

Abstract
Objective
Type I IFNs have emerged as potential activators of the IFN signature and elevated STAT1 expression in RA synovium, but mechanisms that induce synovial IFN expression are unknown. Recently, TNFα was shown to induce a delayed IFN response in macrophages. Thus, we tested whether TNFα, classically thought to activate inflammatory NF-κB target genes in RA, also contributes to the ‘IFN signature’ in RA synovial macrophages.
Methods
Synovial fluid macrophages purified from patients with rheumatoid arthritis (n=24) and spondyloarthopathies (SpA) (n=18) were lysed immediately after isolation or cultured ex vivo in the absence or presence of blockade of endogenous type I IFN or TNFα. Expression of IFN-inducible target genes was measured by qPCR and ELISA.
Results
Expression of an IFN signature and STAT1 in RA synovial macrophages was suppressed when type I IFNs or TNFα were blocked, whereas TNFα blockade did not affect expression of IFN response genes or STAT1 in SpA synovial macrophages. RA synovial fluid suppressed the IFN signature in RA synovial macrophages, and in TNFα-, IFNα- and IFNβ-stimulated control macrophages. Type I IFNs suppressed expression of IL-8 and MMP9 in RA synovial macrophages and in TNFα-stimulated control macrophages.
Conclusions
Our findings identify a new function for TNFα in RA synovitis by implicating TNFα as a major inducer of the RA synovial IFN response. The results suggest that the expression of IFN response genes in RA synovium is regulated by interplay between TNFα and opposing homeostatic factors expressed in the synovial microenvironment.
Keywords: rheumatoid arthritis, TNFα, type I interferon, STAT1
Introduction
Increased expression of IFNγ-inducible genes in rheumatoid arthritis (RA) synovial tissues and macrophages was an early finding of molecular studies of RA pathogenesis (1), and has been confirmed by gene expression profiling (2, 3). IFNγ-inducible genes enhance antigen presentation and inflammatory cytokine production, thus a synovial “IFNγ response” likely contributes to RA pathogenesis. Because IFNγ is expressed at vanishingly low concentrations in RA synovium (1), a conundrum in the field has been to identify the cytokines that drive expression of “IFN-γ-inducible genes” in RA synovitis. The discovery of the Jak-STAT signaling pathway clarified that most of the “IFNγ-inducible genes” expressed in RA synovium are targets of the transcription factor signal transducer and activator of transcription 1 (STAT1) (4). STAT1 protein expression and activation are increased in RA synovial cells compared to normal controls and STAT1 is predominantly localized to the intimal layer that contains activated cells (5-7). Elevated levels of STAT1 mRNA and STAT1 target genes were also found in a subgroup of RA synovial tissues and in RA synovial macrophages (2, 3). STAT1 is activated by, and mediates the effects of, several cytokines that are expressed in RA synovium, specifically IFNγ, type I IFNs, IL-6, IL-10 and IL-27 (8, 9). Thus, these cytokines are candidate activators of STAT1 in RA synovium. As RA synovial IFNγ expression is low, and RA synovial macrophage responses to IL-6, IL-10 and IL-27 are attenuated (3, 10-12), type I IFNs emerge as potential activators of synovial STAT1 in RA.
Type I IFNs are key inducers of an anti-viral response and activate predominantly STAT1 and STAT2. STAT1 and STAT2, together with IRF9, form the ISGF3 complex that induces expression of “IFN-response genes” such as IFIT1, MX1 and OAS (13). In addition, type I IFNs activate STAT1 homodimers that induce expression of many of the same genes as does IFN-γ (13). Thus, an “IFN signature” activated by type I IFNs will contain elements of the “IFNγ-STAT1” response discussed above and expression of ISGF3 target genes (which are more specific to type I IFNs). Genomic profiling has revealed that type I IFNs and ISGF3-dependent IFN-response genes are expressed in peripheral blood of a subset of RA patients (14). One study detected IFNβ protein in synovial tissue from RA patients (15), and increased expression of IFN response genes was observed in synovial fluid macrophages from patients with RA (3, 16). Additionally, there has been growing interest in exploring the relationship between the type I IFN signature in RA patients and their response to therapy. One study found that elevated baseline plasma levels of type I IFN activity were associated with a favorable response to TNF antagonist treatment (17). Another group found that a post-infliximab increase in the IFN response gene activity in PBMCs was associated with a poor clinical response (18). Additionally, a high pre-treatment IFN signature in PBMCs was a biomarker of a poor response to rituximab (19). A separate study showed that patients who develop a type I IFN signature post-treatment experience a better response to rituximab (20). Overall, these studies establish that expression of IFN-inducible genes is elevated in subsets of RA patients and can change with therapy, but their role in disease and in the response to therapy remains obscure.
Type I IFNs are pleiotropic and can contribute to or attenuate autoimmunity and inflammation in RA (21). Type I IFNs may contribute to disease pathogenesis by promoting autoimmunity and the production of chemokines, or they may be involved in homeostatic responses through the inhibition of tissue destruction, neo-angiogenesis, and inflammation (21). Based on these homeostatic functions, IFNβ has been approved for treatment of relapsing forms of multiple sclerosis (22) and several studies have revealed its therapeutic potential in uveitis, Behçet's disease, and animal models of arthritis and colitis (23-29). However, clinical trials of IFNβ-1a therapy in RA patients have not demonstrated significant improvement, although the lack of efficacy may be explained by inadequate dosing or duration of therapy (30, 31). The role of type I IFNs in RA synovium and the mechanisms responsible for their induction in arthritic joints are unclear.
