

Abstract
Basal cell carcinoma (BCC) is the most common human tumor. Mutations in the hedgehog (HH) receptor Patched (PTCH) are the main cause of BCC. Due to their high and increasing incidence, BCC are becoming all the more important for the health care system. Adequate animal models are required for the improvement of current treatment strategies. A good model should reflect the situation in humans (i.e., BCC initiation due to Ptch mutations on an immunocompetent background) and should allow for (i) BCC induction at a defined time point, (ii) analysis of defined BCC stages, and (iii) induction of BCC in 100% of animals. In addition, it should be easy to handle. Here, we compare several currently existing conventional and conditional Ptch knockout mouse models for BCC and their potential use in preclinical research. In addition, we provide new data using conditional Ptch flox/flox mice and the K5-Cre-ER T+/− driver.
1. General Aspects and Current Therapies of BCC
1.1. Epidemiology
BCC is a tumor of the skin and the most prevalent cancer in the Western world. Its incidence is increasing worldwide. Retrospective studies show that the increase in mainland Europe is approximately 1/100,000 persons per year and even 6/100,000 in the UK [1]. It is estimated that the lifetime risk of developing BCC for a child born in 1994 is 28% to 33% [2] and that young people will suffer more and more from this tumor [3]. Risk factors for BCC formation are exposure to ultraviolet radiation (UV) or ionizing radiation (IR), immunosuppression, or a genetic predisposition [4]. Due to their high and increasing incidence, BCC are becoming an important issue for the health care system [5]. In some countries, the cost of care for BCC and other nonmelanoma skin cancers comprises 9% of the costs for all cancers [6].
1.2. Histology
BCC are usually well differentiated and the tumor cells appear histologically similar to basal cells of the epidermis. BCC can be subdivided into two subgroups that show either an indolent or an aggressive growth behavior. The indolent-growth variants comprise nodular/micronodular and superficial BCC. These subtypes occur in 21%/15% and 17% of cases, respectively, and thus are the most common BCC variants [7]. Whereas nodular BCC consist of nests of basaloid cells in the dermis, superficial BCC are characterized by numerous small tumor nests attached to the undersurface of the epidermis by a broad base. The more aggressive tumors are less frequent and include infiltrative, metatypical, morpheaform, or sclerosing BCC (for review see [8]). Although BCC very rarely metastasize, they can result in local tissue destruction due to invasion into deeper layers of the skin, thereby causing significant morbidity [9].
1.3. Molecular Pathogenesis of BCC
BCC are thought to be caused by uncontrolled activation of the hedgehog (HH) signaling pathway. In the majority of cases, this is due to inactivating mutations in the HH receptor and tumor suppressor gene PTCH. PTCH mutations in BCC were first observed in basal cell nevus syndrome (also known as nevoid basal cell carcinoma syndrome or Gorlin-Goltz syndrome), which is a rare familial autosomal-dominant disorder that predisposes the affected individual to developing this tumor. Only a minority of BCC are caused by activating mutations in Smoothened (SMO) (reviewed in [9]).
PTCH normally acts as an inhibitor of HH signaling by repressing the function of SMO. Binding of the HH ligand to PTCH or inactivating PTCH mutations suspend this inhibition, which allows activation of SMO and results in the formation of activator forms of the GLI zinc finger transcription factors GLI2 and GLI3. Activation of GLI2 and GLI3 leads to transcription of GLI1. Thus, the expression level of GLI1 is considered as a reliable indicator of the pathway's activity. Another HH target is PTCH itself, which regulates its expression in a negative feedback (reviewed in [10]). Indeed, nearly all BCC express GLI1 and PTCH, which demonstrates the important role of aberrant HH signaling in these tumors [11].
Several other signaling pathways are presumably involved in BCC tumorigenesis. Mutations of the tumor suppressor gene p53 have been shown in 40% of sporadic BCC [12] and were correlated with aggressive behavior [13–15]. In addition, activation of canonical Wnt/β-catenin signaling seems to play a role in specific histological BCC subtypes. These subtypes include early stages of superficial BCC [16], pilomatricoma (a tumor of the hair follicle [17]) as well as infiltrative BCC variants [18, 19]. Indeed, nuclear β-catenin is found in infiltrative BCC and in superficial BCC [18], but only rarely in human nodular BCC [17, 18, 20]. BCC also express activated AKT [21]. Finally, EGFR signaling seems to be an essential in vivo requirement in HH-driven BCC because EGFR signaling cooperates with the HH pathway to induce genes (e.g., JUN, SOX9, and FGF19) critical for the determination of the oncogenic BCC phenotype [22].
Growth and progression of human BCC is also highly influenced by the tumor microenvironment. For example, tumor-associated macrophages are able to enhance the invasive phenotype and angiogenesis [23]. Furthermore, α-smooth muscle actin positivity of peritumoral fibroblast tends to be greatest in infiltrative tumor areas [24]. In addition, stromal cells of BCC produce high levels of Gremlin1, which is a factor stimulating BCC growth by antagonizing bone morphogenic protein-mediated repression of cell proliferation [25, 26]. Finally, EGFR ligands are increased in the tumor stroma [27], which may influence tumor intrinsic EGFR signaling (see above).
However, whereas all the above-mentioned factors may influence the susceptibility to BCC or the BCC phenotype, deregulation of HH signaling is the central abnormality in all these tumors and seems to play the major role in its formation [9].
1.4. Conventional Treatment Options of BCC
Surgical excision is currently by far the most commonly used treatment of BCC. However, surgery can result in permanent tissue damage and scarring, which is unwanted especially in facial areas. In addition, surgery may be problematic if the tumor is localized around the eye, mouth, or in close vicinity of the cartilage of the nose and ears [9]. This has led to less invasive treatment strategies such as photodynamic therapy or application of imiquimod-containing creams. Photodynamic therapy refers to a technique in which the tumor is treated with a photosensitizing chemical in a cream and is exposed to light several hours later [28]. Imiquimod is an immune response modifier, which stimulates the Toll-like receptor 7 and increases the activity of natural killer cell, macrophages, and the proliferation and differentiation of B lymphocytes [29]. Another option is cryotherapy, which destroys the skin lesion by application of extreme cold such as compressed nitrous oxide [30]. Another agent for topical application is 5-fluorouracil (5% cream), which leads to tumor necrosis. Among the drawbacks of the latter agent is the limited tissue penetration [31].
Although these therapies are associated with moderate morbidity, the outcome is still considered to be unspecific. In addition, these treatments sometimes have side effects such as pain, scarring, and local skin reactions [31]. Together, these data show that the availability of a simplified and more effective treatment would contribute to lower the costs related to this tumor.
1.5. Targeting the HH Signaling Pathway in BCC
The knowledge about the genetic and molecular events involved in BCC pathogenesis has enormously contributed to the establishment of new treatment options. Very successful have been strategies specifically targeting HH signaling. The first small-molecule inhibitor of the HH pathway was the naturally occurring compound cyclopamine that inhibits SMO activity by direct binding [32]. Within the last few years, more potent SMO inhibitors have been developed and are currently being tested in phase I and II clinical trials [33]. Recently, the SMO antagonist vismodegib (Erivedge, GDC-0449) has been approved by the FDA for the treatment of metastasizing and locally destructive BCC [34, 35]. However, although vismodegib shows both remarkable therapeutic and preventive efficacy, the cumulative toxicity of this agent has led to discontinuation of therapy in a substantial fraction of patients [35, 36]. Therefore, it will be necessary to develop strategies that ameliorate some of the common toxicities of this drug [35].
2. Mouse Models of BCC for Preclinical Studies
The establishment of new treatment strategies requires adequate animal models. An ideal model should allow for analysis or modulation of molecular events associated with tumor initiation or tumor progression. It should also permit to evaluate antitumor therapies useful to prevent, inhibit, or even to induce regression of BCC in vivo. To fulfill these requirements, an ideal animal model should allow for analysis of BCC that have reached a defined BCC stage after their initiation in 100% of animals.
