

Abstract
7–β–D-Ribofuranosylxanthine, a previously unreported isomer of xanthosine, was prepared in four steps from 7-benzylxanthine. The procedure, which involves the use of pivaloyloxymethyl groups to protect the xanthine ring, was also applied to preparation of some 1-N-alkyl derivatives of 7-ribosylxanthine. Adenosine receptor affinity for these compounds was determined. 7–β–D-Ribofuranosylxanthine was found to have higher affinity and greater selectivity for the A1 receptor than previously reported xanthine nucleosides, and to be a partial agonist.
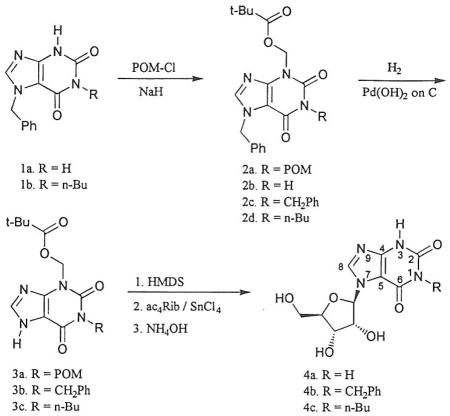
Recently, nucleosides derived from 1,3-dialkylxanthines have been found to be of interest as adenosine receptor ligands.1 To further explore the pharmacological potential of xanthine nucleosides, it was of interest to us to prepare 7-ribosylxanthines unsubstituted at N-3, including the parent compound, 7–β–D-ribofuranosylxanthine, a previously unreported isomer of xanthosine. 1,3-Dialkylxanthine nucleosides have been prepared by Lewis acid catalyzed ribosylation of the heterocycle, a reaction which takes place regio-specifically at N-7 as originally reported for theophylline.2 In contrast to 1,3-dialkylxanthines, ribosylation of xanthine gives the 9-ribosyl derivative, xanthosine, as the only product. The difference in the regiospecificity of these two reactions is most likely due to the steric effect of the alkyl substituent at N-3, which blocks ribosylation at N-9.
One possible approach to xanthine-7-ribosides unsubstituted at N-3 would be to prepare a suitably substituted imidazole nucleoside and to complete the purine ring in subsequent reactions. This approach has been used for the preparation of some other 7-ribosyl purines,3 and the isomeric 1- and 3-ribosyl xanthines were prepared by an analogous approach from pyrimidine nucleosides.4,5 The alternative approach, reported here, is to use protecting groups on the preformed xanthine nucleus to direct ribosylation to N-7. This method was previously reported for preparation of 7-ribosylhypoxanthine.6 The protecting group chosen for this work was the pivaloyloxymethyl (POM) group, which has been used previously to protect N-7 of xanthines.7 This group is easily introduced by alkylation, and is removed along with the sugar protecting groups by treatment with base after the ribosylation step.
Preparation of 7–β–D-ribofuranosylxanthine began with 7-benzylxanthine (1a).8 Reaction with an excess of chloromethyl pivalate (POM-Cl) in the presence of base gave the bis(pivaloyloxymethyl) xanthine 2a. A small amount of 7-benzyl-3-(pivaloyloxymethyl)xanthine (2b) was separated from the fully protected compound by chromatography. Removal of the benzyl group by hydrogenolysis over palladium hydroxide on carbon gave 1, 3-bis(pivaloyloxymethyl)xanthine (3a). Persilylation followed by tin (IV) chloride catalyzed coupling with 1, 2, 3, 5-tetra-O-acetylribofuranose gave the fully protected nucleoside, which was purified by chromatography prior to removal of the protecting groups with ammonium hydroxide. 7–β–D-ribofuranosylxanthine (4a) was isolated as a crystalline solid in 62% yield from 3a. No isomeric nucleosides were observed as products of the glycosylation. The site of attachment of the sugar was confirmed by comparison of the 13C nmr spectrum with those of 7–β–D-ribofuranosyltheophylline and xanthosine. For nucleoside 4a the signals for C4 and C5 appear at 150.0 and 106.0 ppm respectively, comparable to the shifts of 148.7 and 105,7 found for theophylline-7-riboside, and in contrast to the shifts of 135.7 and 116.2 found for xanthosine.