TNFα is a pro-inflammatory cytokine that plays an important role during immune responses to pathogens and in chronic inflammatory diseases like RA (32). In RA, TNFα is chiefly produced by macrophages and induces the production of pro-inflammatory cytokines, chemokines, and tissue degrading enzymes. The importance of TNFα in RA pathogenesis was first suggested when Feldman and colleagues used ex vivo synovial cultures to demonstrate that addition of neutralizing TNFα antibodies to cultures of synovial membranes derived from RA patients reduced the production of pro-inflammatory cytokines including IL-1β, GM-CSF, and IL-6 (33, 34). Based on this finding they proposed that there is a TNFα-dependent cytokine cascade in RA(32). The role of TNFα in RA was confirmed through the effectiveness of anti-TNFα therapy in animal models of arthritis (35-38). Since then, five anti-TNFα biologics have been approved for the treatment of RA and this therapy is effective in about 70% of patients (39).
TNFα induces inflammatory gene expression through activation of mitogen-activated protein kinase (MAPK) and NF-κB signaling pathways and does not directly activate Jak-STAT signaling; thus, TNFα has not been considered as a potential activator of the synovial IFN signature. However, a recent study by Yarilina et al. has shown that longer term TNFα exposure initiates an IFNβ-mediated autocrine loop in blood-derived macrophages (16). Low amounts of IFNβ act in synergy with canonical TNFα-induced MAPK and NF-κB signals to induce the expression of inflammatory genes (including genes encoding chemokines such as CXCL9 and CXCL10) and also classical IFN-response genes, such as IFIT1, MX1 and OAS. We tested whether TNFα also could contribute to an IFN response in RA synovial macrophages (Mϕ), and thus to the ‘IFN signature’ observed in RA. To gain insight into the regulation of cytokine responses in vivo during human disease, we followed the ex vivo culture approach pioneered by Feldman and colleagues (32). We found that the STAT1/IFN-signature apparent in RA synovial fluid (SF) Mϕ increased on ex vivo culture and was dependent on autocrine TNFα, which activated type I IFN responses. Surprisingly, RA SFs suppressed the TNF-mediated IFN signature in RA synovial macrophages, and also suppressed induction of IFN responses by exogenous TNFα, IFNα and IFNβ in control macrophages. Our findings implicate TNFα as a major inducer of the RA synovial IFN response and suggest that the expression of IFN response genes in RA synovium is regulated by interplay between TNFα and opposing homeostatic factors expressed in the synovial microenvironment.
Materials and Methods
Patients
SF from active effusions was obtained (protocol approved by the Hospital for Special Surgery Institutional Review Board) from 24 patients with RA and 18 patients with spondyloarthopathies (SpA). Active effusion was defined as an acute non-infectious inflammatory SF accumulation attributed to a flare of RA or SpA that required arthrocentesis based on medical indications. The diagnosis of RA was based on the 1987 American College of Rheumatology criteria (40). SpA patients included individuals with psoriatic arthritis, ankylosing spondyloarthropathy, reactive arthritis, inflammatory bowel disease related arthritis, and undifferentiated SpA. There was limited information about patients’ medications and correlation of our findings with therapy was not possible.
Macrophage Purification
PBMCs from healthy volunteers and mononuclear cells from the SFs of RA and SpA patients were isolated as previously described (12).
Cell Culture
The CD14+ macrophages were cultured in RPMI 1640 medium (Invitrogen Life Technologies) supplemented with 10% FBS (HyClone). The following inhibitors, isotype controls and cytokines were added: etanercept (10 μg/ml) (AMGEN), hIgG1 (10 μg/ml) (R&D Systems), blocking Ab to IFN α/β Receptor Chain 2 (IFNAR2) (2 μg/ml) (PBL Interferon Source), mIgG2a (2 μg/ml) (R&D Systems), anti-hIFNα IgA (0.5μg/ml) (InvivoGen), hIgA2 (0.5μg/ml) (InvivoGen), LEAF™ Purified anti-hIFNβ (10μg/ml) (BioLegend) and mIgG1 (10μg/ml) (R&D Systems), hTNFα (10-40 ng/ml) (PeproTech), hIFNα (10-10,000 U/ml) (PBL Interferon Source) and hIFNβ (20pg/ml) (PeproTech). Cells were incubated in the presence or absence of SF from RA patients diluted 1:4 in media.
ELISA
SF from patients and culture supernatants from overnight SF macrophages were used. Paired capture and detection Abs to human soluble TNF Receptor 1 (Abcam) and CXCL10 (R&D Systems) were used.
Real-time quantitative RT-PCR (qPCR)
Total RNA was extracted using an RNeasy mini kit (Qiagen) with DNase treatment and 0.5 μg of total RNA was reverse transcribed using a First Strand cDNA Synthesis kit (Fermentas). qPCR was performed as previously described (8).
Results
IFN signature in RA SF Mϕ is dependent on type I IFNs
We reasoned that an ex vivo culture approach that had been successfully used to dissect the cytokine network in RA (33) could be adapted to study regulation of the IFN response in RA synovial macrophages. We isolated SF Mϕ from 11 RA and 12 SpA patients and prepared mRNA from freshly isolated cells, and from cells cultured overnight with or without blocking antibodies against IFNAR2, which blocks signaling by type I IFNs. Mouse IgG2a was used as isotype control. We measured the expression of classical IFN-inducible genes IFIT1, IFIT2, IRF7, OASp71 and MX1 (activated by ISGF3) and CXCL9 and CXCL10 (activated by STAT1 homodimers). We and others had previously shown that expression of these genes is higher in RA than in control macrophages (2, 3, 16). Strikingly, expression of these genes further increased on ex vivo culture of RA macrophages (Figure 1), consistent with endogenous production and autocrine action of a cytokine(s) that activates ISGF3 and/or STAT1. Accordingly, blockade of IFNα/β signaling resulted in nearly complete downregulation of expression of IFIT1, IFIT2, IRF7, OASp71, MX1, and CXCL10, indicating that expression of these genes is dependent on type I IFN in this system. Expression of CXCL9 was not affected by IFN blockade (data not shown); CXCL9 expression may be maintained by NF-κB or alternative mechanisms (41). Mϕ isolated from SFs of SpA patients were used as a disease control. In contrast to RA macrophages, induction of IFN response genes in SpA macrophages was minimal or absent (Fig. 1), although levels of gene expression were suppressed below baseline by IFN blockade. Overall, these results suggest that endogenous type I IFNs drive expression of several IFN response genes in RA synovial macrophages, and that this autocrine regulation is less prominent in SpA macrophages.
