

Abstract
The electrophilic fluorination of several (triphos)Pt-aryl+ establishes the first example of aryl–F coupling from a Pt center.
The demand for organofluorine compounds has stimulated much recent effort to develop metal mediated fluorination reactions.1 Despite the versatility of available C–X (X = C, N, O, S, Cl, Br, I, etc.) coupling methodologies,2 C–F couplings via reductive elimination remain challenging.1d,f Metal catalyzed C–F couplings that utilize fluoride sources encounter additional challenges due to the intrinsically low polarizability and nucleophilicity, pronounced hydration power, and high basicity of F−. Nevertheless, several notable Pd0/II catalyzed nucleophilic fluorinations have been recently reported.3 More fruitful have been recent metal-catalyzed electrophilic fluorination reactions,5–8 wherein high-valent metal fluoro intermediates (e.g. Pd(IV),4 Ag(II)···Ag(II),5 Au(III),6 etc.) are more prone to productive reactivity, including C–H activation, cross-coupling, and C–F reductive elimination.1f,7
To explore Pt analogues of these electrophilic reactions, we recently demonstrated a system that efficiently fluorinates Pt–Csp3 bonds.8b As illustrated in eqn (1), wherein PPP = bis(2-diphenylphosphinoethyl)phenylphosphine (i.e., triphos), the C–F coupling proved to be stereoretentive and was proposed to occur by concerted reductive elimination of a putative dicationic Pt(IV)–F intermediate (A). The reaction was accelerated by increased steric congestion around Pt,8b however, information on the short-lived Pt(IV)–F species was lacking.
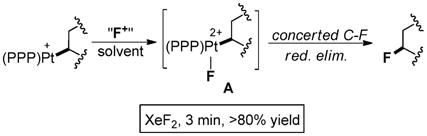 |
(1) |
Sp2-carbon–halogen bond forming reactions from Pt(IV) centers are rare,1g,9,10 with the few known examples restricted to C–I and C–Br couplings.11 Extending our efforts on Pt–C bond fluorination reactions, we have examined the electrophilic fluorination of (triphos)Pt-aryl+ complexes. Herein, we report these reactions and provide evidence that supports the intermediacy of Pt(IV)–F complexes in the C–F reductive coupling reaction.
Complexes 1–4 were synthesized by ligand displacement of (COD)PtAr(X) (COD = cycloocta-1,5-diene, X = Cl, or I) with triphos, followed by salt metathesis with NaBF4.12,13 Complex 5 was prepared by treating chloro(2-phenylpyridine)[2-(2-pyridyl)- phenyl-C,N]Pt14 with triphos, while its dicationic isostere 6 was obtained by reacting [(triphos)Pt(NCC6F5)](BF4)215 with 2-phenylpyridine.12
These compounds were characterized by NMR and HRMS, with the molecular structure of 4§ being verified by X-ray analysis (Fig. 1).12 Consistent with the solid state structure of 4, NOESY analysis suggested that the ortho-substituent in 2, 4–6 preferentially oriented syn to the central P-Ph group of the triphos ligand. While 2, 4, 5 and 6 exist exclusively in this syn-rotamer, both syn- and anti-forms (2.7: 1) were observed for 3.12 The preference for the syn- over the anti-form suggests that the face of the square plane containing the apical P-Ph group is less congested and may be more kinetically accessible.
Fig. 1.
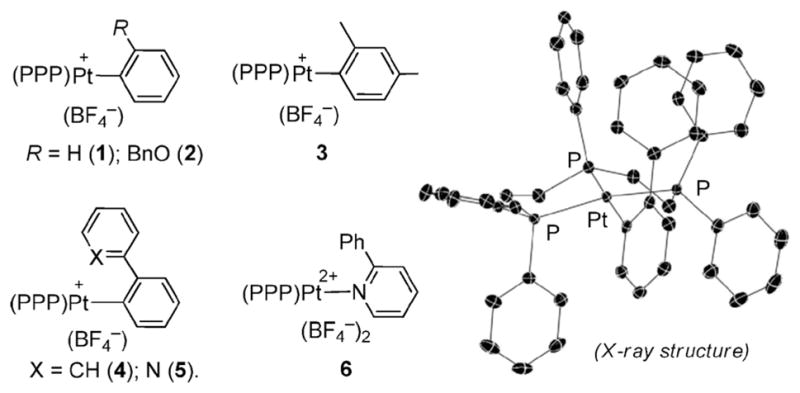
Left: complexes 1–6; right: X-ray structure of 4 (H atoms and BF4− anion are omitted for clarity).