Hitherto, several murine BCC models exist. These include Ptch knockout mice and mice overexpressing Hh, oncogenic Smo, Gli1 or Gli2 specifically in the skin using the keratin (K) 5, 6, or 14 promoters. Depending on the gene and the targeted cell type, the skin tumor subtypes range from follicular hamartoma and trichoepithelioma to nodular or invasive BCC [16, 37–45]. In addition, allografts from BCC-bearing Ptch +/− p53−/− mice or from Shh transgenics can be grown in scid mice [43, 46]. Finally, the cell line ASZ001 generated from a BCC of an irradiated Ptch heterozygous mouse (see below) has been successfully implanted into nude mice and used to study the effects of the EGFR-inhibitor afatinib [22].
Since most human BCC arise due to PTCH mutations and since the stromal microenvironment plays an important role in formation and progression of this tumor (see section “Molecular pathogenesis of BCC”), immunocompetent Ptch mutant mice certainly represent the closest model to the human condition.
3. Ptch Knockout Mouse Models for BCC
3.1. Spontaneous Ptch Mutations in Mice
Two spontaneous Ptch mutant animals have been described. The spontaneous recessive mutation “mesenchymal dysplasia” (mes) is caused by a deletion of 32 bp in the C-terminal cytoplasmic domain of Ptch [47]. Ptc1mes/mes mice are viable and show increased proliferation and hyperplasia of the basal cell layer [48]. However, in spite of these skin anomalies Ptc1mes/mes mice do not develop BCC even after exposure to radiation [49].
The Ptch1D11 is a mutation caused by an aberrant recombination event while producing a Ptch null allele for the generation of Ptch neo12/+ mice (see below). The Ptch1D11 locus presumably results in a weak Ptch allele [50]. Ptch1D11/D11 animals are sterile, but otherwise appear normal [50].
3.2. Conventional Ptch Knockout Mice
So far, two different conventional Ptch knockout mouse models for BCC have been described. These are the Ptch neo12 and Ptch neo67 strains, in which exons 1 and 2 or exons 6 and 7, respectively, are deleted in the germline [51, 52]. Homozygous Ptch neo12/neo12 and Ptch neo67/neo67 embryos die around embryonic day 9.5 due to heart and neural tube closure defects. Heterozygous Ptch neo12/+ and Ptch neo67/+ animals survive and show increased susceptibility to spontaneous formation of rhabdomyosarcoma, medulloblastoma, and tiny epidermal hyperproliferations. To induce BCC, Ptch neo12/+ animals are usually exposed to UV three times per week for up to several months [53–55]. After 12 months of chronic UV exposure, all Ptch neo12/+ mice develop lesions with histologic features of human BCC. Of these lesions, 44% can be classified as superficial, 13% have histologic features of nodular or infiltrating human BCC and 43% have features of trichoblastoma [53]. Chronic UV exposure also results in macroscopic tumors. Of these visible tumors, approximately 20% are BCC or trichoblastomas (tumors with follicular differentiation that share many histologic features with BCC), 30% are squamous cell carcinoma (SCC) or keratoacanthomas (SCC-like tumors), and 50% are fibrosarcomas or fibromas [53].
The situation is somewhat different when inducing BCC-like lesions by IR. As shown by Aszterbaum et al. [53], a single dose of 1–4 Gy applied at 2 months of age results in microscopic trichoblastoma-like tumors in all Ptch neo12/+ mice after 1 year. Another study performed by Mancuso et al. revealed that a dose of 3-4 Gy applied to adult Ptch neo67/+ mice at the age of 2-3 months leads to nodular BCC-like lesions in 21–47% of animals, and in infiltrative lesions in 5–12% [45]. IR exposure never results in fibrosarcomas or SCC [45, 53]. Particularly BCC in the IR-induced model further progress into an aggressive phenotype [45]. Immune surveillance was not impaired in either model [53].
On the molecular level, formation of IR-induced nodular BCC requires Ptch heterozygosity in conjunction with mutations in other molecules such as p53 [45]. Moreover, the progression into an aggressive phenotype seems to be associated with biallelic loss of Ptch [45]. This might be different from human BCC, which in most cases lack aggressiveness [56] and which frequently show loss of heterozygosity at the PTCH locus on chromosome 9q22 already at the nonaggressive stage [57–59]. Thus, it remains to be resolved whether loss of the wildtype Ptch allele in irradiated mice indeed triggers BCC aggressiveness or whether it is just a secondary event due to general irradiation-induced genomic instability.
These differences to human BCC and the fact that BCC in irradiated Ptch heterozygous mice develop at undefined time points and in indefinite areas of the exposed skin render this animal model may complicate the examination of early molecular processes involved in the initiation of BCC. However, these mice are a great tool to evaluate new treatment options of microscopic, macroscopic, and aggressive BCC that are caused by Ptch mutations along with additional irradiation-dependent mutations. Indeed, irradiated Ptch neo12/+ knockouts have been used in several preclinical studies (Table 1), which are described in the following section.
Table 1.
Ptch knockout mouse models for preclinical BCC treatment studies.
| BCC model | Mode of BCC induction | Treatment | Reference |
|---|---|---|---|
| Pt ch neo12/+ | UV | Cyclopamine | [60] |
| Pt ch neo12/+ skin punches | UV | CUR61414 | [61] |
| Pt ch neo12/+ | UV | α-difluoromethylornithine | [62] |
| Pt ch neo12/+ | IR/UV | Tazarotene | [55] |
| Pt ch neo12/+ | UV | Tazarotene, ATRA, AGN195813, AGN194204, AGN194310 | [63] |
| Pt ch neo12/+ | IR/UV | Celecoxib, sulindac, MF-tricyclic | [64] |
| Pt ch neo12/+ | UV | Green/black tea | [54] |
| Pt ch neo12/+ K14-Cre-ER p53fl/fl | IR and conditional p53 ablation | CUR61414 | [65] |
| Pt ch neo12/+ K14-Cre-ER p53fl/fl | IR and conditional p53 ablation | Itraconazole | [66] |
| Pt ch neo12/+ K14-Cre-ER p53fl/fl | IR and conditional p53 ablation | Vitamin D3 | [67] |
| Pt ch flox/flox ER T2+/− | conditional Ptch ablation | Calcitriol | [68] |
UV: ultraviolet radiation; IR: ionizing radiation.
To study the effects of the Hh inhibitor cyclopamine [60], BCC have been induced in Ptch neo12/+ animals by UV exposure 3 times per week from age 6 to 32 weeks. After this time, approximately 50% of the mice had developed one or more macroscopic BCC. For the following 20 weeks the animals were treated with cyclopamine that significantly reduces tumor growth [60]. Regression of microscopic BCC after Hh inhibition has also been shown in skin punches of UV-irradiated Ptch neo12/+ mice, which were kept in cell culture for 6 days and treated for the last 4 days with the small molecule inhibitor of Hh signaling CUR61414 [61].
Pt ch neo12/+ animals have also been used to analyze the antitumoral effects of α-difluoromethylornithine (DFMO) [62]. DFMO is a potent inhibitor of cutaneous ornithine decarboxylase, which is expressed in BCC and is known to promote tumor formation [69, 70]. To analyze its antitumoral effects, Ptch neo12/+ animals were irradiated with UV 3 times per week for 32 weeks [62]. Thereafter, the tumor-bearing animals obtained DFMO in the drinking water for 20 weeks. The results show that DMFO reduced the number of visible BCC and diminished BCC-like microscopic lesions. Furthermore, a reduction of Ptch, Gli1, Gli2, and Gli3 expression in nontumor-bearing skin of these animals was evident [62].
A fourth study analyzed the antitumoral activity of the retinoid tazarotene [55]. Retinoids are ligands of the retinoic acid receptor (RAR) and the retinoid X receptor (RXR) and show tumor-suppressive capacity in several tumor entities [71]. Tazarotene was topically applied to the skin of 1.5 or 2.5 months old Ptch neo12/+ mice for 5 consecutive days/week. Two weeks after onset of treatment, that is, at the age of 2 or 3 months, BCC were induced by exposure to UV (3 times/week) or IR (once), respectively. In order to examine the growth of microscopic BCC, skin biopsies of the UV-treated group were taken at the ages of 7, 9, and 11 months, whereas those of the IR-treated group were taken at the age of 10 months. Tazarotene treatment reduced the number and size of microscopic BCC after UV or IR exposure and also prevented formation of macroscopic BCC in IR-exposed animals at the age of 16 months [55]. A follow-up study showed that tazarotene also inhibited the number and size of preexisting microscopic BCC lesions [63]. For this purpose, animals were exposed to IR at 2 months of age and the tazarotene treatment was started 5 months later for additional 5 months. Efficacy was also shown for other retinoid-related agents such as ATRA (pan-RAR agonist), AGN195813 (RARα agonist), or AGN194204 (pan-RXR agonist), however, to a lesser extent [63].
