

Abstract
The therapeutic principle of allogeneic haematopoietic cell transplantation (allo-HCT) is based on an active donor immune system that eliminates host-derived tumour cells. We hypothesized that in addition to the alloantigen-driven anti-tumour response, disruption of the immunological microenvironment within the tumour is responsible for its elimination after allo-HCT. We observed that induction of graft-versus-host disease (GvHD) significantly reduced the abundance of luc+ FoxP3+ regulatory T (Treg) cells in the tumour tissue, which is indicative of impaired or over-ridden tumour recruitment signals towards Treg cells. Analysis of the intestines and liver revealed chemokines and purine nucleotides as candidates for attracting Treg to these sites of inflammation. Despite its expression on tissue-residing Treg cells, the chemokine receptor CCR3 was not critical for Treg-cell function following allo-HCT. Extracellular ATP can attract immune cells via P2Y2. P2Y2 was found to be expressed on Treg cells, and we found a partial reduction of GvHD prevention when P2Y2−/− rather than P2Y2+/+ Treg cells were given. Exogenous local inflammation reduced Treg-cell accumulation in the tumour, suggesting a potential clinical approach to prevent Treg-cell-mediated tumour escape. In conclusion, we demonstrate that GvHD-related inflammation reduced Treg-cell numbers at the tumour sites, which may in turn help to explain the observation that patients with GvHD have a lower risk of tumour relapse.
Keywords: chemokines, chemotaxis, graft-versus-host disease, regulatory T cell trafficking, tumour microenvironment
Introduction
Local immunosuppression is a critical component of the tumour microenvironment. CD4+ CD25+ regulatory T (Treg) cells were shown to block anti-tumour immunity in different tumour entities.1 However, the process of a tumour fostering its own immunosuppressive milieu can be disrupted by a massive immunological intervention such as allogeneic haematopoietic cell transplantation (allo-HCT). In the context of graft-versus-host disease (GvHD), Treg cells were shown to allow for graft-versus-tumour (GvT) activity in several lymphoma and leukaemia models.2–5 One conceivable explanation for this could be a fundamental change in the tumour microenvironment or in other tissues after cytotoxic conditioning, both before allo-HCT and when GvHD evolves. As Treg cells potently reduce anti-tumour immune responses in the tumour microenvironment, reduction of their number at the tumour site would potently enhance allo-immune responses against the tumour. The suppressive activity of Treg cells is closely linked with their locations in tissue,6,7 indicating that determining Treg-cell trafficking under different conditions after allo-HCT is critical to understanding their different immunomodulatory effects. We have previously shown that Treg-cell recruitment to lymphoma tissue is via the CXCR4/SDF-1 axis following allo-HCT.8 However, the impact of strong inflammation, as is found during GvHD, on tumour-mediated Treg-cell recruitment is currently unclear. Besides chemokines, metabolites such as nucleotides released from damaged cells after allo-HCT9 could function as chemotaxis factors which influence Treg-cell trafficking, because they were shown to modulate chemotaxis in other immune cells.10
Therefore, the goal of this study was to delineate factors impacting on Treg-cell migration and accumulation in the presence or absence of tumours and inflammation. We employed in vivo Treg-cell tracking methodologies based on bioluminescence imaging,11 in combination with the neutralization of different chemokines identified by microarray analysis and genetic deficiency for P2Y2 in the Treg-cell compartment. Our data indicate that chemotaxis signals from inflamed tissues more potently recruit Treg cells compared with the tumour microenvironment and that Treg-cell-mediated suppression is partly dependent on P2Y2.
Methods
Mice
C57BL/6 (H-2Kb, Thy-1.2), FVB/N (H-2Kq, Thy-1.2) and BALB/c (H-2Kd, Thy-1.2) mice were purchased either from Charles River Laboratory (Sulzburg, Germany) or from the local stock of the animal facility at Freiburg University. Mice were used between 6 and 12 weeks of age. Only gender-matched combinations were used for transplant experiments. The luciferase (luc+) transgenic C57BL/6 mice have been previously described.12 P2Y2-deficient mice were kindly provided by Dr Marco Idzko (Freiburg University). All animal protocols (G07–19, G08–8) were approved by the University Committee on the Use and Care of Laboratory Animals at Albert-Ludwigs University Freiburg, Freiburg, Germany.
