

Abstract
Peroxisome-proliferator-activated receptors (PPARs) are ligand-activated nuclear receptors that exert in the liver a transcriptional activity regulating a whole spectrum of physiological functions, including cholesterol and bile acid homeostasis, lipid/glucose metabolism, inflammatory responses, regenerative mechanisms, and cell differentiation/proliferation. Dysregulations of the expression, or activity, of specific PPAR isoforms in the liver are therefore believed to represent critical mechanisms contributing to the development of hepatic metabolic diseases, disorders induced by hepatic viral infections, and hepatocellular adenoma and carcinoma. In this regard, specific PPAR agonists have proven to be useful to treat these metabolic diseases, but for cancer therapies, the use of PPAR agonists is still debated. Interestingly, in addition to previously described mechanisms regulating PPARs expression and activity, microRNAs are emerging as new important regulators of PPAR expression and activity in pathophysiological conditions and therefore may represent future therapeutic targets to treat hepatic metabolic disorders and cancers. Here, we reviewed the current knowledge about the general roles of the different PPAR isoforms in common chronic metabolic and infectious liver diseases, as well as in the development of hepatic cancers. Recent works highlighting the regulation of PPARs by microRNAs in both physiological and pathological situations with a focus on the liver are also discussed.
1. Introduction
Obesity, the metabolic syndrome (MetS), diabetes, hepatitis viruses (HBV/HCV), and abusive alcohol consumption are currently the principal etiological factors favoring the occurrence of hepatocellular adenoma and carcinoma worldwide [1–8]. With obesity, MetS, and diabetes, the liver can develop a wide spectrum of disorders, occurring in individuals in absence of excessive alcohol consumption, or HBV/HCV infections, and ranging from insulin resistance (IR), nonalcoholic fatty liver diseases (NAFLD, including steatosis and insulin resistance), to nonalcoholic steatohepatitis (NASH, inflammation, and fibrosis associated with steatosis), and to cirrhosis (characterized by replacement of liver tissue by extracellular matrix and regenerative nodules) [9, 10]. Hepatocellular adenoma (HCA) and carcinoma (HCC) can then occur as the ultimate stage of these diseases [11–13]. Since obesity and metabolic diseases have reached pandemic proportions worldwide, the incidence of IR/NAFLD/NASH/cirrhosis and HCA/HCC is expected to dramatically increase in the future, likely becoming the most common hepatic diseases worldwide [1, 6]. With hepatitis viruses (HBV/HCV) and abusive alcohol consumption, a very similar spectrum of histological abnormalities is observed in the liver (ranging from IR to hepatic steatosis, steatohepatitis, fibrosis, and cirrhosis) and also precedes HCA/HCC development. However, the molecular mechanisms triggering IR, steatosis, inflammation, fibrosis and cirrhosis are significantly different depending on the etiologies of these hepatic diseases.
Hepatic steatosis consists in the excessive accumulation of neutral lipids (mainly triglycerides and cholesterol esters) in cytoplasmic lipid droplets of the hepatocytes. This abnormal accumulation of cytoplasmic lipid droplets can result from (i) an excessive importation of free fatty acids (FFAs) released by adipocytes, or coming from the diet; (ii) from a diminished hepatic export of FFA (altered synthesis or secretion of VLDL); (iii) an increased de novo lipogenesis; (iv) an impaired β-oxidation of FFA [14, 15]. Steatosis and hepatic IR are closely associated but it is still poorly understood whether it is steatosis, which causes IR, or vice versa. It is clear however that steatosis and IR usually precede inflammation, fibrosis, and cirrhosis of the liver. Recent evidence indicates that progression toward inflammation, fibrosis, and cirrhosis is likely due to the appearance of additional multiple dysregulated mechanisms, including the production of lipotoxic intermediates, oxidative stress (e.g., through alterations of the mitochondrial activity or lipid peroxidation), mitochondrial/peroxisomal abnormality, altered nuclear receptors signaling, deregulated cytokines signalling, gut microbial signalling, hepatocyte apoptosis, and leptin resistance, all of them contributing to various extent to the progression towards inflammation and fibrosis/cirrhosis [14–18]. These are thus multifactorial diseases, for which the precise orchestration of their development and progression still remains unclear. In addition, since not all patients with steatosis develop substantial liver injuries, this varying susceptibility of individuals to progress towards inflammation/fibrosis and cirrhosis further indicates that these disorders involve multifaceted processes also highly dependent on environmental factors and genetic predisposition [16–18].
Steatosis, inflammation, fibrosis, and cirrhosis can also be regarded as preneoplastic states of the liver, since HCA/HCC might occur as an end stage of these diseases [13]. HCC is a cancer with a very poor prognosis and its clinical management has been the object of intensive research efforts. Surgical resection of the tumour and liver transplantation are currently the only treatments with curative potential. However, only few patients are eligible for surgery and critical issues are associated with tumour resection and the efficiency of hepatic regeneration. Recent evidence indicates that physical inactivity and fat-enriched diets, both becoming major health emergencies in our western society, have a significant impact not only on HCC occurrence and progression, but also on the liver regeneration process. Based on these findings, a major challenge today is to understand how specific nutrients, in particular fats, and an abnormal hepatic lipid metabolism, affect major signalling pathways promoting carcinogenesis.
Peroxisome-proliferator-activated receptors (PPARs) are ligand-activated nuclear receptors that exert a transcriptional activity regulating energy homeostasis and other basic cellular processes in multiple metabolically active organs. These receptors are classically ligand activated, with the best characterized ligands being fatty acids and their derivatives. Three major isoforms of PPARs (PPARα, PPARβ/δ, and PPARγ) have been characterized to date [19]. Specific isoforms of these PPARs are expressed in most of the highly metabolically active tissues such as skeletal muscles, heart, adipose tissue, and liver. In the liver specifically, PPARs regulate a whole spectrum of physiological functions including cholesterol and bile acid homeostasis, lipid and glucose metabolism, inflammatory responses, regenerative mechanisms, cell differentiation, and cell cycle [20, 21]. Dysregulations of the expression, or activity, of specific PPAR isoforms are now also well accepted to represent critical mechanisms contributing to the development of a wide range of liver diseases. Indeed, PPARs have been recently implicated in the development of widely spread diseases affecting the liver such as IR, NAFLD/NASH, alcoholic liver diseases (ALD), HBV/HCV infection, and HCA/HCC.
2. Role of Specific PPAR Isoforms in Liver Diseases
2.1. PPARs in Hepatic Insulin Resistance and Steatosis
The role of specific PPARs isoforms in the occurrence and development of hepatic IR and steatosis is still controversial, as well as the potential benefits of specific PPARs agonists to treat these diseases [22]. It is however well accepted that PPARs are tightly implicated in processes regulating the accumulation and storage of triglycerides, lipid oxidation, and insulin sensitivity of hepatocytes [23].
2.1.1. PPARγ
PPARγ is generally increased in steatotic livers of both animal models of obesity and human obese patients [24, 25]. In vivo studies demonstrated that hepatocytes- and macrophage-specific PPARγ knockout mice were protected against high-fat (HF) diet-induced steatosis development [26]. As well, knockdown of PPARγ using in vivo injection of adenoviruses could also improve fatty liver and inflammation in mice fed a high saturated fat diet [27]. However, in vitro or in vivo studies with PPARγ agonists have led to contradictory results regarding steatosis and IR development. For example, thiazolidinediones induce steatosis in human primary hepatocytes and hepatoma cells [28], but reduce in vivo hepatic steatosis potentially through upregulation of adiponectin production and adiponectin receptors expression in the liver and adipose tissue [29–32]. On the other hand, SKLB102 (a barbituric acid-based derivative PPARγ agonist) was shown to activate lipogenesis and to exacerbate steatosis in mice fed a high-fat/high-calorie diet, although this agonist appears to improve the general outcome of NAFLD/NASH, likely by stimulating insulin sensitivity in both mice or hepatoma cells [30]. In human several clinical studies have reported that thiazolidinediones (rosiglitazone and pioglitazone) lead to a decreased hepatic steatosis and a regression of lobular inflammation [33–35]. It is worthy to note that in human, genetic variants of PPARγ (C161T, Pro12Ala) or dominant-negative mutations are specifically associated with NAFLD and progression of NAFLD towards inflammation and fibrosis [36–41].
2.1.2. PPARα
As opposed to PPARγ, the role of PPARα to prevent hepatic steatosis, likely by favoring fatty acid oxidation, is supported by a number of studies. Indeed, PPARα expression is decreased in the liver of rodents with NAFLD [42] and PPARα knockout mice display an increased steatosis, oxidative stress, and inflammation when fed an HF Western diet [43, 44]. Fenofibrates, a class of PPARα agonists, were also shown to improve hepatic steatosis in a mouse model of hereditary fatty liver in absence of obesity or diabetes [45] and in OLETF rats, which spontaneously develop NAFLD [32]. Other PPARα agonists (e.g., Wy 14 643) administered to mice fed an HF diet failed to prevent liver inflammation, but could improve other metabolic parameters such as hyperglycemia and hepatic steatosis [46]. Interestingly, the protective effects of fish oil (n-3 polyunsaturated fatty acids) dietary supplementation in mice fed a choline/methionine-deficient diet, or of aldose reductase inhibition in the diabetic db/db mouse model, on steatosis development were strongly correlated with a significant increase in the expression of PPARα [47, 48]. Finally, as for PPARγ, a human genetic variant of PPARα (Val227Ala) was specifically associated with NAFLD [49]. Although PPARα agonists are used in clinic to treat mixed dyslipidemia and hypertriglyceridemia, few studies have investigated the outcomes of these treatments for NAFLD. Fenofibrate in conjunction with dietary guidelines led to an improved steatosis and liver enzymes in 42% of patients included in the study (n = 62) [50]. On the contrary, in another pilot study by Fernandez-Miranda et al., administration of fenofibrate could partially improve liver enzyme profiles and hepatocellular ballooning in patients with biopsies-confirmed NAFLD, but not steatosis, inflammation, and fibrosis [51]. Therefore, from the current available data, the therapeutic potential of PPARα agonists to treat IR and NAFLD in human remains to be evaluated.