Figure 1. Endogenous type I IFNs promote an IFN signature in RA synovial fluid Mϕ.

CD14+ macrophages isolated from the synovial fluids of patients with RA (n=11) or SpA (n=12) were lysed immediately after purification or cultured overnight (12 hours) in the absence or presence of blocking IFNAR monoclonal antibodies (2 μg/ml) to block endogenous type I IFN. Mouse IgG2a was used as isotype control (not shown). Amounts of IFIT1 mRNA (A), MX1 mRNA (B), IFIT2, OASp71, IRF7 mRNA (C), and CXCL10 mRNA (D) were measured using real-time PCR and normalized relative to GAPDH mRNA. The Friedman test followed by post-hoc analysis with the Wilcoxon matched-pairs signed rank test was used for statistical analysis. (*=p<0.05, **=p<0.01, ***=p<0.001)
TNFα contributes to the IFN signature in RA SF Mϕ
TNFα can initiate an IFNβ-mediated autocrine loop that induces expression of IFN response genes (16) and RA synovial macrophages express low amounts of TNFα on ex vivo culture (42). Thus, we determined whether there is expression of endogenous TNFα that contributes to the observed IFN signature in RA SF Mϕ. Actually, TNFα mRNA was readily detected in RA SF Mϕ by qPCR; the average Ct value was 27, which represented a specific signal as the Ct value dropped to 33 when reverse transcriptase was omitted, indicating that RA SF Mϕ express TNF-α. SF macrophages from 18 RA and 13 SpA patients were cultured in the absence or presence of etanercept (10μg/ml) to block signaling by endogenous TNFα. Human IgG1 (10 μg/ml) was used as an isotype control. Strikingly, expression of genes whose expression was dependent on type I IFNs, IFIT1 and CXCL10, was downregulated by etanercept in RA Mϕ (p<0.05) (Figure 2A and C). A significant downregulation of CXCL10 expression by TNFα blockade was confirmed at the protein level using ELISA to measure CXCL10 in culture supernatants (p<0.01) (Figure 2C, right panel). In contrast to RA, the level of IFIT1 and MX1 expression was not changed significantly by TNFα blockade in SpA-derived Mϕ (Figure 2A-B). These results indicate that TNFα contributes to the IFN signature in RA SF Mϕ.
Figure 2. TNFα contributes to the IFN signature in RA synovial fluid Mϕ.

CD14+ Mϕ from SF of 18 patients with RA and 13 patients with SpA were cultured o/n in the presence of IgG1 or etanercept (10 mg/ml) (A-C). D, Mϕ from blood were cultured with TNFα in the presence or absence of anti-IFNα, anti-IFNβ or both. The expression of IFIT1 mRNA (A), MX1 mRNA (B) and CXCL10 mRNA (C, D) was measured using real-time PCR and normalized relative to GAPDH mRNA. The Friedman test followed by post-hoc analysis with the Wilcoxon matched-pairs signed rank test was used for statistical analysis. C, CXCL10 protein amounts in the culture supernatants of RA SF Mϕ were measured using ELISA. A Wilcoxon matched-pairs signed rank test was used for statistical analysis. (*=p<0.05, **=p<0.01, ***=p<0.001)
The contribution of IFNβ to the TNF-α-induced IFN signature in macrophages has been described recently (16). The potential role of IFNα was investigated by culturing in vitro generated human Mϕ from healthy donors’ peripheral blood with or without TNF-α (40ng/ml) in the presence or absence of anti-IFNα, anti-IFNβ and a combination of anti-IFNα and anti-IFNβ antibodies or isotype controls. Interestingly, the TNF-α-induced IFN signature was attenuated significantly by IFN-blocking antibodies used individually and was abrogated by the combination of the two (Figure 2D), suggesting that both IFNα and IFNβ contribute to the TNF-α-mediated IFN signature in human macrophages.
Type I IFN and TNFα contribute to STAT1 expression in RA SF Mϕ
One mechanism by which TNFα promotes an IFN signature is to increase expression of STAT1 and thereby amplify induction of downstream genes (6, 16). Thus, we analyzed the regulation of STAT1 expression in RA SF macrophages. STAT1 expression in RA synovial macrophages was strongly dependent on type I IFNs (Fig. 3A). STAT1 expression was also diminished after TNFα blockade in a statistically significant manner (Figure 3B, p<0.05), although inhibition of STAT1 expression by TNFα blockade was partial. We reasoned that heterogeneity among subsets of patients with RA may obscure differences in STAT1 expression when all patients were analyzed as one group. Thus, we further analyzed the effects of TNFα blockade on the expression level of STAT1 mRNA in SF-derived Mϕ from each individual RA patient. The addition of etanercept suppressed STAT1 expression in 14 out of 18 RA samples (78%) (p<0.001); STAT1 expression was not decreased by TNFα blockade only in 4 out of 18 (22%) Mϕ samples (Figure 3C). Thus, there is a subset of patients in whom STAT1 expression is not mediated by TNFα. In addition, neither IFN nor TNFα blockade diminished STAT1 expression in the disease control SpA macrophages (Fig. 3). These results suggest that TNFα induces an IFN signature in RA synovial macrophages at least in part by increasing STAT1 expression.