Bone marrow transplantation model, T-cell transfer and histopathology scoring
Bone marrow transplantation experiments were performed as previously described.4 Briefly, recipients were injected intravenously with 5 × 106 wild-type (WT) bone marrow cells after lethal irradiation with 900 cGy. To isolate Treg cells, splenocytes were stained for CD4 and CD25 and the cell-sorting gate was adjusted for the 20% CD4 cell with the highest CD25 expression. The purity of adoptively transferred Treg cells was > 90% CD4+ Foxp3 cells (data not shown). Cell doses are indicated for each experiment. For the subacute GvHD model for Treg-cell tracking the doses were: Treg: 8 × 105, Tconv: 1 × 105. The following transplant models were employed: C57BL/6→BALB/c and FVB/N→C57BL/6. Slides of liver, small bowel and large bowel samples collected on day 7 were stained with haematoxylin & eosin (H & E) and scored according to a previously published histopathology scoring system.13
B-cell lymphoma models
A20 cells were injected subcutaneously into the shaved right flank on the day of allo-HCT after the second irradiation. When indicated, WT or luc+ transgenic A20 B-cell lymphoma (BALB/c background, 5 × 106), was employed. The animals were killed when the tumour reached a diameter > 15 mm or showed ulceration.
In vivo chemokine neutralization experiments
CCR3 inhibitor (SB 328437, TOCRIS CatNo.:3650) was injected intraperitoneally at a dosage of 100 mg/kg per mouse on day 0, which was previously shown to have in vivo activity.14 Control groups received DMSO.
Local inflammation model
For induction of inflammation mice were injected subcutaneously with 20 μl complete Freund's adjuvant H37Ra in oil (Difco Laboratories, Detroit, MI) into the left footpad.
In vivo bioluminescence imaging
In vivo bioluminescence imaging was performed as previously described.11 Briefly, mice were injected intraperitoneally with luciferin (15 μg/g bodyweight). Ten minutes later mice were imaged using an IVIS100 charge-coupled device imaging system (Xenogen, Alameda, CA) for 5 min. Cell expansion was quantified in photons/second/cm2. Imaging data were analysed and quantified with living image 3.0 Software (Calipers, Rüsselsheim, Germany).
Migration assay
For Treg-cell migration studies, 24-well flat-bottomed plates with a pore size of 5 μm (Costar; Corning, Bodenheim, Germany) were employed. RPMI-1640 medium with 2% fetal calf serum was used in the transwells, 100 μl in upper wells and 500 μl in the lower chambers. Attractants were added to the lower chamber. CCL22, CCL1, CCL3, CCL5, CCL8 (R&D Systems, Wiesbaden, Germany) were added to the medium at a concentration of 100 ng/ml and 2 × 105 Treg cells were added to the insert and placed on the transwell. After incubation for 2 hr at 37° the number of cells migrated across the transwell were quantified by 40 seconds FACS measurement.
Preparation of intestines for cell isolation
The intestines were removed, homogenized and digested in RPMI-1640, 20% fetal calf serum and 200 units/ml collagenase (Worthington, Lakewood, NJ) at 37° for 4 hr. The cell suspension was then filtered through a 40-μm nylon mesh (BD Biosciences, Heidelberg, Germany).
Flow cytometry
All antibodies were purchased from BD Pharmingen (Heidelberg, Germany), Biolegend (Fell, Germany) and eBiosciences (Heidelberg, Germany) and used as FITC, phycoerythrin, Alexa647 or eFluor450 conjugates. All unlabelled FACS antibodies were conjugated with allophycocyanin using a LYNX Rapid APC antibody conjugation kit (AbD serotec, Düsseldorf, Germany) according to the manufacturer's instructions. The following antibodies were used for flow cytometric analysis: CD4 (GK 1.5/RM4-5), CD8 (53-6.7), CD25 (PC61), CD11c (HL3/N418), CD19 (6D5), Thy-1.1 (H1S51), Foxp3 (FJK-16s, CCR1 (polyclonal), CCR3 (TG14/CCR3), CCR5 (HM-CCR5), CCR8 (polyclonal), H-Kb (AF6-88.5), H-Kd (GF1 1.1) and CD107a (1D4B). Data were acquired with a CyanADP (Beckman Coulter, Krefeld, Germany) and then analysed with flowjo 7/8 software (Tristar, Ashland, OR).
