

Abstract
Neurogenesis requires negative regulation through differentiation of progenitors or their programmed cell death (PCD). Growth regulation is particularly important in the postnatal cerebellum, where excessive progenitor proliferation promotes medulloblastoma, the most common malignant brain tumor in children. We present evidence that PCD operates alongside differentiation to regulate cerebellar granule neuron progenitors (CGNPs) and to prevent medulloblastoma.
Here we show that genetic deletion of pro-apoptotic Bax disrupts regulation of cerebellar neurogenesis and promotes medulloblastoma formation. In Bax−/− mice, the period of neurogenesis was extended into the third week of postnatal life, and ectopic neurons and progenitors collected in the molecular layer of the cerebellum and adjacent tectum. Importantly, genetic deletion of Bax in medulloblastoma-prone ND2:SmoA1 transgenic mice greatly accelerated tumorigenesis. Bax-deficient medulloblastomas exhibited strikingly distinct pathology, with reduced apoptosis, increased neural differentiation and tectal migration. Comparing Bax+/+ and Bax−/− medulloblastomas, we were able to identify up-regulation of Bcl-2 and nuclear exclusion of p27 as tumorigenic changes that are required to mitigate the tumor suppressive effect of Bax. Studies on human tumors confirmed the importance of modulating Bax in medulloblastoma pathogenesis.
Our results demonstrate that Bax-dependent apoptosis regulates postnatal cerebellar neurogenesis, suppresses medulloblastoma formation, and imposes selective pressure on tumors that form. Functional resistance to Bax-mediated apoptosis, required for medulloblastoma tumorigenesis, may be a tumor-specific vulnerability to be exploited for therapeutic benefit.
Keywords: Bax, CGNPs, cerebellar neurogenesis, medulloblastoma, SmoA1 transgenic mouse
Introduction
Proliferation of neural progenitors must be strictly regulated during cerebellar development and the consequences of failed regulation are significant. A large number of cerebellar granule neurons (CGNs) is required to populate the internal granule cell layer (IGL) of the cerebellum and cerebellar hypoplasia causes impaired neural development and neurologic function (1). Excessive proliferation of cerebellar granule neuron progenitors (CGNPs), however, supports the transformation of CGNPs to give rise to medulloblastoma, an embryonal tumor of CGNP origin that is the most common malignant brain tumor in children (2).
CGNPs arise during embryogenesis from the rhombic lip and migrate tangentially to form a secondary germinal zone called the external granule layer (EGL). In mice, CGNPs proliferate in the EGL for the first 15 days after birth, greatly expanding their population. During this time, they leave the cell cycle in progressively greater numbers and migrate across the adjacent Purkinje cell layer to establish the IGL, where they terminally differentiate into neurons (3).
Positive regulation of CGNP proliferation is accomplished through stimulation of the Sonic Hedgehog (Shh) signaling pathway (4). Diverse molecular mechanisms have been identified that exert an anti-proliferative effect by causing CGNPs to exit the cell cycle. These differentiation-inducing mechanisms include Ren (5), Jsap1 (6), Gpr3 (7), and β-Arrestin (8), all of which converge on the cyclin-dependent kinase inhibitor p27/Kip1, which has been shown to be a potent suppressor of medulloblastoma (9, 10). The observation that p53 mutation increases the frequency of medulloblastoma in both mice (11) and humans (12), suggests that apoptosis may also play a role in suppressing medulloblastoma. Loss of p53, however, may affect many processes in addition to apoptosis. Whether apoptotic mechanisms operate in parallel with differentiation mechanisms to regulate CGNP proliferation remains untested, and the role of Bax in medulloblastoma pathogenesis is unknown.
During the development of the central nervous system, Bax-dependent apoptotic processes limit neural population size by culling differentiated neurons that have migrated or synapsed inappropriately (13), and by winnowing mitotic progenitors of the subventricular zone (14, 15). The relevance of Bax-dependent apoptosis to cerebellar neurogenesis, however, has not been demonstrated; while a previous study of cerebella from Bax−/− mice did not detect any effect on CGN number (16), this study did not examine CGNPs directly.
We have directly compared CGNP proliferation in Bax−/− and wild-type cerebella in order to determine the impact of Bax-dependent apoptosis on postnatal cerebellar growth. Moreover, we have examined the effect of Bax deletion on SmoA1-driven medulloblastoma formation to determine whether disruption of Bax-dependent mechanisms changes the frequency or latency of cerebellar tumorigenesis. Here we show that inactivation of Bax prolongs the period of CGNP proliferation, permanently altering the distribution of CGNs in the resulting cerebella. Additionally, we show that loss of Bax synergizes with Smoothened activation to accelerate medulloblastoma formation.
Results
Down-regulation of CGNP proliferation is delayed in Bax−/− mice
To examine the effect of blocking Bax-mediated apoptosis on the negative regulation of CGNP proliferation, we compared cerebella of Bax+/+ and Bax−/− littermates at P16, when cerebellar neurogenesis has typically waned. As anticipated, immunohistochemistry (IHC) for proliferation marker Ki67 demonstrated a sparse layer of proliferating cells in the EGL of Bax+/+ mice. In contrast, Ki67 staining revealed a comparatively dense layer of proliferation cells in the EGL of Bax−/− mice (Fig. 1a,b). To measure proliferation directly in vivo, we counted CGNPs that incorporated EdU 24 hours after a single intraperitoneal (IP) injection at P15, using 4 replicate Bax+/+ and Bax−/− mice. Quantification of EdU+ cells in a corresponding region of EGL in each of the replicates for each genotype confirmed that Bax-deficient CGNPs continued to proliferate in greater numbers between P15-P16 (Fig. 1c). FACS-based quantification of the proportion of EdU+ cells from dissociated cerebella 3 pairs of Bax+/+ and Bax−/− littermates injected with EdU at P15 and harvested at P16 produced similar results, again demonstrating greater proliferation in Bax−/− cerebella at the time when neurogenesis typically ends (Fig. 1d).
Figure 1. The period of CGNP proliferation is extended in Bax−/− mice.

A,B) IHC for proliferation marker Ki67 (red) at P16 on representative cerebella of Bax+/+ (A) and Bax−/−. (B) littermates demonstrates increased proliferation of Bax-deficient CGNPs when neurogenesis typically wanes. Nuclei are counterstained blue with DAPI. Scale bars represent 500 μm. C) Representative images demonstrating incorporation of thymidine analogue EdU, 24 hours after IP injection at P15 into Bax+/+ and Bax−/− littermates. EdU was visualized by Click-It chemistry with Alexa-488 and EdU+ cells were counted in equivalent regions of EGL in anatomically matched sections from 4 replicate mice of each genotype. Scale bars represent 25 μm D) As an alternative method of quantifying proliferation, cerebella from 3 Bax+/+ and 3 Bax−/− mice were dissociated 24 hours after EdU injection IP at P15 and EdU+ cells counted by FACS. Data presented in the graph are mean proportion of EdU+ cells, normalized to the mean for Bax+/+ mice, ± SEM.