2.1.3. PPARβ/δ
Recent evidence indicates that PPARβ/δ can exert a protecting activity against IR. In vitro studies showed that PPARβ/δ expression in HepG2 hepatoma cells could decrease steatosis and IR induced by oleic acid likely by up-regulating PTEN expression and activity [52]. Consistent with these data, PPARβ/δ knockout mice develop glucose intolerance in part by decreasing the hepatic carbohydrate catabolism [53], but PPARβ/δ-deficient mice seem to have no defect in liver triglyceride and glycogen accumulation [54]. Whether PPARβ/δ positively or negatively regulates lipid metabolism and steatosis development remains to date still controversial. Indeed, expression of PPARβ/δ in the liver of mice through adenoviral infection led to either amelioration of hepatic steatosis in obese db/db mice [55] or exacerbated accumulation of lipids in hepatocytes from HF-fed mice [56]. Several other studies with specific agonists of PPARβ/δ reported also a beneficial effect of this nuclear receptor activation on hepatic IR and steatosis development. For example, the PPARβ/δ agonist GW501516 could prevent cytokine-induced IR in human hepatoma cells and in mice liver cells [57] or could improve IR in db/db mice by suppressing hepatic glucose output [53]. GW501516 also alleviated liver steatosis by increasing fatty acid oxidation in mice fed an HF diet [58] or in mice fed a methionine/choline-deficient diet [59]. Another agonist, NNC61-5920, was able to attenuate hepatic IR and to improve the expression profile of genes involved in the lipid metabolism both in mice and rats fed an HF diet. However, whereas systemic insulin sensitivity was improved in mice fed a HF diet, rats under the same conditions displayed systemic IR, suggesting that PPARβ/δ agonists induced specie-specific metabolic effects [60]. Pharmacological activation of PPARβ/δ by L-165041 in mice rendered obese and diabetic by HF feeding could also decrease IR and hepatic steatosis [61], whereas steatosis and inflammation were improved in LDLR−/− mice fed a Western diet and treated with L-165041 [62]. Clinical trials with PPARβ/δ agonists have recently provided also encouraging perspectives to treat NAFLD. Indeed, healthy volunteers treated with GW501516 displayed reduced plasmatic triglycerides after two weeks [63], whereas, in overweighted subjects, GW501516 led to reduced plasmatic triglycerides and LDL levels and a decreased fat content in the liver [64]. Finally, MBX-8025, a novel PPARβ/δ agonist, decreases liver enzymes, hypercholesterolemia and hypertriglyceridemia in dyslipidemic overweighted patients [65].
2.2. PPARs in Liver Inflammation and Fibrosis
Evidence indicates that defects in signalling associated with all three isoforms of PPARs could further contribute to the progression of hepatic IR and steatosis towards more severe stages of liver diseases.
2.2.1. PPARγ
Several studies indicated that PPARγ deficiency in hepatic stellate cells is associated with a transdifferentiation and activation of these cells leading to excessive formation of fibrotic tissue in the liver [66–68]. Surprisingly however, a recent study by Yamazaki and colleagues concluded that downregulation of PPARγ2 in the liver through adenovirus injection improves inflammation and steatosis in mice fed a high-saturated-fat diet [27]. Available data with PPARγ agonists consistently suggest that activation of PPARγ signalling protects the liver against fibrosis and inflammation in mice and rats. Indeed administration of rosiglitazone to mice or rats fed a choline/methionine-deficient diet prevented fibrosis development [69, 70]. In another study, rosiglitazone was also reported to stimulate antioxidant gene expression, β-oxidation of fatty acids and to suppress inflammation and fibrosis in mouse models of NASH [31]. Finally, in human with NASH, pioglitazone significantly improved the biochemical and histological feature of NASH and discontinuation of the treatment led to a rebound of the diseases [71].
2.2.2. PPARα
Consistent with a protective role of PPARα for IR and steatosis development, PPARα-null mice fed a Western diet develop more steatosis, oxidative stress, and inflammation in the liver than wild-type mice [44]. Deletion of PPARα in apoE2-KI mice fed a Western diet also aggravates liver steatosis and inflammation development. However, agonists of PPARα administered to foz/foz mice (rendered diabetic by feeding with a HF diet) failed to resolve liver inflammation although other histological parameters, including steatosis, hepatocytes ballooning and neutrophils/macrophages recruitment, were improved [46]. Intriguingly, the beneficial effects of vitamin E on NASH in human were correlated with a decreased, and not an increased, expression of PPARα [72].
2.2.3. PPARβ/δ
PPARβ/δ-deficient mice appear to be more sensitive to chemical hepatotoxic agents. For example, PPARβ/δ knockout mice treated with CCL4 developed more liver necrosis, displayed more elevated ALT enzymes, inflammation and profibrotic genes expression than control mice, therefore suggesting that PPARβ/δ protects the liver from inflammation and fibrosis [73]. Consistent with this study, transcriptional genomic analysis with the liver of PPARβ/δ-deficient mice suggested that PPARβ/δ could have an anti-inflammatory action in the liver [54] and the PPARβ/δ agonist GW501516 was shown to improve hepatic inflammation in mice fed a methionine/choline-deficient diet (a widely used model for NASH) [59].
Interestingly, PPARβ/δ seems to be required in Kupffer cells to support the switch between the proinflammatory macrophage M1 to the anti-inflammatory macrophage M2 [74], whereas in stellate cells PPARβ/δ is highly up-regulated following activation [75]. It is thus likely that PPARβ/δ has distinct roles in parenchymal and nonparenchymal hepatic cells, which affect, in a still poorly understood manner, the paracrine dialog between these cells and the development of inflammation and fibrosis.
2.3. PPARs in Alcoholic Liver Diseases and Hepatitis Virus Infections
Although studies focusing on the role of PPARs in alcoholic liver diseases (ALD) and HBV/HCV infections might be sometimes scarce (e.g., regarding the role of PPARβ/δ in these diseases), evidence accumulates suggesting that an unbalanced expression/activation of distinct PPAR isoforms may also contribute to the wide spectrum of liver disorders induced either by excessive alcohol consumption or HBV/HCV.
2.3.1. PPARγ
In the liver of HCV-infected patients, PPARγ expression was found to be highly up-regulated and to contribute to the development of HCV-associated steatosis [25, 76, 77]. Two viral factors, the core protein and the NS5A protein, were suggested to mediate PPARγ upregulation either through transactivation of LXRα [76–78] or by inducing SOCS7 expression [79]. PPARγ is also increased in the liver of HBV-infected patients [80], likely through HBV X protein-dependent mechanisms [81, 82]. However, whether PPARγ confers or not a replicative advantage to HBV still remains a controversial issue [83–85].
2.3.2. PPARα
With excessive alcohol consumption, PPARα gene transcription is inhibited through still unknown mechanisms [86] and may contribute, through an increased oxidative stress and inflammatory response, to the wide spectrum of liver disorders observed with alcoholism [87]. PPARα was also reported to be down-regulated by HCV in the liver of infected patients [88, 89]. Since PPARα blocks the replication and production of infectious viral particles, its downregulation likely confers a replicative advantage to the virus in spite of the resulting metabolic disorders for the host cells [90, 91].
2.3.3. PPARβ/δ
Although virtually none is known about the potential role of PPARβ/δ in HBV/HCV infection, only one study has outlined the potential benefit of PPARβ/δ activation in liver injuries related to alcohol consumption. Indeed, pharmacological activation of PPARβ/δ by L-165041 in rats chronically fed with ethanol attenuated the severity of liver injury by diminishing oxidative stress, lipid peroxidation and by restoring hepatic insulin responsiveness [92].
2.4. PPARs in Liver Cancer
As previously mentioned, HCA and HCC represent the ultimate stages of liver diseases induced by obesity (NAFLD/NASH), alcohol consumption, or hepatitis viruses (HBV/HCV) [11–13]. It is therefore expected that PPARs signalling can either favor, or refrain, carcinogenic processes in the liver. However, data have revealed that PPARs signalling might have a different outcome on carcinogenesis as compared to other metabolic diseases of the liver.
2.4.1. PPARγ
Although the role of PPARγ in the development of liver metabolic diseases with different etiologies has led in some cases to controversial results (see previous sections), there is a general consensus about the fact that PPARγ activity can counteract the occurrence and progression of cancer in the liver. In in vitro studies, PPARγ overexpression induced apoptosis in HCC cell lines [93]. On the other hand, PPARγ agonists administration to HCC cell lines promoted cell cycle arrest, apoptosis, and anoikis [94, 95], sensitized the cells to 5-fluorouracil antitumoural activity [96], and inhibited migration of these cells [97]. Interestingly, in addition to prevent HBV replication in vitro [85], PPARγ agonists triggered also antineoplastic effects on HBV-associated HCC cells [98]. In vivo studies corroborated data obtained in vitro. Indeed, PPARγ-null mice display a higher susceptibility to the development of HCC induced by the carcinogen diethylnitrosamine (DEN). The administration PPARγ agonists (rosiglitazone) also reduced HCC development induced by DEN in rats or by hepatoma cell xenograft in mice [99–101]. Together, these studies indicate that PPARγ may act as a tumour suppressor and that specific agonists could be used in cancer prevention.
2.4.2. PPARα
As opposed to PPARγ, activation of PPARα in the liver leads to carcinogenesis in rodents [102–104]. Indeed, chronic administration of the PPARα agonists Wy-14, 643 or bezafibrate in mice induced HCC with time, an effect that was not observed in PPARα-null mice [102, 105, 106]. Interestingly, the PPARα carcinogenic effect on the liver seems to be age dependent in rodent, suggesting that with ageing the liver become more sensitive to the carcinogenic affects of PPARα transcriptional activity [107]. An aberrant expression of PPARα also contributes to the hepatocarcinogenic events induced by the solvent trichloroethylene (TCE) in rodents [108] and PPARα activation was necessary to induce steatosis and HCC in a model of transgenic mice expressing the HCV core protein [109, 110]. Surprisingly, the carcinogenic effects of PPARα agonists were not confirmed in human patients, suggesting species differences in PPARα signalling [103]. Consistent with this study, mice expressing a human PPARα were less susceptible to develop cancer with agonist administration [111, 112]. These differential susceptibilities to PPARα-induced HCC in rodent versus humans could be related to the inhibition by PPARα, in rodents but not in human, of the expression of the let-7c miRNA, which targets the Myc oncogene [102].