Figure 3. Type I IFNs and TNFα contribute to STAT1 expression in RA synovial fluid Mϕ.

CD14+ macrophages isolated from the synovial fluid of 18 patients with RA and 13 patients with SpA were lysed immediately after purification or cultured overnight (12 hours) in the presence or absence of isotype control or IFNAR antibodies (2 μg/ml) (A) or etanercept (10 μg/ml) (B). A-B, The expression of STAT1 mRNA was measured using real-time PCR and normalized relative to GAPDH expression. C, The RA patient population was divided into two categories depending on whether STAT1 induction was TNFα-dependent (n=14, p=0.0001) or TNFα-independent (n=4, p=0.125). The Friedman test followed by post-hoc analysis with the Wilcoxon matched-pairs signed rank test was used for statistical analysis. (*=p<0.05, **=p<0.01, ***=p<0.001)
Suppression of TNF-induced IFN response and STAT1 expression by RA SFs
The activation state of cells in the RA synovium reflects a balance between the opposing actions of inflammatory and homeostatic factors (9). To test whether RA SFs contain factors that could suppress expression of the IFN signature, we performed ex vivo cultures of arthritic macrophages in the presence of SF versus a media/FBS control. Strikingly, inclusion of SFs in the cultures suppressed expression of IFN response genes IFIT1 and MX1 and of STAT1 (Fig. 4). As expected, there was minimal development of an IFN response in cultures of SpA macrophages, and addition of SpA SFs did not downregulate expression of IFN response genes. These results suggest that the RA SF microenvironment attenuates the expression of a type I IFN response and that this effect is specific for RA.
Figure 4. RA synovial fluids suppress IFN responses in RA synovial fluid Mϕ.

CD14+ macrophages isolated from the synovial fluids of 9 patients with active RA and 8 patients with SpA were lysed immediately after purification or cultured overnight (12 hours) in media or in the same patient's synovial fluid (diluted 1:4 in media). The expression of IFIT1 mRNA (A), MX1 mRNA (B) and STAT1 mRNA (C) was measured using real-time PCR and normalized relative to GAPDH mRNA. The Friedman test followed by post-hoc analysis with the Wilcoxon matched-pairs signed rank test was used for statistical analysis. (*=p<0.05, **=p<0.01, ***=p<0.001)
We next tested whether the inhibitory effects observed with RA SF reflected an intrinsic feature of RA synovial macrophages, or whether RA SFs more broadly attenuate expression of an IFN response. To this end, we tested whether RA SFs could inhibit induction of an IFN signature by exogenous recombinant TNFα in control blood-derived macrophages from healthy donors. We cultured blood-derived macrophages overnight with or without TNFα stimulation, in the presence or absence of RA SFs. In agreement with a recent report by our group (16), TNFα induced an IFN response, as reflected by increased expression of IFIT1, MX1 and STAT1 (Figure 5A). Notably, the presence of RA SFs strongly inhibited TNFα-induced expression of IFN response genes by 91%, 95%, and 82% for IFIT1, CXCL10, and STAT1, respectively (Figure 5A, p<0.0001). Addition of SF alone did not affect baseline gene expression (Figure 5A). To test whether RA SFs globally or selectively affect TNFα responses, we tested the effects of RA SFs on TNFα-induced expression of inflammatory genes IL-8 and MMP9, which are IFN-independent TNFα-inducible genes. RA SFs only partially suppressed TNFα-induced expression of IL-8 and MMP9 by 60% and 55%, respectively (Figure 5B). Thus, RA SFs contain homeostatic factors that oppose the action of TNFα, but preferentially suppress TNFα-induced IFN response genes, while induction of inflammatory genes by TNFα is preserved. We tested whether one such factor could be soluble TNF receptors. Soluble TNF Receptor 1 levels in RA SFs were on average 1.7(±0.4) ng/ml, which is insufficient to block the action of TNFα that was added at 10 ng/ml.
Figure 5. RA SFs inhibit TNFα-induced expression of IFN response genes in blood-derived Mϕ.

CD14+ Mϕ isolated from the peripheral blood were cultured overnight with or without TNFα (10 ng/ml) (A-B), IFNα or IFNβ (20pg/ml) (C-D), in the presence or absence of RA SF. IFIT1 mRNA (A, C and D), CXCL10 mRNA and STAT1 mRNA (A), IL8 mRNA and MMP9 mRNA (B) were measured by real time PCR and normalized relative to GAPDH mRNA. Gene induction by TNFα, IFNα or IFNβ in the absence of SF was set at 100% and mRNA levels for the other conditions were expressed as % of this induction. The Friedman test followed by post-hoc analysis with the Wilcoxon matched-pairs signed rank test was used for analysis. (*=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001)
Next, we investigated whether RA SF attenuate IFN signature by directly inhibiting type I IFN function. To this end, we cultured blood-derived macrophages for 3h with or without exogenous IFNα or IFNβ stimulation (20pg/ml), in the presence or absence of RA SFs. As expected, both IFNα and IFNβ robustly induced target genes including IFIT1 and IFIT2. Notably, RA SFs strongly inhibited the IFNα- and IFNβ-mediated expression of these genes (Figure 5C-D and data not shown), suggesting that RA SF microenvironment suppresses type I IFN function/signaling.