Using Cyclin D2 as a marker of proliferation detectable by Western blot (17), we compared CGNP proliferation in paired Bax-deficient and wild-type littermates at a series of time points spanning the period of cerebellar neurogenesis. We found that proliferation during the later half of the neurogenesis period was consistently, reproducibly higher in Bax−/− mice compared to age-matched Bax+/+ littermates. Cyclin D2 was equally abundant in lysates of whole cerebella from Bax−/− and Bax+/+ mice at P7. From P11-P18, however, Cyclin D2 was consistently more abundant in cerebella from Bax-deficient animals, indicating Bax deletion delayed the down-regulation of CGNP proliferation seen in the wild-type littermates (Fig 2a). We considered whether this change in CGNP regulation caused by Bax deletion might be explained by an increase in either the availability of Shh, or in the responsiveness of CGNPs to Shh. Previous investigations found that Purkinje cell number is increased in Bax−/− mice (16). Since Shh is secreted by Purkinje cells (18, 19), we compared abundance of Shh in cerebella of Bax+/+ and Bax−/− mice. Western blot analysis demonstrated that Shh abundance was not increased by Bax deletion and thus could not account for increased CGNP proliferation (Fig. 2a). To determine whether Bax-deficiency caused an increase in the proliferative response to Shh, we isolated CGNPs from Bax+/+ and Bax−/− littermates, divided CGNPs into replicate wells, and exposed cultured cells to Shh or vehicle for 24 hours. We then compared CGNP proliferation in each well by Western blot for Cyclin D2. Bax+/+ and Bax−/− CGNPs demonstrated equivalent baseline expression of Cyclin D2 and equivalent increase in Cyclin D2 in response to Shh (Fig. 2b).
Figure 2. Bax deficient CGNPs have normal response to Shh but altered response to pro-apoptotic stimulus.

A) A representative Western blot demonstrates temporal expression patterns of indicated proteins in lysates of whole cerebella harvested from Bax+/+ and Bax−/− littermates at the ages indicated, with β-Actin used as a loading control. While Cyclin D2 elevation waned more slowly in Bax−/− mice, Shh abundance remained constant over time and did not vary with genotype. Differential expression of Cyclin D2 was noted in at least 3 paired Bax+/+ and Bax−/− littermates at each time point from P11-P17. B) Comparison of proliferation of CGNPs isolated from Bax+/+ and Bax−/− littermates and cultured in the presence or absence of exogenous Shh, measured by Western blot for Cyclin D2, demonstrated that proliferative response to Shh was not affected by Bax deletion. 3 replicate wells for each condition demonstrated equivalent findings. C) Representative Western blot comparing apoptosis induced by dexamethasone in Bax+/+ and Bax−/− littermates, detected by cC3 in whole cerebellar lysates 24 hours after IP injection of dexamethasone or saline. D) Examination of Bcl-2 family proteins over CGNP development, in Bax+/+ and Bax−/− mice. A SmoA1-induced medulloblastoma is included for comparison.
While Bax deletion did not cause a change in abundance of Shh or in the response of CGNPs to Shh, Bax deletion was sufficient to disrupt apoptosis of CGNPs. Dexamethasone has previously been demonstrated to induce apoptosis specifically in P7 CGNPs within 4 hours after a single IP injection (20, 21). We injected Bax−/− and Bax+/+ littermates at P7 with IP dexamethasone and measured apoptosis in the cerebellum by Western blot for cleaved Caspase 3 (cC3). While dexamethasone induced substantial caspase activation in cerebellum of wild-type mice, Bax deletion markedly reduced cell death in dexamethasone-injected mice, reducing cC3 without affecting total Caspase 3 abundance (Fig. 2c). Western blot on Bax+/− and Bax−/− littermates demonstrated no change in Bak expression caused by Bax deletion (Fig. 2d), indicating that loss of Bax alone was sufficient to prevent dexamethasone-induced apoptosis. Importantly, Bax deletion caused a detectable increase in expression of Bcl-2 and Mcl-1 (Fig. 2d), which may have contributed to decreasing the apoptotic response by interfering with Bak-mediated compensation. These results, along with our analyses of CGNP proliferation, reveal a significant, specific role for Bax-dependent apoptosis in regulating postnatal neurogenesis in the cerebellum.
Abnormal CGNP migration in Bax−/− mice
Along with altering the temporal pattern of CGNP proliferation, Bax deletion caused a change in the spatial distribution of CGNP progeny. During postnatal development, proliferating CGNPs typically migrate across the Purkinje cell layer to the IGL, where they terminally differentiate. Examination of cerebella from Bax-deficient mice at P17, after most CGNPs had migrated from the EGL, revealed distinct populations of ectopic cells: focal collections of differentiated neurons where the EGL had been, and a streak of proliferating progenitors migrating into the adjacent tectum (Fig. 3a, b). IHC for the neuronal marker NeuN and the proliferation marker Ki67 demonstrated that most of the ectopic EGL cells exhibited neuronal differentiation while few cells continued to proliferate (Fig. 3c,d). In contrast, many cells in the ectopic population entering the tectum labelled with Ki67 (Fig. 3e). EdU incorporation 24 hours after IP injection also marked this tectal ectopic population as proliferating progenitors (Fig. 3f). These cells did not express NeuN (data not shown). While this tectal population of ectopic cells was contiguous with the IGL of the first cerebellar folium, the cells resembled CGNPs in their appearance on H&E stained sections, their incorporation of EdU, and their lack of NeuN staining. To confirm that these ectopic cells were CGNPs, and that Bax deletion caused a cell-autonomous effect on CGNP migration pattern, we analysed mice in which Bax was deleted conditionally within the Math1 lineage, which is limited, in the cerebellum, to the CGNPs and their progeny (22). Specifically, we bred mice with a floxed allele of Bax (Baxf/f) with transgenic mice expressing cre recombinase under the Math1 promoter (Math1-cre). In the resulting Math1-cre; Baxf/f mice at P17, we noted collections of ectopic cells in the molecular layer and a streak of ectopic cells migrating into the tectum, as we saw in the Bax−/− mice (Fig. 3g,h). Thus, Bax deficiency acted on CGNPs to disrupt not only the timing of proliferation but also the migration pattern of CGNP-derived cells.
