

1. Disease Characteristics
1.1 Name of the disease (synonyms)
Centronuclear myopathy, myotubular myopathy.1
1.2 OMIM# of the disease
Myotubular myopathy, XLCNM, MTM, XLMTM, CNMX, X-linked recessive centronuclear myopathy, 310400
Centronuclear myopathy 1, CNM1, autosomal dominant, 160150
Centronuclear myopathy 2, CNM2, autosomal recessive, 255200
1.3 Name of the analyzed genes or DNA/chromosome segments
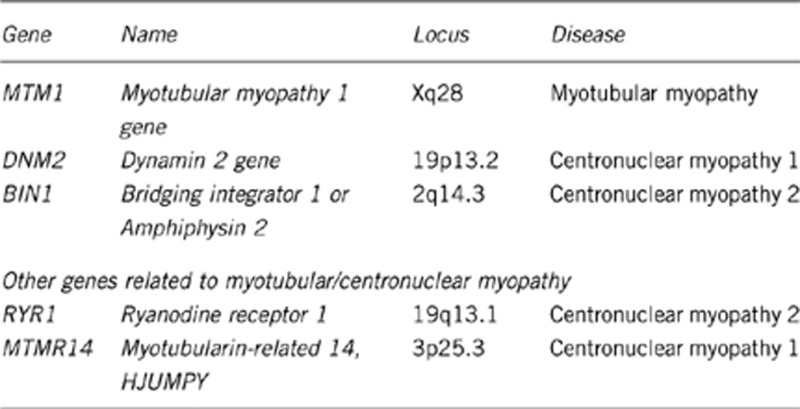
Contrary to what is indicated in OMIM, the reported mutation in MYF6 did not cause a recognized form of centronuclear myopathy.2
RYR1 mutations have been mainly implicated in CNM2 associated with autosomal-recessive inheritance.3, 4 Implication of heterozygous RYR1 mutations in dominant CNM awaits confirmation.
MTMR14/hJUMPY heterozygous inactivating variants have been reported in sporadic cases; whether they are disease-causing or acting as modifier of the phenotype remains to be determined.5
1.4 OMIM# of the gene(s)
MTM1, 300415
DNM2, 602378
BIN1, 601248
RYR1, 180901
MTMR14, 611089
1.5 Mutational spectrum
MTM1: Around 4006, 7, 8, 9, 10, 11, 12 mutations have been reported in all exons of the gene and include point mutations (missense, nonsense and splicing mutations), insertions and small and large deletions. The majority of mutations are clustered in exons 4, 8, 9, 11 and 12. The most common mutation identified is c.1261-10A>G, which has been demonstrated to affect normal splicing, and occurs in ∼7% of patients. Large deletions that affect one or more exons occur in ∼7% of patients. Large deletions including part or the entire upstream MAMLD1 gene cause a contiguous gene syndrome with hypospadias and myotubular myopathy in males.13 A duplication of exon 10 has been recently described in an affected male.14
DNM2: Thirteen heterozygous missense changes or in-frame deletions or insertions have been reported with hotspots at position 368, 369, 465, 522, 618 and 619.5, 15, 16, 17, 18, 19, 20 In the middle domain, the recurrent p.R465W mutation is most common, likely accounting for ∼25% of families, whereas p.E368K and p.R369W are found in ∼20% and ∼10% of families, respectively. The PH domain mutation p.R522H is found in roughly 10% of families, whereas mutations of residues 618 and 619 account for about 15% of families. A distinct subset of mutations cause autosomal-dominant Charcot–Marie–Tooth neuropathy (CMTDIB and CMT2M).21, 22, 23
BIN1: Five homozygous mutations have been reported in patients with CNM2 including three missense changes (p.K35N, p.D151N and p.R154Q) and two nonsense mutations (p.Q573X and K575X).24, 25, 26
RYR1: To date, RYR1-related CNM has been mainly associated with compound heterozygosity for recessive RYR1 mutations, typically a missense mutation and a mutation predicted to reduce the amount of the functional RyR1 protein.3, 4 The mutational spectrum includes missense mutations, frame-shifting small insertions and deletions, and splice-altering mutations. Three recurrent RYR1 mutations associated with CNM have been reported in the South African population due to multiple founder effects.3 Cases of RYR1-related CNM with a single heterozygous RYR1 missense, either of de novo occurrence27 or inherited from an asymptomatic parent,3 have been reported in a number of cases. These findings may reflect autosomal-dominant inheritance with variable penetrance or the presence of a mutation on the second allele not detected by routine sequencing.