When subjected to electrophilic fluorination conditions, these (triphos)Pt-aryl+ complexes were found to be much less reactive than their Pt-alkyl+ analogs.8b When screening common “F+” sources including N-fluorobenzenesulfonimide, several N-fluoropyridinium salts, Selectfluor® and XeF2, only the latter two exhibited reasonable reactivity with 1, for which the optimal solvent was identified to be acetonitrile. 31P and 19F NMR spectroscopy proved most advantageous for in situ monitoring of these reactions and Selectfluor® proved to be cleaner and more productive than XeF2.
With 1, a complex mixture of phenyl Pt(IV)–F species was obtained upon reacting with XeF2 (RT,<20 min). By contrast, Selectfluor® provided one main phenyl Pt(IV)–F complex (RT, ~2 h) (δF = −360.3 ppm, JPt–F = 1453 Hz).12 These Pt(IV)–F species, however, failed to reductively eliminate PhF even after prolonged heating (80 °C, >30 h).
The ortho-substituents considerably slowed down the reactions of 2 and 3 with XeF2 and Selectfluor®, however, their presence proved beneficial for achieving the desired sp2 C–F coupling. In the case of 2, XeF2 provided one major Pt(IV)–F complex (δ = −352.8 ppm, JPt–F = 1442 Hz) in ~75% NMR yield (RT, 12 h).12 However, the precise structure of this product remains unclear, as all attempts to crystallize it failed and spectroscopic data were not conclusive. Heating a freshly prepared reaction mixture containing this Pt(IV)–F complex at 80 °C led to only traces of the aryl–F coupling product (<5% GC-MS yield). Similar results were obtained when directly reacting 2 with XeF2 at 80 °C. In contrast, reactions of 3 with XeF2 (RT, 15 h) directly generated a substantial amount of the aryl–F coupling product 1-fluoro-2,4-dimethylbenzene (~55% NMR yield), along with the corresponding [(triphos)Pt-NCMe]2+ by-product. The formation of a Pt(IV)–F complex (δF=−351.9 ppm, JPt–F=1146 Hz) in~25%NMR yield and other unidentified Pt species was also observed.12 To our knowledge, this reaction represents the first example of aryl–F coupling from a Pt center.
Despite being unreactive at RT, Selectfluor® readily fluorinated 2 and 3 at 80 °C to produce the aryl fluoride;12 no Pt(IV)–F species was observable during in situ monitoring of these reactions. These results are summarized in Table 1.
Table 1.
Fluorination of complexes 1–3 with Selectfluor®a
| Complex | Product | Time | NMR yieldb (%) |
|---|---|---|---|
| 1 | [(PPP)PtIV(Ph)(F)]2+ | <20 min | 60–70 |
| 2 |
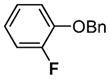
|
1 h | 91 |
| 3 |
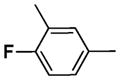
|
2 h | >95 |
Conditions: complexes 1–3 (0.02 mmol), 1.5 equiv. of Selectfluor®, dry CD3CN (0.5 mL), 80 °C.
Mass balance: structurally unidentified organometallic Pt species.
Surprisingly, the reaction of 4 with XeF2 preferentially yielded the ortho-cyclometalated complex 7 (Scheme 1). NMR monitoring of the reaction revealed its gradual conversion to the Pt(IV)–F complex, 7§, which was characterized byNMR, HRMSand X-ray diffraction.12 In contrast to the aforementioned Pt(IV)–F species, this complex exhibits a 19F NMR resonance at δ = −299.9 ppm with a considerably diminished 195Pt–19F coupling (~173 Hz).
Scheme 1.
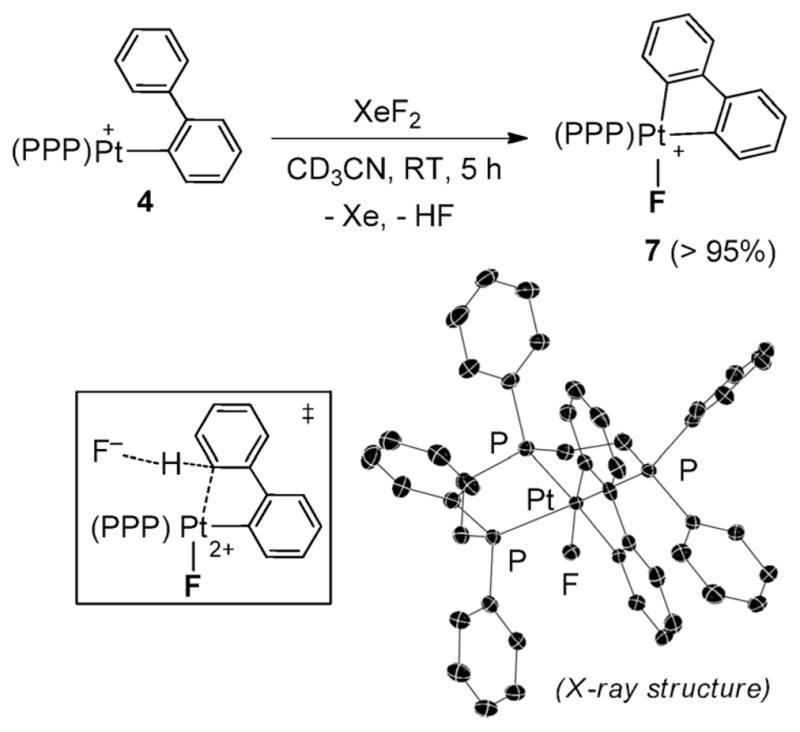
Generation of complex 7; inset: X-ray structure of 7 (H atoms and anion are omitted for clarity).