Type I IFNs suppress inflammatory gene expression in macrophages
Type I IFNs can downregulate inflammatory gene expression in human blood-derived macrophages (43). We investigated the role of type I IFNs and the IFN signature in the regulation of inflammatory gene expression in synovial macrophages. We tested the effects of blocking IFN signaling on the expression of inflammatory genes during ex vivo culture of arthritic macrophages. In contrast to increased expression of IFN response genes (Fig. 1), IL-8 expression significantly decreased with overnight culture of RA macrophages (Figure 6A). Notably, IFN blockade resulted in a significant increase in IL-8 expression (Figure 6A), suggesting that IFNs suppress IL-8 expression in synovial macrophages. Additionally, IFN blockade augmented expression of MMP9 (Figure 6B). Accordingly, exogenous IFNα effectively suppressed TNFα-induced expression of IL-8 and MMP9 in blood-derived macrophages (Figure 6C). Together these results suggest that type I IFNs have homeostatic functions in the regulation of human Mϕ by attenuating pro-inflammatory and tissue destructive responses to TNFα.
Figure 6. IFNα modulates TNFα responses in human Mϕ.
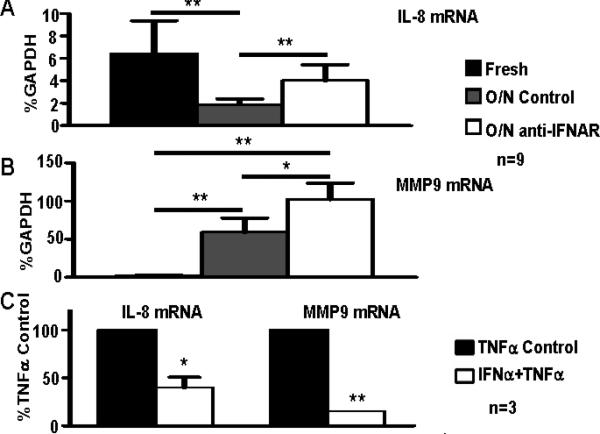
A-B, CD14+ macrophages isolated from the synovial fluids of 9 patients with RA were lysed immediately after purification or cultured overnight (12 hours) in the absence or presence of IFNAR antibodies (2 μg/ml); mouse IgG2a (2 μg/ml) was used as an isotype control. The expression of IL-8 mRNA and MMP9 mRNA was measured using real time PCR and normalized relative to GAPDH mRNA. The Friedman test followed by post-hoc analysis with the Wilcoxon matched-pairs signed rank test was used for statistical analysis. C, CD14+ cells isolated from the peripheral blood of 3 healthy donors were cultured in the presence or absence of IFNα (10,000 U/ml) for 48 hours and were then stimulated with TNFα (10 ng/ml) for 3 hours. The expression of IL8 mRNA and MMP9 mRNA was measured using real time PCR and normalized relative to GAPDH mRNA. Relative mRNA levels are expressed as a percentage of gene induction following TNFα stimulation alone, which was set at 100%. A paired Student's t test was used for statistical analysis. (*=p<0.05, **=p<0.01, ***=p<0.001)
Discussion
An “IFN signature” and elevated STAT1 in peripheral blood and synovial cells has been associated with RA, and can change after therapy with TNF blockers or rituximab (2, 3, 6, 7, 14-20). Mechanisms that regulate the IFN response in RA and its pathophysiological significance are unknown. This study identifies TNFα as a significant inducer of the IFN signature and STAT1 expression in synovial macrophages in a majority of RA patients. These results suggest that in addition to its role in the induction of pro-inflammatory and tissue destructive mediators, TNFα contributes to the IFN/STAT1 signature in RA synovium. This links TNFα to synovial Jak-STAT signaling and identifies a new function of TNFα in RA synovium that may have both pathogenic and protective aspects. Surprisingly, RA SFs preferentially suppressed TNFα-induced expression of IFN response genes and STAT1. Our data suggest that this suppressive effect results at least in part from direct inhibition of type I IFN function/signaling and the expression of an IFN response by RA synovial macrophages is dynamically regulated by opposing inflammatory and homeostatic factors during RA synovitis.
An IFN signature has been described in several autoimmune diseases, notably SLE, Sjogren's syndrome, and RA (14, 44, 45). This peripheral blood IFN signature is thought to reflect elevated systemic type I IFN levels. Although most cell types can produce type I IFNs, plasmacytoid dendritic cells (pDCs) that produce copious amounts of IFNγ have been proposed as major contributors to the peripheral IFN signature in SLE (46). In this model, pDCs are activated by nucleic acid-containing immune complexes that induce signaling by TLR7 and possibly TLR8 and TLR9, leading to IFNα production. Interestingly, pDC-mediated production of IFNα is suppressed by TNFα (47), which provides one explanation for why expression of IFN response genes in peripheral blood cells can rise after TNF blockade therapy. Our results provide an alternative non-mutually exclusive scenario describing regulation of the IFN signature in macrophages at the site of RA synovial inflammation. In this disease, anatomic location, and cell type, TNFα promotes IFNAR-mediated expression of an IFN signature. Thus, the driver of the RA synovial IFN response is an endogenous cytokine (TNFα) rather than a TLR ligand, and TNFα promotes rather than suppresses IFN responses. Our results predict that the synovial IFN signature will diminish, rather than increase, after TNF blockade therapy. These results highlight that differential regulation of the TNFα-IFN-STAT1 axis in various cell types and anatomic locations needs to be considered in order to understand regulation of IFN responses in human autoimmune diseases.
