but also suggested direct interaction of these residues with the substrate diphosphate. Notably, limonene synthase structures were determined in the presence of poorly reactive 2-fluoro analogs of geranyl and linalyl diphosphate. These indicated that the N-terminal pair of arginines did not directly interact with the substrate diphosphate, leading to the suggestion that these exerted indirect effects through their interactions with other conserved residues. However, the 2-fluoro-GPP analog was found to actually have been isomerized to 2-fluoro-LPP during crystallization, leaving some possibility that these arginines might interact more specifically with the diphosphate moiety of GPP. No catalytic base for terminating deprotonation was identified, leaving one to speculate that this function is fulfilled by the diphosphate anion, as suggested in several other cases above.
3.2.2 Bornyl diphosphate synthase
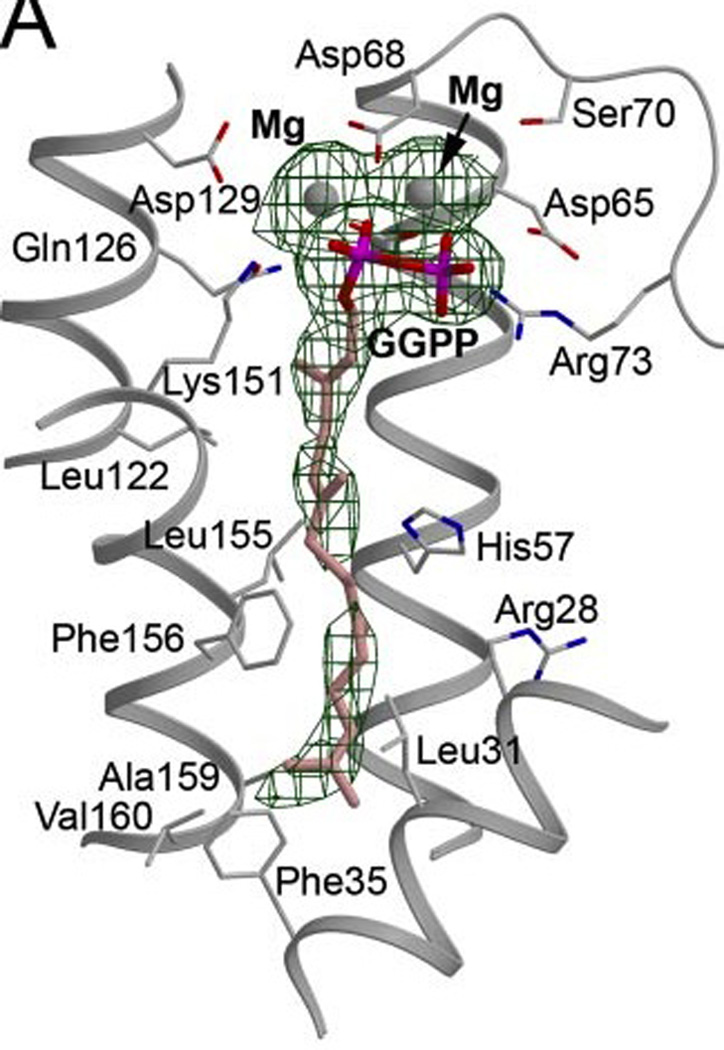
The first mono-TPS to be structurally characterized was actually the bornyl diphosphate synthase from culinary sage (Salvia officinalis).64 Structures were determined in the presence of pyrophosphate-Mg2+3, and in the absence or presence of several different aza analogs of carbocation intermediates as well, revealing tight constraints on binding of the pyrophosphate-Mg2+3 co-product (Fig. 16). This is consistent with the previous finding that bornyl diphosphate is formed by readdition to the same oxygen atom involved in the diphosphate ester bond of GPP.70,71 On the other hand, a 7-aza-7,8-dihydro-limonene analog, bound in the presence of pyrophosphate and Mg2+, is oriented to enable ion pairing of the aza cation and pyrophosphate anion, which is opposite that expected for the catalytic intermediate (Fig. 17). This provides perhaps the most convincing evidence for the hypothesis that TPS catalyzed reactions are under kinetic rather than thermodynamic control. Another noteworthy finding from this series of structures was the consistent presence of a particular water molecule in the active site cavity, which has several potential hydrogen bonding interactions with the enzyme. This indicates that such relatively tightly bound water molecules may help form the contours of the active site, and potentially could act as the catalytic base for the observed production of a number of monoterpene olefins by this enzyme. Indeed, it has been noted that, despite the reactive nature of carbocations, unless positioned in particular orientations, it is more likely that water molecules will add as proton accepting general bases rather than recombine with carbocation intermediates in terpenoid cyclization reactions.72 Structures determined in the presence of the later intermediate analog 2-azabornane, as well as the enzymatic product, bornyl diphosphate, were used to define an active site cavity into which the catalyzed reaction was modeled, revealing “excess” volume that may help explain the observed lack of product fidelity (i.e., production of several monoterpene olefins in addition to bornyl diphosphate73).
Fig. 16.
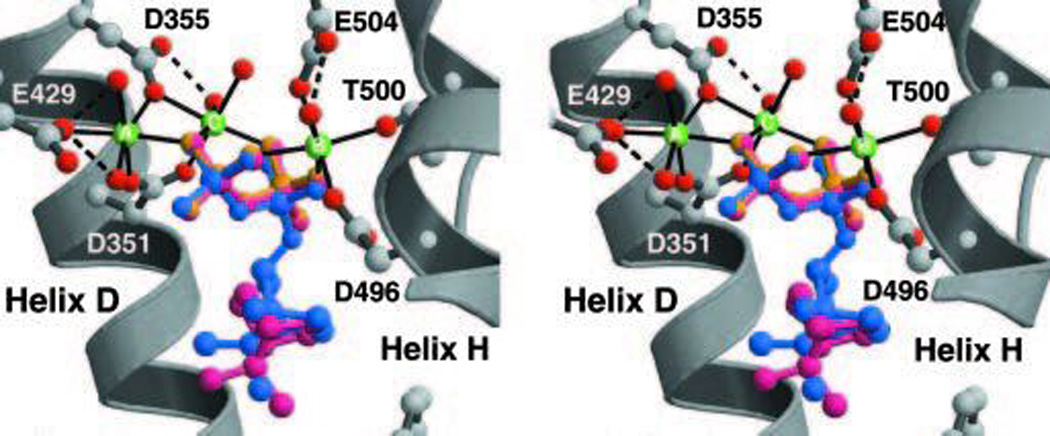
A structural overlay of bornyl diphosphate synthase structures determined with various ligands demonstrating the invariant location of the trio of Mg2+ and, hence diphosphate group. Reprinted with permission from ref. 64. Copyright 2002 National Academy of Sciences, USA
Fig. 17.

The reaction catalyzed by bornyl diphosphate synthase, along with depiction of the relative configuration of aza-analogs of early and late stage reaction intermediates to the Mg2+3-pyrophosphate complex found in the various co-crystal structures, demonstrating the dominant effect of aza-pyrophosphate ion-pairing on binding. Reprinted from ref. 76.
3.2.3 Cineole synthase
Another interesting variant for alternative chemistry in TPS catalysis is provided by cineole synthase, a structure for which has been determined for such an enzyme from Salvia fruticosa.74 Formation of the cyclic ether product is thought to be accomplished by addition of water to the initially cyclized α-terpinyl cation, to form α-terpineol, with subsequent attack of the endo-double bond and proton removal to yield 1,8-cineole. Notably, the structure suggested a water-binding pocket, with a key role for a particular asparagine, which is conserved in the known cineole synthases (Fig. 18). Indeed, previous chimeric analysis of mono-TPSs from Salvia officinalis indicated an important role for this region in specifying the use of such chemistry, including speculation on the importance of this Asn,75 as later verified by structural analysis. In particular, isoleucine substitution for this Asn abolished incorporation of water, with production of 1,8-cineole and α-terpineol (a minor component of the product mixture from the wild-type enzyme) no longer observed. Additional mutational changes, guided by sequence comparison of cineole synthases to closely related sabinene synthases, led to more specific production of sabinene. Moreover, it was possible to induce a sabinene synthase from Salvia pomifera to produce at least small amounts of cineole and α-terpineol via converse mutational alterations, most critically introduction of the key Asn residue.74
Fig. 18.

Cineole synthase. A) A scheme of catalyzed reactions, both the production of cineole as well as sabinine. B) A depiction of the active site of cineole synthase showing the water molecule suggested to be added in the course of the catalyzed reaction, along with 3-aza-2,3-dihydrogeranyl diphosphate substrate analog (from a bornyl diphosphate synthase structure64), as well as important Asn residue. Reprinted with permission from ref. 74. Copyright 2007 The American Society for Plant Biology.
3.3 Hemiterpene synthases
The only known structure for a DMAPP specific (i.e., hemi-)TPS is that recently reported for the isoprene synthase from grey poplar (Populus × canescens).76 The structure was determined as a complex with Mg2+ and the unreactive dimethylallyl-S-thiolodiphosphate analog, which suggested the use of the released diphosphate anion as the catalytic base, much as previously suggested for other class I terpenoid synthases, as described above. In addition, the structure of this enzyme exhibits a shallower cavity relative to even mono-TPSs (Fig. 19), consistent with its specificity for the smaller substrate DMAPP, although it should be noted that GPP acts as non-reactive competitive inhibitor of this hemi-TPS.
Fig. 19.