In a next study, the antitumoral effects of cyclooxygenases (COX) inhibitors have been analyzed [64]. COX inhibitors belong to nonsteroidal anti-inflammatory drugs, which are thought to prevent the formation of SCC in humans [72]. The COX inhibitors sulindac (nonspecific COX inhibitor), MF-tricyclic (COX2-specific inhibitor) or celecoxib (COX2-specific inhibitor) were administered starting at the age of 6 weeks and BCC were induced 2 weeks later by exposure to UV (3 times/week, continued until the age of 12 months) or IR (once). At the age of 9 months, the burden of microscopic BCC was reduced by 35% in celecoxib-treated animals and by 50–60% in sulindac- or MF-tricyclic-treated mice [64].
In just another study, Ptch neo12/+ mice were utilized to assess the effect of tea on BCC formation [54]. The rational for this experiment was the observation that green tea may protect against photocarcinogenesis [73]. Green or black tea was added to the drinking water of Ptch neo12/+ mice beginning from the age of 46 days. UV exposure (3 times/week) was started 2 weeks later. However, neither number nor size of BCC was reduced 5 or 7 months after initial UV exposure [54].
Pt ch neo12/+ mice were also used to analyze the effects of itraconazole, vitamin D3, or CUR61414. Similar to CUR61414, the antifungal compound itraconazole and vitamin D3 derivatives have been shown to inhibit Hh signaling, probably by interaction and inhibition of SMO [61, 66, 74]. In order to accelerate BCC carcinogenesis in these studies, Ptch neo12/+ mice were crossed to K14-Cre-ER p53fl/fl mice and p53 was deleted in the Ptch neo12/+ K14-Cre-ER p53fl/fl offspring at the age of 6 weeks by injection of 100 μg tamoxifen on 3 consecutive days. Two weeks later, the animals were exposed once to IR. This resulted in visible BCC at the age of 5-6 months. CUR61414 was applied topically twice daily to BCC on the dorsal skin 5 days a week for up to 42 days. This decreased the tumor size by 60%, which was accompanied by inhibition of Gli1 expression in tumor tissue [65]. The same was shown for itraconazole. When applied orally twice daily for 18 days, itraconazole led to a significant suppression of tumor growth, which was reversible after drug withdrawal [66]. The treatment with vitamin D3 was also effective. Although an impact on tumor size has not been mentioned by the authors, vitamin D3 blocked proliferation and Hh signaling in visible BCC when applied topically 5 days/week for 30 days [67].
Altogether, these data show that conventional Ptch knockout mice are an extremely valuable tool to analyze the efficacy of new anti-BCC drugs. Still there might be a few drawbacks when using irradiated Ptch +/− animals. First, the onset of tumor formation is variable, with tumors arising at different time points and different sites after exposure to radiation [45]. This may complicate studies, which address the use of drugs in small precursors as opposed to progressed tumors. Second, due to the mode of BCC induction by exposure to IR or UV, the molecular mechanisms responsible for BCC formation are probably very heterogeneous. This heterogeneity may also be reflected by the spectrum of skin tumor, which comprises nodular, superficial as well as infiltrative BCC subtypes, trichoblastomas, and also SCC [45, 53]. These characteristics of irradiated Ptch +/− mice may hamper the evaluation of new treatment strategies designed for targeting specific BCC subtypes.
3.3. Conditional Ptch Knockout Mice
With respect to timing of BCC initiation and to investigate defined BCC stages, conditional Ptch knockout mice may be a more suitable model. Conditional knockouts also allow for induction of the Ptch mutation in specific cell lineages, which is important when seeking for, for example, the identification of the cell of origin of BCC [44, 75].
To our knowledge five different conditional Ptch knockout mouse strains have been generated up to date. Of these, only one has been used in a preclinical study targeting BCC [68].
3.3.1. Conditional Ptch Knockout Mice Targeting Exons 1, 2, or 3 of the Ptch Gene
In Ptch neo/neo mice, exon 3 is flanked by loxP sites [76]. The deletion of exon 3 is expected to lead to a premature stop codon and thus to a truncated Ptch protein. Indeed, embryos with a homozygous deletion of Ptch exon 3 display developmental defects and die at embryonic day 9.5. This is similar to conventional Ptch knockouts, in which the homozygous germline mutation results in embryonal lethality between embryonic day 9.0 and 10.5 [51, 52]. In adult Ptch neo/neo mice, BCC can be induced with tissue-specific Cre drivers. For example, BCC arise in conditional Krt6a Cre Ptch neo/neo mice after activation of the Krt6a promotor by topical application of retinoic acid (RA) [44]. Expression of Krt6aCre results in a loss of Ptch in 40% of interfollicular basal cells and outer root sheath cells of multiple hair follicles. Within 4 weeks, 25% of animals develop basal cell invaginations and after 12 additional weeks 100% of mice suffer from BCC, which show high Hh signaling activity. However, since the Krt6a promotor is also permanently active in the companion cell layer, untreated Krt6a Cre Ptch neo/neo mice develop epidermal hyperproliferations by 9 to 12 months and suffer from hair loss. These hyperproliferations are associated with hair follicles or sebaceous glands and do not progress to BCC.
BCC in Ptch neo/neo mice can also be induced using the skin-specific K14Cre or the MxCre mouse. The latter strain is transgenic for a Cre recombinase controlled by the interferon-inducible promoter Mx.1. Besides heamatopoetic cells, liver, spleen, kidney, lung, gastric epithelium, and other tissues [77], the Mx.1 promotor is also active in basal cells of the skin [78]. K14Ptch1Δ/Δ mice (derived from a cross of K14Cre and Ptch neo/neo mice) develop BCC within 3-4 weeks after birth. In M x Ptch1Δ/Δ animals, BCC occur 8–10 weeks after activation of the MxCre by intraperitoneal injection of the immune stimulator polyinosinic-polycytidylic acid (poly(I:C)) on 3 consecutive days. However, due the widespread activity of the Mx.1 promotor, activation of MxCre in Ptch neo/neo mice also ablates Ptch in other organs. This result in B- and T-cell defects, thymic atrophy, increased numbers of myeloid progenitors, and loss of osteoblasts [78]. Due to these defects, this model is rather unsuitable for preclinical studies using anti-BCC drugs.
In addition to Ptch neo/neo mice, other conditional Ptch knockout mouse models targeting Ptch exons 1, 2, or 3 exist. In the Ptch1c/c mouse model, the Ptch exon harboring the first ATG of the Ptch gene and exon 2 are flanked by loxP sites [79]. According to the provided data and to the precise nomenclature (see [80]), the exon containing the first ATG equates exon 1B. Therefore, the floxed region in the Ptch1c/c mouse model additionally contains the alternative first Ptch exons 1 and 1A [80]. After Cre-mediated excision of these exons, the resulting Ptch1ΔloxP/ΔloxP embryos display the same phenotype as homozygous embryos derived from conventional knockouts. However, embryonic and neonatal lethality is also observed in some Ptch1c/c mice, which probably results from Ptch misexpression due to the insertion of a lacZ gene.
The Ptch1c/c mouse model is similar to a third Ptc1F1-2m conditional mouse model described by Taniguchi and colleagues, which likewise allows for the ablation of the exons 1B, 1, 1A, and 2 [81]. Finally, one recent publication described Ptch1neo(fl)Ex2(fl) mice, which develop BCC-like lesions after activation of the Cre recombinase K5Cre*PR1 by RU486 or of Lgr5-EGFP-IHRES-creERT2 by tamoxifen [75]. Although not explicitly mentioned by the authors, we assume that exon 2 is targeted in Ptch1neo(fl)Ex2(fl) animals.