2.4.3. PPARβ/δ
The role of PPARβ/δ in cancer is currently the subject of intense investigations that have led to controversial hypothesis. Indeed, different studies support a pro-carcinogenic role for PPARβ/δ as a differentiation and anti-inflammatory factor [104]. Regarding liver carcinogenesis, only few studies have provided insights into the role of PPARβ/δ in this process. PPARβ/δ agonists, such as GW501516, were reported to significantly enhance the growth of various hepatoma cells, whereas inhibition of PPARβ/δ expression by siRNAs had the opposite effect [113]. PPARβ/δ transcriptional activity was also involved in the IL-6-induced proliferation of cultured chicken hepatocytes [114]. It thus remains to investigate more in detail how PPARβ/δ activity impacts on liver cancer.
3. MicroRNAs
In addition to PPARs activation triggered by fatty acids and derivatives specific ligands, the transcriptional activity of the various PPARs isoforms in physiological and pathological situations is regulated by other numerous and complex epigenetic, transcriptional, and posttranscriptional mechanisms (Figure 1). These include epigenetic inhibitory mechanisms (e.g., methylation of the PPARs promoter and histone acetylation) [115, 116], positive and negative regulation of PPARs mRNA transcription by various transcription factors [117–120], posttranslational modifications of the proteins, interaction with other cellular factors, which may affect their transcriptional activities, intracellular localization, and stability of the PPARs proteins [19, 121–125]. Interestingly, a further level of complexity in these regulatory mechanisms has recently emerged and involves an epigenetic regulation of PPAR isoforms by microRNAs, which can either degrade or repress PPAR mRNAs at the translational level.
Figure 1.
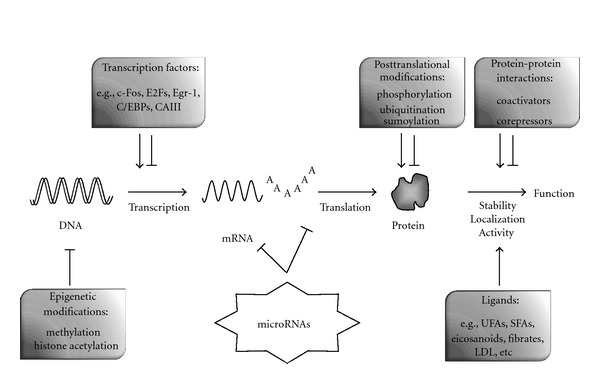
Regulation of PPARs expression and activity. The scheme presents distinct mechanisms reported to regulate the expression and activity of specific PPARs isoforms. Epigenetic mechanisms, including DNA methylation and histone acetylation, may restrict the PPARs promoter activity and several transcription factors have been described to modulate either positively or negatively the transcription of distinct PPARs. As well, posttranslational modifications at the protein level (phosphorylation, ubiquitination, and sumoylation) and interactions with coactivators (e.g., CBP/p300, SRC-1, PGC1α) or corepressors (e.g., RIP-140α, SMRT α) regulate the transcriptional activity, localization, and stability of PPARs isoforms. miRNAs represent a new recently described class of regulators of PPARs transcriptional activity by exerting a control on PPARs mRNA degradation and translation.
miRNAs are small noncoding RNAs of about 20 nucleotides that bind to conserved 3′UTR sequences of their target mRNA, therefore inducing either inhibition of their translation or their degradation [126]. First described in C. elegans [127], 1921 miRNAs have been discovered and registered to date (according to miRBase microRNA database, http://www.mirbase.org/) and more than 30% of proteins-coding genes are supposed to be targeted by miRNAs [128]. Most of the miRNAs are intergenics and can be expressed independently, but a few of them are also located in introns of known protein-coding genes and are co-transcribed with these genes [129]. The biogenesis of miRNAs is a well-conserved mechanism. The RNA polymerase II transcribes most of microRNAs as a long primary transcript enclosing a stem-loop structure [130]. This process can be regulated by several transcription factors that bind directly to the miRNA promoter elements and control their expression [131]. miRNAs maturation occurs in the nucleus where the RNase III Drosha and other cofactors cleave the pri-miRNA bearing the hairpin structure to generate a pre-miRNA product [132]. The precursor pre-miRNA of about 60–70 nucleotides is then exported from the nucleus to the cytoplasm by the exportin-5 [133, 134], a process that is regulated by the Drosha activity [135, 136]. In the cytoplasm, the RNase III Dicer cleaves the miRNA stem loop to generate a mature miRNA of 20–22 nucleotides long [137]. Mature miRNA targeting a mRNA finally necessitates the RNA-induced silencing complex RISC, in which the Argonaute protein (Ago2 for mammals) is the catalytic center [138, 139]. Of note, Drosha, Dicer, and Ago2 are components of miRNAs processing that are essential for life since specific knockout for one of these proteins in mice induces severe developmental defects or death [140].
Finding the targets of specific miRNA currently remains a challenge. Bioinformatic predictions and proteomic analyses are performed to estimate potential targets for a given miRNA [141]. However, there is a great redundancy in miRNAs capable to target a specific mRNA and each miRNA can also target hundreds of different mRNAs [142, 143] thereby increasing tremendously the complexity of this type of global analyses. In addition, miRNAs usually induces only a maximum of twofold downregulation of their target protein, thereby exerting only a fine-tuning control of protein expression. Despite these difficulties to investigate the physiological and pathological potential roles of miRNAs, accumulating evidence indicates that miRNAs play an important role in multiple cellular processes, as well as in the development of several diseases, including the MetS, diabetes, neurodegeneration, cardiovascular and immune diseases [144–148]. Numerous studies also recently outlined miRNAs as critical regulatory factors promoting or inhibiting cancer development [149, 150]. More particularly in the liver, several miRNAs (including miR-21, miR-29, miR-122, miR-132, miR-155, miR-192, and miR-322) were reported to exert a control on hepatocyte differentiation, glucose and lipid metabolism, fatty liver diseases, fibrosis, and HBV/HCV infection [151–160]. In liver cancer, for example, HCC, “omics” analyses have revealed that numerous miRNAs are dysregulated. Importantly, while miR-122 is down-regulated in HCC thereby potentially impairing cell differentiation, many others are up-regulated, including miR-96, miR-221, miR-224, and miR-21, which have all been reported to regulate cell proliferation and apoptosis [161, 162].
4. MicroRNA-Dependent PPARs Regulation
The epigenetic regulation of PPARs expression and activity by miRNAs is a new field of research still in its infancy. However, accumulating evidence now suggests that alterations of the expression/activity of PPARs isoforms by distinct miRNAs could represent critical molecular mechanisms involved in the physiopathology of each organ undergoing a PPARs-dependent control. In this regard, bioinformatics predictions of miRNAs potentially targeting the different PPARs isoforms reveal that hundreds of different miRNAs could participate to the regulation of PPARs expression/activity in different cells/tissues (see Table 1). Currently, most of the studies investigating the role of miRNAs in the regulation of distinct PPARs isoforms have been performed in cultured adipocytes or in the adipose tissue. But recent works have also highlighted miRNAs-dependent regulation of PPARs in the liver and have implicated these regulatory processes in the development of hepatic diseases.
Table 1.
miRNAs potentially targeting PPARγ, PPARα and PPARβ/δ. Four different databases, that is, miRanda (http://www.microRNA.org/), PicTar (http://www.pictar.org/), TargetScanHuman (http://www.targetscan.org/) and MicroCosm Targets (http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets) were scanned to find all predicted miRNAs that possibly regulate PPARγ, PPARα and PPARβ/δ expression by binding to specific seed sequences in the transcribed 3′UTR regions of different PPARs isoforms.
(a)
| PPARα | ||||
|---|---|---|---|---|
| miRanda | TargetScan | MicroCosm targets | PicTar | |
| miR-25 | miR-7 | miR-200 | miR-892 | miR-19 |
| miR-28 | miR-9 | miR-203 | ||
| miR-32 | miR-10 | miR-204 | ||
| miR-92 | miR-17 | miR-211 | ||
| miR-145 | miR-18 | miR-214 | ||
| miR-155 | miR-19 | miR-219 | ||
| miR-181 | miR-20 | miR-223 | ||
| miR-200 | miR-21 | miR-291 | ||
| miR-216 | miR-22 | miR-294 | ||
| miR-217 | miR-23 | miR-295 | ||
| miR-218 | miR-24 | miR-302 | ||
| miR-224 | miR-27 | miR-338 | ||
| miR-342 | miR-34 | miR-351 | ||
| miR-363 | miR-93 | miR-372 | ||
| miR-367 | miR-101 | miR-373 | ||
| miR-374 | miR-105 | miR-427 | ||
| miR-376 | miR-106 | miR-428 | ||
| miR-421 | miR-124 | miR-429 | ||
| miR-425 | miR-125 | miR-449 | ||
| miR-429 | miR-128 | miR-506 | ||
| miR-431 | miR-138 | miR-508 | ||
| miR-491 | miR-141 | miR-518 | ||
| miR-543 | miR-142 | miR-519 | ||
| miR-590 | miR-144 | miR-520 | ||
| miR-615 | miR-150 | miR-548 | ||
| miR-708 | miR-181 | miR-590 | ||
| miR-873 | miR-182 | miR-670 | ||
| miR-761 | ||||
| miR-1378 | ||||
| miR-1420 | ||||
| miR-3619 | ||||
| miR-4262 | ||||
| miR-4319 | ||||
| miR-4735 | ||||
| miR-4782 | ||||
| miR-5127 | ||||
(b)
| PPARγ | ||||
|---|---|---|---|---|
| miRanda | Target scan | MicroCosm targets | Pictar | |
| miR-1 | miR-328 | miR-27 | miR-27 | miR-27 |
| miR-9 | miR-329 | miR-128 | miR-30 | miR-128 |
| miR-18 | miR-338 | miR-130 | miR-33 | miR-130 |
| miR-24 | miR-340 | miR-301 | miR-34 | miR-301 |
| miR-25 | miR-361 | miR-454 | miR-128 | |
| miR-26 | miR-362 | miR-721 | miR-130 | |
| miR-27 | miR-363 | miR-3666 | miR-142 | |
| miR-32 | miR-367 | miR-4295 | miR-144 | |
| miR-33 | miR-370 | miR-193 | ||
| miR-34 | miR-371 | miR-301 | ||
| miR-92 | miR-376 | miR-338 | ||
| miR-96 | miR-411 | miR-409 | ||
| miR-101 | miR-421 | miR-431 | ||
| miR-122 | miR-431 | miR-449 | ||
| miR-128 | miR-448 | miR-454 | ||
| miR-130 | miR-449 | miR-513 | ||
| miR-133 | miR-454 | miR-520 | ||
| miR-137 | miR-485 | miR-526 | ||
| miR-142 | miR-488 | miR-545 | ||
| miR-144 | miR-490 | miR-548 | ||
| miR-145 | miR-505 | miR-559 | ||
| miR-148 | miR-590 | miR-574 | ||
| miR-150 | miR-613 | |||
| miR-152 | miR-653 | |||
| miR-153 | miR-758 | |||
| miR-181 | miR-1271 | |||
| miR-182 | miR-1297 | |||
| miR-185 | ||||
| miR-194 | ||||
| miR-199 | ||||
| miR-204 | ||||
| miR-206 | ||||
| miR-211 | ||||
| miR-224 | ||||
| miR-301 | ||||
| miR-324 | ||||
(c)
| PPARβ/δ | |||