Figure 3. Abnormal migration of CGNPs in Bax−/− mice.

A,B) Paraffin sections of cerebellum in sagittal plane from representative P17 Bax+/+ (A) and Bax−/− (B) mice, stained by H&E. In (B) black arrowheads and box C,D highlight collections of cells remaining in the molecular layer. Yellow arrowhead and boxes E and F highlight a separate population migrating rostrally into the tectum. C) IHC for the NeuN (red) demonstrates neural differentiation in the ectopic collection highlighted in Box C,D of Panel B. D) IHC for Ki67 (red) demonstrates rare proliferative cells in the same collection. E) IHC for Ki67 (red) shows proliferating cells migrating into the tectum that were highlighted in Box E of Panel B. F) Incorporation of EdU (green) confirms that ectopic cells migrating into the tectum, highlighted in Box F of Panel B, continue to proliferate. G) H&E stained sagittal section showing normal histology of cerebellum from a P17 Baxf/f mouse without cre. H) In a representative, comparable section from a Math1-cre; Baxf/f mouse at P17, ectopic cells persist in the molecular layer (black arrowheads) and migrate into the tectum (yellow arrowheads). In C–F, nuclei are counterstained blue with DAPI. Scale bars represent 500 μm (A,B), 100 μm (C–F), or 250 μm (G,H).
Bax deficiency accelerates ND2:SmoA1 induced tumorigenesis
Mechanisms that down-regulate CGNP proliferation through differentiation have consistently been found to exert a tumor-suppressive effect in preventing medulloblastoma. To determine whether Bax-dependent apoptosis functioned as an additional barrier to tumorigenesis, we measured the effect of Bax deficiency on induction of medulloblastoma by SmoA1, a tumorigenic, constitutively active allele of Smoothened (23). We bred ND2:SmoA1 that were Bax+/+,+/− and−/−, then compared time to symptomatic tumor formation in each group. For both genotypes, tumor symptoms began within 4–5 days of the first detection of change in head shape, and mice were euthanized at the onset of symptoms.
Bax deficiency increased the frequency and decreased the latency of tumor formation. Overall tumor incidence in ND2:SmoA1 mice was significantly increased by Bax deletion: 14/15 for Bax−/− vs 7/16 for Bax+/+ (p=0.006; 2-sided Fisher’s exact test). The difference in tumor incidence in Bax haploinsufficient and Bax wild-type ND2:SmoA1 mice was not statistically significant: 11/18 for Bax+/− vs 7/16 for Bax+/+ (p=0.49; 2-sided Fisher’s exact test). Bax−/− mice also developed medulloblastoma significantly faster (p<0.0000001; Log-Rank test) than mice with 1 or 2 functional copies of Bax (Fig. 4). Haploinsufficiency did not cause a discernable change in the rate of tumorigenesis (data not shown). The more frequent and rapid onset of medulloblastoma in Bax−/−; ND2:SmoA1 mice reveals the tumor suppressive effect exerted by Bax when at least 1 functional allele is present.
Figure 4. Deletion of Bax accelerates medulloblastoma tumorigenesis.
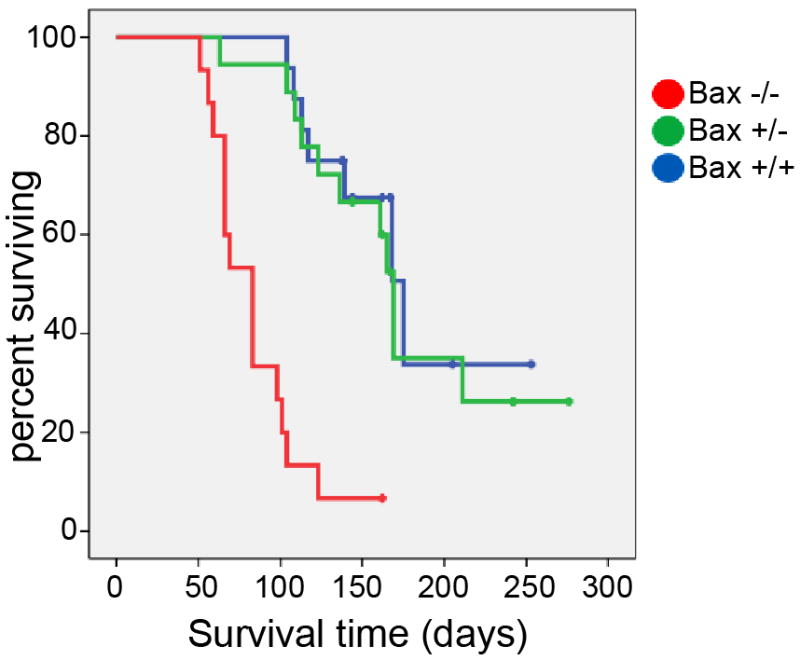
Event-free survival curves for Bax+/+; SmoA1 mice (n=16), Bax+/−; SmoA1 mice (n=18), and Bax−/−; SmoA1 mice (n=15). Deletion of Bax significantly altered the rate of symptomatic tumor formation (P<0.0000001; Log-Rank test), increasing tumor frequency and reducing tumor latency.
Bax-deficient tumors have increased differentiation and reduced apoptosis
In addition to accelerating the frequency and onset of tumorigenesis, Bax deletion also exerted a profound effect on tumor pathology (Fig. 5a,b). ND2:SmoA1-induced medulloblastomas are typically homogeneous, composed of morphologically monotonous cells with molded nuclei and scant cytoplasm (Fig. 5a,c). In contrast, medulloblastomas in Bax−/−; ND2:SmoA1 complex transgenic mice were markedly heterogeneous, containing rests of monotonous tumor cells interspersed with less densely packed cells with round, regular nuclei that resembled neurons in the IGL (Fig. 5b,d). We compared proliferation, apoptosis and differentiation in Bax+/+ and Bax−/− tumors, using IHC for mitotic marker phosphohistone H3 (PH3), cC3 and NeuN. IHC for PH3 demonstrated markedly lower frequency of mitosis in Bax-deficient tumors (Fig. 5e,h). IHC for cC3 consistently demonstrated on-going apoptosis in medulloblastomas with intact Bax (Fig. 5f) and absence of apoptosis in tumors with Bax deletion (Fig. 5i). Expression of NeuN, a marker of neural differentiation was markedly lower in Bax+/+ medulloblastoma compared to Bax-deleted tumors (Fig. 5g,j). We replicated these findings in 3 tumors from Bax+/+ and Bax−/− mice, performed automated cell counts, and found a statisitically robust correlation between reduced PH3+ cells, reduced cC3+ cells and increased NeuN+ cells in medulloblastomas with Bax deletion (Fig. 5k). Thus although Bax−/−; ND2:SmoA1 mice developed tumors and died more rapidly, the resultant tumors demonstrated reduced proliferation and apoptosis, and increased differentiation.