MTMR14: Two inactivating heterozygous variants have been found in patients with sporadic CNM, including one with an additional DNM2 heterozygous mutation.5
Neonatal severe cases should be first tested for all exons of MTM1 and DNM2 exons 8 and 16. In female neonates, MTM1 mutation is associated with skewed X-inactivation or, less frequently, other X-chromosomal abnormalities. Childhood or adult onset is more likely associated with DNM2 and RYR1 mutations, respectively. In cases of clear recessive inheritance, BIN1 and RYR1 should be tested first.
1.6 Analytical methods
Bidirectional sequencing of coding regions of genomic DNA with flanking intronic sequences is currently the method of choice for screening of the MTM1 (NM_000252), DNM2 (isoform 1; NM_001005360) and BIN1 (isoform 8; NM_004305) genes. In complement of sequence analysis, a multiplex ligation-dependent probe amplification (MLPA) kit for detection of dosage anomalies of one or more exons of the MTM1 gene is available (SALSA MLPA kit P309-A1 MTM1, MRC, Holland). Some MTM1 mutations are detected only through RNA and protein analysis28 from cell lines (fibroblasts, lymphoblasts or myoblasts) or biopsies using RT-PCR and western blot, respectively. cDNA/protein analysis using RT-PCR or western blot is also a useful tool in RYR1-related CNM2 where the amount of functional RyR1 protein is often reduced.3 Because of its large size, RYR1 may be better tested by cDNA sequencing from a muscle biopsy. Next-generation sequencing may also be a cost-effective alternative in the near future in cases where RYR1 testing is contemplated.
1.7 Analytical validation
Mutation identification should be performed by de novo amplification and sequencing from the patient's DNA sample. Analytical validation is undertaken by comparison with database entries (mutations and polymorphisms) and published data. Special care is required for interpretation, as novel mutations and variants of unknown clinical significance are identified particularly in large genes such as RYR1. Testing of other affected/unaffected relatives in the family to see if the mutation segregates with the disease, and the study of ethnically matched controls to exclude polymorphisms, may be necessary. Confirmation of the pathogenicity of MTM1 and RYR1 mutations is possible using RT-PCR and/or western blot.3, 28, 29
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence)
The paediatric point prevalence (age <18 years) of centronuclear and myotubular myopathies has been estimated to be <1 in 100 000,30 distributed among MTM1 (45%), DNM2 (15%), RYR1 (10-15%) and BIN1 (<5%). The remaining ∼20% of cases likely have mutations in yet to be identified genes.
1.9 If applicable, prevalence in the ethnic group of investigated person
The various forms of CNM have been found at roughly equal frequencies in all the ethnic groups studied.
1.10 Diagnostic setting

Comment: Mutation testing is used mainly to confirm a suspected clinical diagnosis. A list of laboratories that perform testing can be found on the Orphanet web site (http://www.orpha.net) and GeneTests (www.genetests.org/). Prenatal diagnosis is available for families with known mutations, depending on severity and country regulations. Prenatal diagnosis should only be considered in families where pathogenicity for mutations has been clearly established, and is especially challenging for RYR1 as this gene harbours a large number of variations with uncertain significance.
Preimplantation genetic diagnosis may be offered to families with the same molecular prerequisite as for prenatal diagnosis, depending on the regulatory environment in their country.
Predictive diagnosis for CNM is available for adults from families with known mutations and a risk of late-onset disease. In the context of predictive testing, specific pre-testing genetic counselling should be proposed according to the internationally accepted guidelines. Testing for RYR1 mutations does not only test for CNM and other RYR1-related myopathies but also may be predictive for the malignant hyperthermia susceptibility (MHS) trait, an allelic but not consistently associated complication of RYR1 mutations. The index case and relatives found to harbour RYR1 mutations can be advised directly about their MHS risk if the mutation identified has been previously documented to be MHS-associated or can be referred for appropriate testing.