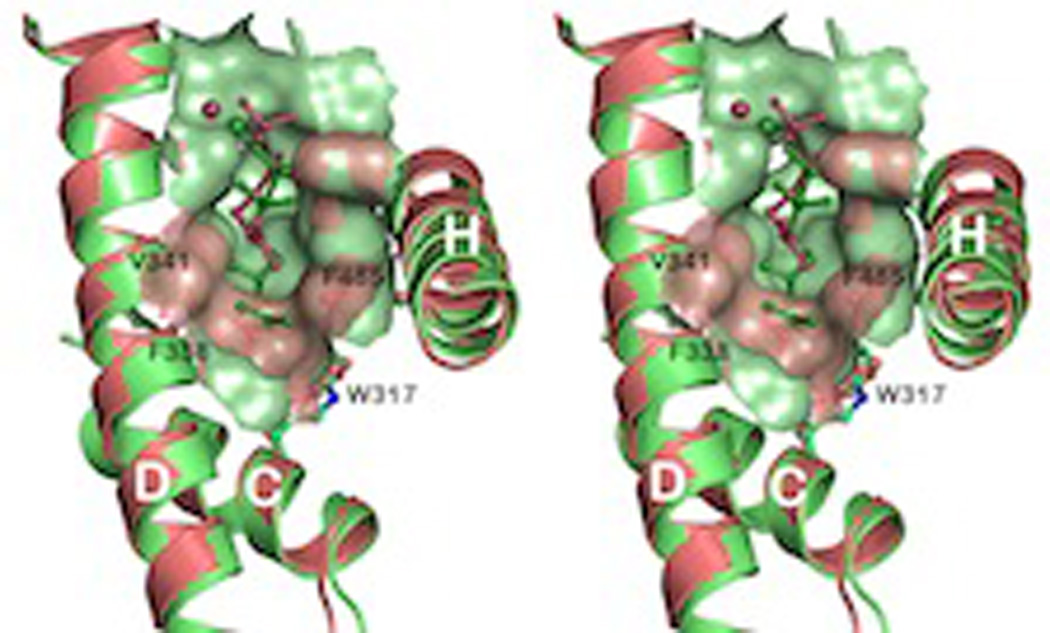
A superposition of isoprene (hemiterpene) and bornyl diphosphate (monoterpene) synthase active site cavities, demonstrating the reduced size of the isoprene synthase (attributed to a pair of Phe). Reprinted with permission from ref. 76. Copyright 2011 Elsevier.
3.4 Diterpene synthases
While GGPP is the general precursor to diterpenoid natural products, a significant fraction of those known (est. 7000 of 12 000) are classified as labdane-related,77 and these are distinguished by their biosynthetic origins via initial (bi)cyclization of GGPP by class II diterpene cyclases (which catalyze protonation-initiated reactions that leave the allylic diphosphate ester linkage intact and proceed via formation of a labdaenyl carbocation intermediate that gave rise to the labdane-related nomenclature used here77). Accordingly, di-TPS can react with either GGPP or such derived bicycles, typically exclusively, including (stereo) specificity78 for particular variants of the GGPP derived bicycles. The two di-TPSs to have had their structures determined provide examples of specificity for GGPP versus a derived bicycle. Both of these enzymes also are from plants, and exhibit the γβα tri-domain architecture that provided the structural homology based evidence for the evolutionary origins of plant terpene synthases described in the Introduction (Fig. 2), and discussed in more detail below.
3.4.1 Taxadiene synthase
The first di-TPS to have its structure determined was the taxadiene synthase from the yew tree Taxus brevifolia, which reacts specifically with GGPP.5 Surprisingly, it was possible to obtain structures of this enzyme bound not only to an analog of its GGPP substrate, 2-fluoro-GGPP, but also 13-aza-13,14-dihydro-copalyl diphosphate (13-aza-CPP) – albeit with two 13-aza-CPP found in the active site, demonstrating some plasticity, despite 13-aza-CPP not being an analog for the catalyzed reaction. Indeed, it was noted that this enzyme exhibits some promiscuity in catalyzed product outcome, consistent with the “excess” volume of the active site, even with the true substrate analog 2-fluoro-GGPP bound, as the taxadiene product could be modeled into this cavity in at least two distinct orientations (Fig. 20). One of these conformations led to the suggestion that diphosphate anion co-product might serve as a catalytic base in this enzyme as well (i.e., alongside all the other such indications for such co-product dependent catalysis discussed above). This plant di-TPS was found to have a tri-domain structure, containing not only the usual plant TPS β and α domains, but also a γ domain, which is essentially inserted between the first and second helices of the β domain. Strikingly, the β and γ domains exhibit structural homology to class II triterpene cyclases, which fold into two double α-barrel domains (as discussed below in Section 4). Because class II diterpene cyclases contain homologous sequence to the γβ domains found in taxadiene synthase this suggested that plant diterpene synthases exhibit a modular architecture, as discussed in more detail below.
Fig. 20.

A depiction of the active site cavity for taxadiene synthase (meshwork enclosure) with bound fluorogeranylgeranyl diphosphate substrate analog (grey) and one of the potential orientations for the taxadiene product (blue). Reprinted by permission from Macmillan Publishers Ltd: Nature, ref. 5, copyright 2011.
3.4.2 Abietadiene synthase
Very recently, we have reported the structure of the abietadiene synthase from grand fir (Abies grandis), which exhibits the same γβα tri-domain architecture noted above for taxadiene synthase, and directly demonstrates the modular nature of plant terpene synthases.79 In particular, abietadiene synthase is a bifunctional enzyme, catalyzing both class II (bi)cyclization of GGPP to copalyl diphosphate (CPP) and subsequent class I cyclization, with these reactions occurring in separate locations,80 although only the class I active site found in the α domain will be discussed here. It should be noted that while this enzyme traditionally has been considered to produce a mixture of double-bond isomers of abietadiene,81 recent results with a closely related enzyme from Norway spruce (Picea abies) indicate that these may actually produce, by capture of water, the unstable tertiary allylic alcohol abieta-8(14)-en-13-ol instead (with this dehydrating during analysis to the observed mixture of olefins).82 The grand fir abietadiene synthase has served as a model di-TPS, with extensive mechanistic and mutational analysis of its class I activity having been previously reported.80,83–87 Of particular interest has been the identification of a single residue change (serine for alanine substitution) that short-circuits the class I reaction catalyzed with the copalyl diphosphate (CPP) product of its class II bicyclization of GGPP, to yield isopimaradienes rather than the rearranged abietane skeleton observed with the wild-type enzyme.87,88 This was based on previous work demonstrating an analogous single residue switch for product outcome in other labdane-related (i.e., CPP specific) di-TPSs,89,90 as well as identification of a closely related isopimaradiene synthase from Norway spruce.91 Nevertheless, determination of a structure for abietadiene synthase enabled docking of a pimaradiene product into the class I active site, which nicely depicts the proximity of this switch position to C8, the location of the carbocation in the key pimarenyl+ intermediate (Fig. 21). This docked conformation further is consistent with the hypothesis that the striking effect of these single residue switches on product outcome is due not only to their interactions with this key intermediate, but the ability of the pyrophosphate anion co-product to drive carbocation migration towards itself, increasing the complexity of the catalyzed reaction.92 Notably, such an effect is consistent with both previous structural analysis of aza analogs of carbocation intermediates, which indicated that aza cation-diphosphate anion pairing was the predominant factor in driving bound orientation – i.e., rather than complimentary fit of the hydrocarbon skeleton to the active site contour (this is most dramatically illustrated by the “backwards” orientation of 7-aza-limonene in bornyl diphosphate synthase, as discussed above in Section 3.2.2 – see Fig. 17), as well as the strong synergistic binding observed between inorganic pyrophosphate and aza analogs of the intermediates formed by carbocation migration in the abietadiene synthase catalyzed reaction.85 This then provides another role for the pyrophosphate anion co-product in TPS catalyzed reactions beyond potentially acting as the catalytic general base – i.e., the pyrophosphate may also drive carbocation migration toward itself, with the effect of increasing reaction complexity, in the absence of other counter-acting electrostatic effects, specifically in the case described here, introduction of a hydroxyl dipole that stabilizes the initially formed pimarenyl carbocation long enough for terminating deprotonation to occur instead of rearrangement.
Fig. 21.

Abietadiene synthase. A) A scheme for the class I reactions catalyzed by the wild-type (with Ala) or mutant (Ser) enzyme. B) A depiction of the sandaracopimaradiene product of mutant enzyme docked into the active site cavity along with modeled pyrophosphate-Mg2+3 co-product, also shown is the side chain of the key alanine residue. Adapted from ref. 79.