TNFα has been placed at the apex of the inflammatory cytokine network in RA synovium (32). As such, TNFα directly induces production of cytokines that are expressed in the synovium and can activate STAT1, such as IL-6 and IL-27 (8, 48), and may indirectly promote production of IL-10. Although a role for TNFα for inducing IFNβ in myeloid cells has recently emerged, TNFα induces low amounts of IFNβ, several orders of magnitude lower than TLRs (16). Although IFNβ is expressed in RA synovium (15), expression is likely not high relative to other STAT1-activating cytokines such as IL-6. Thus, the importance of the TNFα-IFN axis in inducing STAT1 expression and activating RA synovial macrophage IFN response and STAT1-target genes was not anticipated. One explanation for a dominant role for TNF-induced IFNs in synovial macrophage Jak-STAT signaling is that signaling by IL-10 and IL-27 is blocked in RA synovial macrophages (3, 10, 12), and IL-6 signaling in macrophages in an inflammatory environment is blocked by TNFα and IL-1 (49). In contrast, IFNα and/or miniscule amounts of IFNβ (on the order of 2-8 pg/ml) can synergize with conventional inflammatory TNFα signaling to induce IFN responses in macrophages (16). Thus, TNF-induced IFN emerges as an important inducer of STAT-mediated gene expression in RA synovial macrophages in many patients. However, there was a clear subset of patients in whom the IFN-STAT1 response was not dependent on TNFα. In these patients, STAT1 is presumably activated by cytokines whose expression is not dependent on TNFα, possibly T cell-derived cytokines such as IFN-γ. It will be interesting to determine if this subset of patients has a distinct response to TNF blockade therapy.
Homeostatic factors are expressed during synovitis as part of the patients’ attempt to control inflammation (9). Several of these factors, such as IL-10, soluble TNF Receptor 1 and PGEs can inhibit TNFα responses. These factors and other known inhibitors of TNFα signaling broadly suppress TNFα responses and TNFα-induced gene expression. The presence of such homeostatic factors has been documented in RA SFs and likely explains the partial attenuation of TNFα-induced inflammatory gene expression that we observed. In contrast to partial attenuation of inflammatory TNFα signaling, RA SFs nearly completely inhibited TNFα-mediated induction of IFN response genes in synovial or blood-derived macrophages. This suggests that RA SFs contain a factor(s) that either selectively blocks the production of type I IFNs by macrophages, or inhibits the type I IFN receptor or downstream signaling. Interestingly, our experiments suggest that SF factors can attenuate type I IFN function/signaling, and it will be important to identify such factors and their mechanism of action. SF factors that attenuate TNFα and IFN responses likely correspond to regulatory factors produced as part of termination of normal transient immune responses. In the chronic setting of RA synovitis such factors are unable to completely turn off TNFα-induced inflammatory signaling via NF-κB and related pathways and to abrogate TNFα-driven inflammation, and thus inflammation continues unabated (32). However, attenuation of the IFN-mediated component of TNFα responses in RA synovium appears to be more complete, based on our results and previous work showing only a modest IFN signature in RA synovium (2, 3). Whether such attenuation of IFN responses is beneficial or maladaptive depends upon whether type I IFNs have more of a pro- or anti-inflammatory effect on RA synovitis.
Type I IFNs have both pathogenic and homeostatic roles in autoimmune diseases depending on context. Pathogenic functions include promoting autoimmunity by augmenting humoral immunity and antibody production, activation of DCs, breakage of peripheral tolerance to self antigens, promotion of Th1 differentiation, induction of chemokines, and priming of cells for enhanced responses to subsequent stimulation with cytokines or inflammatory factors (21). These immune-activating functions likely contribute to a pathogenic role of type I IFNs in autoimmune diseases including lupus, dermatomyositis, Sjögren's syndrome, and scleroderma. Yet, type I IFNs can also inhibit tissue destruction and inflammation by inhibiting Th17 cells generation, increasing the expression of anti-inflammatory mediators such as IL-10 and IL-1RA, reducing the expression of pro-inflammatory mediators, suppressing angiogenesis, reducing tissue invasion by inflammatory cells, and suppressing osteoclastogenesis (21). The overall pathogenic versus protective role of type I IFNs in the pathogenesis of autoimmune diseases will be determined by the balance between its activating effects on autoimmunity versus its modulating effects on downstream inflammation, thus explaining the disease- and context-dependent effects of type I IFNs. Type I IFNs are protective in multiple sclerosis, but are likely pathogenic in SLE (21). It is likely that in RA type I IFNs exert both pathogenic effects by promoting autoimmunity and protective effects by attenuating inflammatory cytokine production and synoviocyte proliferation in inflamed joints. Our findings indicate that type I IFNs can suppress expression of certain inflammatory mediators in RA synovial macrophages and in TNFα-stimulated blood-derived macrophages, but additional work will be required to elucidate the full profile of IFN effects on disease-relevant synovial cell types. Studies with animal models suggest that type I IFNs are protective in inflammatory arthritis (23, 26-28), but clinical trials showed lack of efficacy in RA patients (30, 31). Longer term studies, perhaps in selected patient sub-populations or in combination with TNF blockade (where exogenously administered IFN would compensate for the predicted drop in synovial IFN response after TNF blockade) may be required to elucidate the role of type I IFNs in RA.
Acknowledgements
We thank the patients and physicians (Dalit Ashani, Anne Bass, Theodore Fields, Susan Goodman, Joseph Markenson and Sergio Schwartzman) of HSS for providing synovial fluids. We thank Xiaoyu Hu for helpful discussions and BaoHong Zhao for critical review of the manuscript.
This work was supported by grants from the NIH (L.B.I) and SLE Lupus Foundation (G.D.K).
Footnotes
Author contributions
R.A.G., G.G. and A.L. designed and performed experiments, interpreted data and wrote the manuscript. G.D.K. designed and supervised experiments, conceptualized the project, interpreted data and wrote the manuscript. L.B.I. conceptualized and oversaw the project and wrote the manuscript. G.D.K. had access to all primary data and is responsible for its integrity.
Conflict of interest
The authors declare no financial conflicts of interest.