Figure 5. Tumors in Bax−/− mice displayed markedly different pathology, with increased heterogeneity, reduced proliferation, absence of apoptosis and increased differentiation.

A,B) Comparison of H&E stained sagittal sections of cerebella from tumor-bearing Bax+/+ (A) and Bax−/− (B) mice. Rostral invasion of tectum (arrowhead) was consistently noted in Bax−/− mice and never in Bax+/+ or Bax+/− mice. C) Bax+/+ tumor exhibited typical, repetitive pathology, with small round cells with little cytoplasm. D) Heterogeneous histology in a Bax−/− tumor included both rests of small round blue cells (red arrows) and regions of cells resembling granule neurons (blue arrows) as well as abundant neuropil (white arrows) E–J) Analysis of Bax+/+ (E–G) and Bax−/− (H–J) medulloblastomas by IHC for PH3 (E,H), cC3 (F,I) and NeuN (G,J) demonstrated reduced proliferation, reduced apoptosis and increased differentiation in tumors with Bax deletion. K) Quantitative comparison of cells expressing PH3, cC3 and NeuN in a set of 3 Bax+/+ and 3 Bax−/− medulloblastomas. L,M) Adjacent paraffin sections of a representative tumor from a Bax−/−; ND2:SmoA1 mouse labeled by IHC with hematoxylin counterstain demonstrate Bax−/− medulloblastoma cells invading the adjacent tectum include both NeuN+ (L) and PCNA+ (M) populations. Scale bars represent 1000 μm (A,B), 100 μm (C–J), or 200 μm (L,M).
Altered tumor-cell migration in Bax-deficient medulloblastomas
Invasion of the tectum by tumor cells was noted in all Bax−/− medulloblastomas and was never noted in tumors with intact Bax. Invading tumor cells followed the rostral migration pattern of ectopic progenitors observed in Bax−/− cerebella at P17 (Fig. 5b, compare to Fig. 3b). As seen throughout the Bax−/− tumors, the population of invading cells was heterogeneous, including differentiated cells that were NeuN+ and undifferentiated proliferating cells that were PCNA+ (Fig. 5l,m). Thus, migration abnormalities seen in Bax−/− cerebella during postnatal development were recapitulated and amplified in Bax−/− medulloblastomas.
Specific tumorigenic adaptations required by presence of Bax
Our survival analysis demonstrated that deletion of Bax reduced the time required for mice to develop symptomatic medulloblastoma. Previous investigators have demonstrated in diverse mouse models of medulloblastoma that during the latent period that precedes tumor detection, hypertrophic CNGPs acquire additional oncogenic changes that mediate that progression from precursor lesion to tumor (23, 24). We theorized that if tumorigenesis typically requires time for the development of resistance to endogenous Bax, hypertrophic Bax−/− CGNPs might progress to medulloblastoma more quickly. To detect tumor-specific adaptations that are necessitated by Bax, we compared expression of anti-apoptotic Bcl-2 family proteins in precursor lesions (Fig. 6a) and tumors from Bax+/+, Bax+/− and Bax−/− mice. In these same samples, we also analyzed expression of p27, because of the central role of this protein in suppressing medulloblastoma through differentiation (9, 10).
Figure 6. Tumorigenesis in the presence of Bax requires specific adaptations that were not observed in Bax-deficient medulloblastomas.

A) H&E stained sagittal sections of cerebella from Bax+/+ and Bax−/− ND2:SmoA1 mice at P22 show hyperproliferative, preneoplastic lesions in the EGL with adjacent areas of normal cerebellum. B) Representative Western blot comparing Bax, Bcl-2, Bcl-XL, Mcl-1, full length Caspase 3, cC3, p-27 and Cyclin D2 in whole cerebella from Bax+/+ and Bax−/− ND2:SmoA1 mice at P22 and in medulloblastomas from ND2:SmoA1 with Bax genotype indicated. β-Actin serves as a loading control and for each protein analyzed, while Cyclin D2 serves as a marker of proliferation. C,D) Representative IHC for Bcl-2 (red) on medulloblastoma sections demonstrated abundant Bcl-2 protein in tumors with intact Bax (C), and absence of Bcl-2 expression throughout Bax−/− tumors (D), with Bcl-2 in white matter adjacent to the tumors (white arrows) serving as an internal positive control. E) Tumors with intact Bax consistently demonstrated reduced p27, demonstrated by IHC (red), with foci in which p27 was localized to the cytoplasm and excluded from the nucleus (white arrows), causing poor correspondence between red p27 staining and blue nuclear stain. F) IHC for p27 (red) demonstrates that p27 protein was not excluded from nuclei in Bax-deficient tumors, causing nuclei to label with both red and blue (DAPI). Scale bars represent 100 μm (A,B), 200 μm (C,D) or 50μm (E,F). G) Quantification of fraction of p27+ cells (left) and p27+ cells with nuclear exclusion of p27 protein (right) in Bax+/+ and Bax−/− tumors.
We found Bcl-2 and Mcl-1 to be specifically up-regulated in the course of tumorigenesis in the presence of Bax. Premalignant lesions in P22 cerebella of ND2:SmoA1 mice expressed relatively low levels of Bcl-2 and Mcl-1 and relatively high levels of Bak, Bcl-XL, and p27 (Fig. 6b). Importantly, tumors with intact Bax consistently up-regulated Bcl-2 and Mcl-1. In contrast Bcl-2 and Mcl-1 were consistently less abundant in Bax−/− tumors. Expression of Bak was also mildly but consistently decreased in Bax−/− tumors. In contrast, Bcl-XL was abundant in all tumors, and was more highly expressed in the Bax−/− subset. Cellular analysis of Bcl-2 expression by IHC in an independent set of 4 Bax+/+ and 4 Bax−/− medulloblatomas demonstrated widespread intra-tumoral expression of Bcl-2 in 4/4 Bax+/+ cases and consistent absence of Bcl-2 in all tumor cells in 4/4 Bax−/− cases (Fig. 6c,d). Bcl-2 protein in white matter adjacent to tumors served as an internal control in Bax−/− sections and also explains the faint Bcl-2 band observed in Western blot of Bax−/− tumors. Thus up-regulation of Bcl-2 and Mcl-1 were consistently observed, tumor-specific changes that were not required in mice with Bax deletion.
