

Abstract
BioA, a pyridoxal 5′-phosphate (PLP) dependent aminotransferase, catalyzes the second step of biotin biosynthesis, converting 7-keto-8-aminopelargonic acid (KAPA) into 7,8-diaminopelargonic acid (DAPA). Amiclenomycin (ACM) isolated from cultures of different Streptomyces strains is a potent mechanism-based inhibitor of BioA that operates via an aromatization mechanism, irreversibly labeling the PLP cofactor. However, ACM is plagued by inherent chemical stability. Herein we describe the synthesis of four inhibitors, inspired by ACM but containing an allylic amine as the chemical warhead, designed to both improve stability and operate via a complementary Michael addition-pathway upon enzymatic oxidation of the allylic amine substrate to an enimine. Acyclic analogue M-1 contains a terminal olefin as the pro-Michael acceptor. The synthesis of M-1 features an alkyne-zipper reaction and the Overman rearrangement as key synthetic operations. The cyclic analogues M-2/3/4 contain either an endocyclic or exocyclic olefin as the pro-Michael acceptor. These were all prepared using a common strategy employing DIBAL reduction of a precursor bicyclic lactam, followed by in situ Horner-Wadsworth-Emmons (HWE) olefination as the key synthetic steps.
INTRODUCTION
Biotin (vitamin H or B7), a structurally simple bicyclic molecule comprised of an imidazol-2-one fused to a tetrahydrothiophene with a pentanoic acid side-chain, is a cofactor required for all organisms (Figure 1). Biotin is covalently attached via an amide linkage to the ε-amino group of a conserved lysine residue of biotin carrier protein domains, which are part of multimeric enzymes involved in carboxy-transfer reactions.1 In acetyl-CoA carboxylase, a representative biotin-dependent enzyme, the biotin cofactor is directly carboxylated at the N-1 position of the imidazol-2-one ring to afford a stable carbamic acid (t1/2 > 100 min at pH 8).1 Subsequent transfer of the activated carboxy group onto acetyl-CoA affords malonyl-CoA, the key monomeric building block for synthesis of fatty acids. Biotin-dependent enzymes are also found in other primary and secondary metabolic pathways including gluconeogenesis, amino acid catabolism, and polyketide synthesis.1
Figure 1.

Conversion of KAPA to DAPA catalyzed by BioA. DAPA is elaborated to biotin by two additional enzymes (BioD and BioB), then covalently attached to biotin carboxylase carrier protein domains (BCCP) by an ATP-dependent biotin protein ligase.
Bacteria, fungi, and plants synthesize biotin de novo, whereas mammals must obtain this vital cofactor either from their diet or from commensal bacteria in the digestive tract. Biotin auxotrophs of Escherichia coli and Mycobacterium tuberculosis that can only survive when biotin is supplemented in the growth medium have been isolated.1,2 The concentration of biotin in serum from humans is approximately 2 nM, which could potentially rescue biotin auxotrophs.3 In the case of M. tuberculosis, Schnappinger and co-workers have shown, using a novel mycobacterial conditional expression system, that exogenous host biotin is not sufficient to support bacterial growth or maintenance in a murine infection model.4
The lack of new antibiotics for M. tuberculosis and other clinically significant Gram negative bacteria coupled with the dramatic increase of multidrug resistant strains requires new lead compounds and exploration of other biochemical pathways outside the conventional antibiotic targets of RNA transcription and DNA-, protein-, and cell-wall synthesis. Based on the confirmed essentiality and inherent bacterial specificity, the biotin biosynthetic pathway represents an attractive target for the development of new antibacterial agents.
BioA, a pyridoxal 5′-phosphate (PLP) dependent aminotransferase, catalyzes the second step of biotin biosynthesis, converting 7-keto-8-aminopelargonic acid (KAPA) into 7,8-diaminopelargonic acid (DAPA) using S-adenosylmethionine (SAM) as the amino donor (Figure 1).5 Amiclenomycin (ACM), isolated from cultures of different Streptomyces strains,6 and its simplified amino-alcohol analog7 (ACM-OH, Figure 2A) are potent inhibitors of BioA.8 Structurally, both ACM and ACM-OH bear a symmetrical cis-1,4-cyclohexadiene ring that acts as the chemical warhead, resulting in irreversible inactivation of the PLP cofactor of BioA via an aromatization mechanism.5b,9 Interestingly, ACM and ACM-OH exhibit selective activity against M. tuberculosis from over 40 bacterial strains evaluated.8
Figure 2.

A) Design of novel Michael addition-based inhibitors; B) Proposed Michael addition-based mechanism of inhibition.
While mechanistically and structurally intriguing, amiclenomycin suffers from inherent poor chemical stability, which results in rapid aromatization to an inactive aniline derivative.8 Consequently, we sought to develop inhibitors wherein the cis-1,4-cyclohexadiene warhead was replaced with a series of acyclic and cyclic warheads containing chemically stable allylic amines capable of inactivating BioA via a Michael addition mechanism (see Figure 2A, M-1/2/3/4). This inhibitor design is based on extensive previous studies by Silverman and others on related mechanism-based inhibitors of PLP-dependent enzymes.10 As illustrated in Figure 2B using M-4 as a representative example, the inhibitor is expected to behave like a normal substrate undergoing transamination with the enzyme bound PLP cofactor to afford the inhibitor–PLP aldimine I. Subsequent redox isomerization (i.e. a 1,3-prototropic shift) mediated by a general base (Lys283 in BioA from M. tuberculosis) provides the electrophilic enimine III via quinonoid intermediate II. Enimine III is a Michael acceptor and can react with the general base to afford adduct IV.
RESULTS AND DISCUSSION
Acyclic analogue M-1 was designed to impart flexibility in the inhibitor scaffold to allow optimal positioning of the β-carbon of the Michael acceptor with the incipient nucleophile. Retrosynthetically, we envisioned M-1 could be derived from a trans-allylic imidate by the Overman rearrangement transform (Scheme 1).11 The palladium-catalyzed version using either (R)- or (S)-COP-Cl would allow access to either enantiomer.11c Further disconnection affords a terminal alkyne that in turn can be prepared by isomerization of commercially available 3-alkyn-1-ol.
Scheme 1.

Synthesis of M-1.
The synthesis of acyclic analogue M-1 began with the Brown–Yamashita alkyne isomerization of 3-octyn-1-ol 1 into the more thermodynamically stable terminal alkyne (as a result of the greater Csp-H versus Csp-Csp3 bond strength) employing ethylenediamine and NaH followed by TBS protection to afford 2 (Scheme 1).12 Deprotonation of the alkyne and addition to formaldehyde provided propargyl alcohol 3. Stereospecific reduction of 3 by Red-Al furnished trans-allylic alcohol 4, which was converted to the corresponding key trichloroacetimidate intermediate 5 by treatment with trichloroacetonitrile and DBU. 1,3-Transposition was accomplished by refluxing 5 in xylene to furnish allylic amine 6.11 Finally, sequential removal of TBS and trichloroacetyl groups provided M-1 in good overall yield (Scheme 1).
Since cyclic analogues M-2/3/4 share common structural features, a general homologation strategy was developed. Retrosynthetically, we envisaged that all analogs could be derived from the corresponding bridged lactams (Figure 3).13 The advantages of utilizing the lactams as starting points include: 1) the stereochemistry at the bridge points ensures the cis configuration once the lactam is opened; 2) there are multiple routes to convert lactams into corresponding amino alcohol derivatives, which allows us to find the optimal conditions for side chain homologation; 3) these lactams are either commercially available or can be synthesized conveniently from reported procedures.13
Figure 3.