Conventional histology and immunohistochemistry
Fresh frozen sections of 5-μm thickness were mounted on positively charged pre-cleaned microscope slides (Superfrost/Plus; R.Langenbrink, Emmendingen, Germany). For morphological analysis the tissues were stained with H & E. Evaluation of the stained tissue sections was performed on a Zeiss Axioplan 2 microscope (Zeiss, Jena, Germany). The standard objectives used were 20×/numerical aperture 0·45 and 40×/numerical aperture 0·60. Digital photos from the microscope were obtained using a Spot digital camera.
Statistical analysis
Differences in animal survival (Kaplan–Meier survival curves) were analysed by log-rank test. For comparison of means between experimental groups, the Student's t-test was used. A P-value < 0·05 was considered statistically significant.
Results
Donor type Treg cells accumulate within the intestines and spleen despite the presence of malignant B-cell lymphomas when acute GvHD is active
A factor that may influence Treg-cell recruitment after allo-HCT is inflammation caused by the preparative regimen, including chemotherapy or irradiation in the early phase and GvHD in the later phase. To study the impact of GvHD-related inflammation, we used our previously described Treg-cell migration model.8 The conventional T cells (Tconv) were given at a dose which allowed for viable B-cell lymphoma tissue (Fig. 1a), yet also caused histologically confirmed GvHD (Fig. 1b). In the presence of GvHD, signal from luc+ donor-type Treg cells could be detected in areas projecting over the spleen and the abdominal area (Fig. 1c, lower panel). To more precisely localize the origin of the Treg signal, the organs were removed surgically after luciferin injection and imaged within 10 min ex vivo. Ex vivo bioluminescence imaging demonstrated that GvHD led to Treg-cell-derived bioluminescence signal within the intestines, spleen and liver, which are each GvHD target organs (Fig. 1d). Conversely, in the absence of GvHD these organs demonstrated modest signal intensity and tumour tissue was infiltrated. Quantification of the Treg-cell-derived photons projecting over the tumour area demonstrated a reduction in signal when GvHD was induced (Fig. 1e). These data indicate that in the absence of GvHD, Treg cells are recruited towards tumour tissue. Conversely, when the GvHD target organs liver, spleen and intestines are inflamed by ongoing GvHD, Treg cells are recruited towards these sites and are then significantly less abundant within the tumour.
Figure 1.
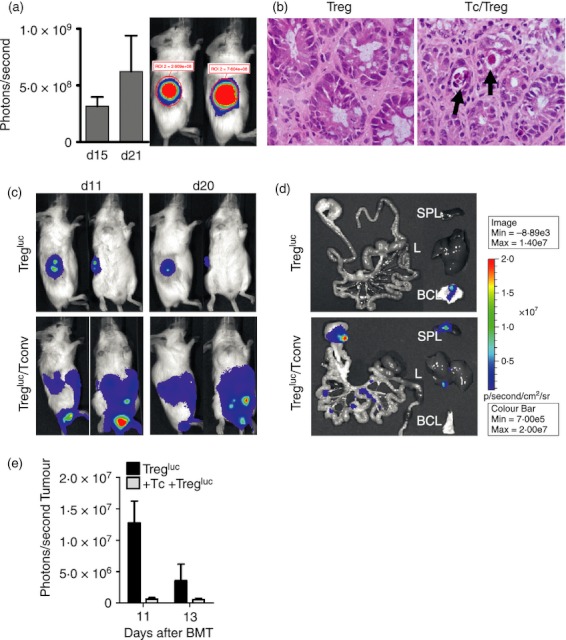
Impact of systemic inflammation due to graft-versus-host disease (GvHD) on regulatory T (Treg) cell migration. (a) Luc expressing A20 (5 × 106) were given subcutaneously in the right flank after irradiation of BALB/c recipients of allogeneic haematopoietic cell transplantation (allo-HCT) (H2-Kd). Conventional T (Tconv) cells (1 × 105, H2-Kb) were given 2 days following allo-HCT to induce acute GvHD. As shown for representative time-points the lymphoma was viable despite T-cell injection. The experiment was performed three times with similar results. (b) Seven days after transplantation, mice from the indicated groups were killed and sections of small bowel and large bowel were stained with haematoxylin & eosin (arrow: crypt abscess). In the presence of Tconv, subacute GvHD with typical histological signs was detected. A representative colon section per group is shown. The experiment was performed three times with similar results. (c) Bioluminescence imaging signal of representative mice is shown. Wild-type A20 (5 × 106) were given subcutaneously in the right flank after irradiation of BALB/c allo-HCT recipients (H2-Kd) and either Tregluc (8 × 105, H2-Kb) alone (n = 10) or Tregluc (8 × 105,H2-Kb) + Tconv (1 × 105,H2-Kb) (n = 15) were injected on day 0. The experiment was performed three times and the resulting numbers are derived from a pooled analysis. (d) 20 days after transplantation, mice from the indicated groups were killed and ex vivo imaging of the gastrointestinal tract (GIT), spleen (SPL), liver (L) and A20 B-cell lymphoma tissue was performed. The experiment was performed twice with similar results. (e) Treg-cell signal in photons derived from the tumour area is for two time-points (n = 3 per group). The experiment was performed twice with similar results.