While Bcl-2 expression was reduced in Bax−/− tumors, p27 was markedly more abundant. Bax haploinsufficiency increased p27 expression, and homozygous Bax deletion caused a further increase (Fig. 6b). IHC confirmed reduced expression of p27 in Bax+/+ medulloblastomas and also revealed differential localization of the p27 protein: while tumors with intact Bax demonstrated translocation of p27 from nucleus to cytoplasm as previously reported (9), Bax-deficient tumors demonstrated robust nuclear p27 (Fig. 6e, f). Thus down-regulation p27 and its exclusion from the nucleus, like up-regulation of Bcl-2, were consistently observed steps in the progression from precursor lesion to medulloblastoma, and these steps were not required when Bax was deleted.
Bax is an obstacle that is mitigated in human medulloblastoma pathogenesis
Because of the impact of Bax on tumorigenesis in our medulloblastoma model, we sought to determine whether Bax exerts an effect on the human disease. In a tissue micro-array (TMA) consisting of 3–6 cores from each of 20 human medulloblastomas, IHC detected widespread expression of Bax in every core, demonstrating that Bax is not commonly silenced nor deleted (Fig. 7a–c). We noted heterogeneous expression of Bcl-2, and p27, heterogeneous tendency to localize p27 to the cytoplasm, and variable rates of apoptosis, demonstrated by IHC for cC3 (Fig. 7a–d). Importantly, the TMA demonstrated an inverse correlation between Bcl-2 and cC3: the mean rate of apoptosis in tumors with <5% of cells Bcl-2+ was 30%, while the rate of apoptosis was 9% in tumors with >5% of cells Bcl-2+ (p=0.00013). This inverse correlation, further demonstrated by linear regression analysis on all samples in the TMA (Fig. 7e), suggests that Bcl-2 is functionally active in opposing an inherent tendency to undergo PCD. A similar linear regression analysis of cytoplasmic p27 demonstrated a tendency for decreased cC3 with increased nuclear exclusion of p27 that approached but did not reach statistical significance (p=0.0524), suggesting a possible role for cytoplasmic p27 in avoidance of PCD that may be more complex than that of Bcl-2. Through this analysis, we were able to confirm the impact of Bax in human medulloblastoma and the continued engagement of the diverse mechanisms through which it is mitigated.
Figure 7. Consistent Expression of Bax, and variable expression of Bcl-2, p27 and cC3 in human medulloblastoma samples.

A) low-magnification images of a representative region of a medulloblastoma TMA, demonstrating expression of indicated protein by IHC in adjacent samples from 6 different patients. Bax, Bcl-2 and cC3 are demonstrated in brown, while p27 is demonstrated in red fluorescence, and nuclei are counterstained blue. Scale bars represent 250 μm. B) Range of expression of Bax, Bcl-2, p27 and cC3 demonstrated in higher-magnification representative images. For Bax, Bcl-2 and cC3, top panel shows a representative sample with relatively high expression and bottom panel shows a representative sample with relatively lower expression of indicated protein. For p27, top panel demonstrates a core with nuclear localization, while the bottom panel shows a sample with cytoplasmic p27 that is excluded from the nucleus. Scale bars represent 200 μm. C) Box and whiskers plots demonstrating mean (line), 25th–75th percentiles (box), and range (error bars) of percent positive cells for each protein. D) Box and whiskers plot of fraction of p27+ cells with nuclear exclusion of p27 protein. E) Expression of Bcl-2 correlated inversely with apoptosis, measured by IHC for cC3.
Discussion
The potential for cerebellar neurogenesis to give rise to medulloblastoma underscores the importance of regulating CGNP proliferation. While previous investigations have examined neuronal PCD in the postnatal cerebellum (25), our study is the first to implicate the intrinsic apoptotic pathway in the regulation of CGNPs, the cells of origin for medulloblastoma. We found that deletion of Bax was sufficient to block apoptosis of CGNPs, causing CGNP proliferation to extend beyond P15. Clearly, multiple mechanisms operate to limit CGNP proliferation, as even in Bax−/− mice CGNPs do not proliferate indefinitely. However, these additional mechanisms, including p27-mediated differentiation, were strikingly ineffective in preventing tumor formation when Bax-dependent apoptosis was blocked.
In ND2:SmoA1 mice, the tumorigenic effect of SmoA1 is balanced by tumor suppressive mechanisms, such that while all mice develop CGNP hypertrophy, not all mice develop tumors. The importance of differentiation for limiting the growth of hypertrophic CGNPs in pre-neoplastic lesions has been demonstrated by lineage tracing experiments (24). However, the increased frequency of medulloblastoma in Bax−/−; ND2:SmoA1 mice demonstrates that when apoptosis is prevented, developmental differentiation mechanisms are insufficient to block progression from hypertrophy to tumor. In fact, Bax−/− medulloblastomas demonstrated marked up-regulation of p27, accompanied by increased differentiation and nevertheless shorter survival times. Considered against previous studies demonstrating that loss of p27 also accelerates medulloblastoma formation (9, 10), our findings reveal that effective tumor suppression depends on both apoptosis and differentiation mechanisms acting in concert.
In addition to demonstrating that Bax-dependent apoptosis functions as a brake on tumorigenesis, we have identified specific mechanisms through which this brake can be overcome. In the progression from hyperplastic precursor lesion to medulloblastoma in SmoA1 mice, we observed consistent up-regulation of Bcl-2 and disruption of p27 through down-regulation and cytoplasmic localization. Strikingly, these mechanisms were never observed and thus clearly not required, in tumors that formed in SmoA1; Bax−/− mice. The marked up-regulation of p27 in Bax−/− tumors, moreover, suggests that high levels of nuclear p27 may exert a pro-apoptotic effect that is only tolerated when apoptosis is blocked. Accordingly, shuttling p27 to the cytoplasm may prevent the ability of nuclear p27 to drive PCD. Thus, our data link both up-regulation of Bcl-2 and displacement of p27 in medulloblastoma to the presence of Bax.
Our analysis of human medulloblastoma confirmed the interrelation of Bax, p27 and Bcl-2 in the human disease. In the patient-derived tumors, as in the SmoA1-induced mouse model, Bax was robustly expressed and consistently accompanied by cytoplasmic p27. Moreover, the expression of Bcl-2 correlated inversely with the rate of apoptosis. While previous investigators have noted Bcl-2 expression in medulloblastoma, a correlation with prognosis has not emerged. Our findings are thus the first to demonstrate that Bcl-2 is functionally active in medulloblastoma, and importantly reveal an inherent tendency toward PCD that Bcl-2 acts to repress.