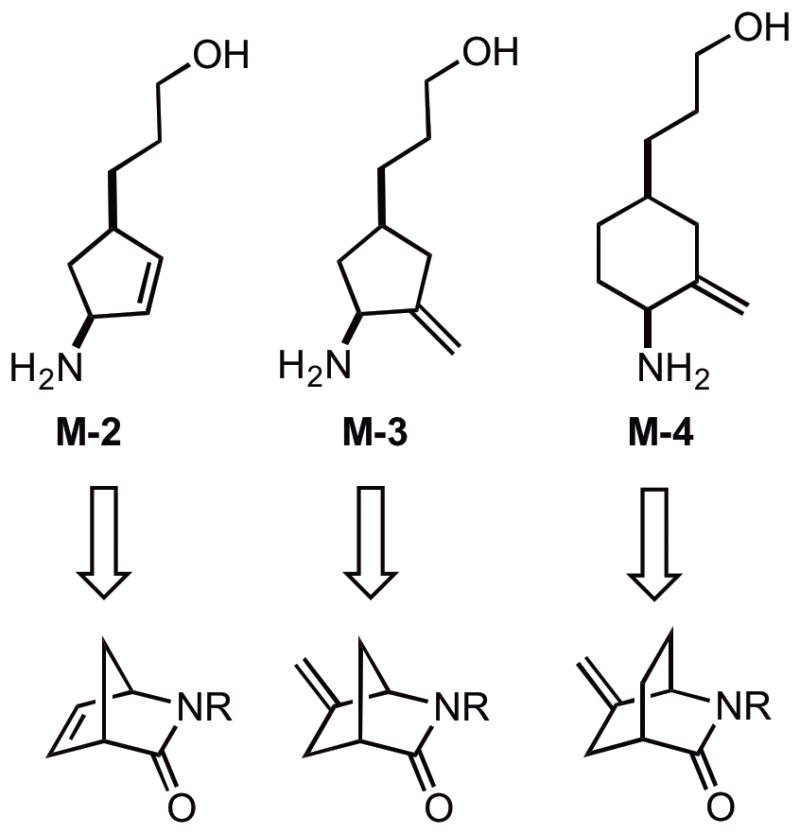
Retrosynthetic analysis of M-2/3/4.
Analogue M-2 can be synthesized from commercially available Vince lactam 7a, which was converted to the N-Boc derivative 7b as reported in 95% yield.14 The Boc group enhances the electrophilicity of the amide enabling ring-opening under substantially milder conditions than 7a. Three homologation strategies were tested using 7b as the starting material (Scheme 2). We first followed the reported procedures to synthesize the substituted malonate ester 8 from 7b in 4 steps.13a LiCl-mediated decarboxylation of 8 did lead to the desired monoester 11, however, the reaction required high temperature (~160 °C), at which loss of the Boc group became a major competitive side reaction. To find a better decarboxylation method, DCC-promoted coupling with Meldrum’s acid followed by reduction was performed.15 Although the subsequent decarboxylation went smoothly under very mild condition with quantitative yield, the coupling reaction of Meldrum’s acid with the amino acid intermediate derived from Vince lactam 7b suffered from epimerization as a result of the highly activated acyl-pyridinium intermediate. Lastly, a Horner-Wadsworth-Emmons (HWE) olefination–conjugate reduction strategy was evaluated for the homologation. DIBAL-H reduction of 7b afforded an intermediate hemiaminal in equilibrium with the open-chained amino-aldehyde, which was reacted with triethyl phosphonoacetate to afford the conjugate ester 10 with exclusive trans configuration in 81% isolated yield over two steps. CuBr-catalyzed conjugate reduction furnished the desired ester intermediate 11 in quantitative yield. As observed previously,16 the use of cyclohexene in large excess was essential to prevent reduction of the isolated alkene. Finally, reduction of ester 11, followed by Boc deprotection gave M-2 as its hydrochloride salt. Overall, the HWE olefination–conjugate reduction strategy required the shortest synthetic route and provided the highest isolated product yield among the three routes evaluated and was subsequently used for the synthesis of both M-3 and M-4.
Scheme 2.

Synthesis of M-2.
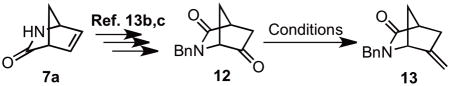
The synthesis of M-3 bearing an exocyclic methylene also started from Vince lactam 7a (Scheme 3). Following a previous report, the 6-keto Vince lactam 12 was synthesized conveniently in 5 steps.13b,c Four sets of reaction conditions were then screened for the olefination step (Table 1). The conventional Wittig (Entry 1) and Peterson (Entry 2) conditions led to incomplete reaction and poor isolated yields. On the other hand, the Tebbe-type olefination conditions (Entries 3 and 4) provided a fairly good yield of 13. Replacement of Zn and CH2Br2 with Nysted’s reagent (Entry 4) both substantially shortened the reaction time and improved the isolated yield to 88%. Therefore, the TiCl4 mediated olefination using Nysted’s reagent (Entry 4) became the standard condition for synthesis of exocyclic methylene analogs M-3/4.
Scheme 3.

Synthesis of M-3.
Table 1.
Optimization of olefination
| ||
|---|---|---|
| Entry | Condition (12→13) | Results |
| 1 |
|
~30% yield, incomplete reaction |
| 2 | TMSCH2MgBr, THF followed by NaH | ~35% yield, incomplete reaction |
| 3 | CH2Br2, Zn, TiCl4 THF-CH2Cl2, | 76% yield, 3 d |
| 4 | Nysted’s Reagent TiCl4, THF-CH2Cl2 | 88% yield, 4 h |
With an efficient and reliable method for the synthesis of 13 established, we now sought to complete the synthesis of M-3 (Scheme 3). First, protecting group exchange was performed (13→14). Chemoselective reductive removal of the N-benzyl protecting group, without disturbing the sensitive exocyclic olefin, was successfully accomplished employing Birch conditions. The resultant intermediate amide was then Boc-protected to afford 14. It should be noted that the relatively harsh reaction conditions required in the preparation of bicyclic lactam 12 necessitate a benzyl protecting group. Bicyclic amide 14 was elaborated to M-3 in analogy to the synthesis of M-2 by sequential DIBAL-H reduction, HWE olefination, conjugate reduction, LAH ester reduction, and deprotection of the Boc group.
The synthesis of M-4 bearing a 6-membered ring and exocyclic methylene proceeded as shown in Scheme 4. 6-Keto lactam 18 was first synthesized following the reported procedures from 2-pyridone 17.13d Methylenation employing Nysted’s reagent afforded 19. Next, the benzyl protecting group was exchanged for a Boc group (19→20). Elaboration of 20 to 22 was accomplished in four steps by sequential DIBAL-H reduction, HWE olefination, conjugate reduction, and LAH ester reduction in 67% overall yield over 4 steps (90.5% average yield per step) with a single purification. Treatment of 22 with 4 N HCl in dioxane furnished M-4, which was isolated as the hydrochloride salt in 89% by recrystallization from MeOH–Et2O.
Scheme 4.

Synthesis of M-4.
Compounds M-1/2/3/4 were evaluated for enzyme inhibition using a continuous coupled assay under initial velocity conditions.17 Unfortunately, only analogue M-2 showed any enzyme inhibition of BioA providing a modest IC50 value of 57 MM (Table 1). However, M-2 did not exhibit time-dependent inhibition as would be expected for an irreversible inhibitor. These results show that BioA is less promiscuous than originally anticipated. Our recently disclosed crystal structure of BioA with a dihydropyridone inhibitor, which acts as an aromatization mechanism-based inactivator,8 suggests that inhibitors should be relatively flat. Indeed, the cyclohexadiene warhead of amiclenomycin (ACM) is planar.
In conclusion, we have synthesized and biochemically characterized a series of compounds designed to inhibit BioA. Overall, the synthesis of M-1 is flexible and redox economical18 using isomerizations to manipulate the functionality. A common strategy was developed for the synthesis of analogues M-2/3/4 featuring DIBAL-H reduction of a bicyclic lactam to an intermediate hemiaminal and subsequent in situ Horner-Wadsworth-Emmons olefination. Introduction of the exocyclic methylene in analogues M-3/4 was optimally performed using the Nysted reagent. Biochemical evaluation of all four inhibitors indicate that planarity of the inhibitor is likely important since only cyclopentene M-2 demonstrated activity.