The 1-butyl and 1-benzyl nucleosides were also prepared from the corresponding 1-alkyl-3-pivaloyloxymethylxanthines, which could be made by either of two methods. The 1-benzyl xanthine 2c was prepared by benzylation of the 3-pivaloyloxymethyl compound 2b. This useful intermediate (2b) could be prepared in larger amounts when required by reaction of 7-benzylxanthine with a smaller excess of chloromethyl pivalate. The 1-butyl xanthine 2d was prepared by reaction of the previously reported 7-benzyl-1-butylxanthine (1b)9 with chloromethyl pivalate. Removal of the 7-benzyl protecting group from these intermediates by hydrogenolysis gave the xanthines 3b and 3c. Ribosylation and deprotection were carried out as before and the nucleosides 4b and 4c were isolated in reasonable yields.
Nucleosides 4a–c were examined for their ability to displace radioligands from adenosine receptors. Results are presented in the table, along with previously published results for 1, 3-dibutylxanthine-7-riboside (4d).1
TABLE.
Affinities of xanthine nucleosides at rat adenosine receptors.
| Nucleoside | A1a | A2Ab | A3c |
|---|---|---|---|
| 4a | 0.787 ± 0.282 μM | 27 ± 12% | 17 ± 1% |
| 4b | 10.5 ± 2.7 μM | < 10% | < 10% |
| 4c | 10.3 ± 3.5 μM | < 10% | < 10 % |
| 1,3-dibutyl1 (4d) | 4.19 μM | 19.5 μM | 6.03 μM |
vs. [3H]R-PIA binding at rat cerebral cortical membranes, expressed as mean Ki ± sem.
% displacement of [3H]CGS21680 from rat striatal membranes at 10−4 M or Ki.
% displacement of [125I]AB-MECA from transfected CHO membranes at 10−5 M or Ki.
Comparison of the 1-butyl compound 4c with the 1,3-dibutyl nucleoside shows that the loss of the 3-butyl group significantly decreases binding to A2A and A3 receptors, while binding to the A1 receptor is only decreased by a factor of about 2. More surprising is the observation that the unsubstituted nucleoside 4a has a higher affinity at the A1 receptor than any of the other alkylxanthine nucleosides studied so far. Comparison of the binding of 4a and 4c at the A1 receptor shows that the 1-butyl group decreases affinity by at least a factor of 10. These results suggest that it is important to examine xanthine nucleosides with different substituents at either N-1 or N-3, rather than just the equivalently substituted compounds which have been reported so far.
The intrinsic activity of 7-β-D-ribofuranosylxanthine (4a) at the A1 receptor was determined in a functional assay. G protein activation initiated through receptor occupation was determined by measurement of [35S]GTP-γ-S binding as previously reported.13 Results are shown in the figure. 7-β-D-Ribofuranosylxanthine (4a) was found to be a partial agonist, with a relative efficacy of ~50% compared with the full agonist, R-N6-(phenylisopropyl)adenosine (R-PIA). 4a did not antagonize activation by R-PIA.
FIGURE.

Stimulation by adenosine A1 agonists of guanine nucleotide binding to G-proteins in rat brain membranes.
1,3-Dialkylxanthine ribosides having equivalent substituents at N1 and N3, such as 4d, were previously shown to be partial agonists at rat A1 receptors.14 Here we have shown that even the unsubstituted derivative 4a behaves similarly, and indeed has greater affinity. Partial A1 adenosine receptor agonists are of interest as potential anti-seizure or anti-lipolytic therapeutic agents, possible displaying fewer side-effects than full agonists.
Experimental
Melting points were determined on a Laboratory Devices Mel-Temp apparatus and are uncorrected. Nmr spectra were recorded in DMSO-d6 on a Varian VXR-300 spectrometer. Chemical shifts are reported in ppm downfield from TMS. Elemental analyses were obtained from Desert Analytics, Tucson, Arizona. The assistance of Dr. Chhabil Dass in obtaining mass spectra of the nucleosides is gratefully acknowledged.