As shown in Scheme 1, the Pt center in 7 adopts an octahedral coordination geometry, with the Pt–F bond (2.099(2) Å) oriented anti to the central P-Ph group of the triphos ligand, and the biphenyl moiety adopting a C,C′-chelating mode. Similar cyclometalation of an ortho sp2-C–H bond was previously noted upon fluorinating (triphos)Pt-CH2Ph+ with XeF2 in melting acetonitrile.8b This reactivity mode apparently reflects the intermediacy of Pt(IV) fluorides in both cases.8b The propensity of Pt(IV) and Pd(IV) centers in metalating aromatic C–H bonds has been demonstrated and exploited recently in several coupling strategies.7,8a
Heating an acetonitrile solution of 7 at 80 °C resulted in slow F− extrusion and the concomitant formation of a dicationic Pt(IV)–MeCN adduct, 8 (eqn (2)). No C–F reductive elimination was observed during the process, and X-ray diffraction§ revealed that the MeCN ligand coordinates syn to the triphos ligand’s central P-Ph group (eqn (2)).12 Consistent with the increase in the net charge of the Pt(IV) center are large downfield shifts of the 31P NMR signals as compared to 7 (e.g., Δδ = +22.7 ppm for the central P) and 1H NMR signals of the biphenyl moiety.
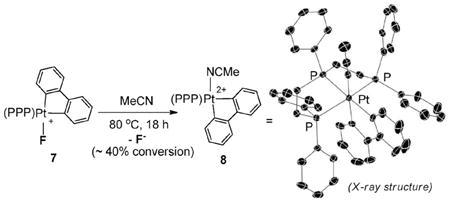 |
(2) |
In addition to 7, reactions of 4 with XeF2 at RT (Scheme 1) also yielded traces of 8 (<5%).12 By contrast, reactions of 4 with Selectfluor® directly provided 8 (85%,~5 h), along with 15% of 2-fluorobiphenyl and the corresponding [(triphos)Pt-NCMe]2+ (Scheme 2).12 We reason that the formation of both 7 and 8 implies the presence of Pt(IV) intermediates.
Scheme 2.

Competitive cyclometalation and C–F coupling pathways.
The contrasting outcomes for reactions of 2–4 with XeF2 and Selectfluor® presumably stem from the presence of a basic fluoride anion in the former case, though a size difference in the “F+” source is also conceivable.16 Shown in Scheme 1 is one way wherein F− could accelerate ortho-metalation vs. reductive elimination. Since the two Pt(II) faces were shown to be sterically different, it is also possible that these reactions evolve differently based on which face F+ attacks.16 Recently, Vigalok and co-workers have also reported an F+ reagent-dependent reaction behavior when fluorinating Pt-aryl complexes.8a
To gain more insights into the Pt(IV)–F species proposed in Schemes 1 and 2, the fluorination of 5 and 6 by XeF2 was examined. In particular, we hoped that the ortho-pyridyl group in 5 could trap the coordinatively unsaturated Pt(IV)–F intermediate. Instead, XeF2 converted 5 into the Pt(II) complex 9 (eqn (3)), whose configuration was deduced from 31P NMR data (e.g., δF = −37.9 ppm, JP1-F = 652 Hz; JPt-P3 = 3745 Hz vs. JPt-P2 = 1863 Hz).12 The formation of this complex presumably occurred via associative displacement of one triphos phosphine arm (P1, eqn (3)) in 5 by the pyridyl ligand, followed by oxidation of the unligated phosphine ligand. We have previously shown that phosphine fluorination by XeF2 is rapid.8b Despite its structural analogy to 4 and 5, complex 6 failed to react with XeF2, indicating that a dicationic Pt(II) center may be too electron deficient to generate a tricationic Pt(IV) structure.