The consistent expression of Bax and the correlation between engagement of Bax-resistance mechanisms and PCD may be highly relevant to the unique responsiveness of medulloblastoma to chemo-radiotherapy. Of all malignant brain tumors, medulloblastoma is the most amenable to treatment, with 5 year survival rates markedly higher than high-grade glioma or supratentorial PNET (26–28). The underlying mechanism for the increased susceptibility of medulloblastoma to cytotoxic therapy may be that apoptosis in medulloblastoma is prevented by dynamic, reversible mechanisms of Bax resistance, in contrast to more static, less reversible mechanisms for avoiding apoptosis in more refractory tumors. Strategies that modulate resistance of medulloblastoma cells to the proapoptotic potential of endogenous Bax may be well suited to medulloblastoma. BH3-mimetic agents are an example of an intervention that might target this dynamic resistance to PCD.
Previous investigations have demonstrated complementation between Bax and Bak in the regulation of neural progenitor proliferation (15, 29), and suggested that Bak may be more relevant to undifferentiated progenitors (30). In contrast, apoptosis in differentiated neurons depends entirely on Bax (31). Our data demonstrate that apoptosis of CGNPs and consequent suppression of medulloblastoma specifically require Bax. Importantly the presence of wild-type Bak was insufficient to maintain the apoptotic response to dexamethasone or to inhibit medulloblastoma formation in ND2:SmoA1 Bax−/− mice.
An important question that remains is how Bax-dependent mechanisms become activated during neurogenesis to limit CGNP proliferation and suppress medulloblastoma. The finding of ectopic CGNP progeny in Bax−/− cerebella suggests that PCD normally acts to remove CGNPs that fail to migrate to appropriate locations. Such localization-dependent survival could be mediated by dependence receptors. Intriguingly, the rostral migration of Bax-deficient CGNPs and medulloblastoma cells into the tectum bears striking similarity to the phenotype of the rcm mutant mouse (32) in which disruption of Unc5h causes CGNPs to migrate into the tectum in the first week of postnatal life. Unc5h has been reported to act as a dependence receptor (33–35) and this phenotypic similarity suggests that Unc5h and Bax may operate within a single molecular mechanism, in which Unc5h functions as a dependence receptor activating Bax-dependent apoptosis in the absence of ligand. In a similar manner, multiple dependence receptors may activate Bax when CGNPs fail to migrate appropriately, and the loss of Bax-dependent PCD could thus account for the diverse abnormal positions of CGNP progeny in cerebella and medulloblastoma in Bax−/− mice.
The altered invasion pattern and distinctive pathology of Bax−/− medulloblastoma demonstrates that Bax-dependent apoptosis remains active after tumorigenic transformation and continues to define tumor phenotype. Medulloblastomas typically express Bax and grow as homogeneous tumors composed entirely of undifferentiated progenitors. Moreover, Bax-mediated PCD continues to cull tumor cells from the population, even in tumors with up-regulation of Bcl-2. In contrast, Bax-deficient medulloblastomas are devoid of apoptosis and harbor substantial populations of tumor cells with greater differentiation potential. The homogeneity of medulloblastoma would thus seem to result from selection pressure exerted by Bax: undifferentiated proliferating cells that are resistant to Bax-dependent cell death persist, while differentiating cells are removed through activation of the intrinsic apoptotic pathway. When Bax is deleted, terminal differentiation becomes the alternate fate choice for differentiating cells that cannot undergo apoptosis. The shortened survival of mice with Bax−/− tumors demonstrates that a more differentiated pathology is not invariably indicative of benign prognosis, but may instead be a marker of impaired apoptosis.
We have found that Bax-dependent apoptosis is an essential force in postnatal neurogenesis in the cerebellum, and that Bax presents an obstacle that is overcome in medulloblastoma pathogenesis. We identified modulation of Bcl-2 and p27 as key mechanisms that mitigate the tumor suppressive effects of Bax. Importantly, the active role played by Bax in shaping tumor pathology and the inverse relationship between Bcl-2 and apoptosis suggest that Bax resistance in medulloblastoma may be dynamic and reversible. Our conclusions underscore the therapeutic potential of targeting mechanisms that impair the function of Bax in medulloblastoma.
Material and Methods
Generation of mice
The breeding and genotyping of Bax−/− mice have been described previously (36). Math-1 cre mice were generously shared by David Rowitch, MD, PhD, UCSF and Robert Wechsler-Reya, PhD, Sanford-Burnham Medical Research Institute, La Jolla, Ca and have been previously described (37). Baxfl/fl mice were obtained as Baxfl/fl; Bak−/− mice from Jackson Laboratories, Bar Harbor, ME and both mutant alleles of Bak were replaced by wild type alleles thorough selective breeding. Medulloblastoma prone NeuroD2:SmoA1 mice (kindly provided by James Olson, MD, PhD, Fred Hutchinson Cancer Research Center, Seattle, WA) were crossed with the Bax−/− mouse line. Genotypes were determined by PCR: DNA was extracted from toes using Tail Lysis Buffer (Allele, cat#ABP-PP-MT01) and then amplified as described previously (23, 31). Tumor-prone mice were monitored daily for head shape and movement abnormalities and all mice were euthanized at the onset of symptomatic tumors. All animal handling and protocols were carried out in accordance with established practices as described in the National Institutes of Health Guide for Care and Use of Laboratory Animals and as approved by the Animal Care and Use Committee of the University of North Carolina (UNC).
CGNP culture
Primary cultures of CGNPs from Bax wild-type and Bax−/− mice were generated as according to published protocols (38). When indicated, CGNPs were maintained continuously in Shh (0.5 μg/ml; R&D Systems cat#464SH).
In vivo apoptosis
For these studies, P7 mouse pups were injected IP with 50 μl Hanks buffered saline (HBSS), with or without Dexamethasone (250 μM, Sigma cat#D9184). Cerebella were harvested after 4 hours and processed for Western blot as described below.
In vivo proliferation studies
Mouse pups at P15 or P16 were injected IP with 50 μl HBSS containing EdU (250 μM, Invitrogen cat# A10044) and sacrificed after 24 hours. For in situ detection, brains were dissected free and incubated in 4% formaldehyde in PBS for 24 hours at 4°C, then processed for histology. For FACS analysis, cerebella were dissected free, and dissociated and triturated as per cell culture protocol. After trituration, cells were fixed with 1:5 dilution of 4% formaldehyde for 15 seconds with vortexing and further incubated for 15 min at room temperature. Fixed cells were washed with PBS and processed for EdU following manufacturer’s protocol (Invitrogen cat# C35002). EdU incorporation was then detected and quantified by flow cytometry using a Cyan Instrument (Beckman-Coulter) and Summit software (Dako). EdU+ cells were determined by comparison to an unstained fraction of cells and each genotype was repeated in triplicate. To determine the statistical significance of differences between Bax+/+ and Bax−/− results, p values were determined using Student’s t test.