4 Class II terpene cyclases
The class II terpene cyclases are characterized by catalysis of protonation-initiated cyclization reactions, which sometimes includes subsequent rearrangement. In particular, these carbocationic cascade reactions are initiated by the addition of a proton to either a carbon–carbon double-bond, or epoxide (leading to ring opening), with addition of the ensuing carbocation to intramolecular carbon–carbon double-bonds, generally in an iterative fashion to form multicyclic structures. Most commonly, such reactions are catalyzed with the triterpene precursors squalene, or the derived oxido-squalene, as well as with the general diterpene precursor GGPP. However, examples of what appear to be class II cyclization reactions can be found in the sesquiterpene drimanes, as well as certain sesterterpenoids, and a role for class II terpene cyclases has been shown for sesquarterpenoid biosynthesis.93 In any case, these enzymes also often catalyze stereochemically complex cyclization reactions – e.g., oxido-squalene cyclases (OSCs) can produce anywhere from one to five rings, often with subsequent rearrangement following initial cyclization. It further should be noted that, like the class I TPSs, the class II terpene cyclases do not necessarily directly deprotonate the final carbocation, as capture of water also has been observed, with subsequent deprotonation to form a hydroxylated product. Finally, again as noted above for the class I TPSs, the class II terpene cyclases also exhibit a range of catalytic promiscuity, while some are quite specific, others will yield a range of products, with oxido-squalene cyclases particularly noted for their plasticity.94
4.1 Squalene-hopene cyclases
The first class II terpene cyclase structure to be determined was that for the squalene-hopene cyclase (SHC) from Alicyclobacillus acidocaldarius.95 This was reported at the same time as that of the first two class I TPSs,45,46 but was distinguished from these not only in enzymatic reaction mechanism, but also distinct protein fold, as it was found to form two domains, each adopting a double α-barrel structure, which have been designated γ and β, with the γ domain essentially inserted between the first and second helices of the β domain (Fig. 22). In addition, it should be noted that this is a monotopic membrane protein, with one α-helix suggested to reside in the membrane, helping frame the entrance to a hydrophobic channel that presumably enables access for the highly hydrophobic squalene substrate and egress of the similarly hydrophobic pentacyclized hopene product.96 The presence of a bound competitive inhibitor unambiguously identified the active site as residing between the γ and β domains. This cavity is largely hydrophobic, although it contains a key DxDD motif at one end, with the “middle” aspartate hypothesized to act as the catalytic acid that protonates the terminal carbon–carbon double-bond of squalene, in part with assistance from a hydrogen-bonded histidine. Notably, the active site is otherwise lined with aliphatic and aromatic residues that serve to fold the substrate for the ensuing carbocation cascade cyclization reaction, as demonstrated by a later determined structure with the early intermediate analog 2-aza-squalene found to be bound in a conformation closely resembling that required for cyclization.97 Moreover, the conservation of these active site residues from this SHC with those found in OSCs follows a gradient, with those near the DxDD motif most conserved and those at the distal end exhibiting less conservation, consistent with the observed divergent biosynthetic capacity (i.e., particularly of the oxido-squalene cyclases). A number of mutational follow up studies have been presented, and reviewed elsewhere.98–100 Briefly, these studies are consistent with a role for various aromatic residues in stabilization of carbocation intermediates. Strikingly, while most of these studies were confounded by the change in steric volume imposed by the usual restriction of substitution with the naturally occurring aliphatic residues, an elegant study using unnatural amino incorporation, particularly fluorinated tyrosines, clearly demonstrated a role for aromatic π stabilization of specific carbocation intermediates.101 In addition, while not observed in any of the determined structures, it seems likely that the catalytic base is actually a bound water molecule, in part based on the production of diplopterol, formed by the addition of water to the final hopanyl carbocation intermediate, as a minor component of the product mixture.99
Fig. 22.

Squalene-hopene cyclase. A) A scheme for the catalyzed reaction. B) A depiction of the structure determined with anti-cholesterol drug Ro 48–8071. Reprinted with permission from ref. 103. Copyright 2002 Elsevier.
4.2 Oxido-squalene cyclases
While OSCs exhibit extensive catalytic diversity (particularly in plants), the only known structure is that for the human lanosterol synthase,102 which is homologous to that of SHC, cementing the suggested status of this γβ didomain architecture as the class II terpene cyclase fold.3 While a structure of SHC bound to a potential anticholesteremic drug had been previously reported,103 that determined with the human lanosterol synthase is obviously more directly relevant (Fig. 23). Comparison of these structures also provided some insight into the structure–function relationships underlying the different product outcomes mediated by these two triterpene cyclases. For example, the absence of certain aromatic residues associated with pentacycle formation in SHC in the tetracycle producing lanosterol synthase. Also, it had already been determined that OSCs contained only the “middle” aspartate of the squalene-hopene cyclase DxDD catalytic motif, consistent with their ability to protonate only the more basic epoxide ring, but not a carbon–carbon double-bond (i.e., SHCs can cyclize oxido-squalene, but lanosterol synthase cannot cyclize squalene). Perhaps more interestingly, the reaction catalyzed by lanosterol synthase provides an example of rearrangements occurring after (tetra)cyclization, and it has been suggested that this was due, in part, to the lack of a catalytic base appropriately positioned for deprotonation of the initially formed tetracyclic carbocation intermediate, leading to rearrangement that forms a carbocation intermediate that can be deprotonated by an active site histidine (or tyrosine activated by this histidine). It was further suggested that such rearrangement is assisted by a π-electron gradient in the active site, with only three aromatic residues within 6 Å of the carbocation formed by initial (tetra)cyclization, relative to seven such residues neighboring the carbocation resulting from rearrangement. However, while mutational analysis of the histidine and tyrosine has been reported, the effect of the π-electron gradient does not appear to have been investigated.99
Fig. 23.

A depiction of the human lanosterol synthase with anti-cholesterol drug Ro 48–8071 bound in the active site (central cavity – defined by meshwork enclosure). Also shown is a potential orientation in the membrane (polar region is light blue and hydrophobic region, light yellow). Reprinted by permission from Macmillan Publishers Ltd: Nature, ref. 102, copyright 2004.
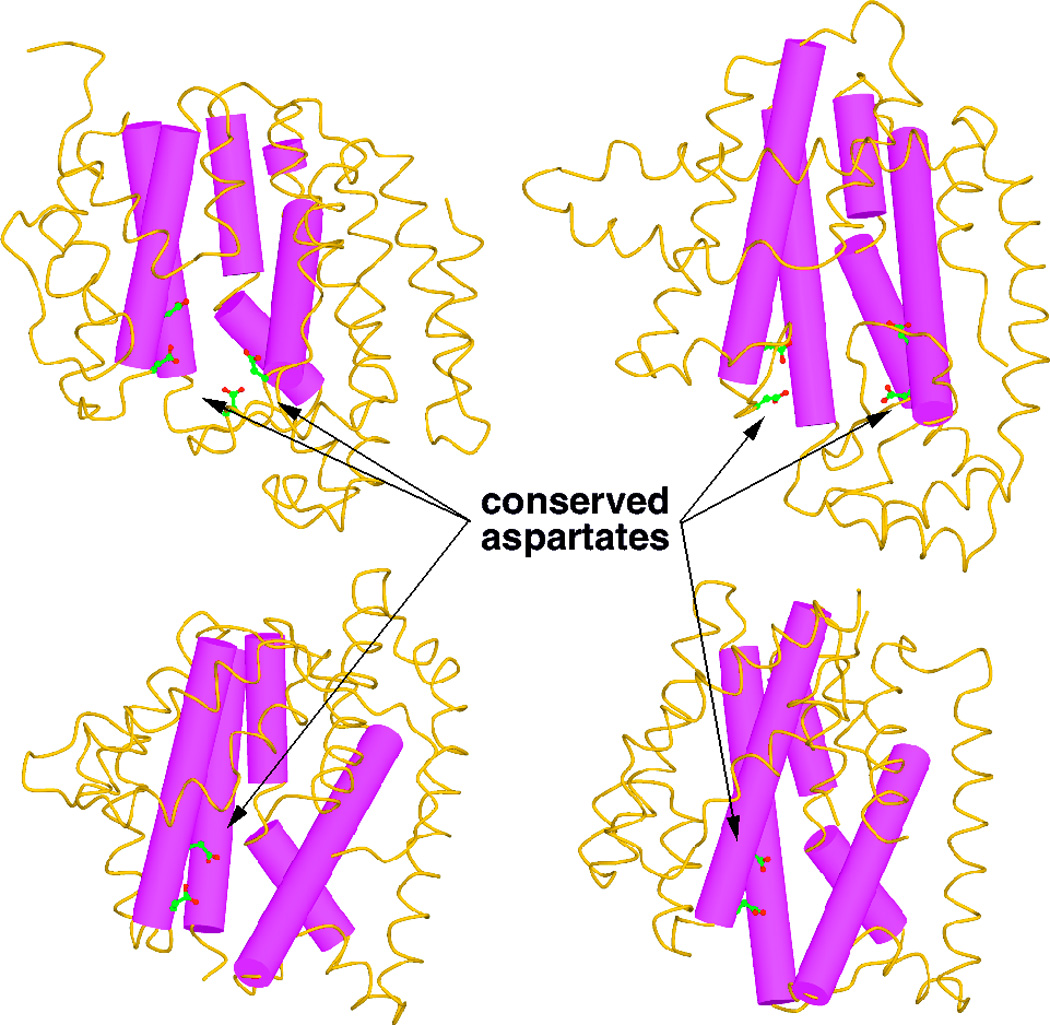
4.3 Class II diterpene cyclases
There are obvious mechanistic similarities between the reactions catalyzed by class II diterpene cyclases (DTCs) and that catalyzed by triterpene cyclases, particularly SHCs, with both using a carbon–carbon double-bond protonation-initiated mechanism. Indeed, the DTCs contain an DxDD motif analogous to that found in SHCs, and in which the “middle” aspartate similarly has been demonstrated to act as the catalytic acid.104 However, consistent with the presence of a diphosphate moiety in their substrates, the DTCs utilize a Mg2+ co-factor, presumably to assist substrate binding and positioning. Nevertheless, as predicted from their mechanistic similarity,104 and later modeled,4 the DTCs also exhibit structural homology to the triterpene cyclases as well.
4.3.1 Copalyl diphosphate synthase
The first structure of a DTC to be determined was that for the ent-copalyl diphosphate synthase from Arabidopsis thaliana.105 This exhibited the same γβα tri-domain architecture noted above for the plant di-TPSs. However, the class II active site lies at the interface between the γβ domains, as indicated by the bound geranylgeranyl thiolodiphosphate substrate analog, as well as presence of the catalytic DxDD motif, while the α domain no longer contains a functional class I active site (e.g., the requisite DDxxD and NSE/DTE motifs have been lost). Strikingly, despite the lack of any readily detectable sequence homology, the γβ domains exhibited structural homology to the triterpene cyclases discussed above – i.e., these are similar double α-barrel domains, although the membrane associated helix is not present, consistent with the use of the soluble GGPP as substrate rather than hydrophobic (oxido-)squalene (as originally shown with the homologous taxadiene synthase;5 Fig. 24; see also evolutionary relationships depicted in Fig. 2). As previously predicted,106 this structural homology includes a similar arrangement of the catalytic DxDD motif as that observed for this same motif in the mechanistically analogous SHCs. Further similarities can be found in the activation of the catalytic “middle” aspartate by hydrogen-bonding to a residue that occupies the same position as the histidine that plays this role in SHCs, albeit this is conserved as an asparagine in the plant class II diterpene cyclases instead. Moreover, there are a number of aromatic residues in the active site that are hypothesized to stabilize carbocation intermediate via aromatic π interactions, several of which are conserved across the plant class II diterpene cyclase family. Unfortunately, no Mg2+ ions were present in the structure, which may explain why the diphosphate moiety of the bound substrate analog was found in two distinct orientations.