EXPERIMENTAL SECTION
General methods
All chemical reactions were performed under an inert atmosphere of dry Ar or N2 in oven-dried (150 °C) glassware. 1H and 13C NMR spectra were recorded on a 600 MHz spectrometer. Proton chemical shifts are reported in ppm from an internal standard of residual chloroform (7.26 ppm) or methanol (3.31 ppm), and carbon chemical shifts are reported using an internal standard of residual chloroform (77.0 ppm) or methanol (49.1 ppm). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad), coupling constant, integration. Melting points were measured on a melting point apparatus and uncorrected. High resolution mass spectra were obtained on a TOF II TOF/MS instrument equipped with either an ESI or APCI interface. TLC analyses were performed on TLC silica gel plates and were visualized with UV light, iodine chamber, 10% sulfuric acid or 10% PMA solution. Purifications were performed by flash chromatography on silica gel (60A). Enzyme inhibition assays were performed on an M5e multi-mode plate reader.
Materials
An anhydrous solvent dispensing system using 2 packed columns of neutral alumina was used for drying THF, Et2O, and CH2Cl2 while 2 packed columns of molecular sieves were used to dry DMF and the solvents were dispensed under argon. 7b14, 813a, 1213b,c, and 1813d were prepared as described.
tert-Butyldimethyl(oct-7-ynyloxy)silane (2)
To ethylene-1,2-diamine (30 mL) at 0 °C, was added NaH (2.54 g, 60 wt% in mineral oil, 64 mmol, 4 equiv) in one portion. The mixture was stirred at 0 °C for 5 min, 1 h at 25 °C, then heated at 65 °C for 1 h. The mixture was cooled to 45 °C and 3-octyn-1-ol (2.0 g, 16 mmol, 1 equiv) was added dropwise over 15 min. After the addition, the reaction was heated at 65 °C and stirred for 1 h. The mixture was cooled down to 0 °C and quenched by the successive cautious addition of H2O (25 mL) and 1 N aqueous HCl (25 mL). The quenched reaction mixture was diluted with 1 N aqueous HCl (25 mL) and extracted with Et2O (3 × 25 mL). The combined organic layers were washed with 1 N aqueous HCl (20 mL), saturated aqueous NaCl (20 mL), dried (MgSO4) and concentrated. Purification by silica gel column chromatography (10:90 EtOAc–hexanes to 20:80 EtOAc–hexanes) afforded 7-octyn-1-ol (1.5 g, 72%) as a colorless oil: Rf = 0.15 (1:4 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.28–1.31 (m, 2H), 1.32–1.38 (m, 2H), 1.40–1.53 (m, 4H), 1.90 (t, J = 2.4 Hz, 1H), 2.12 (td, J = 7.2, 2.4 Hz, 2H), 2.34 (br s, 1H, OH), 3.55 (t, J = 6.6 Hz, 2H); 13C NMR (CDCl3, 150 MHz) δ 18.5, 25.4, 28.6, 28.7, 32.7, 62.8, 68.4, 84.8. All data are consistent with reported literature values.12b
To a solution of 7-octyn-1-ol (1.0 g, 7.9 mmol, 1 equiv) prepared above, DMAP (98 mg, 0.79 mmol, 0.1 equiv) and imidazole (806 mg, 11.8 mmol, 1.5 equiv) in THF (10 mL) at 0 °C, was added a stock solution of tert-butyldimethylsilyl chloride in THF (4.4 M, 2.0 mL, 8.8 mmol, 1.1 equiv) over 5 min. The mixture was stirred for 1 h at 0 °C and quenched by addition of saturated aqueous NH4Cl (15 mL). The aqueous layer was extracted with Et2O (3 × 15 mL). The combined organic layers were washed with H2O (20 mL), saturated aqueous NaCl (20 mL), dried (MgSO4) and concentrated. Purification by silica gel column chromatography (5:95 EtOAc–hexanes to 10:90 EtOAc–hexanes) afforded the title compound (1.8 g, 95%) as a colorless oil: Rf = 0.65 (10:90 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 0.04 (s, 6H), 0.88 (s, 9H), 1.33 (p, J = 7.2 Hz, 2H), 1.40 (p, J = 7.2 Hz, 2H), 1.50–1.54 (m, 4H), 1.93 (t, J = 2.4 Hz, 1H), 2.17 (td, J = 7.2, 2.4 Hz, 2H), 3.60 (t, J = 6.6 Hz, 2H); 13C NMR (CDCl3, 150 MHz) δ −5.3, 18.42, 18.44, 25.3, 26.0, 28.5, 28.6, 32.7, 63.1, 68.1, 84.7; HRMS (APCI+) calcd for C14H29OSi [M + H]+ 241.1982, found 241.1985 (error 1.2 ppm).
9-(tert-Butyldimethylsilyloxy)non-2-yn-1-ol (3)
To a solution of 2 (1.2 g, 5.0 mmol, 1 equiv) in THF (20 mL) at −78 °C, was added n-BuLi (2.5 M in hexanes, 2.4 mL., 6.0 mmol, 1.2 equiv) dropwise over 15 min. The mixture was stirred at −78 °C for 30 min and paraformaldehyde (980 mg, 32.5 mmol, 6.5 equiv) was added in one portion. The reaction was gradually warmed to 25 °C over 3 h. TLC indicated complete conversion of starting material and the reaction was then quenched by addition of saturated aqueous NH4Cl (25 mL). The aqueous layer was extracted with Et2O (3 × 20 mL). The combined organic layers were washed with H2O (20 mL), saturated aqueous NaCl (20 mL), dried (MgSO4) and concentrated. Purification by silica gel column chromatography (5:95 EtOAc–hexanes to 10:90 EtOAc–hexanes) afforded the title compound (1.3 g, 85%) as a colorless oil: Rf = 0.25 (20:80 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 0.03 (s, 6H), 0.88 (s, 9H), 1.30–1.40 (m, 4H), 1.47–1.53 (m, 4H), 1.86 (br s, 1H, OH), 2.19 (tt, J = 7.2, 1.8 Hz, 2H), 3.59 (t, J = 6.6 Hz, 2H), 4.22 (t, J =1.8 Hz, 2H); 13C NMR (CDCl3, 150 MHz) δ –5.1, 18.6, 18.9, 25.5, 26.2, 28.8, 28.9, 32.9, 51.4, 63.4, 78.7, 86.5; HRMS (APCI+) calcd for C15H31O2Si [M + H]+ 271.2088, found 271.2083 (error 1.8 ppm).
(E)-9-(tert-Butyldimethylsilyloxy)non-2-en-1-ol (4)
To a solution of 3 (1.95 g, 7.2 mmol, 1 equiv) in Et2O (40 mL) at 0 °C, was added Red-Al (65 wt% in toluene, 6.5 mL, 19.5 mmol, 3 equiv) dropwise over 15 min. The solution was stirred at 0 °C for 6 h and quenched by the dropwise addition of MeOH (0.5 mL) followed by 1 N aqueous potassium sodium tartrate (25 mL). The mixture was gradually warmed to 25 °C and stirred for 0.5 h. The aqueous layer was then extracted with Et2O (3 × 20 mL) and the combined organic layers were dried (MgSO4) and concentrated. Purification by silica gel column chromatography (5:95 EtOAc–hexanes to 10:90 EtOAc–hexanes) afforded the title compound (1.83 g, 93%) as a colorless oil: Rf = 0.23 (20:80 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 0.03 (s, 6H), 0.88 (s, 9H), 1.29–1.30 (m, 4H), 1.36–1.38 (m, 2H), 1.48–1.50 (m, 2H), 1.67 (br s, 1H, OH), 2.02 (q, J = 6.6 Hz, 2H), 3.58 (t, J = 6.6 Hz, 2H), 4.05 (d, J = 6.0 Hz, 2H), 5.58–5.75 (m, 2H); 13C NMR (CDCl3, 150 MHz) δ −5.4, 18.2, 25.5, 25.9, 28.8, 29.0, 32.0, 32.7, 63.2, 63.6, 128.8, 133.2; HRMS (APCI+) calcd for C15H33O2Si [M + H]+ 273.2244, found 273.2251 (error 2.6 ppm).