Histology and IHC
For histological analysis and IHC, mouse brain and tumor tissue were embedded in paraffin and sectioned. H&E stained sections were prepared using standard techniques. For human tumor studies, cases of newly-diagnosed, untreated classic and nodular/desmoplastic medulloblastoma were obtained from the surgical pathology archives at UNC Hospitals from 21 patients (18 classic, 3 nodular/desmoplastic) and reviewed by a neuropathologist (C.R.M.). Only tumors with >90% viable tumor cell content were included. A TMA was constructed on a TMArrayer (Pathology Devices, Westminster, MD) using 3–6 replicate 1 mm cores from each case. Human tonsil was used as a control. All human specimens were obtained under a protocol approved by the UNC Institutional Review Board and were thoroughly de-identified.
IHC was performed on paraffin embedded sections after deparaffinization in Histoclear and rehydration in a graded ethanol series, heated to boiling in 10 mM Citrate buffer pH 6.0 in a pressure cooker for 15 minutes, then transferred to PBS. After antigen retrieval, IHC was performed as previously described (Gershon et al., 2009), using primary antibodies: PH3 (Cell Signaling cat#9706), cC3 (human: Dako, cat# CP229C; mouse: Cell Signaling, cat# 9664), Ki-67 (Millipore, cat# AB9260MI), NeuN (Millipore, cat# MAB377), PCNA (Cell Signaling, cat# 2586), p27 (human: DAKO, cat# M7203; mouse: Cell Signaling, cat# 3686), Bax (Sigma, cat# AF820) and Bcl-2 (human: DAKO, cat# M0887; mouse: Cell Signaling, cat#3498). IHC-stained human TMA sections were counterstained with hematoxylin, scanned using an Aperio ScanScope XT (Vista, CA), and analyzed using Definiens (Parsippany, New Jersey) Developer XD software. For Bcl-2 detection in mouse tumors, tissue was not embedded in paraffin, but rather was sectioned by Vibratome to 100 μm thickness and stained by IHC without antigen retrieval. For immunofluorescence, nuclei were counterstained with 4′6-diamino-2-phenylindole (DAPI) (Sigma, US), 1μg/ml, for 5 minutes and immunoreactivity was evaluated with a Leica epifluorescence DM5000B microscope (Leica Microsystems, US).
Western blot analysis
Cultured cells, whole cerebella, and tumors were lysed by homogenization in lysis buffer (Cell Signaling, cat# 9803). Protein concentrations were quantified using the Bicinchoninic acid method (Thermo Scientific, cat# 23227) and equal concentrations of protein were resolved on SDS-polyacrylamide gels then transferred to PVDF membranes. Immunologic analysis was performed on a SNAP ID device (Millipore, US) using manufacturer’s protocol with primary antibodies to β-Actin (Cell Signaling, cat# 4970), Bax-N20 (Santa Cruz, sc-493), Bak (Cell Signaling, cat# 9521), Bcl-2 (Cell Signaling, #3498), Bcl-XL (Cell Signaling, cat# 2764), Mcl-1 (Cell Signaling, cat# 5453), full-length Caspase 3 (Cell Signaling, cat# 9662), cC3 (Cell Signaling, cat# 9664), Cyclin D2 (Cell Signaling, #3741), p27 (Cell Signaling, cat# 3686), Shh (Cell Signaling, cat# 2207). Secondary antibodies were anti-rabbit IgG HRP (Cell Signaling, cat# 7074), and anti-mouse IgG HRP (Cell Signaling, cat# 7076). Antibody conjugates were visualized by chemiluminescence (ECL, Amersham Life Science, cat# RPN2106).
Acknowledgments
We thank James Olson, Fred Hutchinson Cancer Research Center for generously sharing ND2:SmoA1 mice, and Mervi Eeva, Jackie Kylander and Stephanie Cohen in the UNC Translational Pathology Laboratory (TPL) for expert technical assistance. The UNC TPL is supported, in part, by grants from the National Cancer Institute (3P30CA016086), National Institute of Environmental Health Sciences (3P30ES010126), Department of Defense (W81XWH-09-2-0042), and the UNC University Cancer Research Fund (UCRF). CRM is supported in part by a Clinical Investigator award from the Damon Runyon Cancer Research Foundation (CI-45-09). TRG is supported by a Scholar Award from the St. Baldrick’s Foundation. Major potions of this work were supported by the Morgan Adams Foundation and the Matthew Larson Brain Tumor Foundation.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Bolduc M-E, Du Plessis AJ, Sullivan N, Khwaja OS, Zhang XUN, Barnes K, et al. Spectrum of neurodevelopmental disabilities in children with cerebellar malformations. Developmental Medicine & Child Neurology. 2011;53(5):409–416. doi: 10.1111/j.1469-8749.2011.03929.x. [DOI] [PubMed] [Google Scholar]
- 2.Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TTT, et al. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132(10):2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- 3.Hatten ME. CENTRAL NERVOUS SYSTEM NEURONAL MIGRATION. Annual Review of Neuroscience. 1999;22(1):511–539. doi: 10.1146/annurev.neuro.22.1.511. [DOI] [PubMed] [Google Scholar]
- 4.Wechsler-Reya RJ, Scott MP. Control of Neuronal Precursor Proliferation in the Cerebellum by Sonic Hedgehog. Neuron. 1999;22(1):103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 5.Di Marcotullio L, Ferretti E, De Smaele E, Argenti B, Mincione C, Zazzeroni F, et al. RENKCTD11 is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2004 Jul 20;101(29):10833–10838. doi: 10.1073/pnas.0400690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato T, Torashima T, Sugihara K, Hirai H, Asano M, Yoshioka K. The scaffold protein JSAP1 regulates proliferation and differentiation of cerebellar granule cell precursors by modulating JNK signaling. Molecular and Cellular Neuroscience. 2008;39(4):569–578. doi: 10.1016/j.mcn.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka S, Shaikh IM, Chiocca EA, Saeki Y. The Gs-Linked Receptor GPR3 Inhibits the Proliferation of Cerebellar Granule Cells during Postnatal Development. PLoS One. 2009;4(6):e5922. doi: 10.1371/journal.pone.0005922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parathath SR, Mainwaring LA, Fernandez-L A, Guldal CG, Nahlé Z, Kenney AM. β-Arrestin-1 links mitogenic sonic hedgehog signaling to the cell cycle exit machinery in neural precursors. Cell Cycle. 2010;9(19):4013–4024. doi: 10.4161/cc.9.19.13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhatia B, Malik A, Fernandez LA, Kenney AM. p27(Kip1), a double-edged sword in Shh-mediated medulloblastoma: Tumor accelerator and suppressor. Cell Cycle. 2010 Nov;9(21):4307–4314. doi: 10.4161/cc.9.21.13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ayrault O, Zindy F, Rehg J, Sherr CJ, Roussel MF. Two Tumor Suppressors, p27Kip1 and Patched-1, Collaborate to Prevent Medulloblastoma. Molecular Cancer Research. 2009 Jan 1;7(1):33–40. doi: 10.1158/1541-7786.MCR-08-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes & Development. 2005 Nov 15;19(22):2656–2667. doi: 10.1101/gad.1368605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfaff E, Remke M, Sturm D, Benner A, Witt H, Milde T, et al. TP53 Mutation Is Frequently Associated With CTNNB1 Mutation or MYCN Amplification and Is Compatible With Long-Term Survival in Medulloblastoma. Journal of Clinical Oncology. 2010 Dec 10;28(35):5188–5196. doi: 10.1200/JCO.2010.31.1670. [DOI] [PubMed] [Google Scholar]