Fig. 24.

A comparison of taxadiene synthase structure with that of squalene-hopene cyclase. Structurally homologous domains are indicated by identical coloring (green and yellow), with N-terminal helix that forms part of the β domain highlighted in pink, and membrane associated helix in the squalene-hopene cyclase highlighted by grey stripes. Reprinted by permission from Macmillan Publishers Ltd: Nature, ref. 5, copyright 2011.
4.3.2 Abietadiene synthase
As noted above, we have very recently reported the structure of the bifunctional abietadiene synthase from grand fir,79 which not only contains a functional class I active site in its α domain, but also a class II active site at the interface between its γβ domains, this later of which will be discussed here. Due to the absence of a substrate analog, this active site is in an “open” conformation, with one loop occupying a distinct position relative to that found in the substrate analog bound “closed” conformation of the ent-copalyl diphosphate synthase structure. Notably, previous mutational analysis in abietadiene synthase of some of the aromatic residues conserved in plant DTCs had already suggested a role in catalysis for one of those found in the active site near the DxDD motif.106 To verify a role for the Asn hydrogen bonded to the “middle” Asp of the DxDD (Fig. 25), this was substituted with alanine, leading to a specific ~100-fold decrease in class II (but not class I) activity. Intriguingly, previous results had led to the hypothesis that certain plant DTCs were inhibited by higher concentrations of their Mg2+ co-factor, which seems to be controlled by a single residue whose identity (histidine versus arginine) dictates susceptibility to such inhibition.107 However, this residue was not actually found in the class II active site of either the ent-copalyl diphosphate or abietadiene synthases. Nevertheless, alanine substitution for this residue in abietadiene synthase exhibited a dramatic ~1000-fold loss of class II activity (although, again, class I activity was essentially unaffected), consistent with previous results with the ent-copalyl diphosphate synthase,107 which had led to the suggestion that this residue would not only be in the active site, but play a catalytic role as well, although this has now been disproven by these structures. Finally, given the absence of a substrate analog in this structure, along with the nonproductive orientation of the substrate analog observed in the ent-copalyl diphosphate synthase structure, we undertook molecular dynamics simulations to examine how catalysis might occur. Interestingly, this computational analysis indicated not only that the “closed” loop conformation prevented bulk water access to the active site, but also the counterintuitive finding that in this “closed” enzyme conformation the substrate actually exhibited more flexibility, critically including achieving catalytically productive orientations.
Fig. 25.
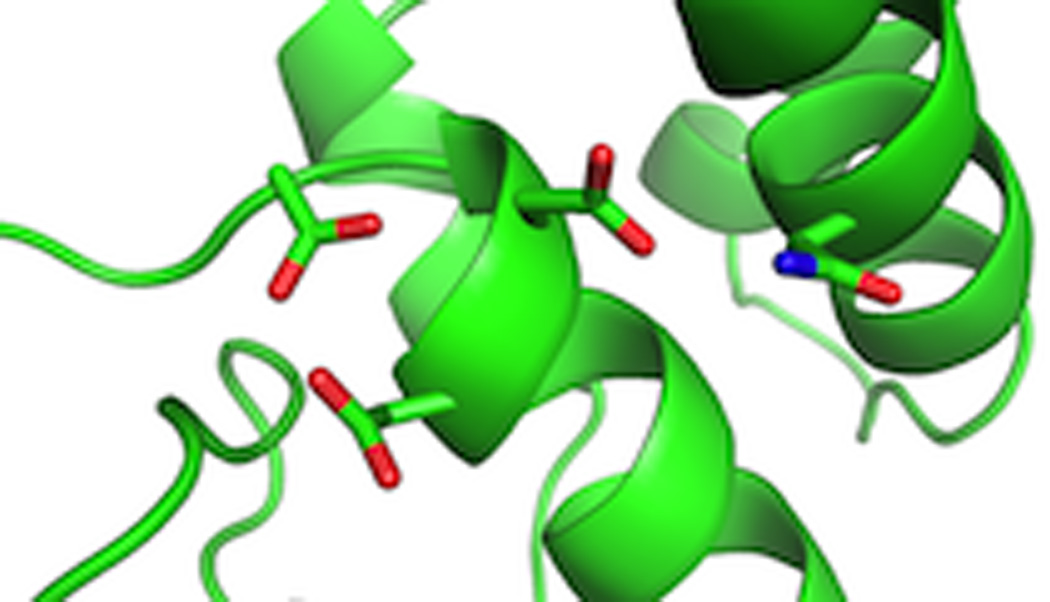
A close-up view of the class II active site of abietadiene synthase showing the aspartates of the DxDD motif and interacting Asn.
5 Structural insights into evolutionary relationships
While we have already mentioned the evolutionary implications of the observed structural homology in the Introduction section (Fig. 2), here we will more fully discuss these evolutionary relationships. The first observation of structural homology was that found between the mechanistically analogous diphosphate-ionization initiated trans-IDSs and TPSs. This distant homology was revealed by determination of the structure of TPSs, and their evident similarity to the previously characterized avian FPP synthase, despite the lack of any readily detectable sequence homology (Fig. 26). The conserved α-helical bundle assemblies have then been designated the class I terpenoid synthase fold.3 Indeed, such structural homology highlighted derivation of the TPS DDxxD and NSE/DTE divalent metal ion binding motifs from the isoprenyl diphosphate synthase aspartate-rich FARM and SARM motifs. Perhaps more interestingly, as noted at the time,45 such homology is consistent with a previously advanced hypothesis that sequentially acting enzymes often share homologous origins,108 which was further bolstered by the observation of similar structural homology between the consecutively acting trans-IDSs and squalene synthases.35
Fig. 26.
A structural comparison of (sesqui)terpene synthases with FPP synthase highlighting conservation of the class I terpenoid synthase fold (blue). Reprinted with permission from ref. 53. Copyright 2000 The American Society for Biochemistry and Molecular Biology.
Structural homology of the class II terpene cyclases is arguably somewhat more surprising. While these were known to have some mechanistic similarity in the use of protonation to initiate cyclization, clear differences were evident – particularly between the triterpene cyclases (i.e., SHCs and OSCs) and DTCs. For example, the use of chemically distinct substrates, the hydrophobic (oxido-)squalene versus the soluble GGPP, respectively, as well as use of Mg2+ as a co-factor by the DTCs (although this presumably is related to its need to bind the diphosphate moiety of its GGPP substrate). Nevertheless, much as observed with the class I enzymes, despite the lack of any readily recognizable sequence homology, the class II tri- and di-terpene cyclases were found to exhibit structural homology. In particular, these fold into a bi-domain structure wherein both domains assume related double α-barrel structures. Nevertheless, there are some telling differences as well – e.g., the membrane associated helix found in the monotopic triterpene cyclases is no longer present in the DTCs that utilize GGPP instead of (oxido-)squalene (Fig. 24). Moreover, the only determined structures of DTCs are from plant derived enzymes, which additionally contain a C-terminal domain homologous in both sequence and structure to the class I TPSs.
Strikingly, the domain composition of plant diterpene synthases indicates that these played a central role in plant terpene synthase evolution (Fig. 2),4 specifically that of the class I TPS and class II DTC families (note that plants also contain expanded families of class II OSCs whose evolution appears to have been completely independent and relatively straightforward109). Of particular importance is the occurrence of bifunctional enzymes, which catalyze consecutive class II and class I reactions, such as abietadiene synthase, and seem to represent fusion of a class II DTC and class I TPS from bacteria – i.e., a modular arrangement. The observed γβα tri-domain architecture of these enzymes are then derived from the γβ di-domains of the bacterial class II DTC, consistent with the noted distant sequence homology (critically, this includes the catalytic DxDD motif), which has been coupled to the α domain of a bacterial class I di-TPS, again as suggested by the noted distant sequence homology.110 This initial suggestion was further supported by more sophisticated sequence comparisons along with protein structure modeling,4 and finally demonstrated by crystallo-graphic structure determination,5 as discussed above. Notably, previous phylogenetic analyses indicated that diterpene synthases are the ancestral form of the plant class I TPSs, specifically those required in all vascular plants for production of the gibberellin phytohormones necessary for normal plant growth and development.111,112 Indeed, non-vascular plants contain bifunctional diterpene synthases that produce the relevant ent-kaurene intermediate.113 On the other hand, vascular plants contain a pair of mono-functional enzymes that serve this purpose, both a class II ent-copalyl diphosphate and class I ent-kaurene synthases, as found in both angiosperms and gymnosperms.114,115 These presumably arose from an ancient gene duplication and sub-functionalization event, although both retain the ancestral γβα tri-domain architecture. The ent-copalyl diphosphate synthase required for gibberellin biosynthesis seems to represent the ancestor for the divergent class II DTCs found in the angiosperms, while the ent-kaurene synthase is the ancestor of the divergent class I di-TPSs (although it should be noted that the gymnosperms have retained bi-functional diterpene synthases in their more specialized metabolism, particularly for resin acid biosynthesis). More broadly, ent-kaurene synthase seems to further represent the ancestor for all plant (class I) TPSs, providing a source for the N-terminal β domains found in this large and functionally diverse enzymatic family, largely from early loss of the γ domain, which seems to have occurred with retention of the N-terminal sequence that folds back and forms part of the class I active site in both the ancestral bifunctional enzymes,106 as well as derived TPSs (as discussed above). Consistent with this scenario, it has been noted that the γ domain does not contact the class I α domain,79 and loss of this domain seems to have occurred more than once in the plant lineage, although much of the observed diversity seems to stem from the early γ domain loss event noted above.116
Given the number of crystal structures now available for class I terpenoid synthases, we wondered how more detailed structural comparisons might impact the evolutionary scenario presented above. This was examined by principal component analysis wherein each of the currently known structures was structurally aligned and their differences treated as “motions”. Notably, examination of the resulting data, specifically a plot of the first and second principal “motions” (i.e., distinguishing structural differences), which together represent 80% of the observed variability, has some interesting implications (Fig. 27). For example, the trans-IDSs, from all organisms, are clustered together, consistent with structural constraints imposed by their common mechanism, which requires only slight adjustment to enable production of different chain lengths as discussed above. By contrast, the TPSs are more scattered, although the plant TPSs do form a distinct cluster. The scatter among these may reflect their more divergent mechanisms, with the clustering of the plant TPSs consistent with their clear homologous origins, which contrasts with the much reduced sequence similarity among the microbial TPSs that also are more scattered in this plot. This structural diversity and lack of sequence similarity among the microbial enzymes may indicate independent and/or more recent derivation of these from the presumably ancestral trans-IDSs. However, it should be noted that the observed structural clustering of plant TPSs may more simply reflect constraints imposed on their class I terpenoid synthase fold α domains by the presence of the co-occurring and interacting β domains. Reasoning that such constraints might be evident from the motions induced by substrate binding, we compared those enzymes for which unliganded and liganded structures had been determined, finding that the plant TPSs seemed to undergo decreased conformational change (Fig. 28), consistent with reduced structural flexibility. Nevertheless, given the variability in substrate and product outcome mediated by the structurally characterized plant enzymes, any such structural constrains do not seem to have significantly restricted their catalytic diversity.