(E)-9-(tert-Butyldimethylsilyloxy)non-2-enyl 2,2,2-trichloroacetimidate (5)
To a solution of 4 (272 mg, 1 mmol, 1 equiv) in CH2Cl2 (10 mL) at 0 °C, were sequentially added DBU (174 ML, 1.15 mmol, 1.15 equiv) and CCl3CN (147 ML, 1.46 mmol, 1.46 equiv) dropwise. The solution was stirred at 0 °C for 2 h. TLC indicated complete conversion and the reaction was concentrated in vacuo. Purification by silica gel column chromatography (hexanes to 10:90 EtOAc–hexanes) afforded the title compound (395 mg, 95%) as a colorless oil: Rf = 0.45 (10:90 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 0.04 (s, 6H), 0.88 (s, 9H), 1.30–1.32 (m, 4H), 1.39–1.41 (m, 2H), 1.49–1.51 (m, 2H), 2.07 (q, J = 7.2 Hz, 2H), 3.59 (t, J = 6.6 Hz, 2H), 4.73 (d, J = 6.6 Hz, 2H), 5.67 (dt, J = 15.0, 6.6 Hz, 1H), 5.86 (dt, J = 15.0, 6.6 Hz, 1H), 8.26 (br s, 1H); 13C NMR (CDCl3, 150 MHz) δ −5.0, 18.6, 25.9, 26.2, 29.0, 29.1, 32.5, 33.0, 63.4, 70.2, 91.8, 123.2, 137.3, 162.8; HRMS (APCI+) calcd for C17H33Cl3NO2Si [M + H]+ 416.1341, found 416.1351 (error 2.4 ppm).
(±)-N-[9-(tert-Butyldimethylsilyloxy)non-1-en-3-yl]-2,2,2-trichloroacetamide (6)
A solution of 5 (300 mg, 0.74 mmol, 1 equiv) in o-xylene (2 mL) was heated at 130 °C for 6 h. TLC indicated complete conversion and the reaction was concentrated in vacuo. Purification by silica gel column chromatography (hexanes to 10:90 EtOAc–hexanes) afforded the title compound (265 mg, 88%) as a colorless oil: Rf = 0.40 (10:90 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 0.04 (s, 6H), 0.88 (s, 9H), 1.27–1.40 (m, 7H), 1.48–1.51 (m, 1H), 1.59–1.61 (m, 1H), 1.63–1.68 (m, 1H), 3.59 (t, J = 6.6 Hz, 2H), 4.40 (p, J = 6.6 Hz, 1H), 5.18 (d, J = 10.8 Hz, 1H), 5.23 (d, J = 16.8 Hz, 1H), 5.79 (ddd, J = 16.8, 10.8, 5.4 Hz, 1H), 6.50 (br d, J = 7.2 Hz, 1H, NH); 13C NMR (CDCl3, 150 MHz) δ −5.0, 18.6, 25.7, 25.8, 26.2, 29.3, 32.9, 34.6, 53.8, 63.3, 93.1, 116.3, 136.9, 161.4; HRMS (ESI−) calcd for C17H31Cl3NO2Si [M – H]− 414.1195, found 414.1198 (error 0.7 ppm).
(±)-7-Aminonon-8-en-1-ol hydrochloride salt (M-1)
To a solution of 6 (215 mg, 0.52 mmol, 1 equiv) in THF (3 mL) at 0 °C, was added HF•Py (1 mL) dropwise. The solution was stirred at 0 °C for 2 h when TLC indicated complete conversion. The reaction was diluted with EtOAc (20 mL) and washed with saturated aqueous NH4Cl (10 mL). The aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried (MgSO4) and concentrated. Purification by silica gel column chromatography (5:95 EtOAc–hexanes to 50:50 EtOAc–hexanes) afforded (±)-2,2,2-trichloro-N-(9-hydroxynon-1-en-3-yl)acetamide (134 mg, 95%) as a colorless oil: Rf = 0.40 (50:50 EtOAc–hexanes); 1H NMR (CD3OD, 600 MHz) δ 1.36 (br s, 6H), 1.52 (t, J = 6.0 Hz, 2H), 1.63–1.67 (m, 2H), 3.53 (t, J = 6.6 Hz, 2H), 4.33 (p, J = 7.2 Hz, 1H), 4.84 (br s, 1H), 5.11 (d, J = 10.8 Hz, 1H), 5.19 (d, J = 16.8 Hz, 1H), 5.85 (ddd, J = 16.8, 10.8, 5.4 Hz, 1H), 8.68 (d, J = 7.2 Hz, 1H); 13C NMR (CD3OD, 150 MHz) δ 26.9, 27.3, 30.2, 33.6, 35.1, 55.7, 63.0, 94.4, 116.0, 138.9, 163.7; HRMS (APCI+) calcd for C11H19Cl3NO2 [M + H]+ 302.0476, found 302.0485 (error 3.0 ppm).
To a solution of (±)-2,2,2-trichloro-N-(9-hydroxynon-1-en-3-yl)acetamide prepared above (134 mg, 0.50 mmol, 1 equiv) in toluene (2 mL) at −78 °C, was added a 1 M DIBAL-H solution in CH2Cl2 (2 mL, 4 equiv) dropwise over 10 min. The solution was stirred at −78 °C for 1 h, then the reaction was quenched by the dropwise addition of MeOH (0.5 mL) and 1 N aqueous potassium sodium tartrate (25 mL). The mixture was gradually warmed to 25 °C and stirred for 30 min. The aqueous layer was extracted with EtOAc (3 × 20 mL) and the combined organic layers were dried (MgSO4) and concentrated. The crude material was acidified with 4 N HCl in dioxane (2 mL) and concentrated again. Re-crystallization from MeOH–Et2O (1:20, 5 mL) afforded the title compound (75 mg, 78%) as an off white solid: mp = 89–91 °C; 1H NMR (CD3OD, 600 MHz) δ 1.35–1.40 (m, 6H), 1.51–1.54 (m, 2H), 1.63–1.66 (m, 1H), 1.73–1.75 (m, 1H); 3.54 (t, J = 6.6 Hz, 2H), 3.68 (q, J = 5.4 Hz, 1H), 5.41 (d, J = 10.8 Hz, 1H), 5.43 (d, J = 17.4 Hz, 1H), 5.82 (ddd, J = 17.4, 10.8, 5.4 Hz, 1H); 13C NMR (CD3OD, 150 MHz) δ 26.4, 26.8, 30.1, 33.5, 34.1, 55.5, 63.0, 121.3, 135.4; HRMS (ESI+) calcd for C9H20NO [M + H]+ 158.1539, found 158.1544 (error 3.2 ppm).
(±)-Ethyl cis-(E)-3-(4-{[(tert-butoxy)carbonyl]amino}cyclopent-2-enyl)acrylate (10)
To a solution of 7b (300 mg, 1.4 mmol, 1 equiv) in toluene (4 mL) at −78 °C, was added dropwise 1 M DIBAL-H in toluene (2.2 mL, 2.2 mmol, 1.5 equiv) over 15 min. The solution was stirred at −78 °C for 1 h and quenched by the addition of MeOH (0.5 mL) and saturated aqueous potassium sodium tartrate (20 mL). The mixture was gradually warmed to 25 °C and stirred for 0.5 h. The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with H2O (20 mL), saturated aqueous NaCl (20 mL), dried (MgSO4) and concentrated to afford (±)-tert-butyl 3-hydroxy-2-azabicyclo[2.2.1]hept-5-ene-2-carboxylate as a mixture of diastereomers epimeric at C-3, which was used directly in the next step without further purification.
To a solution of triethylphosphonoacetate (0.28 mL, 1.68 mmol, 1.2 equiv) in toluene (4 mL) at 0 °C, was added NaH (60 wt% in mineral oil, 72 mg, 1.68 mmol, 1.2 equiv) in one portion. The mixture was stirred at 0 °C for 30 min, then a solution of (±)-tert-butyl 3-hydroxy-2-azabicyclo[2.2.1]hept-5-ene-2-carboxylate prepared above in toluene (2 mL) was added dropwise. The reaction was gradually warmed to 25 °C and stirred for 15 h. TLC indicated complete conversion and the reaction was partitioned between saturated aqueous NH4Cl (20 mL) and EtOAc (20 mL). The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with H2O (20 mL), saturated aqueous NaCl (20 mL), dried (MgSO4) and concentrated. Purification by silica gel column chromatography (hexanes to 10:90 EtOAc–hexanes) afforded the title compound (334 mg, 81%) as a colorless foam: Rf = 0.55 (20:80 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.25 (t, J = 6.6 Hz, 3H), 1.35 (dt, J = 12.0, 6.0 Hz, 1H), 1.40 (s, 9H), 2.61–2.65 (m, 1H), 3.35–3.37 (m, 1H), 4.16 (q, J = 6.6 Hz, 2H), 4.59 (br s, 1H), 4.72 (br s, 1H), 5.72–5.78 (m, 3H), 6.86 (dd, J =15.6, 7.2 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ 14.5, 28.6, 38.5, 46.5, 56.7, 60.6, 79.8, 120.8, 134.0, 134.5, 151.0, 155.3, 166.8; HRMS (APCI+) calcd for C15H24NO4 [M + H]+ 282.1700, found 282.1708 (error 2.8 ppm).