Due to alternative splicing of Ptch exons, the above-mentioned animal models may be somewhat leaky when it comes to a complete deletion of all Ptch transcript variants. As shown by Shimokawa et al. [80], the first Ptch exons 1B, 1 and 1A as well as exons 2–5 can be subjected to alternative splicing. Furthermore, an alternative first exon 1C exists, which is located more than 9 kb upstream of exon 2 and can be spliced into exon 2 or 3 of the Ptch transcript [80]. This has also been shown by Nagao et al. [82, 83], who used a different numbering for the alternative first exons and who named the most upstream exon 1A [82, 83]. Although the role of the various Ptch splice variants is not completely understood, they are expressed in specific tissues and can modulate Hh signaling to various extents [80, 82, 83].
According to Nagao et al., Ptch exons 6 to 9 are not subjected to alternative splicing [83]. Therefore, targeting this region is beneficial in order to obtain a complete loss of regular Ptch transcripts.
3.3.2. Conditional Ptch Knockout Mice Targeting Exons 8 and 9 of the Ptch Gene
We have recently described Ptch flox knockout mice (available at http://www.jax.org/: B6N.129- Ptch1tm1Hahn /J, Stock 012457) permitting the conditional ablation of exons 8 and 9 by introduction of loxP sites into the introns 7 and 9. Ptch flox/flox mice are born at the expected Mendelian ratio and are viable and fertile. Neither the loxP sites nor the neomycin resistance cassette in intron 9 disturb the normal splicing of the Ptch mRNA derived from the Ptch flox allele [84]. As reported by our group, the excision of exons 8 and 9 can be carried out very effectively, thereby generating the Ptch del allele [20, 84–86]. This results in an aberrant Ptch transcript with exon 7 spliced into exon 10 and leads to a frameshift and a premature stop codon. The postulated truncated protein consists of 341 instead of 1093 aminoacids and lacks the sterol sensing domain, the second extracellular loop, and the C-terminus. Due to the lack of appropriate Ptch antibodies, we were not able to detect this protein, but the phenotype of Ptch del/del embryos indicates a complete loss of Ptch function. Indeed, all homozygous Ptch del/del mutants die before embryonic day 10 in utero. Ptch del/+ mice survive and develop malformations at incidences similar to those observed in conventional Ptch knockout mice on the same genetic background [20, 85].
For the induction of BCC, Ptch flox/flox mice can be crossed to Rosa26CreERT2 mice (hereafter ERT2 mice) that express a tamoxifen-inducible Cre recombinase under the control of the ubiquitously active ROSA26 promoter [87]. Activation of ERT2 by a single intramuscular (i.m.) injection of 100 μg tamoxifen results in BCC in 100% of animals. Microscopically, BCC precursors are visible 45 days after tamoxifen induction. The tumors are fully developed by day 90 (Figure 1(a)) [85]. After that time, the tumors start to regress, which is becoming obvious 200 days after tamoxifen-treatment [20]. All BCC in this model have features of the human nodular subtype and are noninvasive. As indicated by abundant Gli1 and Ptch expression, they are characterized by strong Hh signaling activity [20, 85]. The tumors develop preferentially on ears and tails and are very rarely detected in hairy skin. The reason for this preference is unknown but may involve a better blood circulation in ears and tails, resulting in elevated tamoxifen concentrations after i.m. application (discussed in [85]).
Figure 1.
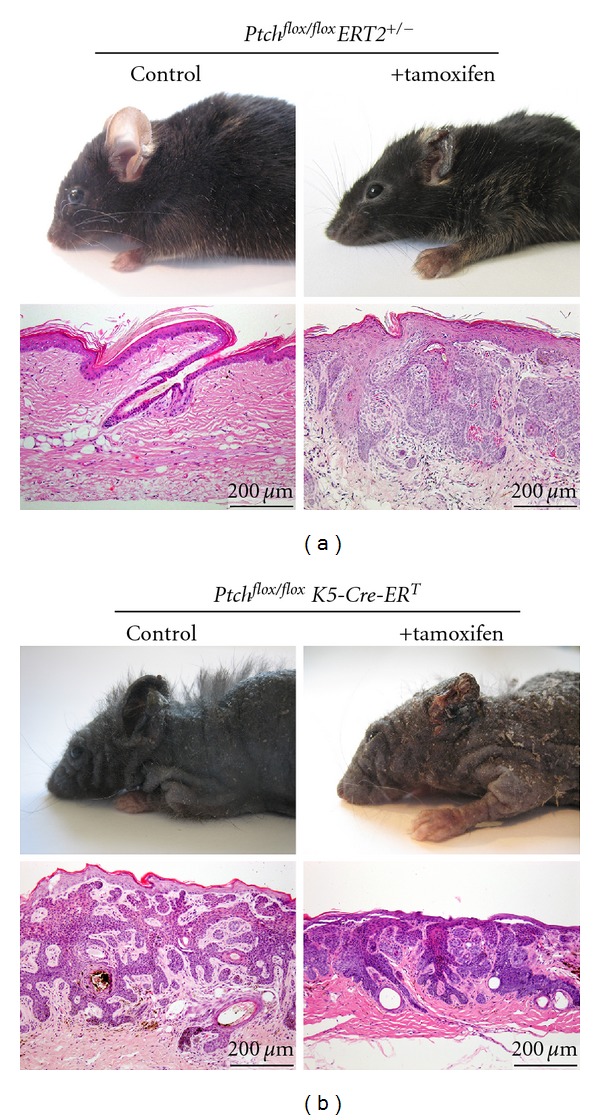
Features of BCC in Ptch flox/flox ER T2+/− and Ptch flox/flox K5-Cre-ER T mice. Ptch flox/flox animals were bred with the mouse lines ERT2 or K5-Cre-ER T. The respective Ptch flox/+ ER T2+/− and Ptch flox/+ K5-Cre-ER T mice were backcrossed to Ptch flox/flox mice to obtain Ptch flox/flox ER T2+/− and Ptch flox/flox K5-Cre-ER T mice. ERT2 or K5-Cre-ER T was activated by one intramuscular (i.m.) injection of 100 μg tamoxifen as described recently [20, 85], or by intraperitoneal injections of 1 mg tamoxifen (10 μg/μL in sterile ethanol/sun flower oil 1 : 25) for 5 consecutive days (see [88]), respectively. Genotyping of the Ptch flox, Ptch del, ERT2, and K5-Cre-ER Talleles was performed as described recently [20, 84, 85]. All mice used in the study were handled in accordance with the German animal protection law. (a) shows the appearance and histology of skin from control and tamoxifen-treated Ptch flox/flox ER T2+/− mice and (b) shows that of control and tamoxifen-treated Ptch flox/flox K5-Cre-ER T mice.
This shows that in Ptch flox/flox ER T2+/− mice BCC can be induced very easily and reliably by one single injection of tamoxifen. Furthermore, in this model, where all mice show full developed BCC 90 days after activation of ERT2, specific antitumor treatments can be commenced at specific time points after tumor induction and at a defined age of the animals.
As indicated in Table 1, we recently examined the antitumor effects of calcitriol, which is the physiologically active form of vitamin D3 [68]. Calcitriol treatment was started either immediately or 60 days after BCC initiation. The treatment was continued until day 90, when all mice were sacrificed. The study revealed that BCC growth was significantly inhibited in mice treated from days 0 to 90, but not in those treated from days 60 to 90 [68]. These data show that conditional Ptch flox/flox ER T2+/− mice are particularly useful to study the preventive or curative effects of a specific anticancer drug. This is due to the reliable BCC initiation and progression to early precursors (after 45 days) and fully developed (after 90 days) tumors.