2. Test Characteristics
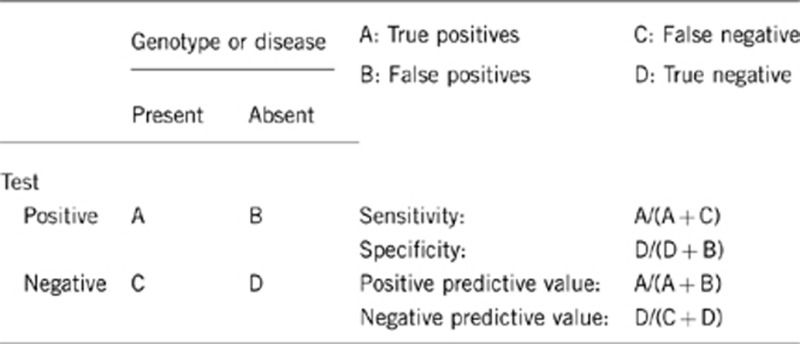
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
The analytical sensitivity depends on the analytical method used. The analytical sensitivity should be indicated in the laboratory report.
The sensitivity for DNA sequencing of genomic PCR products is around 98–100% for small mutations within exons and splice sites, but rare false negatives can be induced by rare polymorphisms in primer sequences causing allele dropout.
Deletions of whole exons or whole genes are not usually detected by sequencing of autosomal genes and mutations in introns, and promoters and enhancers are missed by methods focusing on coding exons. For MTM1 studies, the sensitivity is higher than that for the other genes because deletions of whole exons or the entire gene is easily detected by absence of amplification in males and by MLPA analysis in males and females. An intragenic duplication in the MTM1 gene has been described in a male and has been detected by MLPA analysis. RNA and protein studies may allow detection of mutations outside exons. MLPA testing is also available for RYR1 for a smaller subset of exons.
Thus, a negative result using current methods does not entirely exclude the diagnosis, if clinical features are present. This may depend either on analytical sensitivity or heterogeneity of the phenotype (clinical sensitivity).
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Sequence analysis: ∼100%
MLPA analysis: 98–100%. What appears to be single-exon deletions may be due to a SNP in a primer-binding site and need to be confirmed by sequencing or other quantification methods.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity is around 80% if all known implicated genes are tested. Mutation frequency for the known genes has been provided in section 1.8. Genetic testing is rarely carried out for all known genes; this limitation might be overcome with the introduction of next-generation sequencing.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The proportion is nearly 100%, but no data are available for this measure.
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
The possibility is close to 100% for MTM1 mutations. However, clinical heterogeneity has been reported and incomplete penetrance cannot be excluded for mutations associated with late onset like specific DNM2 mutations. Inter- and intrafamilial clinical variability has to be taken into account in RYR1-related CNM where additional sequence variations running independently in the family may modify the phenotype. Female carriers for a MTM1 mutation do not usually develop overt symptoms31 in the absence of skewed X-inactivation (which can be different comparing muscle and other tissues such as blood) or other X-chromosomal abnormalities, but the true prevalence of subtle features is not well known. For MTMR14, incomplete penetrance or mutations acting as modifier alleles have been suggested.5
In addition, specific DNM2 mutations are found in patients with Charcot–Marie–Tooth neuropathy who do not develop centronuclear myopathies (CMTDIB and CMT2M23).
2.6 Negative clinical predictive value
(probability of not developing the disease if the test is negative)
Index case in that family had been tested:
The negative clinical predictive value is likely to be near 100%.
Index case in that family had not been tested:
Not applicable as other yet undiscovered genes could cause the disease.
3. Clinical Utility
Clinical utility refers to the ability of genetic test results, either positive or negative, to provide information that is of value in the clinical setting.32
3.1 (Differential) diagnosis: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

CNM is defined on the basis of clinical and, most importantly, histopathological assessments. To date, a comprehensive assessment comprising detailed evaluation of clinical, histopathological and, whenever possible, muscle MRI features should be performed prior to genetic testing and therefore represents a prerequisite to genetic testing rather than a diagnostic alternative.16, 33, 34 The diagnosis should then be confirmed at the genetic level by the identification of the mutation. In the future, next-generation sequencing might be proposed before the completion of the detailed clinical and histopathological studies. A genetic diagnosis is important for counselling and recurrence risk purposes.
Biochemistry:
At least 95% of patients with MTM1 mutations show a decreased level of myotubularin by western blot analysis.28, 29 For MTM1, this test can be performed from muscle and non-muscle cells as lymphoblasts or fibroblasts. RYR1 mutations may or may not lead to a detectable decrease in the protein level in muscle sample.