(±)-Ethyl cis-3-(4-{[(tert-butoxy)carbonyl]amino}cyclopent-2-enyl)propanoate (11)
To a solution of 10 (281 mg, 1.0 mmol, 1.0 equiv), CuCl (74 mg, 0.75 mmol, 0.75 equiv) and cyclohexene (0.4 mL, 4.0 mmol, 4.0 equiv) in MeOH (20 mL) at −78 °C, was added NaBH4 (189 mg, 5.0 mmol, 5.0 equiv). The reaction was stirred at −78 °C for 1 h, during which time the solution went from brown to green. MS (APCI+) indicated complete conversion. The reaction was concentrated in vacuo while the flask was still cold. The crude product was then partitioned between saturated aqueous NH4Cl (30 mL) and EtOAc (30 mL). The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with saturated aqueous NaCl, dried (MgSO4) and concentrated. Purification by silica gel column chromatography (5:95 EtOAc–hexanes to 20:80 EtOAc–hexanes) afforded the title compound (280 mg, 99%) as a colorless foam: Rf = 0.55 (20:80 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.07 (dt, J = 12.0, 6.0 Hz, 1H), 1.24 (t, J = 7.2 Hz, 3H), 1.43 (s, 9H), 1.64 (dt, J = 15.6, 7.8 Hz, 1H), 1.76 (dt, J = 15.6, 7.8 Hz, 1H), 2.31 (t, J = 7.8 Hz, 2H), 2.31–2.60 (m, 2H), 4.11 (q, J = 7.2 Hz, 2H), 4.54 (br s, 1H), 4.66 (br s, 1H), 5.65 (d, J = 5.4 Hz, 1H), 5.77 (d, J = 5.4 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ 14.3, 28.6, 31.6, 32.7, 38.6, 44.0, 56.6, 60.6, 79.4, 132.3, 137.1, 155.4, 173.7; HRMS (APCI+) calcd C15H26NO4 [M + H]+ 284.1856, found 284.1857 (error 0.4 ppm).
(±)-cis-3-(-4-Aminocyclopent-2-enyl)propan-1-ol hydrochloride salt (M-2)
To a solution of 11 (140 mg, 0.5 mmol, 1 equiv) in THF (5 mL) at 0 °C, was added LiAlH4 (28.4 mg, 0.75 mmol, 1.5 equiv) in one portion. The reaction was stirred at 0 °C for 0.5 h and quenched by the successive dropwise addition of MeOH (0.5 mL) and saturated aqueous potassium sodium tartrate (20 mL). The mixture was warmed up to 25 °C and stirred for 0.5 h. The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with H2O (20 mL), saturated NaCl solution (20 mL), dried (MgSO4) and concentrated. Purification by silica gel column chromatography (10:90 EtOAc–hexanes to 50:50 EtOAc–hexanes) afforded (±)-tert-butyl cis-4-(3-hydroxypropyl)cyclopent-2-enylcarbamate (100 mg, 83%) as a colorless foam: Rf = 0.35 (50:50 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.06–1.08 (m, 1H), 1.31–1.35 (m, 1H), 1.43 (s, 9H), 1.47–1.61 (m, 3H), 2.57–2.60 (m, 2H), 3.63 (t, J = 6.6 Hz, 2H), 4.53 (br s, 1H), 4.66 (br s, 1H), 5.63 (d, J = 4.8 Hz, 1H), 5.80 (d, J = 4.8 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ 28.7, 31.1, 32.9, 39.1, 44.4, 56.7, 63.1, 79.5, 131.5, 137.9, 155.5; HRMS (APCI+) calcd for C13H24NO3 [M + H]+ 242.1751, found 242.1761 (error 4.1 ppm).
(±)-tert-Butyl cis-4-(3-hydroxypropyl)cyclopent-2-enylcarbamate prepared above (48 mg, 0.2 mmol, 1 equiv) was dissolved in 4 N HCl in dioxane (2 mL, 8.0 mmol, 40 equiv) at 0 °C. The solution was stirred at 0 °C for 1 h, then concentration in vacuo. Recrystallization from MeOH–Et2O (1:20, 5 mL) afforded the title compound (31 mg, 89%) as an off white solid: mp = 123–125 °C; 1H NMR (CD3OD, 600 MHz) δ 1.35 (p, J = 6.6 Hz, 1H), 1.43–1.46 (m, 1H), 1.47–1.64 (m, 3H), 2.62–2.67 (m, 1H), 2.76–2.78 (m, 1H), 3.57 (t, J = 6.0 Hz, 2H), 4.25–4.26 (m, 1H), 5.76 (d, J = 5.4 Hz, 1H), 6.11 (d, J = 5.4 Hz, 1H); 13C NMR (CD3OD, 150 MHz) δ 31.9, 33.3, 36.6, 46.5, 58.1, 62.9, 127.9, 143.2; HRMS (ESI+) calcd for C8H16NO [M + H]+ 142.1226, found 142.1220 (error 4.2 ppm).
(±)-2-Benzyl-6-methylene-2-azabicyclo[2.2.1]heptan-3-one (13)
To a suspension of Nysted reagent (20 wt%, 8.4 g, 3.7 mmol, 3.0 equiv) at 0 °C, was added a 1 M solution of TiCl4 in CH2Cl2 (3.6 mL, 3.6 mmol, 3.0 equiv) dropwise over 10 min. The mixture was stirred at 0 °C for additional 1 h then a solution of 12 (264 mg, 1.2 mmol, 1.0 equiv) in THF (4 mL) was added dropwise over 10 min. The reaction was gradually warmed to 25 °C and stirred for 3 h at 25 °C when TLC indicated complete conversion. The reaction was cooled to 0 °C and quenched by the careful dropwise addition of 1 N aqueous HCl (20 mL). The aqueous layer was extracted with EtOAc (3 × 20 mL) and the combined organic layers were washed with H2O (20 mL), saturated aqueous NH4Cl (20 mL), dried (MgSO4) and concentrated. Purification by silica gel column chromatography (10:90 EtOAc–hexanes to 50:50 EtOAc–hexanes) afforded the title compound (230 mg, 88%) as a colorless foam: Rf = 0.45 (50:50 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.54 (d, J = 9.6 Hz, 1H), 1.96–1.99 (m, 1H), 2.21 (dt, J = 16.2, 2.4 Hz, 1H), 2.40 (dt, J = 16.2, 2.4 Hz, 1H), 2.89 (d, J = 2.4 Hz, 1H), 3.70 (d, J = 15.0 Hz, 1H), 3.72–3.73 (m, 1H), 4.75 (d, J = 15.0 Hz, 1H), 4.93 (br s, 1H), 5.03 (t, J = 2.4 Hz, 1H), 7.22–7.27 (m, 3H), 7.30–7.33 (m, 2H); 13C NMR (CDCl3, 150 MHz) δ 31.8, 40.4, 44.3, 45.7, 64.4, 107.5, 127.7, 128.3, 128.8, 137.4, 146.1, 178.6; All data are consistent with reported literature values.13c
(±)-tert-Butyl 6-methylene-3-oxo-2-azabicyclo[2.2.1]heptane-2-carboxylate (14)
To a solution of t-BuOH (1.0 mL, 10.6 mmol, 5.6 equiv) in THF (10 mL) at −78 °C, was condensed NH3 (approximately 10 mL). Na0 (200 mg, 8.7 mmol, 4.6 equiv) was added to the solution in portions to afford a dark blue solution. Next, a solution of 13 (400 mg, 1.88 mmol, 1 equiv) in THF (10 mL) was added dropwise over 20 min. After addition, the reaction was stirred at −78 °C for 15 min and −30 °C for 30 min. The reaction was then cooled to −78 °C and quenched by the dropwise addition of AcOH (1 mL). The mixture was gradually warmed to 25 °C. The reaction was filtered over Celite and concentrated to afford crude 6-methylene-2-azabicyclo[2.2.1]heptan-3-one, which was used directly in the next step without further purification.