|---|---|---|---|
| miRanda | Target scan | MicroCosm targets | Pictar |
| miR-19 | miR-9 | miR-17 | none |
| miR-24 | miR-17 | miR-20 | |
| miR-129 | miR-20 | miR-24 | |
| miR-133 | miR-29 | miR-29 | |
| miR-138 | miR-34 | mi R-33 |
|
| miR-140 | miR-93 | miR-93 | |
| miR-149 | miR-106 | miR-106 | |
| miR-185 | miR-129 | miR-136 | |
| miR-196 | miR-138 | miR-138 | |
| miR-218 | miR-148 | miR-143 | |
| miR-223 | miR-150 | miR-149 | |
| miR-326 | miR-214 | miR-219 | |
| miR-330 | miR-223 | miR-220 | |
| miR-342 | miR-427 | miR-222 | |
| miR-487 | miR-449 | miR-302 | |
| miR-590 | miR-518 | miR-373 | |
| miR-599 | miR-519 | miR-378 | |
| miR-653 | miR-761 | miR-483 | |
| miR-874 | miR-3619 | miR-492 | |
| miR-5127 | miR-512 | ||
| miR-513 | |||
| miR-519 | |||
| miR-520 | |||
| miR-542 | |||
| miR-544 | |||
| miR-550 | |||
| miR-564 | |||
| miR-576 | |||
| miR-631 | |||
| miR-640 | |||
| miR-657 | |||
| miR-874 | |||
| miR-885 | |||
| miR-921 | |||
MicroRNA-Dependent PPARs Regulation in the Liver —
At least 5 distinct miRNAs have been reported to directly, or indirectly, affect PPAR expression in liver cells, mostly in pathological situations, in particular NAFLD (see Table 2). For example, Zheng and coworkers performed miRNA microarrays with human hepatocyte cell line L02 exposed or not to high concentration of fatty acids. They could find more than 30 miRNA either up- or down-regulated in cells challenged with excess of fatty acids. Among them, miR-10b was up-regulated. Further analyses revealed that miR-10b induces steatosis in these cells by directly targeting PPARα and preventing its expression [163]. PPARα was also reported to be specifically regulated by two other miRNAs, miR-21 and miR-27b, in the liver. Indeed, expression of precursors or antisense nucleotides for miR-21, or miR-27b, in Huh-7 hepatoma cells could significantly modulate the expression of PPARα protein, but not its mRNA, suggesting a blockade of PPAR mRNA translation by miR-21/-27b. Interestingly, no correlations were found between PPARα protein and mRNA expression in human livers (n = 24). However, in the same human samples, PPARα protein expression was inversely correlated with miR-21 expression [164]. A potential role for miR-21-dependent regulation of PPARs was further supported by our own observations indicating that PPARα protein expression is up-regulated in primary hepatocytes isolated from miR-21 knockout mice, whereas PPARγ is down-regulated in the liver of mice overexpressing miR-21 (our unpublished data). miR-122, the most highly expressed miRNA in the liver, was also reported to specifically target PPARβ/δ in the liver of mice. Interestingly miR-122 expression appears to be circadian thereby providing an interesting link between miR-122, PPARβ/δ, and the well-known circadian metabolic control occurring in the liver [165]. Finally, PPARγ activity appears also to be under the indirect control of miR-132. Indeed, Mann and coworkers could demonstrate that, in stellate cells of mice treated with CCl4, downregulation of the miR-132 promoted the expression of MeCP2 (a target of miR-132), which in turn binds to PPARγ and promotes the formation of an epigenetic repressor complex inhibiting PPARγ expression and therefore promoting liver fibrosis in this mice model [116].
Table 2.
miRNAs reported to regulate PPARγ, PPARα, and PPARβ/δ expression and activity in the liver.
MicroRNA-Dependent PPARs Regulation in Other Tissues —
Numerous other reports have implicated an miRNA-dependent regulation of distinct PPARs isoforms in non-hepatic tissues that affects various cellular processes including lipid metabolism, inflammation, cell differentiation, carcinogenesis, or wound skin repair.
In the adipose tissue, two studies have unveiled a physiopathologically relevant regulation of PPARγ expression by the miR-27a/b. Indeed, Karbiener et al. reported a significant downregulation of miR-27b, which directly targets PPARγ, during adipogenesis of human multipotent adipose-derived stem (hMADS) cells. They further demonstrated that inhibiting induction of PPARγ in this process by overexpressing miR-27b represses adipogenic marker gene expression and triglyceride accumulation in hMADS [166]. In a second study, levels of miR-27a were inversely correlated with those of PPARγ in mature adipocyte and miR-27a expression in the adipose tissue was down-regulated in obese mice as compared to lean mice. In vitro studies with 3T3-L1 preadipocytes further showed that miR-27a ectopic expression prevents adipocyte differentiation by inhibiting PPARγ through a direct binding to its 3′-UTR sequence [167]. Finally, differentiation of human preadipocytes into adipocytes was also shown to be highly dependent of miR-130 expression. Indeed, miR-130 was shown to directly target PPARγ and thereby to enhance adipogenesis. Consistent with this, expression of miR-130 in the adipose tissue of obese women was lower compared to lean women [168]. Together, these studies suggest that induction of PPARγ by alterations in miR-27a/b and/or miR-130 expressions might be linked to the development of obesity in rodents and humans. miRNAs microarray analyses of the subcutaneous adipose tissue in nondiabetic but severely obese adults also identified miR-519d as an overexpressed miRNAs that targets PPARα. The miR-519d-dependent decrease in PPARα translation was further shown to increase lipid accumulation during pre-adipocyte differentiation suggesting that PPARα loss through miR-519d overexpression importantly contributed to adipocytes hypertrophy observed with obesity [169].
Finally, recent evidence also indicates that alterations of PPARγ and PPARα expression by specific miRNAs occur in other tissues and can contribute to the development of specific disorders. For example, downregulation of PPARγ by an aberrant expression of miR-27b in cardiomyocytes was associated with cardiac hypertrophy in mice [170], whereas miR-27b-dependent PPARγ downregulation in macrophages favors the inflammatory response to LPS [171]. In human osteoarthritic chondrocytes, aberrant expression of miR-22 specifically down-regulates PPARα expression, which in turn promotes IL1-dependent inflammatory processes [172]. Then in endothelial cells of cultured human umbilical vein under oscillatory shear stress, levels of miR-21 were found to be increased and to induce an inhibition of PPARα translation, which favor monocytes adhesion and atherosclerosis formation [173]. Finally, PPARα was reported to be also a target of miR-506, which is increased in human colon cancers and contribute to chemotherapy resistance by down-regulating PPARα expression [174].
5. Conclusion
Although the therapeutic potential of PPARβ/δ for metabolic diseases such as insulin resistance, dyslipidemia, and other associated liver pathologies deserves further investigations, agonists of PPARγ and PPARα have been developed as relevant drugs to treat these disorders. For cancer therapies, the use of PPAR agonists is more debated, in particular for PPARα, which induces liver and bladder cancers in mice, but apparently not in humans. However, the chronic administration of PPAR agonists in human for the treatment of metabolic diseases may importantly increase the risk of developing cardiovascular diseases (e.g., for PPARγ agonists) and specific cancers (e.g., for PPARα agonists). For these reasons, recommendations have been issued for a restricted and invigilated clinical use of PPAR agonists and some of them (e.g., PPARγ agonists) have even been withdrawn from the market in some countries. An attractive therapeutic alternative to systemic administration of PPAR agonists to treat metabolic diseases and/or cancers would be to exert more fine tuning of specific PPAR isoforms expressions/activities in the organ of interest, thereby minimizing the deleterious side effects of chronic systemic administration of these agonists. In this regard, miRNAs represent an interesting class of molecules that could be pharmacologically modulated, for example, with antagomirs, to prevent pathological alterations of PPARs expressions and activities. There might be several advantages associated with this type of therapeutic approaches. First, the aberrant expression of specific miRNAs, for example, inhibiting the expression of PPARs in diseases, is often tissue specific. Therefore, the administration of miRNA-targeting drugs should affect principally the injured organs and secondary systemic effects could be minimized as compared to systemic administration of PPAR agonists. Secondly, as mentioned previously, miRNAs can only modestly modulate (more or less two fold) the expression of their target mRNAs. However, preventing this pathological miRNA-mediated dysregulations of PPARs expression should help to recover a physiological PPAR transcriptional activity in contrast to the ectopic overactivation of PPARs induced by administration of agonists. Finally, based on bioinformatics predictions (see Table 1), there is a high number of miRNAs predicted to be able to affect the expression of PPARs in pathological situations. It is likely that many other miRNAs would be identified in the future as important PPARs regulators in metabolic diseases and cancer, thereby multiplying the miRNA-based therapeutic targets available to treat these diseases. Based on these considerations, additional studies are now required to further assess the pertinence of miRNA-based therapies to enhance specifically the activity of PPAR isoforms as therapeutic weapons in metabolic diseases and cancer.
Acknowledgments
This work was supported by the Swiss National Science Foundation (Grant no. 310030-135727/1), the Desirée and Niels Yde Foundation, the Gertrude von Meissner Foundation, the Fondation Romande pour la Recherche sur le Diabète, the Swiss Cancer League, and the EFSD Research Programme in Diabetes and Cancer to M. Foti.
Abbreviations
- ALD:
Alcoholic liver diseases
- FFAs:
Free fatty acids
- IR:
Insulin resistance
- HCA:
Hepatocellular adenoma
- HBV:
Hepatitis virus B
- HCV:
Hepatitis virus C
- HCC:
Hepatocellular carcinoma
- MetS:
Metabolic syndrome
- PPARs:
Peroxisome-proliferator-activated receptors
- NAFLD:
Nonalcoholic fatty liver disease
- NASH:
Nonalcoholic steatohepatitis
- miRNA:
MicroRNA.