Although the Ptch flox/flox ER T2+/− BCC model is very easy to handle (i.e., application of one single dose of tamoxifen) and is solid with respect to induction of a specific BCC subtype (i.e., 100% of animals develop the nodular subtype 90 days after BCC induction), it also may have some disadvantages. Due to the ubiquitous expression of ERT2, the i.m. application of even a low dose of tamoxifen may cause Ptch deletion in other cell lineages or tissues. Even though we have not found any evidence for Ptch-ablation in other organs than the epidermis and the injected muscle [20, 85], we now have crossed Ptch flox/flox animals to K5-Cre-ER T mice, which express the tamoxifen-inducible ER T selectively in cells of the basal layer of the skin [89–92]. Activation of ER T by 4-hydroxy-tamoxifen (the active metabolite of tamoxifen) is ~10-fold less efficient than that of ERT2 [89]. Therefore, a cumulative dose of 5 mg has been used to activate K5-Cre-ER T in 10-weeks-old Ptch flox/flox K5-Cre-ER T animals. Untreated Ptch flox/flox K5-Cre-ER T mice served as controls. All Ptch flox/flox K5-Cre-ER T mice developed BCC on tails and ears after tamoxifen injection (Figure 1(b)). They also suffered from BCC in hairy skin. Importantly, BCC also developed in Ptch flox/flox K5-Cre-ER T untreated control mice. Some control mice even suffered from complete hair loss (Figure 1(b)). Histological examination revealed that 86% of the controls (12/14) have developed BCC at the age of 87–172 days (Table 2). After 200–246 days, all control mice have developed tumors even without K5-Cre-ER T activation (Table 2, Figure 1(b)). The leakiness of K5-Cre-ER T was also demonstrated on molecular level. Thus, the amount of recombined Ptch alleles in DNA isolated from skin derived from untreated controls was up to 83% (n = 5, mean 62%). This was almost identical to tamoxifen-treated Ptch flox/flox K5-Cre-ER T animals (amount of recombined Ptch allels 91%, n = 6) (Figure 2(a)). Consistent with these data, untreated Ptch flox/flox K5-Cre-ER T mice also showed high expression of Ptch del transcripts in the skin (Figure 2(b)).
Table 2.
BCC formation in Ptch flox/flox flox ER T2+/− and Ptch flox/flox K5Cre ER T +/− mice.
| Genotype | Age at tamoxifen application | n | Age range (days) | Mice with BCC | Healthy |
|---|---|---|---|---|---|
| Pt ch flox/flox flox ER T2+/− | — | 13 | 56–293 | 0 | 13 |
| *Ptch flox/flox flox ER T2+/− | 42–56 days | 10 | 93–365 | 10 | 0 |
| Pt ch flox/flox K5Cre ER T +/− | — | 14 | 87–172 | 12 | 2 |
| Pt ch flox/flox K5Cre ER T +/− | — | 14 | 200–246 | 14 | 0 |
| Pt ch flox/flox K5Cre ER T +/− | 55–82 days | 9 | 132–170 | 9 | 0 |
*Data already published in [85].
Figure 2.
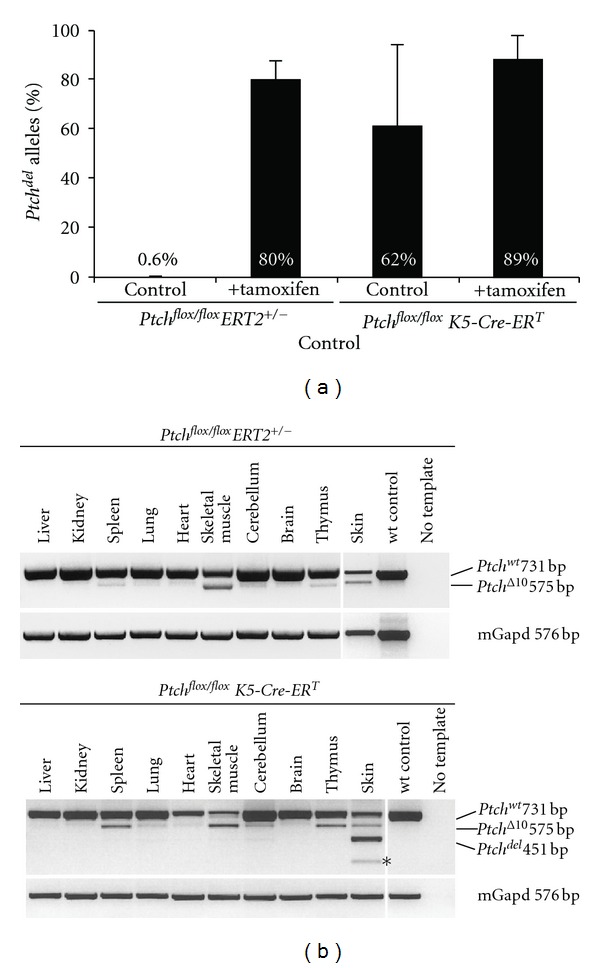
Ptch recombination and expression in Ptch flox/flox ER T2+/− and Ptch flox/flox K5-Cre-ER T mice. (a) The efficiency of loxP recombination at the Ptch locus in DNA derived from tail skin from untreated and tamoxifen treated mice was determined by allele-specific real-time PCR as described in [85]. (b) Ptch transcripts in different tissues were analyzed by RT-PCR. The transcripts derived from the Ptch flox and the Ptch del locus (the latter equates to the floxed Ptch locus after Cre-mediated excision) were analyzed by semiquantitative RT-PCR as described in [85]. In the skin, untreated Ptch flox/flox ER T2+/− mice only expressed Ptch wt transcripts and the normally occurring Ptch Δ10 transcript lacking exon 10. In contrast, untreated Ptch flox/flox K5-Cre-ER T mice expressed Ptch del transcripts and Ptch del transcripts lacking exon 10 (asterisk) in the skin.
This is considerably different to untreated Ptch flox/flox ER T2+/− mice, which do not develop any skin tumors within up to 293 days (n = 13) (Table 2), and which do not show recombination at the floxed Ptch locus in the absence of tamoxifen. Thus, whereas the recombination is 89% in tamoxifen-treated mice (n = 4), it is only 0.6% in untreated Ptch flox/flox ER T2+/− animals (n = 7) and Ptch del transcripts were never detected in any of the examined tissues (Figure 2(a)).
Although BCC in both the Ptch flox/flox ER T2+/− and Ptch flox/flox K5-Cre-ER T model are identical based on histology and also at the level of Hh signaling activity (i.e., BCC of both models express Gli1 and Ptch), K5-Cre-ER T is highly leaky resulting in BCC formation even without Cre activation. Therefore, K5-Cre-ER T should not be used in combination with Ptch flox/flox mice if exact timing of BCC induction is of interest. However, since leakiness of CreER lines can differ between conditional mouse strains [93] it remains to be elucidated whether K5-Cre-ER T leakiness is also seen in other conditional Ptch models.
4. Conclusion
BCC is the most common cancer in humans. Due to their high and increasing incidence, the improvement of current treatment options and the development of new treatment approaches are of great importance. Based on the essential role of HH signaling in formation of BCC, targeting this pathway is currently being put forward (for a review on 36 HH inhibitory compounds see [94]). The preclinical evaluation of these anti-BCC drugs requires good animal models. General requirements for such a model are a close relationship to the human situation (i.e., BCC caused by Ptch mutations on an immunocompetent background), reliable induction of BCC, defined BCC growth, and easy handling.
We have compared several Ptch knockout mouse models suitable for preclinical studies. So far, most studies have been conducted in UV- or IR-exposed conventional heterozygous Ptch knockout mice. Whereas UV-exposure leads to both superficial and nodular BCC and several other tumors, IR-exposure results in nodular and infiltrative BCC. Although the UV- or IR-related BCC models are valuable tools to analyze the antitumoral response of BCC, the responsiveness of defined BCC stages (i.e., early-stage or fully developed) or subtypes (e.g., nodular or superficial) in these models is hard to analyze due to heterogeneous BCC growth. For this purpose, conditional inactivation of Ptch by inducible and cell-specific Cre drivers may be advantageous. Five different conditional Ptch knockout mouse strains are currently available. However, preclinical studies on anti-BCC drugs have only been carried out in one of them. As revealed by this study, conditional Ptch ablation indeed enables the investigator to accurately induce BCC at a defined time point. In addition, conditional Ptch ablation results in a homogeneous BCC histology, which may be due to omission of irradiation. Therefore, conditional Ptch knockout mice are a valuable tool to study the curative or preventive effects of a certain drug on defined BCC subtypes and stages.