Fig. 27.

A principal component analysis of the all the known class I terpenoid synthase folds. Blue, red, green and purple represent those enzymes from animal, bacteria, fungi and plant, respectively. Where available, both ligand-free and ligand-bound structures are shown, whereas for other terpene synthase structure, only one representative structure was analyzed. All structures were aligned to FPP synthase structure (PDB ID 1FPS) and coordinates of C-alpha atom from all structural equivalent residues served as input for principal component analysis. The projection of each structure onto a plane composed by principal motion 1 (PC1) and principal motion 2 (PC2), which accounted for 70% and 10% of the observed variability, respectively, are depicted below. These are derived from PDB entries 1FPS and 1YV5 for FPP synthase from animal, 1EZF for squalene synthase from animal, 1PS1 for pentalene synthase from bacteria, 1RTR and 1RQI for FPP synthase from bacteria, 1V4E for octaprenyl pyrophosphate synthase from bacteria, 2AZJ for hexaprenyl diphosphate synthase from Sulfolobus solfataricus (bacteria), 3AQB for hexaprenyl diphosphate synthase from Micrococcus luteus (bacteria), 3KBK and 3KB9 for epi-isozizaene synthase from bacteria, 1JFA and 1JFG for trichodiene synthase from fungi, 2DH4 for GGPP synthase from fungi, 2E4O and 2OA6 for aristolochene synthase from fungi, 1N1B and 1N20 for bornyl disphosphate synthase from plant, 2J1O for GGPP synthase from plant, 2J5C for 1,8-cineole synthase from plant, 2ONG for limonene synthase from plant, 3APZ and 3AQ0 for GPP synthase from plant, 3G4D and 3G4F for delta-cadinene synthase from plant, 3N0F and 3N0G for isoprene synthase from plant, 3P5P for taxadiene synthase from plant, 3S9V for abietadiene synthase from plant, 3SAE and 3SDQ for alpha-bisabolene synthase from plant, 5EAS and 5EAT for 5-epi-aristolochene synthase from plant.
Fig. 28.

A comparison of class I terpenoid synthases that have had both unliganded and ligand-bound structures determined. The depicted enzymes are indicated along with the RMSD for the overlaid structures. Cyan represents the ligand-free and magenta represents the ligand bound structures of each. Substrate/product/pyrophosphate and their analogs are shown in thick lines and magnesium are shown as spheres. The PDB ID are 1FPS and 1YV5 for FPP synthase from animal, 1RTR and 1RQI for FPP synthase from bacteria, 3APZ and 3AQ0 for GPP synthase, 3KBK and 3KB9 for epi-isozizaene synthase, 2E4O and 2OA6 for aristolochene synthase, 1JFA and 1JFG for trichodiene synthase, 5EAS and 5EAT for 5-epi-aristolochene Synthase, 3G4D and 3G4F for deltacadinene synthase, 1N1B and 1N20 for bornyl disphosphate synthase, 3N0F and 3N0G for isoprene synthase, 3SAE and 3SDQ for alpha-bisabolene synthase.
6 Delving deeper into enzymatic structure–function relationships
It was recognized early on that the terpenoid synthases must orient their substrate(s) to bring the relevant carbon–carbon double-bonds into proximity in the correct orientation for formation of the new carbon–carbon bonds observed in their products,6 and the structural analyses discussed here have borne out these early insights. In the case of the terpene synthases and cyclases, this further offers potential anchimeric assistance – i.e., these intramolecular π bonds act as participating neighboring groups to increase at least the relative rate of certain alternative routes to favor the corresponding bond formation. Indeed, even in the case of the prenylelongases where this is no longer formally an example of neighboring group participation, it seems likely that such effects may increase the overall catalytic rate as well, favoring formation of the initial carbocation (either by ionization or protonation). Moreover, it has been suggested that similar effects also are exerted by the enzymes, particularly via aromatic side-chain mediated quadrapole-carbocation stabilizing interactions. This has been most convincingly demonstrated for SHCs, including correlation of specific aromatic residues for stabilization of discrete carbocation intermediates (e.g., by the alteration in product outcome upon their substitution99,100). Strikingly, perhaps due in part to the smaller size of their substrates, as well as arguably often increased complexity of the catalyzed reactions, the evidence for such effects in TPSs is generally less definitive despite the similar availability of high-resolution crystal structures.
The active site of TPSs must in some sense act as templates to fold their substrates in a product-like conformation – e.g., to orient their carbon-carbon double-bond π orbitals for carbocationic (cyclo)addition,117 with rearrangement at least partially dependent on exact conformation of substituents surrounding the carbocation.118 Given the early example of the structurally characterized tobacco 5-epi-aristolochene synthase and comparison to the closely related premnaspirodiene synthase, it has long been clear that this template can be influenced by not only the residues directly lining the active site, but residues behind these – i.e., the “second layer”.46 While it was possible to identify a set of these second layer residues whose exchange was sufficient to interconvert product outcome between 5-epi-aristolochene and premnaspirodiene synthases,50 more detailed follow-up analysis did not provide an obvious evolutionary path, nor a clear mechanistic understanding of how these changes led to redirection of the related reactions mediated by these enzymes.119 Indeed, efforts directed at engineering class I terpene synthase product outcome have relied on either random mutagenesis of first and second layer residues,120,121 or comparison of closely related but functionally distinct enzymes, as reviewed elsewhere.122 However, the results from these studies largely do not seem to be more generally applicable, nor provide specific insight(s) into the underlying structure-function relationships.
This lack of generalization may be due in part to the complex nature of the catalyzed reactions, which can include not only cyclization, but also a wide variety of rearrangements. The corresponding carbocation intermediates along the various reaction pathways generally represent branchpoints from which different routes can be accessed, leading to alternative product outcome, and TPSs are notorious for infidelity – i.e., the production of a range of products from the same substrate. While some of these mechanistic choices in reaction route will be largely, if not entirely, dictated by initial substrate conformation (i.e., preorganization), it seems likely that the enzymes exert additional levels of control on the catalyzed reaction. As reviewed elsewhere,123 such investigations may be assisted by the emerging application of quantum chemical calculations to these reactions. Notably, experimental verification for at least some results from such computational investigation has been recently reported,124 increasing confidence in their relevance. Of particular interest here is the use of electrostatic interactions, specifically those exerted by the enzymes. Strikingly, a primary effect seems to be that mediated by the released pyrophosphate anion, which is tightly bound and held in place by the enzyme. For example, early labeling studies with bornyl diphosphate synthases demonstrated the use of the same oxygen in the ester linkage of both substrate and product, indicating that the pyrophosphate anion does not move significantly during the reaction.70,71,125 Furthermore, comparison of computational analyses of the reaction leading to bornyl diphosphate by Tantillo, which included only the carbocations and pyrophosphate,126 relative to that by Major, which encompassed the complete enzyme system,127 indicate a dominant role for the released pyrophosphate anion in stabilization/formation of the final bornyl+ intermediate, which is of necessity a secondary carbocation that is intrinsically less stable than the preceding tertiary carbocation intermediate. The reported structural analysis of bornyl diphosphate synthase further supports a dominant electrostatic role for the pyrophosphate anion co-product, as this revealed mechanistically non-relevant counter-ion pairing between aza-analogs of early stage carbocation intermediates and pyrophosphate.64 Also consistent with this is the greater synergy exhibited between pyrophosphate and aza-analogs of late, relative to early, stage carbocation intermediates.92 Perhaps more critically, these results further imply that these TPS catalyzed reactions are under kinetic rather than thermodynamic control – i.e., carbocationic intermediates do not reorient in the active site to ion-pair with the pyrophosphate and are only transiently present, with their movement restricted by the enzymatic active site.61
Nevertheless, it seems likely that the pyrophosphate co-product exerts an electrostatic influence on the catalyzed reaction. Specifically, steering carbocation migration, via alkene cycloaddition(s) and/or rearrangement(s), towards itself to achieve counter ion pairing, although other than in the case of bornyl diphosphate this does not result in readdition of the pyrophosphate group.92 This is perhaps most strikingly observed in certain di-TPSs, where the presence of an inert aliphatic residue enables more complex reactions to proceed towards carbocation intermediates proximal to the pyrophosphate (without readdition), while the presence of a hydroxyl side-chain containing residue short-circuits the reaction at an earlier stage (e.g., see Fig. 21A).87–90 Notably, this effect is position dependent,79 and these results have been interpreted as indicating a role for these specific hydroxyl dipoles in stabilization of early carbocation intermediates long enough for deprotonation to occur, rather than these residues acting as the catalytic base themselves.92 This then provides an intriguing contrast to the role of aromatic residues in class II terpene cyclases, where quadrapole-carbocation stabilization seems to promote further cyclization. However, this may be a reflection of the extended nature of the ring system formed in these cyclization reactions relative to the often more compact and complex reaction sequences catalyzed by TPSs. As has been noted elsewhere,2 there are differences in the exact interactions by which TPSs bind their requisite trio of magnesium ions. In turn, these divalent ions ligate the diphosphate moiety of the substrate, leading us to speculate that the resulting subtle differences in electrostatic environment exerted by the ensuing pyrophosphate-Mg2+3 complex may affect the catalyzed reaction.