References
- 1.Williams R. Global challenges in liver disease. Hepatology. 2006;44(3):521–526. doi: 10.1002/hep.21347. [DOI] [PubMed] [Google Scholar]
- 2.Yip WW, Burt AD. Alcoholic liver disease. Seminars in Diagnostic Pathology. 2006;23(3-4):149–160. doi: 10.1053/j.semdp.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Marchesini G, Babini M. Nonalcoholic fatty liver disease and the metabolic syndrome. Minerva Cardioangiologica. 2006;54(2):229–239. [PubMed] [Google Scholar]
- 4.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264.e1–1273.e1. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shire AM, Roberts LR. Prevention of hepatocellular carcinoma: progress and challenges. Minerva Gastroenterologica e Dietologica. 2012;58(1):49–64. [PubMed] [Google Scholar]
- 6.McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: present and future. Clinics in Liver Disease. 2011;15(2):223–243. doi: 10.1016/j.cld.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang C, Wang X, Gong G, et al. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: a systematic review and meta-analysis of cohort studies. International Journal of Cancer. 2012;130(7):1639–1648. doi: 10.1002/ijc.26165. [DOI] [PubMed] [Google Scholar]
- 8.Cornellà H, Alsinet C, Villanueva A. Molecular pathogenesis of hepatocellular carcinoma. Alcoholism. 2011;35(5):821–825. doi: 10.1111/j.1530-0277.2010.01406.x. [DOI] [PubMed] [Google Scholar]
- 9.Contos MJ, Choudhury J, Mills AS, Sanyal AJ. The histologic spectrum of nonalcoholic fatty liver disease. Clinics in Liver Disease. 2004;8(3):481–500. doi: 10.1016/j.cld.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 10.Adams LA, Angulo P. Recent concepts in non-alcoholic fatty liver disease. Diabetic Medicine. 2005;22(9):1129–1133. doi: 10.1111/j.1464-5491.2005.01748.x. [DOI] [PubMed] [Google Scholar]
- 11.Caldwell SH, Crespo DM, Kang HS, Al-Osaimi AMS. Obesity and hepatocellular carcinoma. Gastroenterology. 2004;127(5) supplement 1:S97–S103. doi: 10.1053/j.gastro.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 12.El-Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clinical Gastroenterology and Hepatology. 2006;4(3):369–380. doi: 10.1016/j.cgh.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nature Reviews Cancer. 2006;6(9):674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 14.Vanni E, Bugianesi E, Kotronen A, De Minicis S, Yki-Järvinen H, Svegliati-Baroni G. From the metabolic syndrome to NAFLD or vice versa? Digestive and Liver Disease. 2010;42(5):320–330. doi: 10.1016/j.dld.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Larter CZ, Chitturi S, Heydet D, Farrell GC. A fresh look at NASH pathogenesis. Part 1: the metabolic movers. Journal of Gastroenterology and Hepatology. 2010;25(4):672–690. doi: 10.1111/j.1440-1746.2010.06253.x. [DOI] [PubMed] [Google Scholar]
- 16.Malaguarnera M, Di Rosa M, Nicoletti F, Malaguarnera L. Molecular mechanisms involved in NAFLD progression. Journal of Molecular Medicine. 2009;87(7):679–695. doi: 10.1007/s00109-009-0464-1. [DOI] [PubMed] [Google Scholar]
- 17.Lewis JR, Mohanty SR. Nonalcoholic fatty liver disease: a review and update. Digestive Diseases and Sciences. 2010;55(3):560–578. doi: 10.1007/s10620-009-1081-0. [DOI] [PubMed] [Google Scholar]
- 18.Rombouts K, Marra F. Molecular mechanisms of hepatic fibrosis in non-alcoholic steatohepatitis. Digestive Diseases. 2010;28(1):229–235. doi: 10.1159/000282094. [DOI] [PubMed] [Google Scholar]
- 19.Michalik L, Wahli W. PPARs mediate lipid signaling in inflammation and cancer. PPAR Research. 2008;23(7):351–363. doi: 10.1155/2008/134059.134059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li G, Guo GL. Role of class II nuclear receptors in liver carcinogenesis. Anti-Cancer Agents in Medicinal Chemistry. 2011;11(6):529–542. doi: 10.2174/187152011796011064. [DOI] [PubMed] [Google Scholar]
- 21.Dharancy S, Louvet A, Hollebecque A, Desreumaux P, Mathurin P, Dubuquoy L. Nuclear receptor PPAR and hepatology: pathophysiological and therapeutical aspects. Gastroenterologie Clinique et Biologique. 2008;32(3):339–350. doi: 10.1016/j.gcb.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 22.Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochimica et Biophysica Acta. 2012;1821(5):809–818. doi: 10.1016/j.bbalip.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 23.Kallwitz ER, McLachlan A, Cotler SJ. Role of peroxisome proliferators-activated receptors in the pathogenesis and treatment of nonalcoholic fatty liver disease. World Journal of Gastroenterology. 2008;14(1):22–28. doi: 10.3748/wjg.14.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pettinelli P, Videla LA. Up-regulation of PPAR-γ mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. Journal of Clinical Endocrinology and Metabolism. 2011;96(5):1424–1430. doi: 10.1210/jc.2010-2129. [DOI] [PubMed] [Google Scholar]
- 25.Lima-Cabello E, García-Mediavilla MV, Miquilena-Colina ME, et al. Enhanced expression of pro-inflammatory mediators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liver disease and hepatitis C. Clinical Science. 2011;120(6):239–250. doi: 10.1042/CS20100387. [DOI] [PubMed] [Google Scholar]
- 26.Morán-Salvador E, López-Parra M, García-Alonso V, et al. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. The FASEB Journal. 2011;25(8):2538–2550. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- 27.Yamazaki T, Shiraishi S, Kishimoto K, Miura S, Ezaki O. An increase in liver PPARγ2 is an initial event to induce fatty liver in response to a diet high in butter: PPARγ2 knockdown improves fatty liver induced by high-saturated fat. Journal of Nutritional Biochemistry. 2011;22(6):543–553. doi: 10.1016/j.jnutbio.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 28.Moya M, José Gómez-Lechón M, Castell JV, Jover R. Enhanced steatosis by nuclear receptor ligands: a study in cultured human hepatocytes and hepatoma cells with a characterized nuclear receptor expression profile. Chemico-Biological Interactions. 2010;184(3):376–387. doi: 10.1016/j.cbi.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 29.Liu S, Wu HJ, Zhang ZQ, et al. The ameliorating effect of rosiglitazone on experimental nonalcoholic steatohepatitis is associated with regulating adiponectin receptor expression in rats. European Journal of Pharmacology. 2011;650(1):384–389. doi: 10.1016/j.ejphar.2010.09.082. [DOI] [PubMed] [Google Scholar]
- 30.Zheng H, Li S, Ma L, et al. A novel agonist of PPAR-γ based on barbituric acid alleviates the development of non-alcoholic fatty liver disease by regulating adipocytokine expression and preventing insulin resistance. European Journal of Pharmacology. 2011;659(2-3):244–251. doi: 10.1016/j.ejphar.2011.03.033. [DOI] [PubMed] [Google Scholar]
- 31.Gupte AA, Liu JZ, Ren Y, et al. Rosiglitazone attenuates age- and diet-associated nonalcoholic steatohepatitis in male low-density lipoprotein receptor knockout mice. Hepatology. 2010;52(6):2001–2011. doi: 10.1002/hep.23941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seo YS, Kim JH, Jo NY, et al. PPAR agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. Journal of Gastroenterology and Hepatology. 2008;23(1):102–109. doi: 10.1111/j.1440-1746.2006.04819.x. [DOI] [PubMed] [Google Scholar]
- 33.Lutchman G, Promrat K, Kleiner DE, et al. Changes in serum adipokine levels during pioglitazone treatment for nonalcoholic steatohepatitis: relationship to histological improvement. Clinical Gastroenterology and Hepatology. 2006;4(8):1048–1052. doi: 10.1016/j.cgh.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 34.Ratziu V, Giral P, Jacqueminet S, et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135(1):100–110. doi: 10.1053/j.gastro.2008.03.078. [DOI] [PubMed] [Google Scholar]
- 35.Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. The New England Journal of Medicine. 2010;362(18):1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savage DB, Tan GD, Acerini CL, et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-γ . Diabetes. 2003;52(4):910–917. doi: 10.2337/diabetes.52.4.910. [DOI] [PubMed] [Google Scholar]
- 37.Hui Y, Yu-yuan L, Yu-qiang N, et al. Effect of peroxisome proliferator-activated receptors-γ and co-activator-1α genetic polymorphisms on plasma adiponectin levels and susceptibility of non-alcoholic fatty liver disease in Chinese people. Liver International. 2008;28(3):385–392. doi: 10.1111/j.1478-3231.2007.01623.x. [DOI] [PubMed] [Google Scholar]
- 38.Rey JW, Noetel A, Hardt A, et al. Pro12Ala polymorphism of the peroxisome proliferatoractivated receptor γ2 in patients with fatty liver diseases. World Journal of Gastroenterology. 2010;16(46):5830–5837. doi: 10.3748/wjg.v16.i46.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gawrieh S, Gawrieh S, Marion MC, et al. Genetic variation in the peroxisome proliferator activated receptor-gamma gene is associated with histologically advanced NAFLD. Digestive Diseases and Sciences. 2012;57(4):952–957. doi: 10.1007/s10620-011-1994-2. [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, Wen J, Li Q, et al. PPARG gene Pro12Ala variant contributes to the development of non-alcoholic fatty liver in middle-aged and older Chinese population. Molecular and Cellular Endocrinology. 2012;348(1):255–259. doi: 10.1016/j.mce.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Gupta AC, Chaudhory AK, Sukriti, et al. Peroxisome proliferators-activated receptor γ2 Pro12Ala variant is associated with body mass index in non-alcoholic fatty liver disease patients. Hepatology International. 2011;5(1):575–580. doi: 10.1007/s12072-010-9225-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jong EY, Kyung MC, Sei HB, et al. Reduced expression of peroxisome proliferator-activated receptor-α may have an important role in the development of non-alcoholic fatty liver disease. Journal of Gastroenterology and Hepatology. 2004;19(7):799–804. doi: 10.1111/j.1440-1746.2004.03349.x. [DOI] [PubMed] [Google Scholar]