Treg-cell infiltration in the inflamed intestines is not attributable to CCR3 signalling
It is conceivable that a certain subset of Treg cells is recruited to the GvHD target organs based on variations in chemokine receptor profiles, so we analysed Foxp3+ Treg cells before adoptive transfer and compared these with the Treg cells that infiltrated liver or intestine as GvHD target organs, as well as spleen and mesenteric lymph nodes (Fig. 2a). To determine the expression of different chemokine receptors on the surface of Treg cells, we isolated Treg cells from naive mice, which represent Treg cells in status because they were injected into transplanted mice and were therefore named ‘injected’. These Treg cells were then compared with Treg cells derived from the liver, spleen, mesenteric lymph nodes and intestines of allo-HCT recipients. We observed that the expression of CCR3 and CCR5 was significantly higher on Treg cells isolated from both lymphoid organs and GvHD target organs after allo-HCT, compared with Treg cells isolated from the spleen of an untreated mouse before the adoptive transfer (Fig. 2a). In agreement with this finding, the role of CCR5 for the protective function and migration potential of Treg cells towards target organs after allo-HCT has been described.15 Conversely, the expression of CCR8 was significantly decreased on the surface of Treg cells isolated from both liver and intestine. In vitro migration analysis of Treg cells towards a chemokine gradient revealed that CCL1, CCL3, CCL5 and CCL8 did not enhance migratory activity in vitro (Fig. 2b). Based on the increased expression of CCR3 on activated Treg cells in inflamed tissues we performed in vivo studies with a neutralizing antibody against CCR3. The biological activity of the previously described CCR3 inhibitor14 was tested in a transwell migration assay using conventional T cells and CXCL12 as chemoattractant. A significantly reduced migration of CD4+ conventional T cells was observed when the CCR3 inhibitor was included (data not shown). As migration of Treg cells towards the lymphoid compartment and the GvHD target tissue is critical for their function, we next evaluated if blocking CCR3 would reduce Treg-cell-mediated protection. In the absence of Treg cells, treatment with anti-CCR3 did not significantly prolong survival (Fig. 2b). Adoptive transfer of Treg cells, together with the conventional T cells, protected the mice from GvHD. This protection from GvHD-related death by Treg cells was also seen in allo-HCT recipients that received anti-CCR3 treatment along with the Treg cells (Fig. 3). These data indicate that CCR3 is not required for Treg-cell recruitment towards inflamed GvHD tissues or their suppressive activity against allo-responses.
Figure 2.