7-Benzyl-1,3-bis(pivaloyloxymethyl)xanthine(2a)
Sodium hydride (60% dispersion, 1.0 g, 24 mmol) was added to a solution of 7-benzylxanthine (2.4 g, 10 mmol) in DMF (100 mL). After 15 min, chloromethyl pivalate (3.5 mL, 24 mmol) was added and the mixture was stirred at ambient temperature for 4 h. The mixture was added to dilute hydrochloric acid (150 mL) and extracted with chloroform. The combined extracts were washed with water and brine, dried over magnesium sulfate, and concentrated. The residue was purified by flash chromatography on silica, eluting with 1% methanol in chloroform. The major product, 1-benzyl-1,3-bis(pivaloyloxymethyl)xanthine, was isolated as a white solid (3.7 g, 80%), and was recrystallized from ethanol, mp 123–124°C; 1H nmr: 1.09 (s, 9H, t-Bu), 1.10 (s, 9H, t-Bu), 5.49 (s, 2H, N7CH2), 5.85 (s, 2H, N1CH2), 5.96 (s, 2H, N3CH2), 7.3–7.4 (m, 5H, Ph), 8.40 (s, 1H, H-8); 13C nmr: 26.5, 38.1, 38.2, 49.2, 64.7, 66.4, 105.6, 127.6, 128.0, 128.6, 136.3, 143.4, 147.7, 149.7, 153.1, 176.4. Anal. Calcd. For C24H30N4O6: C, 61.26; H, 6.43; N, 11.91; found: C, 60.93; H, 6.39; N, 11.79.
A minor product, 7-benzyl-3-(pivaloyloxymethyl)xanthine, was isolated upon further elution of the column.
7-Benzyl-3-(pivaloyloxymethyl)xanthine (2b)
To a solution of 7-benzylxanthine (2.42 g, 10mmol) in DMF (50 mL) were added solid potassium carbonate (2.8 g) and chloromethyl pivalate (2.2 mL, 15 mmol). After stirring at room temperature for 48 h, water was added and the products were extracted into chloroform. The organic phase was washed with brine, dried over magnesium sulfate, and concentrated. The residue was fractionated by chromatography giving 2a (2.1 g, 46%) and 2b (1.2 g, 35%). The latter was crystallized from ethanol, mp 201–202°C. 1H nmr: 1.09 (s, 9H, t-Bu), 5.45 (s, 2H, N7CH2), 5.89 (s, 2H, N3CH2), 7.3–7.4 (m, 5H, Ph), 8.27 (s, 1H, H-8), 11.41 (s, 1H, H-1); 13C nmr: 26.5, 38.2, 49.0, 65.8, 106.3, 127.6, 127.9, 128.6, 136.6, 142.5, 148.5, 150.1, 154.6, 176.6. Anal. Calcd. for C18H20N4O4: C, 60.66; H, 5.66; N, 15.72; found: C, 60.49; H, 5.62; N, 15.71.
1,7-Dibenzyl-3-(pivaloyloxymethyl)xanthine (2c)
To a solution of 7-benzyl-3-(pivaloyloxymethyl)xanthine (0.42 g, 1.2 mmol) in DMF (10 mL) were added potassium carbonate (0.5 g) and benzyl bromide (0.2 mL, 1.4 mmol). After stirring at 50°C for 5 h, the mixture was cooled, diluted with water, and extracted with chloroform. The extracts were dried over magnesium sulfate and concentrated. The product was purified by flash chromatography on silica yielding a white solid (0.4 g, 76%), which was recrystallized from ethanol, mp 105–106°C. 1H nmr: 1.08 (s, 9H, t-Bu), 5.05 (s, 2H, N1CH2), 5.50 (s, 2H, N7CH2), 5.99 (s, 2H, N3CH2), 7.3–7.4 (m, 10H, 2 Ph), 8.34 (s, 1H, H-8); 13C nmr: 26.4, 38.1, 43.6, 49.0, 66.5, 105.9, 127.0, 127.4, 127.6, 127.9, 128.1, 128.5, 136.5, 137.0, 142.9, 147.2, 150.1, 154.0, 176.4. Anal. Calcd. for C25H26N4O4: C, 67.25; H, 5.87; N, 12.55; found: C, 67.41; H, 5.84; N, 12.50.