Acknowledgments
The authors thank Susann Peter and Stefan Wolf for animal care, Anton Berns (Netherlands Cancer Institute, Amsterdam, The Netherlands) for Rosa26CreERT2 mice and Pierre Chambon and Daniel Metzger (University Strasbourg, Strasbourg, France) for the K5-Cre-ER T mouse strain. This work was supported by the Grant HA 2197/5-1 FOR942 from Deutsche Forschungsgemeinschaft to H. Hahn.
Abbreviations
- Ptch:
Patched
- BCC:
Basal cell carcinoma.
References
- 1.Lomas A, Leonardi-Bee J, Bath-Hextall F. A systematic review of worldwide incidence of nonmelanoma skin cancer. British Journal of Dermatology. 2012;166(5):1069–1080. doi: 10.1111/j.1365-2133.2012.10830.x. [DOI] [PubMed] [Google Scholar]
- 2.Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. Journal of the American Academy of Dermatology. 1994;30(5):774–778. doi: 10.1016/s0190-9622(08)81509-5. [DOI] [PubMed] [Google Scholar]
- 3.De Vries E, Van De Poll-Franse LV, Louwman WJ, De Gruijl FR, Coebergh JWW. Predictions of skin cancer incidence in the Netherlands up to 2015. British Journal of Dermatology. 2005;152(3):481–488. doi: 10.1111/j.1365-2133.2005.06386.x. [DOI] [PubMed] [Google Scholar]
- 4.Kwasniak LA, Garcia-Zuazaga J. Basal cell carcinoma: evidence-based medicine and review of treatment modalities. International Journal of Dermatology. 2011;50(6):645–658. doi: 10.1111/j.1365-4632.2010.04826.x. [DOI] [PubMed] [Google Scholar]
- 5.Roewert-Huber J, Lange-Asschenfeldt B, Stockfleth E, Kerl H. Epidemiology and aetiology of basal cell carcinoma. British Journal of Dermatology. 2007;157(supplement 2):47–51. doi: 10.1111/j.1365-2133.2007.08273.x. [DOI] [PubMed] [Google Scholar]
- 6.Staples MP, Elwood M, Burton RC, Williams JL, Marks R, Giles GG. Non-melanoma skin cancer in Australia: the 2002 national survey and trends since 1985. Medical Journal of Australia. 2006;184(1):6–10. doi: 10.5694/j.1326-5377.2006.tb00086.x. [DOI] [PubMed] [Google Scholar]
- 7.Sexton M, Jones DB, Maloney ME. Histologic pattern analysis of basal cell carcinoma: study of a series of 1039 consecutive neoplasms. Journal of the American Academy of Dermatology. 1990;23(6):1118–1126. doi: 10.1016/0190-9622(90)70344-h. [DOI] [PubMed] [Google Scholar]
- 8.Crowson AN. Basal cell carcinoma: biology, morphology and clinical implications. Modern Pathology. 2006;19(supplement 2):S127–S147. doi: 10.1038/modpathol.3800512. [DOI] [PubMed] [Google Scholar]
- 9.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nature Reviews Cancer. 2008;8(10):743–754. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends in Pharmacological Sciences. 2009;30(6):303–312. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 11.Dahmane N, Lee J, Robins P, Heller P, Ruiz I Altaba A. Activation of the transcription factor Gli1 and the sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389(6653):876–881. doi: 10.1038/39918. [DOI] [PubMed] [Google Scholar]
- 12.Reifenberger J, Wolter M, Knobbe CB, et al. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. British Journal of Dermatology. 2005;152(1):43–51. doi: 10.1111/j.1365-2133.2005.06353.x. [DOI] [PubMed] [Google Scholar]
- 13.Auepemkiate S, Boonyaphiphat P, Thongsuksai P. p53 expression related to the aggressive infiltrative histopathological feature of basal cell carcinoma. Histopathology. 2002;40(6):568–573. doi: 10.1046/j.1365-2559.2002.01393.x. [DOI] [PubMed] [Google Scholar]
- 14.Bolshakov S, Walker CM, Strom SS, et al. p53 mutations in human aggressive and nonaggressive basal and squamous cell carcinomas. Clinical Cancer Research. 2003;9(1):228–234. [PubMed] [Google Scholar]
- 15.Ansarin H, Daliri M, Soltani-Arabshahi R. Expression of p53 in aggressive and non-aggressive histologic variants of basal cell carcinoma. European Journal of Dermatology. 2006;16(5):543–547. [PubMed] [Google Scholar]
- 16.Yang SH, Andl T, Grachtchouk V, et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/β-catenin signaling. Nature Genetics. 2008;40(9):1130–1135. doi: 10.1038/ng.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doglioni C, Piccinin S, Demontis S, et al. Alterations of β-catenin pathway in non-melanoma skin tumors: loss of α-ABC nuclear reactivity correlates with the presence of β-catenin gene mutation. American Journal of Pathology. 2003;163(6):2277–2287. doi: 10.1016/s0002-9440(10)63585-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Bahrawy M, El-Masry N, Alison M, Poulsom R, Fallowfield M. Expression of β-catenin in basal cell carcinoma. British Journal of Dermatology. 2003;148(5):964–970. doi: 10.1046/j.1365-2133.2003.05240.x. [DOI] [PubMed] [Google Scholar]
- 19.Pálmer HG, Anjos-Afonso F, Carmeliet G, Takeda H, Watt FM. The vitamin D receptor is a Wnt effector that controls hair follicle differentiation and specifies tumor type in adult epidermis. PLoS ONE. 2008;3(1) doi: 10.1371/journal.pone.0001483.e1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nitzki F, Zibat A, König S, et al. Tumor stroma-derived Wnt5a induces differentiation of basal cell carcinoma of PTCH-mutant mice via caMKII. Cancer Research. 2010;70(7):2739–2748. doi: 10.1158/0008-5472.CAN-09-3743. [DOI] [PubMed] [Google Scholar]
- 21.Hafner C, Landthaler M, Vogt T. Activation of the PI3K/AKT signalling pathway in non-melanoma skin cancer is not mediated by oncogenic PIK3CA and AKT1 hotspot mutations. Experimental Dermatology. 2010;19(8):e222–e227. doi: 10.1111/j.1600-0625.2009.01056.x. [DOI] [PubMed] [Google Scholar]
- 22.Eberl M, Klingler S, Mangelberger D, et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Molecular Medicine. 2012;4(3):218–233. doi: 10.1002/emmm.201100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tjiu JW, Chen JS, Shun CT, et al. Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. Journal of Investigative Dermatology. 2009;129(4):1016–1025. doi: 10.1038/jid.2008.310. [DOI] [PubMed] [Google Scholar]
- 24.Dingemans KP, Ramkema MD, Koopman G, Van Der Wal AC, Das PK, Pals ST. The expression of CD44 glycoprotein adhesion molecules in basal cell carcinomas is related to growth pattern and invasiveness. British Journal of Dermatology. 1999;140(1):17–25. doi: 10.1046/j.1365-2133.1999.02602.x. [DOI] [PubMed] [Google Scholar]
- 25.Sneddon JB, Zhen HH, Montgomery K, et al. Bone morphogenetic protein antagonist gremlin 1 is wideley expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(40):14842–14847. doi: 10.1073/pnas.0606857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valin A, Barnay-Verdier S, Robert T, et al. PTCH 1+/- dermal fibroblasts isolated from healthy skin of Gorlin syndrome patients exhibit features of carcinoma associated fibroblasts. PLoS ONE. 2009;4(3) doi: 10.1371/journal.pone.0004818.e4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rittié L, Kansra S, Stoll SW, et al. Differential ErbB1 signaling in squamous cell versus basal cell carcinoma of the skin. American Journal of Pathology. 2007;170(6):2089–2099. doi: 10.2353/ajpath.2007.060537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee Y, Baron ED. Photodynamic therapy: current evidence and applications in dermatology. Seminars in Cutaneous Medicine and Surgery. 2011;30(4):199–209. doi: 10.1016/j.sder.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Wenzel J, Uerlich M, Haller O, Bieber T, Tueting T. Enhanced type I interferon signaling and recruitment of chemokine receptor CXCR3-expressing lymphocytes into the skin following treatment with the TLR7-agonist imiquimod. Journal of Cutaneous Pathology. 2005;32(4):257–262. doi: 10.1111/j.0303-6987.2005.00297.x. [DOI] [PubMed] [Google Scholar]