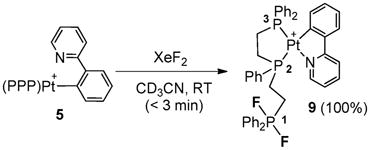 |
(3) |
In summary, we report the first sp2 C–F coupling from a Pt center. Like Pt–Csp3 bonds, steric congestion is a key factor, as is F+ source. We have also demonstrated that ortho-metalation may be competitive with C–F reductive elimination. The intermediacy of Pt(IV)–F complexes, the product of direct F+ addition to Pt(II), is supported by the direct spectroscopic observation of several Pt(IV)–F species and the isolation of ortho-metalation products.
Supplementary Material
Acknowledgments
We acknowledge the generous support of the NIH (GM-60578) and Army Research Office Staff Research Program. SZ thanks NSERC of Canada for a Postdoctoral Fellowship.
Footnotes
Electronic supplementary information (ESI) available: Experimental details, characterization data, and complete X-ray diffraction data. CCDC 838071–838073. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1cc15006e
X-Ray structure data: complex 4 (CCDC 838071), C47H44BCl2F4P3Pt, M = 1054.53, monoclinic, space group P21/c, a = 11.1844(10) Å, b = 15.4199(2) Å, c = 24.7809(3) Å, α = γ = 90°, β = 91.93(1)°, V = 4271.35(8) Å3, Z = 4, T = 100(2) K, 36 354 collected reflections, 8252 unique reflections (Rint = 0.0182); R1 = 0.0241, wR2 = 0.0604 for data with I > 2σ(I), and R1 = 0.0245, wR2 = 0.0607 for all unique data. Complex 7 (CCDC 838072), C49H48BF5NO2P3Pt, M = 1076.69, monoclinic, space group C2/c, a = 31.7695(19) Å, b = 10.0332(6) Å, c = 33.988(3) Å, α = γ = 90°, β = 116.680(1)°, V = 9680.2(12) Å3, Z = 8, T = 180(2) K, 18 908 collected reflections, 9401 unique reflections (Rint = 0.0255); R1 = 0.0302, wR2 = 0.0685 for data with I > 2σ(I), and R1 = 0.0374, wR2 = 0.0707 for all unique data. Complex 8 (CCDC 838073), C48H46.75BF9.50NO1.38P3.50Pt, M = 1154.41, triclinic, space group P1̄, a = 13.666(1) Å, b = 17.484(2) Å, c = 22.340(2) Å, α = 95.002(1)°, β = 113.180(1)°, γ = 96.172(1)°, V = 4829.3(8) Å3, Z = 4, T = 180(2) K, 18 829 collected reflections, 18 829 unique reflections (Rint = 0.0454); R1 = 0.0339, wR2 = 0.0841 for data with I > 2σ(I), and R1 = 0.0483, wR2 = 0.0876 for all unique data.
Notes and references
- 1.(a) Hiyama T. Organofluorine Compounds. Springer; Berlin: 2000. [Google Scholar]; (b) Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (c) Kirk KL. Org Process Res Dev. 2008;12:305. [Google Scholar]; (d) Grushin VV. Acc Chem Res. 2010;43:160. doi: 10.1021/ar9001763. [DOI] [PubMed] [Google Scholar]; (e) Furuya T, Kuttruff CA, Ritter T. Curr Opin Drug Discovery Dev. 2008;11:803. [PubMed] [Google Scholar]; (f) Furuya T, Kamlet AS, Ritter T. Nature. 2011;473:470. doi: 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Vigalok A. Organometallics. 2011;30:4802. [Google Scholar]
- 2.Hartwig JF. Organometallic Metal Chemistry. ch 19 University Science Books; Sausalit, CA: 2010. [Google Scholar]
- 3.For recent examples of metal-catalyzed nucleophilic fluorinations, see: Watson DA, Su M, Teverovskiy G, Zhang Y, García-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661. doi: 10.1126/science.1178239.Katcher MH, Doyle AG. J Am Chem Soc. 2010;132:17402. doi: 10.1021/ja109120n.Hollingworth C, Hazari A, Hopkinson MN, Tredwell M, Benedetto E, Huiban M, Gee AD, Brown JM, Gouverneur V. Angew Chem, Int Ed. 2011;50:2613. doi: 10.1002/anie.201007307.