Histology:
Centronuclear myopathies are characteristically defined by the presence of central nuclei placed in rows in the muscle fibres, but there often exist a number of fibres with one or more internalized nuclei. The central nuclei are only one of the morphological features, which characterize centronuclear myopathies, thus, there are a number of structural abnormalities of the muscle fibres that could provide guidance about the type of CNM.34 For instance, a triad of morphological abnormalities is usually found in DNM2-related CNM: increased numbers of central and internal nuclei, radiating sarcoplasmic strands with oxidative enzyme reactions (so called ‘spokes on a wheel') and type 1 muscle fibre predominance and hypotrophy.15, 35, 36 In MTM1-related CNM, an increased number of centralized nuclei and type 1 muscle fibre predominance and hypotrophy are reported and may be accompanied by the presence of necklace fibers, at least in adult cases.37 To get the best of the morphological study of CNM, one should analyze the muscle biopsies with both histoenzymology and electron microscopy. Indeed, at the ultrastructural level, there is a significant difference between the group of DNM2-, BIN1- and MTM1-CNM and RYR1-related myopathies, because despite of the ultrastructural particularities found in this first group of diseases, the sarcomeres appear preserved with a normal sequential structure, whereas in the RYR1-related myopathies, wide areas of sarcomeric disorganization with loss of the regular sarcomeric sequence are constantly observed.4, 34
3.1.2 Describe the burden of alternative diagnostic methods to the patient
MRI, EMG and DNA sampling are acceptable.
The first suggestion of any primary muscle disease, including centronuclear myopathy, necessitates histological analysis of a muscle biopsy that is an invasive sampling.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Unknown. The future application of next-generation sequencing may position the genetic approach before histological confirmation.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe)
Medical risk such as MH should be considered in patients with RYR1 mutation.
Discussion about recurrence risk for future pregnancies should be offered according to the mode of transmission of the mutation.
If the test result is negative (please describe)
Tests of other disease-causing genes causing similar phenotypes should be pursued.
3.2.2 Which options in view of lifestyle and prevention does a person at risk have if no genetic test has been done (please describe)?
Specialist follow-up is indispensable.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, in all cases where proven pathogenic mutations validate the segregation of the disease.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Family members can be screened for the mutations in the index patient, and mutation carriers can be spared the need for muscle biopsy and other diagnostic procedures.
Depending on the mode of inheritance, family members may also be found not to be at risk and thus spared any need for testing.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes; depending on severity and country regulations. See paragraph 1.10.
4. If applicable, further consequences of testing
Yes, for the patient and the family. A molecular confirmation of the diagnosis will limit unnecessary further diagnostic investigations, which can be invasive (and expensive). Patients and family may find encouragement and support by becoming members of associations that welcome affected families. A molecular diagnosis enables carriers to make informed reproductive decisions.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant agreement number 261469), the European Society of Human Genetics, the Institut National de la Santé et de la Recherche Médicale (INSERM), the Centre National de la Recherche Scientifique (CNRS), University of Strasbourg (UdS), Collège de France, Agence Nationale de la Recherche, Association Française contre les Myopathies (AFM), the Muscular Dystrophy Association (USA), the Myotubular Trust (UK). AHB was supported by NIH R01 AR044345, the Muscular Dystrophy Association (USA) 201302, the Lee and Penny Anderson Family Foundation and the Joshua Frase Foundation. HJ was supported by a grant from the Guy's and St Thomas' Charitable Foundation (Grant number 070404).
The authors declare no conflict of interest.