It seems likely that the dynamics of terpenoid synthase structures also may affect product outcome. For example, even protein motions that are significantly slower than the catalyzed reaction will affect the reaction to the extent that different initial substrate conformations leading to different product outcome are allowed and/or imposed by the accessible different states of the enzyme. Arguably more intriguing would be a role for enzyme dynamics during the course of the reaction. For example, it seems possible that the vibrational modes of the enzymes might help push carbocation intermediates along certain reaction paths. This might even be coupled to the chemistry itself, as these reactions are highly exothermic, which may be translated into motion. However, a role for protein dynamics in these reactions remains conjectural at this time.
7 Conclusions
Over the last two decades, high-resolution crystal structures have become available for terpenoid synthases, which have led to a number of intriguing insights. This includes revelation of unexpected evolutionary relationships, leading to the unifying class I terpenoid synthase designation for the allylic diphosphate ionization-initiated trans-IDSs and downstream acting squalene/phytoene synthases, as well as TPSs, along with unifying the mechanistically distinct protonation-initiated tri- and di- terpene cyclases designated as class II, with fusion and subsequent domain loss of these observed in the plant kingdom. More detailed mechanistic understanding of the catalyzed reactions also has resulted from determination of these structures. An early, if arguably somewhat trivial insight into the pre-nylelongases was the finding that size of the active site cavity clearly affects product chain length, with the more interesting result that structural characterization directly led to the ability to engineer this aspect of catalysis. Determination of triterpene cyclase structures also led to some insight into how product outcome is controlled, largely the effect exerted by aromatic residues, which seem to promote cyclization by stabilizing quadrapole-cation interactions. However, these reactions are still incompletely understood, particularly the enzymatic determinants for rearrangement following initial cyclization. While it has been suggested that this may be specified by a lack of suitably positioned base for deprotonation and/or π-electron gradient leading back to a rearranged carbocation, this remains somewhat uncertain (e.g., how then are alternative product outcomes mediated by wild-type and mutant enzymes, and how does initial cyclization proceed up the π-electron gradient?). The enzymatic structure–function relationships underlying product outcome for the complex reactions catalyzed by TPSs are even less clear. While it is evident that this is controlled to a large degree by the initial substrate conformation imposed by the enzymatic active site, even this is not fully understood. Moreover, the effect of the diphosphate anion co-product in steering carbocation migration has only recently been appreciated, and other than some initial studies on the effect of hydroxyl dipoles, the role of electrostatic interactions has not been examined, with essentially no studies on the potential role protein dynamics might play in these reactions. Thus, the structural characterization of terpenoid synthases reported to-date represents an incomplete view, with some just serving to highlight our incomplete understanding of the enzymatic structure–function relationships underlying the more complex reactions catalyzed by these enzymes. Nevertheless, it seems likely that further investigations, although perhaps now including the use of NMR and explicit examination (via both experimental and computational approaches) of the role of dynamics, will lead to increased understanding, towards the ultimate goal of enabling rational engineering of catalytic activity.
Supplementary Material
Acknowledgements
Support for work on isoprenyl diphosphate and terpene synthases/cyclases in the corresponding author’s laboratory from the NIH (GM076324) is gratefully acknowledged. Y.G. was supported by NIH grant NS010546 to R.B.H.
Biographies

Reuben J. Peters received his B.S. from U.C. San Diego in 1992, and a Ph.D. with David Agard from U.C. San Francisco in 1998. After working as a Postdoctoral Fellow of the Jane Coffin Childs Memorial Fund for Medical Research with Rodney Croteau at Washington State University, Peters joined the faculty at Iowa State University in 2002, where he is now a full professor. His research interests focus on elucidation of the enzymatic mechanisms and metabolic pathways underlying biosynthesis of labdane-related diterpenoid natural products, as well as characterization of their physiological and other, potentially useful functions.

Yang Gao received his B.S. in 2007 from the University of Science and Technology of China, and has been a graduate student in Prof. Richard B. Honzatko’s research group at Iowa State University since then. His thesis research has focused on investigating structure–function relationships in fructose-1,6-bisphosphatase and class I terpene synthases.

Richard B. Honzatko received his B.S. from the University of Michigan in 1976, and a Ph.D. with William Lipscomb from Harvard University in 1982. After working as a National Research Council Associate at the Naval Research Laboratory with Wayne Hendrickson, Honzatko joined the faculty at Iowa State University in 1983, where he is now a full professor. His research interests focus on enzyme mechanisms, allosteric regulation of enzymes and protein structure.
Footnotes
This paper is part of an NPR themed issue on Structural Aspects of Biosynthesis.
Electronic supplementary information (ESI) available. See DOI:10.1039/c2np20059g
References
- 1.Croteau R, Kutchan TM, Lewis NG. In: Biochemistry & Molecular Biology of Plants. Buchanan B, Gruissem W, Jones R, editors. Rockville, MD, USA: Am. Soc. Plant Biologists; 2000. pp. 1250–1318. [Google Scholar]
- 2.Christianson DW. Curr. Opin. Chem. Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wendt KU, Schulz GE. Structure. 1998;6:127–133. doi: 10.1016/s0969-2126(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 4.Cao R, Zhang Y, Mann FM, Huang C, Mukkamala D, Mudock MP, Mead ME, Prisic S, Wang K, Lin K-Y, Chang T-K, Peters RJ, Oldfield E. Proteins: Struct., Funct., Bioinf. 2010;78:2417–2432. doi: 10.1002/prot.22751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Köksal M, Jin Y, Coates RM, Croteau R, Christianson DW. Nature. 2011;469:116–120. doi: 10.1038/nature09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruzicka L, Eschenmoser A, Heusser H. Experientia. 1953;IX:357–367. [Google Scholar]
- 7.Christianson DW. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 8.Kuzuyama T, Noel JP, Richard SB. Nature. 2005;435:983–987. doi: 10.1038/nature03668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metzger U, Keller S, Stevenson CE, Heide L, Lawson DM. J. Mol. Biol. 2010;404:611–626. doi: 10.1016/j.jmb.2010.09.067. [DOI] [PubMed] [Google Scholar]
- 10.Metzger U, Schall C, Zocher G, Unsold I, Stec E, Li SM, Heide L, Stehle T. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14309–14314. doi: 10.1073/pnas.0904897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tarshis LC, Yan M, Poulter CD, Sacchettini JC. Biochemistry. 1994;33:10871–10877. doi: 10.1021/bi00202a004. [DOI] [PubMed] [Google Scholar]
- 12.Joly A, Edwards PA. J. Biol. Chem. 1993;268:3044–3048. [PubMed] [Google Scholar]
- 13.Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, Poulter CD. Proc. Natl. Acad. Sci. U. S. A. 1996;93:15018–15023. doi: 10.1073/pnas.93.26.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. J. Biol. Chem. 2004;279:8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 15.Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH, Rogers MJ, Russell RG, Oppermann U. Proc. Natl. Acad. Sci. U. S. A. 2006;103:7829–7834. doi: 10.1073/pnas.0601643103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang PH. Biochemistry. 2009;48:6562–6570. doi: 10.1021/bi900371p. [DOI] [PubMed] [Google Scholar]
- 17.Rondeau JM, Bitsch F, Bourgier E, Geiser M, Hemmig R, Kroemer M, Lehmann S, Ramage P, Rieffel S, Strauss A, Green JR, Jahnke W. ChemMedChem. 2006;1:267–273. doi: 10.1002/cmdc.200500059. [DOI] [PubMed] [Google Scholar]