7-Benzyl-1-butyl-3-(pivaloyloxymethyl)xanthine(2d)
To a solution of 7-benzyl-1-butylxanthine9 (3.0 g, 10 mmol) and sodium hydride (60%, 0.5 g, 12 mmol) in DMF (100 mL) was added chloromethyl pivalate (1.8 mL, 12 mmol) and the solution was stirred at ambient temperature for 4 h. The mixture was poured into saturated aqueous sodium bicarbonate, and the product was extracted into chloroform, purified by flash chromatography, and crystallized from ethanol yielding 3.5 g (84%), mp 107–108°C. 1H nmr: 0.86 (t, 3H, CH3), 1.09 (s, 9H, t-Bu), 1.26 (m, 2H, MeCH2), 1.50 (m, 2H, EtCH2), 3.85 (t, 2H, NlCH2), 5.49 (s, 2H, N7CH2), 5.96 (s, 2H, N3CH2), 7.3–7.4 (m, 5H, Ph), 8.31 (s, 1H, H-8); 13C nmr: 13.5, 19.4, 26.4, 29.3, 38.1, 39.1, 40.2, 49.0, 66.5, 105.8, 127.6, 127.9, 128.5, 136.6, 142.6, 146.9, 149.9, 154.0, 176.4. Anal. Calcd. for C22H28N4O4: C, 64.06; H, 6.84; N, 13.58; found: C, 64.17; H, 6.96; N, 13.61.
General procedure for debenzylation
The 7-benzylxanthine was dissolved in acetic acid (10 mL per mmol), 20% palladium hydroxide on carbon (0.5 g per mmol) was added, and the mixture was shaken under hydrogen (48 psi) for 20 h. The filtrate was evaporated and the product was recrystallized from ethanol.
1,3-bis(pivaloyloxymethyl)xanthine(3a)
93% yield, mp 183–184°C; 1H nmr: 1.09 (s, 18H, 2 t-Bu), 5.88 (s, 2H, N1CH2), 5.99 (s, 2H, N3CH2), 8.15 (s, 1H, H-8); 13C nmr: 26.4, 26.5, 38.1, 38.2, 64.9, 66.6, 106.1, 141.6, 147.2, 149.9, 153.1, 176.4, 176.5. Anal. Calcd. for C17H24N4O6: C, 53.68; H, 6.36; N, 14.73; found: C, 53.64; H, 6.42; N, 14.83.
1-Benzyl-3-(pivaloyloxymethyl)xanthine (3b)
86% yield, mp 186–187°C; 1H nmr: 1.08 (s, 9H, t-Bu), 5.09 (s, 2H, N1CH2), 6.00 (s, 2H, N3CH2), 7.3–7.4 (m, 5H, Ph), 8.11 (s, 1H, H-8); 13C nmr: 26.5, 38.2, 43.7, 66.8, 106.5, 127.0, 127.3, 128.2, 137.2, 141.1, 146.7, 150.4, 154.1, 176.5. Anal. Calcd. for C18H20N4O4: C, 60.66; H, 5.66; N, 15.72; found: C, 60.80; H, 5.65; N, 15.61.
1-Butyl-3-(pivaloyloxymethyl)xanthine(3c)
84% yield, mp 152–153°C; 1H nmr: 0.86 (t, 3H, CH3), 1.08 (s, 9H, t-Bu), 1.28 (m, 2H, MeCH2), 1.51 (m, 2H, EtCH2), 3.86 (t, 2H, N1CH2), 5.97 (s, 2H, N3CH2), 8.04 (s, 1H, H-8); 13C nmr: 13.6, 19.5, 26.5, 29.5, 38.2, 40.3, 66.7, 106.6, 140.8, 146.4, 150.2, 154.1, 176.5. Anal. Calcd. for C15H22N4O4: C, 55.89; H, 6.88; N, 17.38; found: C, 55.88; H, 6.79; N, 17.44.