References
- Jungbluth H, Wallgren-Pettersson C, Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. 2008;3:26. doi: 10.1186/1750-1172-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerst B, Mennerich D, Schuelke M, et al. Heterozygous myogenic factor 6 mutation associated with myopathy and severe course of Becker muscular dystrophy. Neuromuscul Disord. 2000;10:572–577. doi: 10.1016/s0960-8966(00)00150-4. [DOI] [PubMed] [Google Scholar]
- Wilmshurst JM, Lillis S, Zhou H, et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol. 2010;68:717–726. doi: 10.1002/ana.22119. [DOI] [PubMed] [Google Scholar]
- Bevilacqua JA, Monnier N, Bitoun M, et al. Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol Appl Neurobiol. 2011;37:271–284. doi: 10.1111/j.1365-2990.2010.01149.x. [DOI] [PubMed] [Google Scholar]
- Tosch V, Rohde HM, Tronchere H, et al. A novel PtdIns3P and PtdIns(3,5)P2 phosphatase with an inactivating variant in centronuclear myopathy. Hum Mol Genet. 2006;15:3098–3106. doi: 10.1093/hmg/ddl250. [DOI] [PubMed] [Google Scholar]
- Laporte J, Guiraud-Chaumeil C, Vincent MC, et al. Mutations in the MTM1 gene implicated in X-linked myotubular myopathy. ENMC International Consortium on Myotubular Myopathy. European Neuro-Muscular Center. Hum Mol Genet. 1997;6:1505–1511. doi: 10.1093/hmg/6.9.1505. [DOI] [PubMed] [Google Scholar]
- de Gouyon BM, Zhao W, Laporte J, Mandel JL, Metzenberg A, Herman GE. Characterization of mutations in the myotubularin gene in twenty six patients with X-linked myotubular myopathy. Hum Mol Genet. 1997;6:1499–1504. doi: 10.1093/hmg/6.9.1499. [DOI] [PubMed] [Google Scholar]
- Tanner SM, Schneider V, Thomas NS, Clarke A, Lazarou L, Liechti-Gallati S. Characterization of 34 novel and six known MTM1 gene mutations in 47 unrelated X-linked myotubular myopathy patients. Neuromuscul Disord. 1999;9:41–49. doi: 10.1016/s0960-8966(98)00090-x. [DOI] [PubMed] [Google Scholar]
- Laporte J, Biancalana V, Tanner SM, et al. MTM1 mutations in X-linked myotubular myopathy. Hum Mutat. 2000;15:393–409. doi: 10.1002/(SICI)1098-1004(200005)15:5<393::AID-HUMU1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Herman GE, Kopacz K, Zhao W, Mills PL, Metzenberg A, Das S. Characterization of mutations in fifty North American patients with X-linked myotubular myopathy. Hum Mutat. 2002;19:114–121. doi: 10.1002/humu.10033. [DOI] [PubMed] [Google Scholar]
- Biancalana V, Caron O, Gallati S, et al. Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet. 2003;112:135–142. doi: 10.1007/s00439-002-0869-1. [DOI] [PubMed] [Google Scholar]
- Tsai TC, Horinouchi H, Noguchi S, et al. Characterization of MTM1 mutations in 31 Japanese families with myotubular myopathy, including a patient carrying 240kb deletion in Xq28 without male hypogenitalism. Neuromuscul Disord. 2005;15:245–252. doi: 10.1016/j.nmd.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Hu LJ, Laporte J, Kress W, et al. Deletions in Xq28 in two boys with myotubular myopathy and abnormal genital development define a new contiguous gene syndrome in a 430 kb region. Hum Mol Genet. 1996;5:139–143. doi: 10.1093/hmg/5.1.139. [DOI] [PubMed] [Google Scholar]
- Trump N, Cullup T, Verheij JB, et al. X-linked myotubular myopathy due to a complex rearrangement involving a duplication of MTM1 exon 10. Neuromuscul Disord. 2011;22:384–388. doi: 10.1016/j.nmd.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Bitoun M, Maugenre S, Jeannet PY, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 2005;37:1207–1209. doi: 10.1038/ng1657. [DOI] [PubMed] [Google Scholar]
- Schessl J, Medne L, Hu Y, et al. MRI in DNM2-related centronuclear myopathy: evidence for highly selective muscle involvement. Neuromuscul Disord. 2007;17:28–32. doi: 10.1016/j.nmd.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Echaniz-Laguna A, Nicot AS, Carre S, et al. Subtle central and peripheral nervous system abnormalities in a family with centronuclear myopathy and a novel dynamin 2 gene mutation. Neuromuscul Disord. 2007;17:955–959. doi: 10.1016/j.nmd.2007.06.467. [DOI] [PubMed] [Google Scholar]
- Bitoun M, Bevilacqua JA, Prudhon B, et al. Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann Neurol. 2007;62:666–670. doi: 10.1002/ana.21235. [DOI] [PubMed] [Google Scholar]