- 18.Chang TH, Guo RT, Ko TP, Wang AH, Liang PH. J. Biol. Chem. 2006;281:14991–15000. doi: 10.1074/jbc.M512886200. [DOI] [PubMed] [Google Scholar]
- 19.Kavanagh KL, Dunford JE, Bunkoczi G, Russell RG, Oppermann U. J. Biol. Chem. 2006;281:22004–22012. doi: 10.1074/jbc.M602603200. [DOI] [PubMed] [Google Scholar]
- 20.Kloer DP, Welsch R, Beyer P, Schulz GE. Biochemistry. 2006;45:15197–15204. doi: 10.1021/bi061572k. [DOI] [PubMed] [Google Scholar]
- 21.Guo RT, Cao R, Liang PH, Ko TP, Chang TH, Hudock MP, Jeng WY, Chen CK, Zhang Y, Song Y, Kuo CJ, Yin F, Oldfield E, Wang AH. Proc. Natl. Acad. Sci. U. S. A. 2007;104:10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang TH, Hsieh FL, Ko TP, Teng KH, Liang PH, Wang AH. Plant Cell. 2010;22:454–467. doi: 10.1105/tpc.109.071738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burke CC, Wildung MR, Croteau R. Proc. Natl. Acad. Sci. U. S. A. 1999;96:13062–13067. doi: 10.1073/pnas.96.23.13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burke C, Croteau R. J. Biol. Chem. 2002;277:3141–3149. doi: 10.1074/jbc.M105900200. [DOI] [PubMed] [Google Scholar]
- 25.Hsieh FL, Chang TH, Ko TP, Wang AH. Plant Physiol. 2011;155:1079–1090. doi: 10.1104/pp.110.168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo RT, Kuo CJ, Chou CC, Ko TP, Shr HL, Liang PH, Wang AH. J. Biol. Chem. 2004;279:4903–4912. doi: 10.1074/jbc.M310161200. [DOI] [PubMed] [Google Scholar]
- 27.Guo RT, Kuo CJ, Ko TP, Chou CC, Liang PH, Wang AH. Biochemistry. 2004;43:7678–7686. doi: 10.1021/bi036336d. [DOI] [PubMed] [Google Scholar]
- 28.Sun HY, Ko TP, Kuo CJ, Guo RT, Chou CC, Liang PH, Wang AH. J. Bacteriol. 2005;187:8137–8148. doi: 10.1128/JB.187.23.8137-8148.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sasaki D, Fujihashi M, Okuyama N, Kobayashi Y, Noike M, Koyama T, Miki K. J. Biol. Chem. 2011;286:3729–3740. doi: 10.1074/jbc.M110.147991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujihashi M, Zhang YW, Higuchi Y, Li XY, Koyama T, Miki K. Proc. Natl. Acad. Sci. U. S. A. 2001;98:4337–4342. doi: 10.1073/pnas.071514398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ko TP, Chen YK, Robinson H, Tsai PC, Gao YG, Chen AP, Wang AH, Liang PH. J. Biol. Chem. 2001;276:47474–47482. doi: 10.1074/jbc.M106747200. [DOI] [PubMed] [Google Scholar]
- 32.Guo RT, Ko TP, Chen AP, Kuo CJ, Wang AH, Liang PH. J. Biol. Chem. 2005;280:20762–20774. doi: 10.1074/jbc.M502121200. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Dong C, McNeil M, Kaur D, Mahapatra S, Crick DC, Naismith JH. J. Mol. Biol. 2008;381:129–140. doi: 10.1016/j.jmb.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noike M, Ambo T, Kikuchi S, Suzuki T, Yamashita S, Takahashi S, Kurokawa H, Mahapatra S, Crick DC, Koyama T. Biochem. Biophys. Res. Commun. 2008;377:17–22. doi: 10.1016/j.bbrc.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ., Jr J. Biol. Chem. 2000;275:30610–30617. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- 36.Epstein WW, Poulter CD. Phytochemistry. 1973;12:737–747. [Google Scholar]
- 37.Rivera SB, Swedlund BD, King GJ, Bell RN, Hussey CE, Jr, Shattuck-Eidens DM, Wrobel WM, Peiser GD, Poulter CD. Proc. Natl. Acad. Sci. U. S. A. 2001;98:4373–4378. doi: 10.1073/pnas.071543598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hemmerlin A, Rivera SB, Erickson HK, Poulter CD. J. Biol. Chem. 2003;278:32132–32140. doi: 10.1074/jbc.M213045200. [DOI] [PubMed] [Google Scholar]
- 39.Erickson HK, Poulter CD. J. Am. Chem. Soc. 2003;125:6886–6888. doi: 10.1021/ja034520g. [DOI] [PubMed] [Google Scholar]
- 40.Thulasiram HV, Erickson HK, Poulter CD. Science. 2007;316:73–76. doi: 10.1126/science.1137786. [DOI] [PubMed] [Google Scholar]
- 41.Thulasiram HV, Erickson HK, Poulter CD. J. Am. Chem. Soc. 2008;130:1966–1971. doi: 10.1021/ja0771282. [DOI] [PubMed] [Google Scholar]
- 42.Sallaud C, Rontein D, Onillon S, Jabes F, Duffe P, Giacalone C, Thoraval S, Escoffier C, Herbette G, Leonhardt N, Causse M, Tissier A. Plant Cell. 2009;21:301–317. doi: 10.1105/tpc.107.057885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schilmiller AL, Schauvinhold I, Larson M, Xu R, Charbonneau AL, Schmidt A, Wilkerson C, Last RL, Pichersky E. Proc. Natl. Acad. Sci. U. S. A. 2009;106:10865–10870. doi: 10.1073/pnas.0904113106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steele CL, Crock J, Bohlmann J, Croteau RB. J. Biol. Chem. 1998;273:2078–2089. doi: 10.1074/jbc.273.4.2078. [DOI] [PubMed] [Google Scholar]
- 45.Lesburg CA, Zhai G, Cane DE, Christianson DW. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 46.Starks CM, Back K, Chappell J, Noel JP. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 47.Seemann M, Zhai G, de Kraker JW, Paschall CM, Christianson DW, Cane DE. J. Am. Chem. Soc. 2002;124:7681–7689. doi: 10.1021/ja026058q. [DOI] [PubMed] [Google Scholar]
- 48.Seemann M, Zhai G, Umezawa K, Cane D. J. Am. Chem. Soc. 1999;121:591–592. [Google Scholar]
- 49.Back K, Chappell J. Proc. Natl. Acad. Sci. U. S. A. 1996;93:681–6845. doi: 10.1073/pnas.93.13.6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenhagen BT, O’Maille PE, Noel JP, Chappell J. Proc. Natl. Acad. Sci. U. S. A. 2006;103:9826–9831. doi: 10.1073/pnas.0601605103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rising KA, Starks CM, Noel JP, Chappell J. J. Am. Chem. Soc. 2000;122:1861–1866. [Google Scholar]
- 52.Noel JP, Dellas N, Faraldos JA, Zhao M, Hess BA, Jr, Smentek L, Coates RM, O’Maille PE. ACS Chem. Biol. 2010;5:377–392. doi: 10.1021/cb900295g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caruthers JM, Kang I, Rynkiewicz MJ, Cane DE, Christianson DW. J. Biol. Chem. 2000;275:25533–25539. doi: 10.1074/jbc.M000433200. [DOI] [PubMed] [Google Scholar]
- 54.Shishova EY, Costanzo LD, Cane DE, Christianson DW. Biochemistry. 2007;46:1941–1951. doi: 10.1021/bi0622524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shishova EY, Yu F, Miller DJ, Faraldos JA, Zhao Y, Coates RM, Allemann RK, Cane DE, Christianson DW. J. Biol. Chem. 2008;283:15431–15439. doi: 10.1074/jbc.M800659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Felicetti B, Cane DE. J. Am. Chem. Soc. 2004;126:7212–7221. doi: 10.1021/ja0499593. [DOI] [PubMed] [Google Scholar]
- 57.Deligeorgopoulou A, Allemann RK. Biochemistry. 2003;42:7741–7747. doi: 10.1021/bi034410m. [DOI] [PubMed] [Google Scholar]
- 58.Deligeorgopoulou A, Taylor SE, Forcat S, Allemann RK. Chem. Commun. 2003:2162–2163. doi: 10.1039/b306867f. [DOI] [PubMed] [Google Scholar]
- 59.Rynkiewicz MJ, Cane DE, Christianson DW. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rynkiewicz MJ, Cane DE, Christianson DW. Biochemistry. 2002;41:1732–1741. doi: 10.1021/bi011960g. [DOI] [PubMed] [Google Scholar]
- 61.Vedula LS, Cane DE, Christianson DW. Biochemistry. 2005;44:12719–12727. doi: 10.1021/bi0510476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vedula LS, Jiang J, Zakharian T, Cane DE, Christianson DW. Arch. Biochem. Biophys. 2008;469:184–194. doi: 10.1016/j.abb.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vedula LS, Rynkiewicz MJ, Pyun H-J, Coates RM, Cane DE, Christianson DW. Biochemistry. 2005;44:6153–6163. doi: 10.1021/bi050059o. [DOI] [PubMed] [Google Scholar]
- 64.Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau RB, Christianson DW. Proc. Natl. Acad. Sci. U. S. A. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gennadios HA, Gonzalez V, Di Costanzo L, Li A, Yu F, Miller DJ, Allemann RK, Christianson DW. Biochemistry. 2009;48:6175–6183. doi: 10.1021/bi900483b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aaron JA, Lin X, Cane DE, Christianson DW. Biochemistry. 2010;49:1787–1797. doi: 10.1021/bi902088z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McAndrew RP, Peralta-Yahya PP, DeGiovanni A, Pereira JH, Hadi MZ, Keasling JD, Adams PD. Structure. 2011;19:1876–1884. doi: 10.1016/j.str.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 68.Hyatt DC, Youn B, Zhao Y, Santhamma B, Coates RM, Croteau RB, Kang C. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5360–5365. doi: 10.1073/pnas.0700915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williams DC, McGarvey DJ, Katahira EJ, Croteau R. Biochemistry. 1998;37:12213–12220. doi: 10.1021/bi980854k. [DOI] [PubMed] [Google Scholar]
- 70.Cane DE, Saito A, Croteau R, Shaskus J, Felton M. J. Am. Chem. Soc. 1982;104:5831–5833. [Google Scholar]