General procedure for preparation of nucleosides
A solution of the 3-(pivaloyloxymethyl)xanthine in hexamethyldisilazane (7 mL per mmol) containing ammonium sulfate (1 mg/mL) was heated under reflux for 2 h, then cooled and concentrated. The residue was dissolved in 1,2-dichloroethane (20 mL per mmol) with 1, 2, 3, 5-tetra-O-acetylribose (1.5 equivalents). The solution was cooled in an ice-bath and tin (IV) chloride (1.5 mL per mmol) was added dropwise. The solution was allowed to warm to ambient temperature and stirred for 22 h, then poured into saturated aqueous sodium bicarbonate solution. The product was extracted into dichloromethane and the extracts were washed with brine, dried over magnesium sulfate, and concentrated. Purification by flash chromatography gave the fully protected nucleoside. This was dissolved in a mixture of methanol (10 mL per mmol) and concentrated aqueous ammonia (10 mL per mmol). After standing at ambient temperature for 24 h, solvents were evaporated and the residue was crystallized from ethanol.
7-(β-D-ribofuranosyl)xanthine(4a)
62% yield, mp 233–235°C; 1H nmr: 3.60 (m, 2H, H-5′), 3.90 (m, 1H, H-4′), 4.08 (m, 1H, H-3′), 4.32 (m, 1H, H-2′), 5.02 (t, 1H, 5′-OH), 5.14 (d, 1H3′-OH), 5.44 (d, 1H, 2′-OH), 6.01 (d, J 4.8, 1H, H-l′), 8.35 (s, 1H, H-8), 10.96 (s, 1H, H-1), 11.66 (br s, 1H, H-3). 13C nmr: 60.8, 69.4, 74.6, 85.1, 89.1, 106.0, 141.4, 150.0, 151.0, 155.3. Anal. Calcd. for C10H12N4O6.1/2H2O; C, 40.95; H, 4.47; N, 19.11; found: C, 41.30; H, 4.14; N, 18.77.
1-Benzyl-7-(β-D-ribofuranosyl)xanthine(4b)
73% yield, mp 177–179°C; 1H nmr: 3.60 (m, 2H, H-5′), 3.91 (m, 1H, H-4′), 4.10 (m, 1H, H-3′), 4.33 (m, 1H, H-2′), 5.03 (s, 2H, N1CH2), 5.08 (t, 1H, 5′-OH), 5.16 (br, 1H, 3′-OH), 5.50 (d, 1H, 2′-OH), 6.01 (d, J 4.5, 1H, H-1′), 7.20–7.30 (m, 5H, Ph), 8.43 (s, 1H, H-8), 12.10 (br s, 1H, H-3). 13C nmr: 43.0, 60.6, 69.2, 74.6, 84.9, 89.2, 105.7, 127.0, 127.4, 128.2, 137.6, 141.4, 148.2, 150.8, 154.7. Anal. Calcd. for C17H18N4O6.1/2H2O: C, 53.29; H, 5.00; N, 14.62; found: C, 53.84; H, 4.59; N, 14.71.
1-Butyl-7-(β-D-ribofuranosyl)xanthine(4c)
69% yield, mp 194–196°C; 1H nmr: 0.90 (t, 3H, CH3), 1.29 (m, 2H, MeCH2), 1.51 (m, 2H, EtCH2), 3.62 (m, 2H, H-5′), 3.82 (t, 2H, N1CH2), 3.91 (m, 1H, H-4′), 4.08 (m, 1H, H-3′), 4,32 (m, 1H, H-2′), 5.05 (t, 1H, 5′-OH), 5.15 (d, 1H, 3′-OH), 5.48 (d, 1H, 2′-OH), 6.09 (d, J 4.3, 1H, H-1′), 8.39(s, 1H, H-1), 11.96 (s, 1H, H-3). 13C nmr: 13.6, 19.6, 29.6, 39.5, 60.5, 69.2, 74.6, 84.8, 89.1, 105.6, 141.1, 147.9, 150.6, 154.7. Anal. Calcd. for C14H20N4O6.1/2H2O: C, 48.13; H, 6.06; N, 16.04; found: C, 48.52; H, 5.54; N, 15.90.