To a solution of 6-methylene-2-azabicyclo[2.2.1]heptan-3-one obtained from the previous step and DMAP (114 mg, 0.94 mmol, 0.5 equiv) in CH3CN (10 mL) at 25 °C, was added (Boc)2O (615 mg, 2.8 mmol, 1.5 equiv) in one portion. The mixture was stirred at 25 °C for 2 h, then concentrated in vacuo. Purification by silica gel column chromatography (5:95 EtOAc–hexanes to 20:80 EtOAc–hexanes) afforded the title compound (326 mg, 78%) as a colorless foam: Rf = 0.35 (20:80 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.49 (s, 9H), 1.58 (dt, J = 10.6, 1.2 Hz, 1H), 2.04–2.07 (m, 1H), 2.35–2.38 (m, 1H), 2.46–2.49 (m, 1H), 2.91 (br d, J = 2.4 Hz, 1H), 4.65 (d, J = 1.8 Hz, 1H), 4.96 (t, J = 1.8 Hz, 1H), 5.25 (d, J = 1.8 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ 27.9, 30.8, 38.5, 46.5, 64.0, 82.5, 108.1, 145.2, 148.9, 175.1; All data are consistent with reported literature values.13c
(±)-Ethyl cis-(E)-3(3-{[(tert-butoxy)carbonyl]amino}-4-methylenecyclopentyl)acrylate (15)
The title compound was prepared analogously to compound 10 from 14 (1.0 equiv.) employing DIBAL-H (1.5 equiv) and triethylphosphonoacetate (1.2 equiv) and obtained in 81% isolated yield over 2 steps: Rf = 0.35 (20:80 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.23–1.29 (m, 1H), 1.28 (t, J = 7.2 Hz, 3H), 1.43 (s, 9H), 2.23–2.28 (m, 1H), 2.32 – 2.35 (m, 1H), 2.61–2.69 (m, 2H), 4.18 (q, J = 7.2 Hz, 2H), 4.46 (br s, 1H), 4.55 (br s, 1H), 4.98–5.01 (m, 2H), 5.81 (d, J = 15.6 Hz, 1H), 6.88 (dd, J = 15.6, 7.2 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ 14.2, 28.3, 36.6, 38.2, 39.8, 54.5, 60.2, 79.4, 107.2, 120.6, 150.3, 150.6, 155.6, 166.5; HRMS (APCI+) calcd for C16H26NO4 [M + H]+ 296.1856, found 296.1862 (error 2.0 ppm).
(±)-Ethyl cis-3-[3-{[(tert-butoxy)carbonyl]amino}-4-methylenecyclopentyl]propanoate (16)
The title compound was prepared analogously to compound 11 from 15 (1.0 equiv), CuCl (0.75 equiv), NaBH4 (5.0 equiv) and cyclohexene (4.0 equiv), and obtained in quantitative isolated yield: Rf = 0.35 (20:80 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 0.92–1.02 (m, 1H), 1.24 (t, J = 7.2 Hz, 3H), 1.43 (s, 9H), 1.64–1.69 (m, 2H), 1.85–1.89 (m, 1H), 1.94–2.03 (m, 1H), 2.29 (t, J = 7.2 Hz, 2H), 2.28–2.33 (m, 1H), 2.57 (dd, J = 16.8, 7.2 Hz, 1H), 4.11 (q, J = 7.2 Hz, 2H), 4.36 (br s, 1H), 4.51 (br s, 1H), 4.92 (br s, 1H), 4.95 (br s, 1H); 13C NMR (CDCl3, 150 MHz) δ 14.2, 28.3, 30.6, 32.9, 35.1, 37.2, 40.4, 54.7, 60.3, 79.2, 106.3, 151.6, 155.7, 173.4; HRMS (APCI+) calcd for C16H28NO4 [M + H]+ 298.2013, found 298.2010 (error 1.0 ppm).
(±)-cis-3-(3-Amino-4-methylenecyclopentyl)propan-1-ol hydrochloride salt (M-3)
The title compound was prepared analogously to compound M-2 in 2 steps. First, 16 (1.0 equiv) was reduced with LiAlH4 to afford (±)-tert-butyl cis-4-(3-hydroxypropyl)-2-methylenecyclopentylcarbamate in 83% isolated yield as a colorless foam: Rf = 0.35 (50:50 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 0.97 (q, J = 10.8 Hz, 1H), 1.35– 1.41 (m, 2H), 1.43 (s, 9H), 1.52–1.57 (m, 2H), 1.81–1.96 (m, 2H), 2.29–2.31 (m, 1H), 2.56 (dd, J = 16.8, 7.2 Hz, 1H), 3.60 (t, J = 6.6 Hz, 2H), 4.34 (br d, J = 6.6 Hz, 1H), 4.55 (br d, J = 6.6 Hz, 1H), 4.90 (br s, 1H), 4.92 (br s, 1H); 13C NMR (CDCl3, 150 MHz) δ 28.4, 31.2, 31.8, 35.5, 37.6, 40.7, 54.8, 62.8, 79.2, 106.1, 152.0, 155.8; HRMS (APCI+) calcd for C14H26NO3 [M + H]+ 256.1907, found 256.1917 (error 3.9 ppm).
(±)-tert-Butyl cis-4-(3-hydroxypropyl)-2-methylenecyclopentylcarbamate prepared above was converted to the title compound by treatment with 4 N HCl in dioxane in 89% isolated yield as a white solid: mp 153–155 °C; 1H NMR (CD3OD, 600 MHz) δ 1.30 (q, J = 12.0 Hz, 1H), 1.45–1.58 (m, 4H), 1.96–2.00 (m, 1H), 2.07–2.12 (m, 1H), 2.35–2.41 (m, 1H), 2.61 (dd, J = 16.2, 6.6 Hz, 1H), 3.54 (t, J = 6.6 Hz, 2H), 3.99 (t, J = 7.8 Hz, 1H), 5.21 (br s, 2H); 13C NMR (CDCl3, 150 MHz) δ 32.3, 32.8, 38.3, 39.5, 39.6, 54.9, 63.1, 110.9, 150.1; HRMS (ESI+) calcd for C9H18NO [M + H]+ 156.1383, found 156.1390 (error 4.5 ppm).
(±)-2-Benzyl-7-methylene-2-azabicyclo[2.2.2]octan-3-one (19)
The title compound was prepared analogously to compound 13 from 18 (1.0 equiv) employing Nysted reagent (3.0 equiv) and TiCl4 (3.0 equiv), and obtained in 84% isolated yield: Rf = 0.45 (20:80 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.65–1.74 (m, 3H), 1.86–1.91 (m, 1H), 2.41 (dq, J = 16.8, 2.4 Hz, 1H), 2.57 (dq, J = 16.8, 2.4 Hz, 1H), 2.75 (t, J = 3.0 Hz, 1H), 3.74 (t, J = 3.0 Hz, 1H), 4.37 (d, J =15.0 Hz, 1H), 4.70–4.76 (m, 3H), 7.24–7.26 (m, 3H), 7.28–7.31 (m, 2H); 13C NMR (CDCl3, 150 MHz) δ 23.6, 28.0, 32.3, 39.4, 47.4, 59.9, 107.1, 127.3, 128.0, 128.3, 137.2, 144.8, 174.8; HRMS (APCI+) calcd for C15H18NO [M + H]+ 228.1383, found 228.1375 (error 3.5 ppm).