- 71.Cane DE. Arch. Biochem. Biophys. 2006;448:117–122. doi: 10.1016/j.abb.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 72.Kolesnikova MD, Obermeyer AC, Wilson WK, Lynch DA, Xiong Q, Matsuda SP. Org. Lett. 2007;9:2183–2186. doi: 10.1021/ol070709b. [DOI] [PubMed] [Google Scholar]
- 73.Wise ML, Savage TJ, Katahira E, Croteau R. J. Biol. Chem. 1998;273:14891–14899. doi: 10.1074/jbc.273.24.14891. [DOI] [PubMed] [Google Scholar]
- 74.Kampranis SC, Ioannidis D, Purvis A, Mahrez W, Ninga E, Katerelos NA, Anssour S, Dunwell JM, Degenhardt J, Makris AM, Goodenough PW, Johnson CB. Plant Cell. 2007;19:1994–2005. doi: 10.1105/tpc.106.047779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peters RJ, Croteau RB. Arch. Biochem. Biophys. 2003;417:203–211. doi: 10.1016/s0003-9861(03)00347-3. [DOI] [PubMed] [Google Scholar]
- 76.Koksal M, Zimmer I, Schnitzler JP, Christianson DW. J. Mol. Biol. 2010;402:363–373. doi: 10.1016/j.jmb.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peters RJ. Nat. Prod. Rep. 2010;27:1521–1530. doi: 10.1039/c0np00019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morrone D, Hillwig ML, Mead ME, Lowry L, Fulton DB, Peters RJ. Biochem. J. 2011;435:589–595. doi: 10.1042/BJ20101429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou K, Gao Y, Hoy JA, Mann FM, Honzatko RB, Peters RJ. J. Biol. Chem. 2012;287:6840–6850. doi: 10.1074/jbc.M111.337592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peters RJ, Ravn MM, Coates RM, Croteau RB. J. Am. Chem. Soc. 2001;123:8974–8978. doi: 10.1021/ja010670k. [DOI] [PubMed] [Google Scholar]
- 81.Peters RJ, Flory JE, Jetter R, Ravn MM, Lee H-J, Coates RM, Croteau RB. Biochemistry. 2000;39:15592–15602. doi: 10.1021/bi001997l. [DOI] [PubMed] [Google Scholar]
- 82.Keeling CI, Madilao LL, Zerbe P, Dullat HK, Bohlmann J. J. Biol. Chem. 2011;286:21145–21153. doi: 10.1074/jbc.M111.245951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ravn MM, Coates RM, Flory J, Peters RJ, Croteau R. Org. Lett. 2000;2:573–576. doi: 10.1021/ol991230p. [DOI] [PubMed] [Google Scholar]
- 84.Ravn MM, Coates RM, Jetter R, Croteau R. Chem. Commun. 1998;1998:21–22. [Google Scholar]
- 85.Ravn MM, Peters RJ, Coates RM, Croteau RB. J. Am. Chem. Soc. 2002;124:6998–7006. doi: 10.1021/ja017734b. [DOI] [PubMed] [Google Scholar]
- 86.Peters RJ, Croteau RB. Proc. Natl. Acad. Sci. U. S. A. 2002;99:580–584. doi: 10.1073/pnas.022627099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilderman PR, Peters RJ. J. Am. Chem. Soc. 2007;129:15736–15737. doi: 10.1021/ja074977g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Keeling CI, Weisshaar S, Lin RPC, Bohlmann J. Proc. Natl. Acad. Sci. U. S. A. 2008;105:1085–1090. doi: 10.1073/pnas.0709466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morrone D, Xu M, Fulton DB, Determan MK, Peters RJ. J. Am. Chem. Soc. 2008;130:5400–5401. doi: 10.1021/ja710524w. [DOI] [PubMed] [Google Scholar]
- 90.Xu M, Wilderman PR, Peters RJ. Proc.Natl. Acad.Sci. U. S. A. 2007;104:7397–7401. doi: 10.1073/pnas.0611454104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martin DM, Faldt J, Bohlmann J. Plant Physiol. 2004;135:1908–1927. doi: 10.1104/pp.104.042028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou K, Peters RJ. Chem. Commun. 2011;47:4074–4080. doi: 10.1039/c0cc02960b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sato T, Yoshida S, Hoshino H, Tanno M, Nakajima M, Hoshino T. J. Am. Chem. Soc. 2011;133:9734–9737. doi: 10.1021/ja203779h. [DOI] [PubMed] [Google Scholar]
- 94.Lodeiro S, Xiong Q, Wilson WK, Kolesnikova MD, Onak CS, Matsuda SP. J. Am. Chem. Soc. 2007;129:11213–11222. doi: 10.1021/ja073133u. [DOI] [PubMed] [Google Scholar]
- 95.Wendt KU, Poralla K, Schulz GE. Science. 1997;277:1811–1815. doi: 10.1126/science.277.5333.1811. [DOI] [PubMed] [Google Scholar]
- 96.Wendt KU, Lenhart A, Schulz GE. J. Mol. Biol. 1999;286:175–187. doi: 10.1006/jmbi.1998.2470. [DOI] [PubMed] [Google Scholar]
- 97.Reinert DJ, Balliano G, Schulz GE. Chem. Biol. 2004;11:121–126. doi: 10.1016/j.chembiol.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 98.Wendt KU, Schulz GE, Corey EJ, Liu DR. Angew. Chem., Int. Ed. 2000;39:2812–2833. [PubMed] [Google Scholar]
- 99.Wendt KU. Angew. Chem., Int. Ed. 2005;44:3966–3971. doi: 10.1002/anie.200500804. [DOI] [PubMed] [Google Scholar]
- 100.Hoshino T, Sato T. Chem. Commun. 2002:291–301. doi: 10.1039/b108995c. [DOI] [PubMed] [Google Scholar]
- 101.Morikubo N, Fukuda Y, Ohtake K, Shinya N, Kiga D, Sakamoto K, Asanuma M, Hirota H, Yokoyama S, Hoshino T. J. Am. Chem. Soc. 2006;128:13184–13194. doi: 10.1021/ja063358p. [DOI] [PubMed] [Google Scholar]
- 102.Thoma R, Schulz-Gasch T, D’Arcy B, Benz J, Aebi J, Dehmlow H, Henning M, Stihle M, Ruf A. Nature. 2004;432:118–122. doi: 10.1038/nature02993. [DOI] [PubMed] [Google Scholar]
- 103.Lenhart A, Weihofen WA, Pleschke AE, Schulz GE. Chem. Biol. 2002;9:639–645. doi: 10.1016/s1074-5521(02)00138-2. [DOI] [PubMed] [Google Scholar]
- 104.Prisic S, Xu J, Coates RM, Peters RJ. ChemBioChem. 2007;8:869–874. doi: 10.1002/cbic.200700045. [DOI] [PubMed] [Google Scholar]
- 105.Köksal M, Hu H, Coates RM, Peters RJ, Christianson DW. Nat. Chem. Biol. 2011;7:431–433. doi: 10.1038/nchembio.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Peters RJ, Croteau RB. Biochemistry. 2002;41:1836–1842. doi: 10.1021/bi011879d. [DOI] [PubMed] [Google Scholar]
- 107.Mann FM, Prisic S, Davenport EK, Determan MK, Coates RM, Peters RJ. J. Biol. Chem. 2010;285:20558–20563. doi: 10.1074/jbc.M110.123307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Reardon D, Farber GK. FASEB J. 1995;9:497–503. doi: 10.1096/fasebj.9.7.7737457. [DOI] [PubMed] [Google Scholar]
- 109.Xue Z, Duan L, Liu D, Guo J, Ge S, Dicks J, Osbourn OMPA, Qi X. New Phytol. 2012;193:1022–1038. doi: 10.1111/j.1469-8137.2011.03997.x. [DOI] [PubMed] [Google Scholar]
- 110.Morrone D, Chambers J, Lowry L, Kim G, Anterola A, Bender K, Peters RJ. FEBS Lett. 2009;583:475–480. doi: 10.1016/j.febslet.2008.12.052. [DOI] [PubMed] [Google Scholar]
- 111.Bohlmann J, Meyer-Gauen G, Croteau R. Proc. Natl. Acad. Sci. U. S. A. 1998;95:4126–4133. doi: 10.1073/pnas.95.8.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Trapp SC, Croteau RB. Genetics. 2001;158:811–832. doi: 10.1093/genetics/158.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hayashi K, Kawaide H, Notomi M, Sakigi Y, Matsuo A, Nozaki H. FEBS Lett. 2006;580:6175–6181. doi: 10.1016/j.febslet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 114.Keeling CI, Dullat HK, Yuen M, Ralph SG, Jancsik S, Bohlmann J. Plant Physiol. 2010;152:1197–1208. doi: 10.1104/pp.109.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen F, Tholl D, Bohlmann J, Pichersky E. Plant J. 2011;66:212–229. doi: 10.1111/j.1365-313X.2011.04520.x. [DOI] [PubMed] [Google Scholar]
- 116.Hillwig ML, Xu M, Toyomasu T, Tiernan MS, Gao W, Cui G, Huang L, Peters RJ. Plant J. 2011;68:1051–1060. doi: 10.1111/j.1365-313X.2011.04756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cane D. Acc. Chem. Res. 1985;18:220–226. [Google Scholar]
- 118.Hess BA, Jr, Smentek L, Noel JP, O’Maille PE. J. Am. Chem. Soc. 2011;133:12632–12641. doi: 10.1021/ja203342p. [DOI] [PubMed] [Google Scholar]
- 119.O’Maille PE, Malone A, Dellas N, Andes Hess B, Jr, Smentek L, Sheehan I, Greenhagen BT, Chappell J, Manning G, Noel JP. Nat. Chem. Biol. 2008;4:617–623. doi: 10.1038/nchembio.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yoshikuni Y, Ferrin TE, Keasling JD. Nature. 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- 121.Yoshikuni Y, Martin VJJ, Ferrin TE, Keasling JD. Chem. Biol. 2006;13:91–98. doi: 10.1016/j.chembiol.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 122.Degenhardt J, Kollner TG, Gershenzon J. Phytochemistry. 2009;70:1621–1637. doi: 10.1016/j.phytochem.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 123.Tantillo DJ. Nat. Prod. Rep. 2011;28:1035–1053. doi: 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]
- 124.Zu L, Xu M, Lodewyk MW, Cane DE, Peters RJ, Tantillo DJ. J. Am. Chem. Soc. 2012;134:11369–11371. doi: 10.1021/ja3043245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Croteau RB, Shaskus JJ, Renstrom B, Felton NM, Cane DE, Saito A, Chang C. Biochemistry. 1985;24:7077–7085. doi: 10.1021/bi00346a009. [DOI] [PubMed] [Google Scholar]
- 126.Hong YJ, Tantillo DJ. Org. Biomol. Chem. 2010;8:4589–4600. doi: 10.1039/c0ob00167h. [DOI] [PubMed] [Google Scholar]
- 127.Weitman M, Major DT. J. Am. Chem. Soc. 2010;132:6349–6360. doi: 10.1021/ja910134x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.