Radioreceptor binding assays
Binding of [3H]R-N6-(phenylisopropyl)adenosine ([3H]R-PIA) to A1 receptors from rat cerebral cortex membranes10 and of [3H]-2-[4-[(2-carboxyethyl)phenyl]ethyl-amino]-5′-N-ethylcarbamoyladenosine ([3H]CGS 21680) to A2A receptors from rat striatal membranes was performed as described previously.11 Adenosine deaminase (3 units/mL) was present during the preparation of the brain membranes, in a pre-incubation of 30 min at 30 °C, and during the incubation with the radioligands.
Binding of [125I]N6-(4-amino-3-iodobenzyl)-5′-N-methylcarbamoyladenosine ([125I]AB-MECA) to membranes prepared from CHO cells stably expressing the rat A3 receptor was performed as described.12 The assay medium consisted of a buffer containing 10 mM Mg2+, 50 mM Tris, and 1 mM EDTA, at pH 8.0. The glass incubation tubes contained 100 mL of the membrane suspension (0.3 mg protein/mL), 50 mL of [125I]AB-MECA (final concentration 0.3 nM), and 50 mL of a solution of the xanthine riboside. Nonspecific binding was determined in the presence of 100 mM R-PIA.
Binding of [35S]GTP-γ-S
The binding of [35S]GTP-γ-S (Amersham, Chicago IL, specific activity 1275 Ci/mmol) was carried out using rat cerebral cortical membranes by the general method of Lorenzen et al.13 Membranes were suspended in a buffer containing 50 mM Tris, 3 units/mL adenosine deaminase, 100 mM NaCl, and 10 mM MgCl2, pH 7.4 at a protein concentration of 1–10 μg per tube. The membrane suspension was preincubated with 0.5 μM GDP, R-PIA and/or riboflavin in a final volume of 450 μl buffer at 30° C for 20 min and then transferred to ice for 20 min. [35S]GTP-γ-S was added to a final concentration of 0.1 nM in a total volume of 500 μL and the mixture was incubated for 30 min at 30°C. Nonspecific binding was determined in the presence of 10 μM GTP-γ-S (Sigma, St. Louis MO). Incubation of the reaction mixture was terminated by filtration over GF/B glass fibers using a Brandel cell harvester and washed with the same buffer.
References
- 1.Kim HO, Ji X, Melman N, Olah ME, Stiles GL, Jacobson KA. J Med Chem. 1994;37:4020–4030. doi: 10.1021/jm00049a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vorbrüggen H, Krolikiewicz K, Bennua B. Chem Ber. 1981;114:1234–1255. [Google Scholar]
- 3.Rousseau RJ, Robins RK, Townsend LB. J Amer Chem Soc. 1968;90:2661–2668. doi: 10.1021/ja01012a035. [DOI] [PubMed] [Google Scholar]
- 4.Winkley MW. J Chem Soc (C) 1970:1869–1874. doi: 10.1039/j39700001869. [DOI] [PubMed] [Google Scholar]
- 5.Lipkin D, Cori CT, Rabi JA. J Het Chem. 1969;6:995–996. [Google Scholar]
- 6.Thomas HJ, Montgomery JA. J Org Chem. 1966;31:1413–1416. [Google Scholar]
- 7.Hu MW, Singh P, Ullman EF. J Org Chem. 1980;45:1711–1713. [Google Scholar]
- 8.Bridson PK, Richmond G, Yeh F. Synth Comm. 1990;20:2459–2467. [Google Scholar]
- 9.Bridson PK, Wang X. Synthesis. 1995:855–858. [Google Scholar]
- 10.Schwabe U, Trost T. Naunyn-Schmiedeberg’s Arch Pharmacol. 1980;313:179–187. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]
- 11.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. J Pharmacol Exp Therap. 1989;251:888–893. [PubMed] [Google Scholar]
- 12.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 13.Lorenzen A, Guerra L, Vogt H, Schwabe U. Mol Pharmacol. 1996;49:915–926. [PubMed] [Google Scholar]
- 14.IJzerman AP, van der Wenden EM, von Frijtag Drabbe Künzel JK, Mathôt RAA, Danhof M, Borea PA, Varani K. Naunyn-Schmiedeberg’s Arch Pharmacol. 1994;350:638–645. doi: 10.1007/BF00169369. [DOI] [PubMed] [Google Scholar]