(±)-2-[(tert-Butoxy)carbonyl]-7-methylene-2-azabicyclo[2.2.2]octane-3-one (20)
The title compound was prepared analogously to compound 14 from 19 in 2 steps using identical reagent stoichiometry and obtained in 71% isolated yield: Rf = 0.45 (50:50 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.50 (s, 9H), 1.67–1.70 (m, 1H), 1.72– 1.81 (m, 1H), 1.87–1.94 (m, 2H), 2.38 (dd, J = 17.4, 2.4 Hz, 1H), 2.55 (dd, J = 17.4, 2.4 Hz, 1H), 2.65 (t, J = 2.4 Hz, 1H), 4.77 (br s, 1H), 4.85 (br s, 1H), 4.99 (br s, 1H); 13C NMR (CDCl3, 150 MHz) δ 22.6, 27.1, 28.0, 31.4, 41.2, 57.3, 83.0, 109.2, 143.1, 150.0, 173.8; HRMS (APCI+) calcd C13H20NO3 [M + H]+ 238.1438, found 238.1445 (error 2.9 ppm).
(±)-tert-Butyl cis-4-(3-hydroxypropyl)-2-methylenecyclohexylcarbamate (22)
To a solution of 20 (237 mg, 1.0 mmol, 1 equiv) in toluene (4 mL) at −78 °C, was added dropwise 1 M DIBAL-H in toluene (1.5 mL, 1.5 mmol, 1.5 equiv) over 15 min. The solution was stirred at −78 °C for 1 h and quenched by the addition of MeOH (0.5 mL) and saturated aqueous potassium sodium tartrate (20 mL). The mixture was gradually warmed to 25 °C and stirred for 30 min. The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with H2O (20 mL), saturated aqueous NaCl (20 mL), dried (MgSO4) and concentrated to afford the desired hemiaminal intermediate, which was used directly in the next step without further purification.
To a solution of triethylphosphonoacetate (0.20 mL, 1.2 mmol, 1.2 equiv) in toluene (4 mL) at 0 °C, was added NaH (60 wt% in mineral oil, 48 mg, 1.2 mmol, 1.2 equiv) in one portion. The mixture was stirred at 0 °C for 30 min, then a solution of the hemiaminal intermediate from the previous step in toluene (2 mL) was added dropwise. The reaction was gradually warmed to 25 °C and stirred for 15 h. TLC indicated complete conversion and the reaction was partitioned between saturated aqueous NH4Cl (20 mL) and EtOAc (20 mL). The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with H2O (20 mL), saturated aqueous NaCl (20 mL), dried (MgSO4) and concentrated to afford 21, which was used directly in the next step without further purification.
To a solution of 21 prepared above (309 mg, 1.0 mmol, 1.0 equiv), CuCl (74 mg, 0.75 mmol, 0.75 equiv) and cyclohexene (0.4 mL, 4.0 mmol, 4.0 equiv) in MeOH (20 mL) at −78 °C, was added NaBH4 (189 mg, 5.0 mmol, 5.0 equiv). The reaction was stirred at −78 °C for 1 h. The reaction was concentrated in vacuo while the flask was still cold. The crude product was then partitioned between saturated aqueous NH4Cl (30 mL) and EtOAc (30 mL). The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with saturated aqueous NaCl, dried (MgSO4) and concentrated to afford (±)-Ethyl cis-3-(4-{[(tert-butoxy)carbonyl]amino}-3-methylenecyclohexyl)propanoate, which was used in the next step without further purification.
To a solution of crude (±)-ethyl cis-3-(4-{[(tert-butoxy)carbonyl]amino}-3-methylenecyclohexyl)propanoate prepared above (311 mg, 1.0 mmol, 1 equiv) in THF (5 mL) at 0 °C, was added LiAlH4 (57 mg, 1.5 mmol, 1.5 equiv) in one portion. The reaction was stirred at 0 °C for 30 min and quenched by the successive dropwise addition of MeOH (0.5 mL) and saturated aqueous potassium sodium tartrate (20 mL). The mixture was warmed to 25 °C and stirred for 30 min. The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with H2O (20 mL), saturated NaCl solution (20 mL), dried (MgSO4) and concentrated. Purification by silica gel column chromatography (10:90 EtOAc–hexanes to 50:50 EtOAc–hexanes) afforded the title compound (180 mg, 67% isolated yield in 4 steps) as a colorless foam: Rf = 0.20 (50:50 EtOAc–hexanes); 1H NMR (CDCl3, 600 MHz) δ 1.25–1.35 (m, 2H), 1.44 (s, 9H), 1.52–1.71 (m, 7H), 2.03 (dd, J = 13.2, 7.2 Hz, 1H), 2.25 (dd, J = 13.2, 7.2 Hz, 1H), 3.63 (t, J = 6.6 Hz, 2H), 4.07 (br s, 1H), 4.64 (br s, 1H), 4.73 (br s, 1H), 4.85 (br s, 1H); 13C NMR (CDCl3, 150 MHz) δ 28.2, 28.4, 29.9, 30.3, 30.9, 36.4, 38.2, 53.0, 63.1, 79.3, 108.6, 146.5, 155.2; HRMS (APCI+) calcd for C15H28NO3 [M + H]+ 270.2064, found 270.2061 (error 1.1 ppm).
(±)-cis-3-(4-Amino-3-methylenecyclohexyl)propan-1-ol hydrochloride (M-4)
The title compound was prepared from 22 analogously to compound M-2 and obtained as an off white solid in 89% isolated yield: mp 156–158 °C; 1H NMR (CD3OD, 600 MHz) δ 1.35–1.39 (m, 2H), 1.55–1.57 (m, 2H), 1.62–1.65 (m, 2H), 1.65–1.85 (m, 3H), 2.15 (dd, J = 13.8, 4.2 Hz, 1H), 2.39 (dd, J = 13.8, 4.2 Hz, 1H); 3.55 (t, J = 6.6 Hz. 2H), 3.80–3.82 (m, 1H); 4.97 (br s, 1H), 5.04 (br s, 1H); 13C NMR (CD3OD, 150 MHz) δ 28.1, 30.0, 31.1, 31.2, 37.8, 38.7, 54.6, 63.1, 112.6, 144.2; HRMS (ESI+) calcd for C10H20NO [M + H]+ 170.1539, found 170.1544 (error 3.0 ppm).
BioA Enzyme Assay to Determine IC50 values
We used an established coupled continuous assay that we previously reported to evaluate M-1/2/3/4 for enzyme inhibition under initial velocity conditions in a final assay volume of 100 ML.17 All reactions were performed in triplicate and repeated on two independent days. Inhibitors were dissolved in DMSO and dispensed (1 ML of a 100× DMSO stock solution) into black 96-well plates to provide a final DMSO concentation of 1%. A master mix of 320 nM BioD, 50 nM BioA, 185 nM streptavidin, 20 nM fluorescence tagged dethiobiotin derivative,17 and 5 mM SAM in reaction buffer (100 mM Bicine, 5 mM ATP, 50 mM NaHCO3, 1 mM MgCl2, 0.1 mM PLP, 0.0025% Igepal CA630, 1 mM TCEP, pH 8.6) was prepared, and 98 ML was dispensed into the inhibitor and control wells and the plates were incubated for 10 min at 25 °C. Positive controls (100% inhibition) did not contain the substrate KAPA and negative controls (0% inhibition) did not contained any inhibitor, but just 1 ML of DMSO. Preincubation of the plates from 10 min to 3 h did not affect the observed enzyme activity. Assays were initiated by the addition of KAPA (1 ML of a 1.25 mM stock solution in H2O) providing a final concentation of 12.5 MM. The plate was read for 20 min using an excitation of 485 nm, emission at 530 nm and cutoff at 530 nm. The initial velocity (v0) was determined by the time to produce 90 nM dethiobiotin. The data were fit to the standard four-parameter Hill equation using GraphPad Prism (version 4.0) to provide an IC50 value.
Supplementary Material
Table 2.
Inhibition Activity of M-1–4 against BioA.
| Compound | Structure | IC50 (μM) |
|---|---|---|
| M-1 |
|
>100 |
| M-2 |
|
57.0 ± 1.9 |
| M-3 |
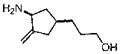
|
>100 |
| M-4 |
|
>100 |
Acknowledgments
This research was supported by a grant from the Bill and Melinda Gates foundation and the Wellcome Trust through the Grand Challenges in Global Health Initiative (to Douglas Young, Imperial College) and the National Institutes of Health (AI091790). We thank Daniel Wilson for performing the inhibition assays.