- 43.Lalloyer F, Wouters K, Baron M, et al. Peroxisome proliferator-activated receptor-α gene level differently affects lipid metabolism and inflammation in apolipoprotein E2 knock-in mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31(7):1573–1579. doi: 10.1161/ATVBAHA.110.220525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARα expression protects male mice from high fat-induced nonalcoholic fatty liver. Journal of Nutrition. 2011;141(4):603–610. doi: 10.3945/jn.110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harano Y, Yasui K, Toyama T, et al. Fenofibrate, a peroxisome proliferator-activated receptor α agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver International. 2006;26(5):613–620. doi: 10.1111/j.1478-3231.2006.01265.x. [DOI] [PubMed] [Google Scholar]
- 46.Larter CZ, Yeh MM, van Rooyen DM, Brooling J, Ghatora K, Farrell GC. Peroxisome proliferator-activated receptor-α agonist, Wy 14643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. Journal of Gastroenterology and Hepatology. 2012;27(2):341–350. doi: 10.1111/j.1440-1746.2011.06939.x. [DOI] [PubMed] [Google Scholar]
- 47.Qiu L, Lin J, Xu F, et al. Inhibition of aldose reductase activates hepatic peroxisome proliferator-activated receptor-α and ameliorates hepatosteatosis in diabetic db/db mice. Experimental Diabetes Research. 2012;2012 doi: 10.1155/2012/789730.789730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larter CZ, Yeh MM, Cheng J, et al. Activation of peroxisome proliferator-activated receptor α by dietary fish oil attenuates steatosis, but does not prevent experimental steatohepatitis because of hepatic lipoperoxide accumulation. Journal of Gastroenterology and Hepatology. 2008;23(2):267–275. doi: 10.1111/j.1440-1746.2007.05157.x. [DOI] [PubMed] [Google Scholar]
- 49.Chen S, Li Y, Li S, Yu C. A Val227Ala substitution in the peroxisome proliferator activated receptor alpha (PPAR alpha) gene associated with non-alcoholic fatty liver disease and decreased waist circumference and waist-to-hip ratio. Journal of Gastroenterology and Hepatology. 2008;23(9):1415–1418. doi: 10.1111/j.1440-1746.2008.05523.x. [DOI] [PubMed] [Google Scholar]
- 50.Athyros VG, Mikhailidis DP, Didangelos TP, et al. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: a randomised study. Current Medical Research and Opinion. 2006;22(5):873–883. doi: 10.1185/030079906X104696. [DOI] [PubMed] [Google Scholar]
- 51.Fernández-Miranda C, Pérez-Carreras M, Colina F, López-Alonso G, Vargas C, Solís-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Digestive and Liver Disease. 2008;40(3):200–205. doi: 10.1016/j.dld.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 52.Wu HT, Chen W, Cheng KC, Ku PM, Yeh CH, Cheng JT. Oleic acid activates peroxisome proliferator-activated receptor delta to compensate insulin resistance in steatotic cells. doi: 10.1016/j.jnutbio.2011.07.006. The Journal of nutritional biochemistry. In press. [DOI] [PubMed] [Google Scholar]
- 53.Lee CH, Olson P, Hevener A, et al. PPARδ regulates glucose metabolism and insulin sensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(9):3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanderson LM, Boekschoten MV, Desvergne B, Müller M, Kersten S. Transcriptional profiling reveals divergent roles of PPARα and PPARβ/δ in regulation of gene expression in mouse liver. Physiological Genomics. 2010;41(1):42–52. doi: 10.1152/physiolgenomics.00127.2009. [DOI] [PubMed] [Google Scholar]
- 55.Sharma R, Torka P. Peroxisome proliferator-activated receptor-δ induces insulin-induced gene-1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology. 2008;48(6):432–441. doi: 10.1002/hep.22602. [DOI] [PubMed] [Google Scholar]
- 56.Liu S, Hatano B, Zhao M, et al. Role of peroxisome proliferator-activated receptor δ/β in hepatic metabolic regulation. Journal of Biological Chemistry. 2011;286(2):1237–1247. doi: 10.1074/jbc.M110.138115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serrano-Marco L, Barroso E, El Kochairi I, et al. The peroxisome proliferator-activated receptor (PPAR) beta/delta agonist GW501516 inhibits IL-6-induced signal transducer and activator of transcription 3 (STAT3) activation and insulin resistance in human liver cells. Diabetologia. 2012;55(3):743–751. doi: 10.1007/s00125-011-2401-4. [DOI] [PubMed] [Google Scholar]
- 58.Barroso E, Rodríguez-Calvo R, Serrano-Marco L, et al. The PPARβ/δ activator GW501516 prevents the down-regulation of AMPK caused by a high-fat diet in liver and amplifies the PGC-1α-lipin 1-PPARα pathway leading to increased fatty acid oxidation. Endocrinology. 2011;152(5):1848–1859. doi: 10.1210/en.2010-1468. [DOI] [PubMed] [Google Scholar]
- 59.Nagasawa T, Inada Y, Nakano S, et al. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARδ agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. European Journal of Pharmacology. 2006;536(1-2):182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 60.Ye JM, Tid-Ang J, Turner N, et al. PPARδ agonists have opposing effects on insulin resistance in high fat-fed rats and mice due to different metabolic responses in muscle. British Journal of Pharmacology. 2011;163(3):556–566. doi: 10.1111/j.1476-5381.2011.01240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu HT, Chen CT, Cheng KC, Li YX, Yeh CH, Cheng JT. Pharmacological activation of peroxisome proliferator-activated receptor delta improves insulin resistance and hepatic steatosis in high fat diet-induced diabetic mice. Hormone and Metabolic Research. 2011;43(9):631–635. doi: 10.1055/s-0031-1280781. [DOI] [PubMed] [Google Scholar]
- 62.Lim HJ, Park JH, Lee S, Choi HE, Lee KS, Park HY. PPARδ ligand L-165041 ameliorates Western diet-induced hepatic lipid accumulation and inflammation in LDLR−/− mice. European Journal of Pharmacology. 2009;622(1–3):45–51. doi: 10.1016/j.ejphar.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 63.Sprecher DL, Massien C, Pearce G, et al. Triglyceride: high-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor δ agonist. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(2):359–365. doi: 10.1161/01.ATV.0000252790.70572.0c. [DOI] [PubMed] [Google Scholar]
- 64.Risérus U, Sprecher D, Johnson T, et al. Activation of peroxisome proliferator-activated receptor (PPAR)δ promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes. 2008;57(2):332–339. doi: 10.2337/db07-1318. [DOI] [PubMed] [Google Scholar]
- 65.Bays HE, Schwartz S, Littlejohn III T, et al. MBX-8025, a novel peroxisome proliferator receptor-delta agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. Journal of Clinical Endocrinology and Metabolism. 2011;96(9):2889–2897. doi: 10.1210/jc.2011-1061. [DOI] [PubMed] [Google Scholar]
- 66.Miyahara T, Schrum L, Rippe R, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. Journal of Biological Chemistry. 2000;275(46):35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 67.Marra F, Efsen E, Romanelli RG, et al. Ligands of peroxisome proliferator-activated receptor γ modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119(2):466–478. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- 68.Zhang F, Lu Y, Zheng S. Peroxisome proliferator-activated receptor-gamma cross-regulation of signaling events implicated in liver fibrogenesis. Cell Signal. 2012;24(3):596–605. doi: 10.1016/j.cellsig.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 69.Nan YM, Fu N, Wu WJ, et al. Rosiglitazone prevents nutritional fibrosis and steatohepatitis in mice. Scandinavian Journal of Gastroenterology. 2009;44(3):358–365. doi: 10.1080/00365520802530861. [DOI] [PubMed] [Google Scholar]
- 70.Tahan V, Eren F, Avsar E, et al. Rosiglitazone attenuates liver inflammation in a rat model of nonalcoholic steatohepatitis. Digestive Diseases and Sciences. 2007;52(12):3465–3472. doi: 10.1007/s10620-007-9756-x. [DOI] [PubMed] [Google Scholar]
- 71.Lutchman G, Modi A, Kleiner DE, et al. The effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitis. Hepatology. 2007;46(2):424–429. doi: 10.1002/hep.21661. [DOI] [PubMed] [Google Scholar]
- 72.Yakaryilmaz F, Guliter S, Savas B, et al. Effects of vitamin e treatment on peroxisome proliferator-activated receptor-α expression and insulin resistance in patients with non-alcoholic steatohepatitis: results of a pilot study. Internal Medicine Journal. 2007;37(4):229–235. doi: 10.1111/j.1445-5994.2006.01295.x. [DOI] [PubMed] [Google Scholar]
- 73.Shan W, Nicol CJ, Ito S, et al. Peroxisome proliferator-activated receptor-β/δ protects against chemically induced liver toxicity in mice. Hepatology. 2008;47(1):225–235. doi: 10.1002/hep.21925. [DOI] [PubMed] [Google Scholar]
- 74.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metabolism. 2008;7(6):485–495. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hellemans K, Michalik L, Dittie A, et al. Peroxisome proliferator-activated receptor-β signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology. 2003;124(1):184–201. doi: 10.1053/gast.2003.50015. [DOI] [PubMed] [Google Scholar]
- 76.Kim K, Kim KH, Ha E, Park JY, Sakamoto N, Cheong J. Hepatitis C virus NS5A protein increases hepatic lipid accumulation via induction of activation and expression of PPARgamma. FEBS Letters. 2009;583(17):2720–2726. doi: 10.1016/j.febslet.2009.07.034. [DOI] [PubMed] [Google Scholar]
- 77.Kim KH, Hong SP, Kim K, Park MJ, Kim KJ, Cheong J. HCV core protein induces hepatic lipid accumulation by activating SREBP1 and PPARγ . Biochemical and Biophysical Research Communications. 2007;355(4):883–888. doi: 10.1016/j.bbrc.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 78.García-Mediavilla MV, Pisonero-Vaquero S, Lima-Cabello E, et al. Liver X receptor alpha-mediated regulation of lipogenesis by core and NS5A proteins contributes to HCV-induced liver steatosis and HCV replication. Laboratory Investigation. 2012;92(8):1191–1202. doi: 10.1038/labinvest.2012.88. [DOI] [PubMed] [Google Scholar]
- 79.Pazienza V, Vinciguerra M, Andriulli A, Mangia A. Hepatitis C virus core protein genotype 3a increases SOCS-7 expression through PPAR-γ in Huh-7 cells. Journal of General Virology. 2010;91:1678–1686. doi: 10.1099/vir.0.020644-0. [DOI] [PubMed] [Google Scholar]
- 80.Niu YH, Yi RT, Liu HL, Chen TY, Zhang SL, Zhao YR. Expression of COX-2 and PPARg in the livers of patients with acute on chronic HBV-related liver failure and their relationship with clinic parameters. Zhonghua Gan Zang Bing Za Zhi. 2011;19(7):511–516. doi: 10.3760/cma.j.issn.1007-3418.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 81.Kim KH, Shin HJ, Kim K, et al. Hepatitis B virus X protein induces hepatic steatosis via transcriptional activation of SREBP1 and PPARgamma. Gastroenterology. 2007;132(5):1955–1967. doi: 10.1053/j.gastro.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 82.Choi YH, Kim HI, Seong JK, et al. Hepatitis B virus X protein modulates peroxisome proliferator-activated receptor γ through protein-protein interaction. FEBS Letters. 2004;557(1–3):73–80. doi: 10.1016/s0014-5793(03)01449-2. [DOI] [PubMed] [Google Scholar]