- Susman RD, Quijano-Roy S, Yang N, et al. Expanding the clinical, pathological and MRI phenotype of DNM2-related centronuclear myopathy. Neuromuscul. 2010;20:229–237. doi: 10.1016/j.nmd.2010.02.016. [DOI] [PubMed] [Google Scholar]
- Bohm J, Biancalana V, Dechene ET, et al. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum Mutat. 2012;33:949–959. doi: 10.1002/humu.22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Noureddine M, Kennerson M, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005;37:289–294. doi: 10.1038/ng1514. [DOI] [PubMed] [Google Scholar]
- Fabrizi GM, Ferrarini M, Cavallaro T, et al. Two novel mutations in dynamin-2 cause axonal Charcot-Marie-Tooth disease. Neurology. 2007;69:291–295. doi: 10.1212/01.wnl.0000265820.51075.61. [DOI] [PubMed] [Google Scholar]
- Bitoun M, Stojkovic T, Prudhon B, et al. A novel mutation in the dynamin 2 gene in a Charcot-Marie-Tooth type 2 patient: clinical and pathological findings. Neuromuscul Disord. 2008;18:334–338. doi: 10.1016/j.nmd.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Nicot AS, Toussaint A, Tosch V, et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39:1134–1139. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- Claeys KG, Maisonobe T, Bohm J, et al. Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology. 2010;74:519–521. doi: 10.1212/WNL.0b013e3181cef7f9. [DOI] [PubMed] [Google Scholar]
- Bohm J, Yis U, Ortac R, et al. Case report of intrafamilial variability in autosomal recessive centronuclear myopathy associated to a novel BIN1 stop mutation. Orphanet. 2010;5:35. doi: 10.1186/1750-1172-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth H, Zhou H, Sewry CA, et al. Centronuclear myopathy due to a de novo dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord. 2007;17:338–345. doi: 10.1016/j.nmd.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Tosch V, Vasli N, Kretz C, et al. Puissant H, Romero NB, Bonnemann CG, Heller B, Duval G, Biancalana V, Laporte J: Novel molecular diagnostic approaches for X-linked centronuclear (myotubular) myopathy reveal intronic mutations. Neuromuscul disord. 2010;20:375–381. doi: 10.1016/j.nmd.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Laporte J, Kress W, Mandel JL. Diagnosis of X-linked myotubular myopathy by detection of myotubularin. Ann Neurol. 2001;50:42–46. doi: 10.1002/ana.1033. [DOI] [PubMed] [Google Scholar]
- Amburgey K, McNamara N, Bennett LR, McCormick ME, Acsadi G, Dowling JJ. Prevalence of congenital myopathies in a representative pediatric united states population. Ann Neurol. 2011;70:662–665. doi: 10.1002/ana.22510. [DOI] [PubMed] [Google Scholar]
- Kristiansen M, Knudsen GP, Tanner SM, et al. X-inactivation patterns in carriers of X-linked myotubular myopathy. Neuromuscul Disord. 2003;13:468–471. doi: 10.1016/s0960-8966(03)00067-1. [DOI] [PubMed] [Google Scholar]
- Javaher P, Kaariainen H, Kristoffersson U, et al. based testing for heritable disorders in Europe. Community Genet. 2008;11:75–120. doi: 10.1159/000111984. [DOI] [PubMed] [Google Scholar]
- Fischer D, Herasse M, Bitoun M, et al. Characterization of the muscle involvement in dynamin 2-related centronuclear myopathy. Brain. 2006;129:1463–1469. doi: 10.1093/brain/awl071. [DOI] [PubMed] [Google Scholar]
- Romero NB. Centronuclear myopathies: a widening concept. Neuromuscul. 2010;20:223–228. doi: 10.1016/j.nmd.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Jeub M, Bitoun M, Guicheney P, et al. Dynamin 2-related centronuclear myopathy: clinical, histological and genetic aspects of further patients and review of the literature. Clin Neuropathol. 2008;27:430–438. doi: 10.5414/npp27430. [DOI] [PubMed] [Google Scholar]
- Bitoun M, Durieux AC, Prudhon B, et al. Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum Mutat. 2009;7:7. doi: 10.1002/humu.21086. [DOI] [PubMed] [Google Scholar]
- Bevilacqua JA, Bitoun M, Biancalana V, et al. ‘Necklace' fibers, a new histological marker of late-onset MTM1-related centronuclear myopathy. Acta Neuropathol. 2008;16:16. doi: 10.1007/s00401-008-0472-1. [DOI] [PubMed] [Google Scholar]
- Melberg A, Kretz C, Kalimo H, et al. Adult course in dynamin 2 dominant centronuclear myopathy with neonatal onset. Neuromuscul Disord. 2009;20:53–56. doi: 10.1016/j.nmd.2009.10.006. [DOI] [PubMed] [Google Scholar]