- 13.Dicou E, Perez-Polo JR. Bax--an emerging role in ectopic cell death. International Journal of Developmental Neuroscience. 2009;27(4):299–304. doi: 10.1016/j.ijdevneu.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Lindsten T, Golden JA, Zong WX, Minarcik J, Harris MH, Thompson CB. The proapoptotic activities of Bax and Bak limit the size of the neural stem cell pool. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003 Dec 3;23(35):11112–11119. doi: 10.1523/JNEUROSCI.23-35-11112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindsten T, Zong W-X, Thompson CB. Defining the Role of the Bcl-2 Family of Proteins in the Nervous System. The Neuroscientist. 2005;11(1):10–15. doi: 10.1177/1073858404269267. [DOI] [PubMed] [Google Scholar]
- 16.Fan H, Favero M, Vogel MW. Elimination of bax expression in mice increases cerebellar purkinje cell numbers but not the number of granule cells. The Journal of Comparative Neurology. 2001;436(1):82–91. [PubMed] [Google Scholar]
- 17.Kenney AM, Rowitch DH. Sonic hedgehog Promotes G1 Cyclin Expression and Sustained Cell Cycle Progression in Mammalian Neuronal Precursors. Mol Cell Biol. 2000 Dec 1;20 (23):9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valerie AW. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Current Biology. 1999;9(8):445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- 19.Dahmane N, Ruiz-i-Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999 Jul 15;126(14):3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 20.Noguchi KK, Walls KC, Wozniak DF, Olney JW, Roth KA, Farber NB. Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death & Differentiation. 2008;15(10):1582–1592. doi: 10.1038/cdd.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heine VM, Rowitch DH. Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11βHSD2-dependent mechanism. J Clin Invest. 2009;119(2):267–277. doi: 10.1172/JCI36376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Machold R, Fishell G. Math1 Is Expressed in Temporally Discrete Pools of Cerebellar Rhombic-Lip Neural Progenitors. Neuron. 2005;48(1):17–24. doi: 10.1016/j.neuron.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 23.Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, et al. The SmoA1 Mouse Model Reveals That Notch Signaling Is Critical for the Growth and Survival of Sonic Hedgehog-Induced Medulloblastomas. Cancer Research. 2004 Nov 1;64(21):7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 24.Kessler JD, Hasegawa H, Brun SN, Emmenegger BA, Yang Z-J, Dutton JW, et al. N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes & Development. 2009;23 (2):157–170. doi: 10.1101/gad.1759909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung AR, Kim TW, Rhyu IJ, Kim H, Lee YD, Vinsant S, et al. Misplacement of Purkinje cells during postnatal development in Bax knock-out mice: a novel role for programmed cell death in the nervous system? The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008 Mar 12;28(11):2941–2948. doi: 10.1523/JNEUROSCI.3897-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Packer RJ. Childhood brain tumors: accomplishments and ongoing challenges. Journal of Child Neurology. 2008 Oct;23(10):1122–1127. doi: 10.1177/0883073808320758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gershon TR, Becher OJ. Medulloblastoma: therapy and biologic considerations. Curr Neurol Neurosci Rep. 2006 May;6(3):200–206. doi: 10.1007/s11910-006-0006-y. [DOI] [PubMed] [Google Scholar]
- 28.Preusser M, de Ribaupierre S, Wohrer A, Erridge SC, Hegi M, Weller M, et al. Current concepts and management of glioblastoma. Ann Neurol. 2011 Jul;70(1):9–21. doi: 10.1002/ana.22425. [DOI] [PubMed] [Google Scholar]
- 29.Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000 Dec;6(6):1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim WR, Kim Y, Eun B, Park O-h, Kim H, Kim K, et al. Impaired Migration in the Rostral Migratory Stream But Spared Olfactory Function after the Elimination of Programmed Cell Death in Bax Knock-Out Mice. The Journal of Neuroscience. 2007 Dec 26;27(52):14392–14403. doi: 10.1523/JNEUROSCI.3903-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Snider WD, Jr, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996 Sep;17(3):401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 32.Ackerman SL, Kozak LP, Przyborski SA, Rund LA, Boyer BB, Knowles BB. The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature. 1997;386(6627):838–842. doi: 10.1038/386838a0. [DOI] [PubMed] [Google Scholar]
- 33.Guenebeaud C, Goldschneider D, Castets M, Guix C, Chazot G, Delloye-Bourgeois C, et al. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol Cell. 2010 Dec 22;40(6):863–876. doi: 10.1016/j.molcel.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 34.Llambi F, Lourenco FC, Gozuacik D, Guix C, Pays L, Del Rio G, et al. The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. The EMBO journal. 2005 Mar 23;24(6):1192–1201. doi: 10.1038/sj.emboj.7600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang H, Ozaki T, Shamim Hossain M, Nakamura Y, Kamijo T, Xue X, et al. A newly identified dependence receptor UNC5H4 is induced during DNA damage-mediated apoptosis and transcriptional target of tumor suppressor p53. Biochemical and Biophysical Research Communications. 2008 Jun 13;370(4):594–598. doi: 10.1016/j.bbrc.2008.03.152. [DOI] [PubMed] [Google Scholar]
- 36.Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ celldeath. Science. 1995 Oct 6;270(5233):96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 37.Matei V, Pauley S, Kaing S, Rowitch D, Beisel KW, Morris K, et al. Smaller inner ear sensory epithelia in Neurog1 null mice are related to earlier hair cell cycle exit. Developmental Dynamics. 2005;234(3):633–650. doi: 10.1002/dvdy.20551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003 Jan 1;130(1):15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]