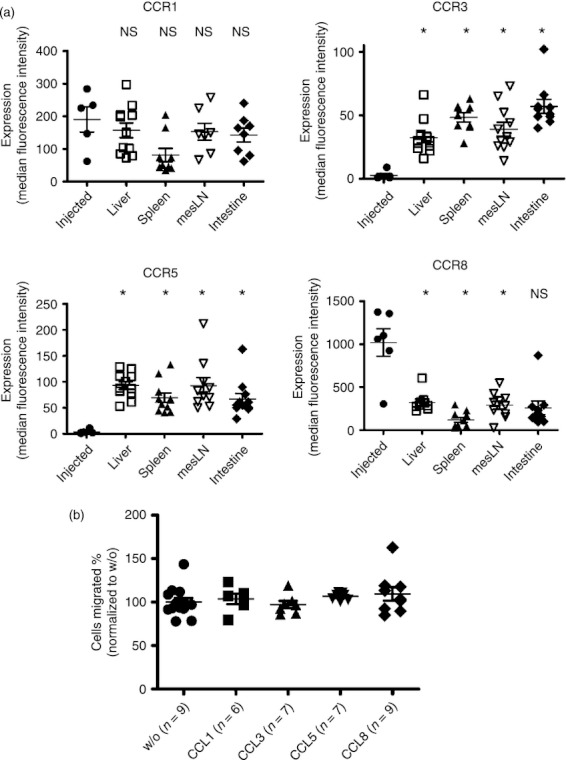
Expression of chemokine receptors on regulatory T (Treg) cells isolated from different tissues. (a) Surface expression of the indicated receptors, after gating on CD4+ FoxP3+ cells is displayed for freshly isolated Treg cells from spleen of naive animals or cells isolated from the organs liver, spleen, mesenteric lymph nodes or intestines of transplanted animals. The Treg cells that were isolated from naive donor mice were termed ‘injected’, the Treg cells isolated from the different organs were named according to the organ they were isolated from. Each data-point represents an individual donor mouse (n = 9; *P < 0·0001). The experiment was performed twice and the results were pooled. (b) In vitro migration of Treg cells was assessed in a transwell chamber with or without CCL1, CCL3, CCL5 and CCL8 as a stimulus (*P < 0·05). The experiment was performed three times with similar results.
Figure 3.
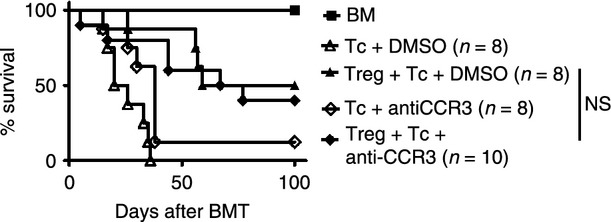
Regulatory T (Treg) cell function is independent of CCR3 in the setting of graft-versus-host disease (GvHD). Allogeneic haematopoietic cell transplantation (allo-HCT) was performed as described for the C57BL/6 into BALB/c combination. Treg cells were given on day 0 and T cells on day 2 after allo-HCT. The CCL3 inhibitor (100 mg/kg) or DMSO were given on day 0. Survival of BALB/c allo-HCT recipients (H2-Kd) receiving bone marrow alone or with conventional T cells ( + Tconv) (3 × 105, H2-Kb) or + Tconv and Treg (8 × 105, H2-Kb). The numbers of animals in each group are indicated in the figure, *P < 0·05. The experiment was performed twice and the results were pooled.
Nucleotides are released from inflamed sites
To study the amount of extracellular ATP released from GvHD sites, a previously described plasma membrane-targeted luciferase (PME) reporter cell line containing luciferase on the cell surface that specifically detects extracellular ATP,16 was applied in the allo-HCT model. We observed increased levels of extracellular ATP in the intestines as GvHD evolved (Fig. 4a), which is an indicator for local cell damage. P2Y2 was previously shown to be involved in immune cell recruitment in response to ATP and other nucleotides.17,18 We found expression of P2Y2 on conventional and regulatory T cells at the protein level indicating that this receptor could have a functional relevance for Treg-cell recruitment (Fig. 4b). Therefore, we next transferred Treg cells that were genetically deficient for P2Y2. The numbers of Treg cells were similar in mice that had received WT rather than P2Y2 Treg cells (data not shown). To evaluate if P2Y2-deficient Treg cells were less potent regulators of GvHD we next transferred luc transgenic effector T cells and monitored their in vivo expansion. The addition of Treg reduced the expansion of luc+ Tconv (Fig. 5a), an effect that was seen in both WT and P2Y2-deficient Treg cells. Consistent with this observation, allo-HCT recipients receiving either WT or P2Y2-deficient Treg cells were both protected from GvHD-related mortality (Fig. 5b). However, the P2Y2−/− cells were less protective than WT Treg cells and survival of the mice receiving P2Y2−/− Treg cells was significantly lower compared with the group receiving WT Treg cells (Fig. 5b). We did not find a difference of transferred WT compared with P2Y2−/− Treg cells with respect to an impact on the growth kinetics of A20 cells (data not shown). These data indicate that P2Y2 has a role in Treg-cell function that could be via attraction towards inflamed GvHD target organs via ATP.