References
- 1.Firestein GS, Alvaro-Gracia JM, Maki R. Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol. 1990;144(9):3347–53. [PubMed] [Google Scholar]
- 2.van der Pouw Kraan TC, van Gaalen FA, Kasperkovitz PV, Verbeet NL, Smeets TJ, Kraan MC, et al. Rheumatoid arthritis is a heterogeneous disease: evidence for differences in the activation of the STAT-1 pathway between rheumatoid tissues. Arthritis Rheum. 2003;48(8):2132–45. doi: 10.1002/art.11096. [DOI] [PubMed] [Google Scholar]
- 3.Antoniv TT, Ivashkiv LB. Dysregulation of interleukin-10-dependent gene expression in rheumatoid arthritis synovial macrophages. Arthritis Rheum. 2006;54(9):2711–21. doi: 10.1002/art.22055. [DOI] [PubMed] [Google Scholar]
- 4.Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 5.Kasperkovitz PV, Verbeet NL, Smeets TJ, van Rietschoten JG, Kraan MC, van der Pouw Kraan TC, et al. Activation of the STAT1 pathway in rheumatoid arthritis. Ann Rheum Dis. 2004;63(3):233–9. doi: 10.1136/ard.2003.013276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu X, Herrero C, Li WP, Antoniv TT, Falck-Pedersen E, Koch AE, et al. Sensitization of IFN-gamma Jak-STAT signaling during macrophage activation. Nat Immunol. 2002;3(9):859–66. doi: 10.1038/ni828. [DOI] [PubMed] [Google Scholar]
- 7.Walker JG, Ahern MJ, Coleman M, Weedon H, Papangelis V, Beroukas D, et al. Changes in synovial tissue Jak-STAT expression in rheumatoid arthritis in response to successful DMARD treatment. Ann Rheum Dis. 2006;65(12):1558–64. doi: 10.1136/ard.2005.050385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalliolias GD, Ivashkiv LB. IL-27 activates human monocytes via STAT1 and suppresses IL-10 production but the inflammatory functions of IL-27 are abrogated by TLRs and p38. J Immunol. 2008;180(9):6325–33. doi: 10.4049/jimmunol.180.9.6325. [DOI] [PubMed] [Google Scholar]
- 9.Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev. 2008;226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ji JD, Tassiulas I, Park-Min KH, Aydin A, Mecklenbrauker I, Tarakhovsky A, et al. Inhibition of interleukin 10 signaling after Fc receptor ligation and during rheumatoid arthritis. J Exp Med. 2003;197(11):1573–83. doi: 10.1084/jem.20021820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sengupta TK, Chen A, Zhong Z, Darnell JE, Jr., Ivashkiv LB. Activation of monocyte effector genes and STAT family transcription factors by inflammatory synovial fluid is independent of interferon gamma. J Exp Med. 1995;181(3):1015–25. doi: 10.1084/jem.181.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalliolias GD, Zhao B, Triantafyllopoulou A, Park-Min KH, Ivashkiv LB. Interleukin-27 inhibits human osteoclastogenesis by abrogating RANKL-mediated induction of nuclear factor of activated T cells c1 and suppressing proximal RANK signaling. Arthritis Rheum. 62(2):402–13. doi: 10.1002/art.27200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–64. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 14.van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, Voskuyl AE, Rustenburg F, Baggen JM, et al. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis. 2007;66(8):1008–14. doi: 10.1136/ard.2006.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Holten J, Smeets TJ, Blankert P, Tak PP. Expression of interferon beta in synovial tissue from patients with rheumatoid arthritis: comparison with patients with osteoarthritis and reactive arthritis. Ann Rheum Dis. 2005;64(12):1780–2. doi: 10.1136/ard.2005.040477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008;9(4):378–87. doi: 10.1038/ni1576. [DOI] [PubMed] [Google Scholar]
- 17.Mavragani CP, La DT, Stohl W, Crow MK. Association of the response to tumor necrosis factor antagonists with plasma type I interferon activity and interferon-beta/alpha ratios in rheumatoid arthritis patients: a post hoc analysis of a predominantly Hispanic cohort. Arthritis Rheum. 62(2):392–401. doi: 10.1002/art.27226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Baarsen LG, Wijbrandts CA, Rustenburg F, Cantaert T, van der Pouw Kraan TC, Baeten DL, et al. Regulation of IFN response gene activity during infliximab treatment in rheumatoid arthritis is associated with clinical response to treatment. Arthritis Res Ther. 12(1):R11. doi: 10.1186/ar2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thurlings RM, Boumans M, Tekstra J, van Roon JA, Vos K, van Westing DM, et al. Relationship between the type I interferon signature and the response to rituximab in rheumatoid arthritis patients. Arthritis Rheum. 62(12):3607–14. doi: 10.1002/art.27702. [DOI] [PubMed] [Google Scholar]
- 20.Vosslamber S, Raterman HG, van der Pouw Kraan TC, Schreurs MW, von Blomberg BM, Nurmohamed MT, et al. Pharmacological induction of interferon type I activity following treatment with rituximab determines clinical response in rheumatoid arthritis. Ann Rheum Dis. 70(6):1153–9. doi: 10.1136/ard.2010.147199. [DOI] [PubMed] [Google Scholar]
- 21.Kalliolias GD, Ivashkiv LB. Overview of the biology of type I interferons. Arthritis Res Ther. 12(Suppl 1):S1. doi: 10.1186/ar2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buttmann M, Rieckmann P. Interferon-beta1b in multiple sclerosis. Expert Rev Neurother. 2007;7(3):227–39. doi: 10.1586/14737175.7.3.227. [DOI] [PubMed] [Google Scholar]
- 23.Yarilina A, DiCarlo E, Ivashkiv LB. Suppression of the effector phase of inflammatory arthritis by double-stranded RNA is mediated by type I IFNs. J Immunol. 2007;178(4):2204–11. doi: 10.4049/jimmunol.178.4.2204. [DOI] [PubMed] [Google Scholar]