- 4.For recent examples of C–F couplings through Pd(IV) intermediates, see: Hull KL, Anani WQ, Sanford MS. J Am Chem Soc. 2006;128:7134. doi: 10.1021/ja061943k.Furuya T, Kaiser HM, Ritter T. Angew Chem, Int Ed. 2008;47:5993. doi: 10.1002/anie.200802164.Furuya T, Ritter T. J Am Chem Soc. 2008;130:10060. doi: 10.1021/ja803187x.Ball ND, Sanford MS. J Am Chem Soc. 2009;131:3796. doi: 10.1021/ja8054595.Wang X, Mei TS, Yu JQ. J Am Chem Soc. 2009;131:7520. doi: 10.1021/ja901352k.Wu T, Yin G, Liu G. J Am Chem Soc. 2009;131:16354. doi: 10.1021/ja9076588.Furuya T, Benitez D, Tkatchouk E, Strom AE, Tang P, Goddard WA, III, Ritter T. J Am Chem Soc. 2010;132:3793. doi: 10.1021/ja909371t.Wang W, Jasinski J, Hammond GB, Xu B. Angew Chem, Int Ed. 2010;49:7247. doi: 10.1002/anie.201003593.
- 5.For Ag(I)-catalyzed electrophilic fluorination reactions, see: Furuya T, Strom AE, Ritter T. J Am Chem Soc. 2009;131:1662. doi: 10.1021/ja8086664.Furuya T, Ritter T. Org Lett. 2009;11:2860. doi: 10.1021/ol901113t.Tang P, Furuya T, Ritter T. J Am Chem Soc. 2010;132:12150. doi: 10.1021/ja105834t.Xu T, Mu X, Peng H, Liu G. Angew Chem, Int Ed. 2011;50:8176. doi: 10.1002/anie.201103225.
- 6.Akana JA, Bhattacharyya KX, Müller P, Sadighi JP. J Am Chem Soc. 2007;129:7736. doi: 10.1021/ja0723784. [DOI] [PubMed] [Google Scholar]
- 7.For a review on the use of F+ to enable coupling and activation reactions from high-valent metal centers, see: Engle KM, Mei TS, Wang X, Yu JQ. Angew Chem, Int Ed. 2011;50:1478. doi: 10.1002/anie.201005142.
- 8.(a) Kaspi AW, Goldberg I, Vigalok A. J Am Chem Soc. 2010;132:10626. doi: 10.1021/ja101436w. [DOI] [PubMed] [Google Scholar]; (b) Zhao SB, Becker JJ, Gagné MR. Organometallics. 2011;30:3926. doi: 10.1021/om200515f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Vigalok A. Chem–Eur J. 2008;14:5102. doi: 10.1002/chem.200701738. [DOI] [PubMed] [Google Scholar]; (b) Kaspi AW, Vigalok A. Top Organomet Chem. 2010;31:19. [Google Scholar]
- 10.For recent examples of sp3-carbon–halogen bond forming reactions from Pt(IV) centers, see: Goldberg KI, Yan J, Winter EL. J Am Chem Soc. 1994;116:1573.Goldberg KI, Yan J, Breitung EM. J Am Chem Soc. 1995;117:6889.Zhao SB, Wang RY, Wang S. Organometallics. 2009;28:2572.Oblad PF, Bercaw JE, Hazari N, Labinger JA. Organometallics. 2010;29:789.
- 11.(a) Ettorre R. Inorg Nucl Chem Lett. 1969;5:45. [Google Scholar]; (b) Yahav-Levi A, Goldberg I, Vigalok A, Vedernikov AN. J Am Chem Soc. 2008;130:724. doi: 10.1021/ja077258a. [DOI] [PubMed] [Google Scholar]; (c) Yahav-Levi A, Goldberg I, Vigalok A, Vedernikov AN. Chem Commun. 2010;46:3324. doi: 10.1039/c001487g. [DOI] [PubMed] [Google Scholar]
- 12.See ESI† for details.
- 13.Although not discussed herein, the reaction of (COD)Pt(mesityl)(I) with triphos triggers migratory insertion of the Pt–mesityl bond into the COD moiety; the product was crystallographically characterized, see: Zhao S-B, Wang R-Y, Gagné MR. Acta Crystallogr, Sect E: Struct Rep Online. 2011;E67:m972. doi: 10.1107/S1600536811023853.
- 14.Godbert N, Pugliese T, Aiello I, Bellusci A, Crispini A, Ghedini M. Eur J Inorg Chem. 2007;32:5105. [Google Scholar]
- 15.Koh JH, Gagné MR. Angew Chem, Int Ed. 2004;43:3459. doi: 10.1002/anie.200453913. [DOI] [PubMed] [Google Scholar]
- 16.Being linear, XeF2 is significantly smaller than Selectfluor®.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.