Figure 4.
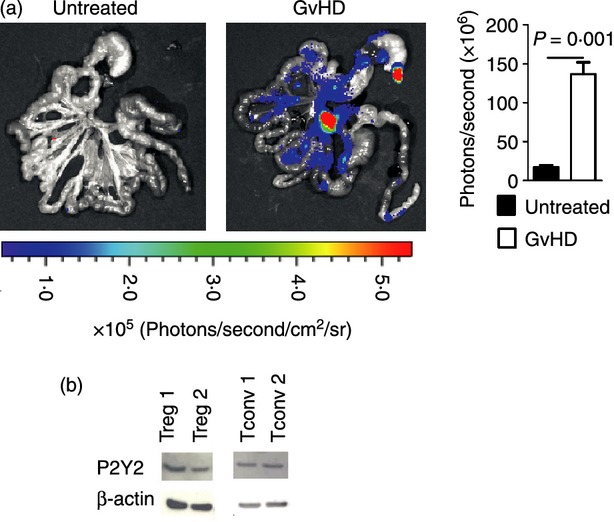
ATP is released from inflamed intestines and P2Y2 is found on regulatory T (Treg) cells. (a) Extracellular ATP is detected by PME cells. Left paned: representative bioluminescence imaging of the isolated gastrointestinal tract with foci of luminescence due to extracellular ATP. Right panel: The photons derived from extracellular ATP are quantified. Experiments were performed three times with n = 3. (b) P2Y2 is found on conventional T (Tconv) cells (CD4 MACS enriched, 92% CD4 positive) and sorted Treg (> 90% Foxp3 positive).
Figure 5.
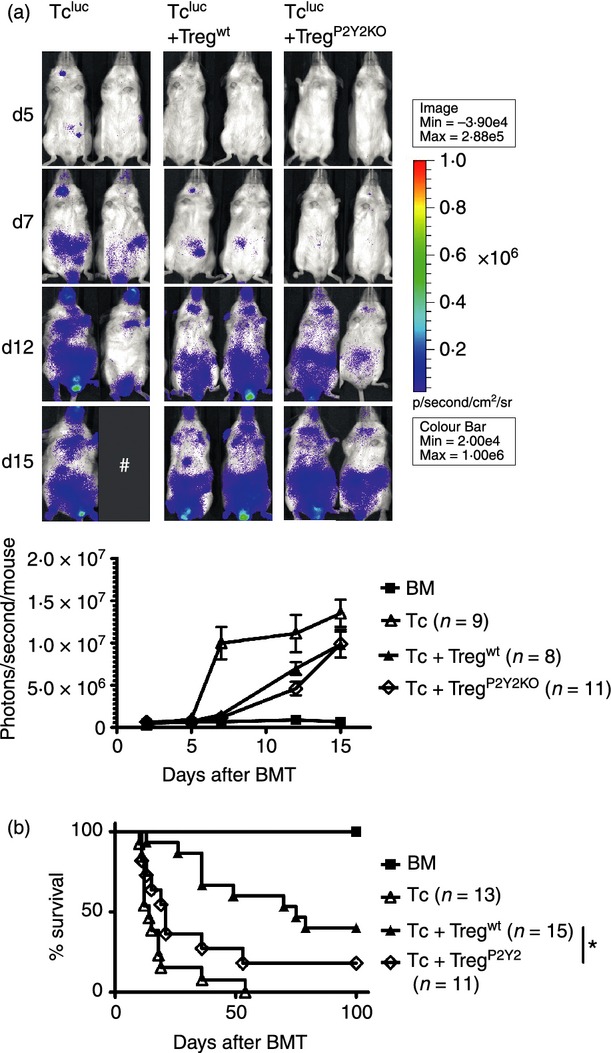
The role of P2Y2 for regulatory T (Treg) cell function after llogeneic haematopoietic cell transplantation (allo-HCT). Allo-HCT was performed as described for the C57BL/6 into BALB/c combination with luc transgenic conventional T (Tconv) cells. P2Y2−/− or wild-type (WT) Treg were given as indicated. (a) Upper panel: Tconv expansion on representative time-points after allo-HCT. Lower panel: quantification of the bioluminescence imaging signal derived from expanding Tconv cells at serial time-points. The experiment was performed twice with similar results. (b) Lower panel: Survival of BALB/c recipients receiving bone marrow alone or +Tconv or +Tconv/Treg (WT versus P2Y2−/−) is displayed, *P < 0·05. The experiment was performed twice and the results were pooled.