- 30.Moesen I, Duncan M, Cates C, et al. Nitrous oxide cryotherapy for primary periocular basal cell carcinoma: outcome at 5 years follow-up. British Journal of Ophthalmology. 2011;95(12):1679–1681. doi: 10.1136/bjo.2009.173021. [DOI] [PubMed] [Google Scholar]
- 31.Ceilley RI, Del Rosso JQ. Current modalities and new advances in the treatment of basal cell carcinoma. International Journal of Dermatology. 2006;45(5):489–498. doi: 10.1111/j.1365-4632.2006.02673.x. [DOI] [PubMed] [Google Scholar]
- 32.Talpale J, Chen JK, Cooper MK, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406(6799):1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 33.Goppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. Journal of Skin Cancer. 2011;2011:8 pages. doi: 10.1155/2011/650258.650258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dlugosz A, Agrawal S, Kirkpatrick P. Vismodegib. Nature Reviews Drug Discovery. 2012;11(6):437–438. doi: 10.1038/nrd3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudin CM. Vismodegib. Clinical Cancer Research. 2012;18(12):3218–3222. doi: 10.1158/1078-0432.CCR-12-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. New England Journal of Medicine. 2012;366(23):2180–2188. doi: 10.1056/NEJMoa1113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hutchin ME, Kariapper MST, Grachtchouk M, et al. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: conditional skin tumorigenesis recapitulates the hair growth cycle. Genes and Development. 2005;19(2):214–223. doi: 10.1101/gad.1258705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grachtchouk M, Mo R, Yu S, et al. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nature Genetics. 2000;24(3):216–217. doi: 10.1038/73417. [DOI] [PubMed] [Google Scholar]
- 39.Grachtchouk V, Grachtchouk M, Lowe L, et al. The magnitude of hedgehog signaling activity defines skin tumor phenotype. EMBO Journal. 2003;22(11):2741–2751. doi: 10.1093/emboj/cdg271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nilsson M, Undèn AB, Krause D, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(7):3438–3443. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Youssef KK, Van Keymeulen A, Lapouge G, et al. Identification of the cell lineage at the origin of basal cell carcinoma. Nature Cell Biology. 2010;12(3):299–305. doi: 10.1038/ncb2031. [DOI] [PubMed] [Google Scholar]
- 42.Sheng H, Goich S, Wang A, et al. Dissecting the oncogenic potential of Gli2: deletion of an NH2-terminal fragment alters skin tumor phenotype. Cancer Research. 2002;62(18):5308–5316. [PubMed] [Google Scholar]
- 43.Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH, Scott MP. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276(5313):817–821. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- 44.Adolphe C, Hetherington R, Ellis T, Wainwright B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Research. 2006;66(4):2081–2088. doi: 10.1158/0008-5472.CAN-05-2146. [DOI] [PubMed] [Google Scholar]
- 45.Mancuso M, Pazzaglia S, Tanori M, et al. Basal cell carcinoma and its development: insights from radiation-induced tumors in Ptch1-deficient mice. Cancer Research. 2004;64(3):934–941. doi: 10.1158/0008-5472.can-03-2460. [DOI] [PubMed] [Google Scholar]
- 46.Wang GY, So PL, Wang L, Libove E, Wang J, Epstein EH. Establishment of murine Basal cell carcinoma allografts: a potential model for preclinical drug testing and for molecular analysis. Journal of Investigative Dermatology. 2011;131(11):2298–2305. doi: 10.1038/jid.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Makino S, Masuya H, Ishijima J, Yada Y, Shiroishi T. A spontaneous mouse mutation, mesenchymal dysplasia (mes), is caused by a deletion of the most C-terminal cytoplasmic domain of Patched (ptc) Developmental Biology. 2001;239(1):95–106. doi: 10.1006/dbio.2001.0419. [DOI] [PubMed] [Google Scholar]
- 48.Nieuwenhuis E, Barnfield PC, Makino S, Hui CC. Epidermal hyperplasia and expansion of the interfollicular stem cell compartment in mutant mice with a C-terminal truncation of Patched1. Developmental Biology. 2007;308(2):547–560. doi: 10.1016/j.ydbio.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 49.Li ZJ, Hui CC. BCC and the secret lives of Patched: insights from Patched mouse models. In: Madan V, editor. Basal Cell Carcinoma. 2012. [Google Scholar]
- 50.Oro AE, Higgins K. Hair cycle regulation of Hedgehog signal reception. Developmental Biology. 2003;255(2):238–248. doi: 10.1016/s0012-1606(02)00042-8. [DOI] [PubMed] [Google Scholar]
- 51.Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse Patched mutants. Science. 1997;277(5329):1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 52.Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nature Medicine. 1998;4(5):619–622. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- 53.Aszterbaum M, Epstein J, Anthony O, et al. Ultraviolet and ionizing radiation enhance the growth of BCC and trichoblastomas in Patched heterozygous knockout mice. Nature Medicine. 1999;5(11):1285–1291. doi: 10.1038/15242. [DOI] [PubMed] [Google Scholar]
- 54.Hebert JL, Khugyani F, Athar M, Kopelovich L, Epstein EH, Aszterbaum M. Chemoprevention of basal cell carcinomas in the ptc1+/- mouse-green and black tea. Skin Pharmacology and Applied Skin Physiology. 2001;14(6):358–362. doi: 10.1159/000056369. [DOI] [PubMed] [Google Scholar]
- 55.So PL, Lee K, Hebert J, et al. Topical tazarotene chemoprevention reduces basal cell carcinoma number and size and Ptch 1+/- mice exposed to ultraviolet or ionizing radiation. Cancer Research. 2004;64(13):4385–4389. doi: 10.1158/0008-5472.CAN-03-1927. [DOI] [PubMed] [Google Scholar]
- 56.Miller SJ. Biology of basal cell carcinoma. Journal of the American Academy of Dermatology. 1991;24(1):1–13. doi: 10.1016/0190-9622(91)70001-i. [DOI] [PubMed] [Google Scholar]
- 57.Gailani MR, Stahle-Backdahl M, Leffell DJ, et al. The role of the human homologue of Drosophila Patched in sporadic basal cell carcinomas. Nature Genetics. 1996;14(1):78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- 58.Undén AB, Zaphiropoulos PG, Bruce K, Toftgård R, Ståhle-Bäckdahl M. Human Patched (Ptch) mRNA is overexpressed consistently in tumor cells of both familial and sporadic basal cell carcinoma. Cancer Research. 1997;57(12):2336–2340. [PubMed] [Google Scholar]
- 59.Teh MT, Blaydon D, Chaplin T, et al. Genomewide single nucleotide polymorphism microarray mapping in basal cell carcinomas unveils uniparental disomy as a key somatic event. Cancer Research. 2005;65(19):8597–8603. doi: 10.1158/0008-5472.CAN-05-0842. [DOI] [PubMed] [Google Scholar]
- 60.Athar M, Li C, Tang X, et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of fas expression and apoptosis. Cancer Research. 2004;64(20):7545–7552. doi: 10.1158/0008-5472.CAN-04-1393. [DOI] [PubMed] [Google Scholar]