- 83.Ondracek CR, McLachlan A. Role of peroxisome proliferator-activated receptor gamma coactivator 1alpha in AKT/PKB-mediated inhibition of hepatitis B virus biosynthesis. Journal of Virology. 2011;85(22):11891–11900. doi: 10.1128/JVI.00832-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoon S, Jung J, Kim T, et al. Adiponectin, a downstream target gene of peroxisome proliferator-activated receptor γ, controls hepatitis B virus replication. Virology. 2011;409(2):290–298. doi: 10.1016/j.virol.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 85.Wakui Y, Inoue J, Ueno Y, et al. Inhibitory effect on hepatitis B virus in vitro by a peroxisome proliferator-activated receptor-γ ligand, rosiglitazone. Biochemical and Biophysical Research Communications. 2010;396(2):508–514. doi: 10.1016/j.bbrc.2010.04.128. [DOI] [PubMed] [Google Scholar]
- 86.Crabb DW, Galli A, Fischer M, You M. Molecular mechanisms of alcoholic fatty liver: role of peroxisome proliferator-activated receptor alpha. Alcohol. 2004;34(1):35–38. doi: 10.1016/j.alcohol.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 87.Moriya T, Naito H, Ito Y, Nakajima T. ‘Hypothesis of seven balances’: molecular mechanisms behind alcoholic liver diseases and association with PPARα . Journal of Occupational Health. 2009;51(5):391–403. doi: 10.1539/joh.k9001. [DOI] [PubMed] [Google Scholar]
- 88.Wu C, Gilroy R, Taylor R, et al. Alteration of hepatic nuclear receptor-mediated signaling pathways in hepatitis C virus patients with and without a history of alcohol drinking. Hepatology. 2011;54(6):1966–1974. doi: 10.1002/hep.24645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dharancy S, Malapel M, Perlemuter G, et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology. 2005;128(2):334–342. doi: 10.1053/j.gastro.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 90.Goldwasser J, Cohen PY, Lin W, et al. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. Journal of Hepatology. 2011;55(5):963–971. doi: 10.1016/j.jhep.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rakic B, Sagan SM, Noestheden M, et al. Peroxisome proliferator-activated receptor α antagonism inhibits hepatitis C virus replication. Chemistry and Biology. 2006;13(1):23–30. doi: 10.1016/j.chembiol.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 92.Pang M, de la Monte SM, Longato L, et al. PPARδ agonist attenuates alcohol-induced hepatic insulin resistance and improves liver injury and repair. Journal of Hepatology. 2009;50(6):1192–1201. doi: 10.1016/j.jhep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu J, Shen B, Chu ESH, et al. Inhibitory role of peroxisome proliferator-activated receptor gamma in hepatocarcinogenesis in mice and in vitro. Hepatology. 2010;51(6):2008–2019. doi: 10.1002/hep.23550. [DOI] [PubMed] [Google Scholar]
- 94.Schaefer KL, Wada K, Takahashi H, et al. Peroxisome proliferator-activated receptor γ inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Research. 2005;65(6):2251–2259. doi: 10.1158/0008-5472.CAN-04-3037. [DOI] [PubMed] [Google Scholar]
- 95.Yoshizawa K, Cioca DP, Kawa S, Tanaka E, Kiyosawa K. Peroxisome proliferator-activated receptor γ ligand troglitazone induces cell cycle arrest and apoptosis of hepatocellular carcinoma cell lines. Cancer. 2002;95(10):2243–2251. doi: 10.1002/cncr.10906. [DOI] [PubMed] [Google Scholar]
- 96.Cao LQ, Wang XL, Wang Q, et al. Rosiglitazone sensitizes hepatocellular carcinoma cell lines to 5-fluorouracil antitumor activity through activation of the PPARγ signaling pathway. Acta Pharmacologica Sinica. 2009;30(9):1316–1322. doi: 10.1038/aps.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee HJ, Su Y, Yin PH, Lee HC, Chi CW. PPARγ/PGC-1α pathway in E-cadherin expression and motility of HepG2 cells. Anticancer Research. 2009;29(12):5057–5063. [PubMed] [Google Scholar]
- 98.Shim J, Kim BH, Kim YI, et al. The peroxisome proliferator-activated receptor γ ligands, pioglitazone and 15-deoxy-Δ(12,14)-prostaglandin J(2), have antineoplastic effects against hepatitis B virus-associated hepatocellular carcinoma cells. International Journal of Oncology. 2010;36(1):223–231. [PubMed] [Google Scholar]
- 99.Borbath I, Leclercq I, Moulin P, Sempoux C, Horsmans Y. The PPARgamma agonist pioglitazone inhibits early neoplastic occurrence in the rat liver. European Journal of Cancer. 2007;43(11):1755–1763. doi: 10.1016/j.ejca.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 100.Yu J, Qiao L, Zimmermann L, et al. Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo. Hepatology. 2006;43(1):134–143. doi: 10.1002/hep.20994. [DOI] [PubMed] [Google Scholar]
- 101.Hsu MC, Huang CC, Chang HC, Hu TH, Hung WC. Overexpression of Jab1 in hepatocellular carcinoma and its inhibition by peroxisome proliferator-activated receptorγ ligands in vitro and in vivo. Clinical Cancer Research. 2008;14(13):4045–4052. doi: 10.1158/1078-0432.CCR-07-5040. [DOI] [PubMed] [Google Scholar]
- 102.Shah YM, Morimura K, Yang Q, Tanabe T, Takagi M, Gonzalez FJ. Peroxisome proliferator-activated receptor α regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Molecular and Cellular Biology. 2007;27(12):4238–4247. doi: 10.1128/MCB.00317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gonzalez FJ, Shah YM. PPARα: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology. 2008;246(1):2–8. doi: 10.1016/j.tox.2007.09.030. [DOI] [PubMed] [Google Scholar]
- 104.Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nature Reviews. 2012;12(3):181–195. doi: 10.1038/nrc3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Peters JM, Cattley RC, Gonzalez FJ. Role of PPARα in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18(11):2029–2033. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- 106.Hays T, Rusyn I, Burns AM, et al. Role of peroxisome proliferator-activated receptor-α (PPARα) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis. 2005;26(1):219–227. doi: 10.1093/carcin/bgh285. [DOI] [PubMed] [Google Scholar]
- 107.Youssef J, Badr M. Enhanced hepatocarcinogenicity due to agonists of peroxisome proliferator-activated receptors in senescent rats: role of peroxisome proliferation, cell proliferation, and apoptosis. TheScientificWorldJournal. 2002;2:1491–1500. doi: 10.1100/tsw.2002.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Corton JC. Evaluation of the role of peroxisome proliferator-activated receptor α (PPARα) in mouse liver tumor induction by trichloroethylene and metabolites. Critical Reviews in Toxicology. 2008;38(10):857–875. doi: 10.1080/10408440802209796. [DOI] [PubMed] [Google Scholar]
- 109.Tanaka N, Moriya K, Kiyosawa K, Koike K, Aoyama T. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor α in transgenic mice: implications for HCV-associated hepatocarcinogenesis. International Journal of Cancer. 2008;122(1):124–131. doi: 10.1002/ijc.23056. [DOI] [PubMed] [Google Scholar]
- 110.Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARα activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. Journal of Clinical Investigation. 2008;118(2):683–694. doi: 10.1172/JCI33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Morimura K, Cheung C, Ward JM, Reddy JK, Gonzalez FJ. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor α to Wy-14,643-induced liver tumorigenesis. Carcinogenesis. 2006;27(5):1074–1080. doi: 10.1093/carcin/bgi329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cheung C, Akiyama TE, Ward JM, et al. Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor α . Cancer Research. 2004;64(11):3849–3854. doi: 10.1158/0008-5472.CAN-04-0322. [DOI] [PubMed] [Google Scholar]
- 113.Xu L, Han C, Lim K, Wu T. Cross-talk between peroxisome proliferator-activated receptor δ and cytosolic phospholipase A(2)α/cyclooxygenase-2/prostaglandin E(2) signaling pathways in human hepatocellular carcinoma cells. Cancer Research. 2006;66(24):11859–11868. doi: 10.1158/0008-5472.CAN-06-1445. [DOI] [PubMed] [Google Scholar]
- 114.Suh HN, Lee SH, Lee MY, Lee YJ, Lee JH, Han HJ. Role of interleukin-6 in the control of DNA synthesis of hepatocytes: involvement of PKC, p44/42 MAPKs, and PPARδ . Cellular Physiology and Biochemistry. 2008;22(5-6):673–684. doi: 10.1159/000185551. [DOI] [PubMed] [Google Scholar]
- 115.Leader JE, Wang C, Fu M, Pestell RG. Epigenetic regulation of nuclear steroid receptors. Biochemical Pharmacology. 2006;72(11):1589–1596. doi: 10.1016/j.bcp.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 116.Mann J, Chu DCK, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138(2):705.e4–714.e4. doi: 10.1053/j.gastro.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhou Y, Jia X, Zhou M, Liu J. Egr-1 is involved in the inhibitory effect of leptin on PPARγ expression in hepatic stellate cell in vitro. Life Sciences. 2009;84(15-16):544–551. doi: 10.1016/j.lfs.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 118.Xiao H, LeBlanc SE, Wu Q, et al. Chromatin accessibility and transcription factor binding at the PPARγ2 promoter during adipogenesis is protein kinase A-dependent. Journal of Cellular Physiology. 2011;226(1):86–93. doi: 10.1002/jcp.22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mitterberger MC, Kim G, Rostek U, Levine RL, Zwerschke W. Carbonic anhydrase III regulates peroxisome proliferator-activated receptor-gamma2. Experimental Cell Research. 2012;318(8):877–886. doi: 10.1016/j.yexcr.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Farmer SR. Transcriptional control of adipocyte formation. Cell Metabolism. 2006;4(4):263–273. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lobo GP, Amengual J, Li HNM, et al. β,β-carotene decreases peroxisome proliferator receptor γ activity and reduces lipid storage capacity of adipocytes in a β,β-carotene oxygenase 1-dependent manner. Journal of Biological Chemistry. 2010;285(36):27891–27899. doi: 10.1074/jbc.M110.132571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wadosky KM, Willis MS. The story so far: post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. American Journal of Physiology. 2012;302(3):H515–H526. doi: 10.1152/ajpheart.00703.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Juge-Aubry CE, Hammar E, Siegrist-Kaiser C, et al. Regulation of the transcriptional activity of the peroxisome proliferator-activated receptor by phosphorylation of a ligand-independent trans-activating domain. Journal of Biological Chemistry. 1999;274(15):10505–10510. doi: 10.1074/jbc.274.15.10505. [DOI] [PubMed] [Google Scholar]