- 24.Mackensen F, Max R, Becker MD. Interferons and their potential in the treatment of ocular inflammation. Clin Ophthalmol. 2009;3:559–66. doi: 10.2147/opth.s3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotter I, Hamuryudan V, Ozturk ZE, Yazici H. Interferon therapy in rheumatic diseases: state-of-the-art 2010. Curr Opin Rheumatol. 22(3):278–83. doi: 10.1097/BOR.0b013e3283368099. [DOI] [PubMed] [Google Scholar]
- 26.Triantaphyllopoulos KA, Williams RO, Tailor H, Chernajovsky Y. Amelioration of collagen-induced arthritis and suppression of interferon-gamma, interleukin-12, and tumor necrosis factor alpha production by interferon-beta gene therapy. Arthritis Rheum. 1999;42(1):90–9. doi: 10.1002/1529-0131(199901)42:1<90::AID-ANR12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 27.van Holten J, Reedquist K, Sattonet-Roche P, Smeets TJ, Plater-Zyberk C, Vervoordeldonk MJ, et al. Treatment with recombinant interferon-beta reduces inflammation and slows cartilage destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res Ther. 2004;6(3):R239–49. doi: 10.1186/ar1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corr M, Boyle DL, Ronacher L, Flores N, Firestein GS. Synergistic benefit in inflammatory arthritis by targeting I kappaB kinase epsilon and interferon beta. Ann Rheum Dis. 2009;68(2):257–63. doi: 10.1136/ard.2008.095356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115(3):695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Genovese MC, Chakravarty EF, Krishnan E, Moreland LW. A randomized, controlled trial of interferon-beta-1a (Avonex(R)) in patients with rheumatoid arthritis: a pilot study [ISRCTN03626626]. Arthritis Res Ther. 2004;6(1):R73–R77. doi: 10.1186/ar1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Holten J, Pavelka K, Vencovsky J, Stahl H, Rozman B, Genovese M, et al. A multicentre, randomised, double blind, placebo controlled phase II study of subcutaneous interferon beta-1a in the treatment of patients with active rheumatoid arthritis. Ann Rheum Dis. 2005;64(1):64–9. doi: 10.1136/ard.2003.020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feldmann M. Translating molecular insights in autoimmunity into effective therapy. Annu Rev Immunol. 2009;27:1–27. doi: 10.1146/annurev-immunol-082708-100732. [DOI] [PubMed] [Google Scholar]
- 33.Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2(8657):244–7. doi: 10.1016/s0140-6736(89)90430-3. [DOI] [PubMed] [Google Scholar]
- 34.Haworth C, Brennan FM, Chantry D, Turner M, Maini RN, Feldmann M. Expression of granulocyte-macrophage colony-stimulating factor in rheumatoid arthritis: regulation by tumor necrosis factor-alpha. Eur J Immunol. 1991;21(10):2575–9. doi: 10.1002/eji.1830211039. [DOI] [PubMed] [Google Scholar]
- 35.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. Embo J. 1991;10(13):4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piguet PF, Grau GE, Vesin C, Loetscher H, Gentz R, Lesslauer W. Evolution of collagen arthritis in mice is arrested by treatment with anti-tumour necrosis factor (TNF) antibody or a recombinant soluble TNF receptor. Immunology. 1992;77(4):510–4. [PMC free article] [PubMed] [Google Scholar]
- 37.Thorbecke GJ, Shah R, Leu CH, Kuruvilla AP, Hardison AM, Palladino MA. Involvement of endogenous tumor necrosis factor alpha and transforming growth factor beta during induction of collagen type II arthritis in mice. Proc Natl Acad Sci U S A. 1992;89(16):7375–9. doi: 10.1073/pnas.89.16.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci U S A. 1992;89(20):9784–8. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smolen JS, Aletaha D, Koeller M, Weisman MH, Emery P. New therapies for treatment of rheumatoid arthritis. Lancet. 2007;370(9602):1861–74. doi: 10.1016/S0140-6736(07)60784-3. [DOI] [PubMed] [Google Scholar]
- 40.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 41.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122(5):707–21. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brennan FM, Chantry D, Jackson AM, Maini RN, Feldmann M. Cytokine production in culture by cells isolated from the synovial membrane. J Autoimmun. 1989;2(Suppl):177–86. doi: 10.1016/0896-8411(89)90129-7. [DOI] [PubMed] [Google Scholar]
- 43.Sharif MN, Sosic D, Rothlin CV, Kelly E, Lemke G, Olson EN, et al. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med. 2006;203(8):1891–901. doi: 10.1084/jem.20051725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mavragani CP, Crow MK. Activation of the type I interferon pathway in primary Sjogren's syndrome. J Autoimmun. 35(3):225–31. doi: 10.1016/j.jaut.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 45.Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003;36(8):481–90. doi: 10.1080/08916930310001625952. [DOI] [PubMed] [Google Scholar]
- 46.Ronnblom L, Eloranta ML, Alm GV. Role of natural interferon-alpha producing cells (plasmacytoid dendritic cells) in autoimmunity. Autoimmunity. 2003;36(8):463–72. doi: 10.1080/08916930310001602128. [DOI] [PubMed] [Google Scholar]
- 47.Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-alpha in autoimmune diseases. Proc Natl Acad Sci U S A. 2005;102(9):3372–7. doi: 10.1073/pnas.0408506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalliolias GD, Gordon RA, Ivashkiv LB. Suppression of TNF-alpha and IL-1 signaling identifies a mechanism of homeostatic regulation of macrophages by IL-27. J Immunol. 185(11):7047–56. doi: 10.4049/jimmunol.1001290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmed ST, Ivashkiv LB. Inhibition of IL-6 and IL-10 signaling and Stat activation by inflammatory and stress pathways. J Immunol. 2000;165(9):5227–37. doi: 10.4049/jimmunol.165.9.5227. [DOI] [PubMed] [Google Scholar]