- 61.Williams JA, Guicherit OM, Zaharian BI, et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(8):4616–4621. doi: 10.1073/pnas.0732813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang X, Kim AL, Feith DJ, et al. Ornithine decarboxylase is a target for chemoprevention of basal and squamous cell carcinomas in Ptch 1+/- mice. Journal of Clinical Investigation. 2004;113(6):867–875. doi: 10.1172/JCI20732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.So PL, Fujimoto MA, Epstein EH., Jr Pharmacologic retinoid signaling and physiologic retinoic acid receptor signaling inhibit basal cell carcinoma tumorigenesis. Molecular Cancer Therapeutics. 2008;7(5):1275–1284. doi: 10.1158/1535-7163.MCT-07-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang JY, Aszterbaum M, Athar M, et al. Basal cell carcinoma chemoprevention with nonsteroidal anti-inflammatory drugs in genetically predisposed PTCH 1+/- humans and mice. Cancer Prevention Research. 2010;3(1):25–34. doi: 10.1158/1940-6207.CAPR-09-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang T, Tang JY, Li D, et al. Targeting superficial or nodular basal cell carcinoma with topically formulated small molecule inhibitor of smoothened. Clinical Cancer Research. 2011;17(10):3378–3387. doi: 10.1158/1078-0432.CCR-10-3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17(4):388–399. doi: 10.1016/j.ccr.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang JY, Xiao TZ, Oda Y, et al. Vitamin D3 inhibits hedgehog signaling and proliferation in murine basal cell carcinomas. Cancer Prevention Research. 2011;4(5):744–751. doi: 10.1158/1940-6207.CAPR-10-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uhmann A, Niemann H, Lammering B, et al. Antitumoral effects of calcitriol in basal cell carcinomas involve inhibition of hedgehog signaling and induction of vitamin D receptor signaling and differentiation. Molecular Cancer Therapeutics. 2011;10(11):2179–2188. doi: 10.1158/1535-7163.MCT-11-0422. [DOI] [PubMed] [Google Scholar]
- 69.Gökmen SS, Aygit AC, Ayhan MS, Yorulmaz F, Gülen Ş. Significance of arginase and ornithine in malignant tumors of the human skin. Journal of Laboratory and Clinical Medicine. 2001;137(5):340–344. doi: 10.1067/mlc.2001.114543. [DOI] [PubMed] [Google Scholar]
- 70.Smith MK, Trempus CS, Gilmour SK. Co-operation between follicular ornithine decarboxylase and v-Ha-ras induces spontaneous papillomas and malignant conversion in transgenic skin. Carcinogenesis. 1998;19(8):1409–1415. doi: 10.1093/carcin/19.8.1409. [DOI] [PubMed] [Google Scholar]
- 71.Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nature Reviews Cancer. 2001;1(3):181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- 72.Butler GJ, Neale R, Green AC, Pandeya N, Whiteman DC. Nonsteroidal anti-inflammatory drugs and the risk of actinic keratoses and squamous cell cancers of the skin. Journal of the American Academy of Dermatology. 2005;53(6):966–972. doi: 10.1016/j.jaad.2005.05.049. [DOI] [PubMed] [Google Scholar]
- 73.Ahmad N, Mukhtar H. Cutaneous photochemoprotection by green tea: a brief review. Skin Pharmacology and Applied Skin Physiology. 2001;14(2):69–76. doi: 10.1159/000056336. [DOI] [PubMed] [Google Scholar]
- 74.Bijlsma MF, Spek CA, Zivkovic D, van de Water S, Rezaee F, Peppelenbosch MP. Repression of smoothened by Patched-dependent (pro-)vitamin D3 secretion. PLoS biology. 2006;4(8):p. e232. doi: 10.1371/journal.pbio.0040232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kasper M, Jaks V, Are A, et al. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(10):4099–4104. doi: 10.1073/pnas.1014489108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ellis T, Smyth I, Riley E, et al. Patched 1 conditional null allele in mice. Genesis. 2003;36(3):158–161. doi: 10.1002/gene.10208. [DOI] [PubMed] [Google Scholar]
- 77.Schneider A, Zhang Y, Guan Y, Davis LS, Breyer MD. Differential, inducible gene targeting in renal epithelia, vascular endothelium, and viscera of Mx1Cre mice. American Journal of Physiology. 2003;284(2):F411–F417. doi: 10.1152/ajprenal.00235.2002. [DOI] [PubMed] [Google Scholar]
- 78.Siggins SL, Nguyen NYN, McCormack MP, et al. The Hedgehog receptor Patched1 regulates myeloid and lymphoid progenitors by distinct cell-extrinsic mechanisms. Blood. 2009;114(5):995–1004. doi: 10.1182/blood-2009-03-208330. [DOI] [PubMed] [Google Scholar]
- 79.Mak KK, Chen MH, Day TF, Chuang PT, Yang Y. Wnt/β-catenin signaling interacts differentially with Ihh signaling in controlling endochondral bone and synovial joint formation. Development. 2006;133(18):3695–3707. doi: 10.1242/dev.02546. [DOI] [PubMed] [Google Scholar]
- 80.Shimokawa T, Rahnama F, Zaphiropoulos PG. A novel first exon of the Patched1 gene is upregulated by Hedgehog signaling resulting in a protein with pathway inhibitory functions. FEBS Letters. 2004;578(1-2):157–162. doi: 10.1016/j.febslet.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 81.Taniguchi E, Min JC, Arenkiel BR, et al. Bortezomib reverses a post-translational mechanism of tumorigenesis for Patched1 haploinsufficiency in medulloblastoma. Pediatric Blood and Cancer. 2009;53(2):136–144. doi: 10.1002/pbc.21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nagao K, Toyoda M, Takeuchi-Inoue K, Fujii K, Yamada M, Miyashita T. Identification and characterization of multiple isoforms of a murine and human tumor suppressor, Patched, having distinct first exons. Genomics. 2005;85(4):462–471. doi: 10.1016/j.ygeno.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 83.Nagao K, Togawa N, Fujii K, et al. Detecting tissue-specific alternative splicing and disease-associated aberrant splicing of the PTCH gene with exon junction microarrays. Human Molecular Genetics. 2005;14(22):3379–3388. doi: 10.1093/hmg/ddi369. [DOI] [PubMed] [Google Scholar]
- 84.Uhmann A, Dittmann K, Nitzki F, et al. The Hedgehog receptor Patched controls lymphoid lineage commitment. Blood. 2007;110(6):1814–1823. doi: 10.1182/blood-2007-02-075648. [DOI] [PubMed] [Google Scholar]
- 85.Zibat A, Uhmann A, Nitzki F, et al. Time-point and dosage of gene inactivation determine the tumor spectrum in conditional PTCH knockouts. Carcinogenesis. 2009;30(6):918–926. doi: 10.1093/carcin/bgp068. [DOI] [PubMed] [Google Scholar]
- 86.Nitzki F, Zibat A, Frommhold A, et al. Uncommitted precursor cells might contribute to increased incidence of embryonal rhabdomyosarcoma in heterozygous Patched1-mutant mice. Oncogene. 2011 doi: 10.1038/onc.2011.157. [DOI] [PubMed] [Google Scholar]
- 87.Hameyer D, Loonstra A, Eshkind L, et al. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiological Genomics. 2007;31(1):32–41. doi: 10.1152/physiolgenomics.00019.2007. [DOI] [PubMed] [Google Scholar]
- 88.Metzger D, Chambon P. Site- and time-specific gene targeting in the mouse. Methods. 2001;24(1):71–80. doi: 10.1006/meth.2001.1159. [DOI] [PubMed] [Google Scholar]
- 89.Indra AK, Warot X, Brocard J, et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Research. 1999;27(22):4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oskarsson T, Essers MAG, Dubois N, et al. Skin epidermis lacking the c-myc gene is resistant to Ras-driven tumorigenesis but can reacquire sensitivity upon additional loss of the p21 Cip1 gene. Genes and Development. 2006;20(15):2024–2029. doi: 10.1101/gad.381206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ehrenreiter K, Kern F, Velamoor V, et al. Raf-1 addiction in Ras-induced skin carcinogenesis. Cancer Cell. 2009;16(2):149–160. doi: 10.1016/j.ccr.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 92.Dumortier A, Durham AD, Piazza MD, et al. Atopic dermatitis-like disease and associated lethal myeloproliferative disorder arise from loss of notch signaling in the murine skin. PLoS ONE. 2010;5(2) doi: 10.1371/journal.pone.0009258.e9258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu Y, Suckale J, Masjkur J, et al. Tamoxifen-independent recombination in the RIP-CreER mouse. PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013533.e13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peukert S, Miller-Moslin K. Small-molecule inhibitors of the hedgehog signaling pathway as cancer therapeutics. ChemMedChem. 2010;5(4):500–512. doi: 10.1002/cmdc.201000011. [DOI] [PubMed] [Google Scholar]