Footnotes
Supporting information Available. 1H NMR and 13C NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.(a) Knowles JR. Annu Rev Biochem. 1989;58:195. doi: 10.1146/annurev.bi.58.070189.001211. [DOI] [PubMed] [Google Scholar]; (b) Moss J, Lane MD. Adv Enzymol Relat Areas Mol Biol. 1971;35:321. doi: 10.1002/9780470122808.ch7. [DOI] [PubMed] [Google Scholar]; (c) Marquet A, Bui BTS, Florentin D. Vit Horm. 2001;61:51. doi: 10.1016/s0083-6729(01)61002-1. [DOI] [PubMed] [Google Scholar]
- 2.a) Dey S, Lane JM, Lee RE, Rubin EJ, Sacchettini JC. Biochemistry. 2010;49:6746. doi: 10.1021/bi902097j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sassetti CM, Rubin EJ. Proc Natl Acad Sci USA. 2003;100:12989. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayakawa K, Oizumi J. J Chromatogr. 1987;413:247. doi: 10.1016/0378-4347(87)80234-7. [DOI] [PubMed] [Google Scholar]
- 4.Park SW, Klotzsche M, Wilson DJ, Boshoff HI, Eoh H, Manjunatha U, Blumenthal A, Rhee K, Barry CE, 3rd, Aldrich CC, Ehrt S, Schnappinger D. PloS Pathog. 2011;7:e1002264. doi: 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Mann S, Ploux O. Biochim Biophys Acta. 2011;1814:1459. doi: 10.1016/j.bbapap.2010.12.004. [DOI] [PubMed] [Google Scholar]; (b) Mann S, Ploux O. FEBS J. 2006;273:4778. doi: 10.1111/j.1742-4658.2006.05479.x. [DOI] [PubMed] [Google Scholar]; (c) Mann S, Colliandre L, Labesse G, Ploux O. Biochimie. 2009;91:826. doi: 10.1016/j.biochi.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 6.(a) Okami Y, Kitahara T, Hamada M, Naganawa H, Kondo S. J Antibiot. 1974;27:656. doi: 10.7164/antibiotics.27.656. [DOI] [PubMed] [Google Scholar]; (b) Kitahara T, Hotta K, Yoshida M, Okami Y. J Antibiot. 1975;28:215. doi: 10.7164/antibiotics.28.215. [DOI] [PubMed] [Google Scholar]; (c) Hotta K, Kitahara T, Okami Y. J Antibiot. 1975;28:222. doi: 10.7164/antibiotics.28.222. [DOI] [PubMed] [Google Scholar]; (d) Poetsch M, Zahner H, Werner RG, Kern A, Jung G. J Antibiot. 1985;38:312. doi: 10.7164/antibiotics.38.312. [DOI] [PubMed] [Google Scholar]
- 7.Mann S, Carillon S, Breyne O, Marquet A. Chem Eur J. 2002;8:439. doi: 10.1002/1521-3765(20020118)8:2<439::AID-CHEM439>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Shi C, Geders TW, Park SW, Wilson DJ, Boshoff HI, Abayomi O, Barry CE, 3rd, Schnappinger D, Finzel B, Aldrich CC. J Am Chem Soc. 2011;133:18194. doi: 10.1021/ja204036t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Sandmark J, Mann S, Marquet A, Schneider G. J Biol Chem. 2002;277:43352. doi: 10.1074/jbc.M207239200. [DOI] [PubMed] [Google Scholar]; (b) Mann S, Florentin D, Lesage D, Drujon T, Ploux O, Marquet A. Helv Chim Acta. 2003;86:3836. [Google Scholar]
- 10.(a) Olson GT, Fu M, Lau S, Rinehart KL, Silverman RB. J Am Chem Soc. 1998;120:2256. [Google Scholar]; (b) Fu M, Nikolic D, Van Breemen RB, Silverman RB. J Am Chem Soc. 1999;121:7751. [Google Scholar]; (c) Fu M, Silverman RB. Bioorg Med Chem. 1999;7:1581. doi: 10.1016/s0968-0896(99)00081-4. [DOI] [PubMed] [Google Scholar]; (d) Fu M, Silverman RB. Bioorg Med Chem Lett. 2004;14:203. doi: 10.1016/j.bmcl.2003.09.065. [DOI] [PubMed] [Google Scholar]; (e) Wang Z, Yuan H, Nikolic D, Van Breemen RB, Silverman RB. Biochemistry. 2006;45:14513. doi: 10.1021/bi061592m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liu D, Pozharski E, Lepore BW, Fu M, Silverman RB, Petsko GA, Ringe D. Biochemistry. 2007;46:10517. doi: 10.1021/bi700663n. [DOI] [PubMed] [Google Scholar]; (g) Lepore BW, Liu D, Peng Y, Fu M, Yasuda C, Manning JM, Silverman RB, Ringe D. Biochemistry. 2010;49:3138. doi: 10.1021/bi902052x. [DOI] [PubMed] [Google Scholar]; (h) Liu D, Pozharski E, Fu M, Silverman RB, Ringe D. Biochemistry. 2010;49:10507. doi: 10.1021/bi101325z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Overman LE. J Am Chem Soc. 1974;96:597. [Google Scholar]; (b) Overman LE. J Am Chem Soc. 1976;98:2901. [Google Scholar]; (c) Anderson CE, Overman LE. J Am Chem Soc. 2003;125:12412. doi: 10.1021/ja037086r. [DOI] [PubMed] [Google Scholar]
- 12.(a) Brown CA, Yamashita A. J Am Chem Soc. 1975;97:891. [Google Scholar]; (b) Denmark SE, Yang SM. J Am Chem Soc. 2002;124:2102. doi: 10.1021/ja0178158. [DOI] [PubMed] [Google Scholar]
- 13.(a) Polla MO, Tottie L, Norden C, Linschoten M, Musil D, Trumpp-Kallmeyer S, Aukrust IR, Ringom R, Holm KH, Neset SM, Sandberg M, Thurmond J, Yu P, Hategan G, Anderson H. Bioorg Med Chem. 2004;12:1151. doi: 10.1016/j.bmc.2003.12.039. [DOI] [PubMed] [Google Scholar]; (b) Palmer CF, Parry KP, Roberts SM, Sik V. J Chem Soc, Perkin Trans 1. 1991;8:2051. [Google Scholar]; (c) Pan Y, Qiu J, Silverman RB. J Med Chem. 2003;46:5292. doi: 10.1021/jm034162s. [DOI] [PubMed] [Google Scholar]; (d) Bowman WR, Bridge CF. Synth Commun. 1999;29:4051. [Google Scholar]; (e) Katagiri N, Muto M, Kaneko C. Tetrahedron Lett. 1989;30:1645. [Google Scholar]; (f) Rassum G, Auzza L, Zambrano V, Burreddu P, Pinna L, Battistini L, Zanardi F, Casiraghi G. J Org Chem. 2004;69:1625. doi: 10.1021/jo0357216. [DOI] [PubMed] [Google Scholar]
- 14.Taylor SJC, McCague R, Wisdom R, Lee C, Dickson K, Ruecroft G, O’Brien F, Littlechild J, Bevan J, Roberts JM, Evans CT. Tetrahedron: Asymmetry. 1993;4:1117. [Google Scholar]
- 15.Hin B, Majer P, Tsukamoto T. J Org Chem. 2002;67:7365. doi: 10.1021/jo026101s. [DOI] [PubMed] [Google Scholar]
- 16.Baird LJ, Timmer MSM, Teedale-Spittle PH, Harvey JE. J Org Chem. 2009;74:2271. doi: 10.1021/jo802561s. [DOI] [PubMed] [Google Scholar]
- 17.Wilson DJ, Shi C, Duckworth BP, Muretta JM, Manjunatha U, Sham YY, Thomas DD, Aldrich CC. Anal Biochem. 2011;416:27. doi: 10.1016/j.ab.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns NZ, Baran PS, Hoffmann RW. Angew Chem Int Ed. 2009;48:2854. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.