- 124.Gilde AJ, Van Bilsen M. Peroxisome proliferator-activated receptors (PPARS): regulators of gene expression in heart and skeletal muscle. Acta Physiologica Scandinavica. 2003;178(4):425–434. doi: 10.1046/j.1365-201X.2003.01161.x. [DOI] [PubMed] [Google Scholar]
- 125.Yu S, Reddy JK. Transcription coactivators for peroxisome proliferator-activated receptors. Biochimica et Biophysica Acta. 2007;1771(8):936–951. doi: 10.1016/j.bbalip.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 126.Grimson A, Farh KKH, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Molecular Cell. 2007;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 128.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature Reviews Genetics. 2008;9(2):102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 129.Saini HK, Griffiths-Jones S, Enright AJ. Genomic analysis of human microRNA transcripts. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(45):17719–17724. doi: 10.1073/pnas.0703890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee Y, Kim M, Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. The EMBO Journal. 2004;23(20):4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Breving K, Esquela-Kerscher A. The complexities of microRNA regulation: mirandering around the rules. International Journal of Biochemistry and Cell Biology. 2010;42(8):1316–1329. doi: 10.1016/j.biocel.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 132.Zeng Y, Cullen BR. Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. Journal of Biological Chemistry. 2005;280(30):27595–27603. doi: 10.1074/jbc.M504714200. [DOI] [PubMed] [Google Scholar]
- 133.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 134.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes and Development. 2003;17(24):3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303(5654):95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 136.Melo SA, Esteller M. A precursor microRNA in a cancer cell nucleus: get me out of here! Cell Cycle. 2011;10(6):922–925. doi: 10.4161/cc.10.6.15119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118(1):57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 138.Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123(4):631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 139.Kawamata T, Seitz H, Tomari Y. Structural determinants of miRNAs for RISC loading and slicer-independent unwinding. Nature Structural and Molecular Biology. 2009;16(9):953–960. doi: 10.1038/nsmb.1630. [DOI] [PubMed] [Google Scholar]
- 140.Park CY, Choi YS, McManus MT. Analysis of microRNA knockouts in mice. Human Molecular Genetics. 2010;19(R2):R169–R175. doi: 10.1093/hmg/ddq367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455(7209):64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sood P, Krek A, Zavolan M, Macino G, Rajewsky N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8):2746–2751. doi: 10.1073/pnas.0511045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gao Y, Schug J, McKenna LB, Le Lay J, Kaestner KH, Greenbaum LE. Tissue-specific regulation of mouse microRNA genes in endoderm-derived tissues. Nucleic Acids Research. 2011;39(2):454–463. doi: 10.1093/nar/gkq782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Thum T. MicroRNA therapeutics in cardiovascular medicine. EMBO Molecular Medicine. 2012;4(1):3–14. doi: 10.1002/emmm.201100191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kantharidis P, Wang B, Carew RM, Lan HY. Diabetes complications: the microRNA perspective. Diabetes. 2011;60(7):1832–1837. doi: 10.2337/db11-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nature Reviews Molecular Cell Biology. 2012;13(4):239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sonntag KC. MicroRNAs and deregulated gene expression networks in neurodegeneration. Brain Research. 2010;1338:48–57. doi: 10.1016/j.brainres.2010.03.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nature Reviews Immunology. 2010;10(2):111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 149.Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467(7311):86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- 150.Costinean S, Zanesi N, Pekarsky Y, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in Eμ-miR155 transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(18):7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xu H, He JH, Xiao ZD, et al. Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology. 2010;52(4):1431–1442. doi: 10.1002/hep.23818. [DOI] [PubMed] [Google Scholar]
- 152.Krützfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with “antagomirs”. Nature. 2005;438(7068):685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 153.Castoldi M, Spasic MV, Altamura S, et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. Journal of Clinical Investigation. 2011;121(4):1386–1396. doi: 10.1172/JCI44883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ferland-McCollough D, Ozanne SE, Siddle K, Willisand AE, Bushell M. The involvement of microRNAs in type 2 diabetes. Biochemical Society Transactions. 2010;38(6):1565–1570. doi: 10.1042/BST0381565. [DOI] [PubMed] [Google Scholar]
- 155.Haybaeck J, Zeller N, Heikenwalder M. The parallel universe: microRNAs and their role in chronic hepatitis, liver tissue damage and hepatocarcinogenesis. Swiss Medical Weekly. 2011;141 doi: 10.4414/smw.2011.13287.w13287 [DOI] [PubMed] [Google Scholar]
- 156.Bala S, Szabo G. MicroRNA signature in alcoholic liver disease. International Journal of Hepatology. 2012;2012 doi: 10.1155/2012/498232.498232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Waidmann O, Bihrer V, Pleli T, et al. Serum microRNA-122 levels in different groups of patients with chronic hepatitis B virus infection. Journal of Viral Hepatitis. 2012;19(2):e58–e65. doi: 10.1111/j.1365-2893.2011.01536.x. [DOI] [PubMed] [Google Scholar]
- 158.Henke JI, Goergen D, Zheng J, et al. MicroRNA-122 stimulates translation of hepatitis C virus RNA. The EMBO Journal. 2008;27(24):3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jiang X, Tsitsiou E, Herrick SE, Lindsay MA. MicroRNAs and the regulation of fibrosis. FEBS Journal. 2010;277(9):2015–2021. doi: 10.1111/j.1742-4658.2010.07632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. Journal of the American Society of Nephrology. 2012;23(3):458–469. doi: 10.1681/ASN.2011050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ladeiro Y, Couchy G, Balabaud C, et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47(6):1955–1963. doi: 10.1002/hep.22256. [DOI] [PubMed] [Google Scholar]
- 162.Wang Y, Lee ATC, Ma JZI, et al. Profiling microRNA expression in hepatocellular carcinoma reveals microRNA-224 up-regulation and apoptosis inhibitor-5 as a microRNA-224-specific target. Journal of Biological Chemistry. 2008;283(19):13205–13215. doi: 10.1074/jbc.M707629200. [DOI] [PubMed] [Google Scholar]
- 163.Zheng L, Lv GC, Sheng J, Yang YD. Effect of miRNA-10b in regulating cellular steatosis level by targeting PPAR-α expression, a novel mechanism for the pathogenesis of NAFLD. Journal of Gastroenterology and Hepatology. 2010;25(1):156–163. doi: 10.1111/j.1440-1746.2009.05949.x. [DOI] [PubMed] [Google Scholar]
- 164.Kida K, Nakajima M, Mohri T, et al. PPARalpha is regulated by miR-21 and miR-27b in human liver. Pharmaceutical Research. 2011;28(10):2467–2476. doi: 10.1007/s11095-011-0473-y. [DOI] [PubMed] [Google Scholar]
- 165.Gatfield D, Le Martelot G, Vejnar CE, et al. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes and Development. 2009;23(11):1313–1326. doi: 10.1101/gad.1781009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Karbiener M, Fischer C, Nowitsch S, et al. MicroRNA miR-27b impairs human adipocyte differentiation and targets PPARγ . Biochemical and Biophysical Research Communications. 2009;390(2):247–251. doi: 10.1016/j.bbrc.2009.09.098. [DOI] [PubMed] [Google Scholar]
- 167.Kim SY, Kim AY, Lee HW, et al. MiR-27a is a negative regulator of adipocyte differentiation via suppressing PPARgamma expression. Biochemical and Biophysical Research Communications. 2010;392(3):323–328. doi: 10.1016/j.bbrc.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 168.Lee EK, Lee MJ, Abdelmohsen K, et al. MiR-130 suppresses adipogenesis by inhibiting peroxisome proliferator-activated receptor γ expression. Molecular and Cellular Biology. 2011;31(4):626–638. doi: 10.1128/MCB.00894-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Martinelli R, Nardelli C, Pilone V, et al. MiR-519d overexpression is associated with human obesity. Obesity. 2010;18(11):2170–2176. doi: 10.1038/oby.2009.474. [DOI] [PubMed] [Google Scholar]
- 170.Wang J, Song Y, Zhang Y, et al. Cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and dysfunction in mice. Cell Research. 2012;22(3):516–527. doi: 10.1038/cr.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jennewein C, Von Knethen A, Schmid T, Brüne B. MicroRNA-27b contributes to lipopolysaccharide-mediated peroxisome proliferator-activated receptor γ (PPARγ) mRNA destabilization. Journal of Biological Chemistry. 2010;285(16):11846–11853. doi: 10.1074/jbc.M109.066399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Iliopoulos D, Malizos KN, Oikonomou P, Tsezou A. Integrative MicroRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS ONE. 2008;3(11) doi: 10.1371/journal.pone.0003740.e3740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhou J, Wang KC, Wu W, et al. MicroRNA-21 targets peroxisome proliferators-activated receptor-α in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(25):10355–10360. doi: 10.1073/pnas.1107052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tong JL, Zhang CP, Nie F, et al. MicroRNA 506 regulates expression of PPAR alpha in hydroxycamptothecin-resistant human colon cancer cells. FEBS Letters. 2011;585(22):3560–3568. doi: 10.1016/j.febslet.2011.10.021. [DOI] [PubMed] [Google Scholar]