Local inflammation interferes with Treg-cell recruitment into the tumour microenvironment
We next evaluated the impact of local inflammation on Treg-cell migration to determine if the effects of GvHD on Treg-cell accumulation were the result of inflammatory recruitment signals. When Complete Freunds’ adjuvant (CFA) was injected into the left foot of allo-HCT-recipient mice, Treg-cell-derived signal was found at this location. At the same time the Treg signal within the A20 B-cell lymphoma area was reduced (Fig. 6a). Significantly fewer photons were emitted from luc+ Treg cels in the A20 B-cell lymphoma region after day 12 in the presence of CFA than in the absence of CFA (Fig. 6b). These data indicate that strong local stimuli at the site of inflammation (Fig. 6c) may counteract tumour-induced Treg-cell recruitment. Tumour progression was reduced by the injection of CFA compared with PBS injection (Fig. 6d). The predominance of the inflammatory signal over tumour-tissue-derived recruitment signals alludes to a hierarchical model for Treg-cell migration after allo-HCT.
Figure 6.
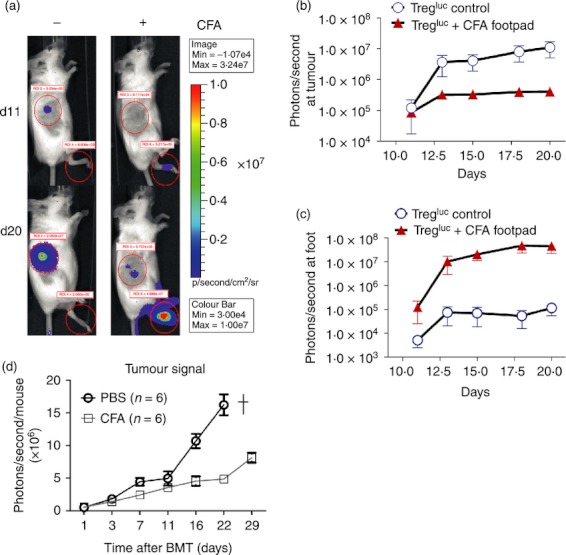
Impact of local inflammation by complete Freunds’ adjuvant (CFA) on regulatory T (Treg) cell migration. (a) Tregluc trafficking was monitored as described in Fig. 1. The recipient received PBS (left column, n = 5) or CFA (n = 6) (right column) injected into the left footpad. Signal is derived from migrating Treg as shown for representative time-points. The experiment was performed three times with similar results. (b) Treg signal in photons derived from the tumour area is plotted against time. The experiment was performed three times and the results were pooled. (c) Treg signal in photons derived from the foot area is plotted against time. The experiment was performed three times and the results were pooled.
Discussion
It is a well-established concept that Treg cells recruited towards lymph nodes draining tumour sites19 or the tumour itself20 interfere with anti-tumour immune responses. In contrast, the role of Treg-cell recruitment in the presence of both tumour and GvHD after allo-HCT is poorly understood. We tracked luciferase transgenic Treg cells in vivo and observed their migration to and accumulation in B-cell lymphoma tissues after allo-HCT, suggesting their potentially immunomodulatory effect at this site. Conversely, Treg-cell accumulation was interrupted when the liver and intestines were inflamed because of ongoing GvHD. Besides ongoing GvHD, Treg-cell migration and accumulation were also impacted by CFA-induced local inflammation. Local CFA injection reduced tumour growth which can be explained by different mechanisms that are not mutually exclusive. The local inflammation clearly caused Treg-cell recruitment away from the tumour, which may reduce the suppressive activity of this cell population in the tumour microenvironment. Besides the effects on Treg-cell recruitment, the CFA-induced inflammation may also cause activation of conventional T cells that exert anti-tumour effects.
We identified increased CCL1 expression in the intestines when GvHD evolved, but could not show enhanced migration of Treg cells towards this chemokine in vitro. Treg cells isolated from inflamed GvHD target tissues displayed higher CCR5 and CCD3 expression. As the role of CCR5 for Treg-cell-mediated GvHD protection has already been shown,15 we turned towards the role of CCR3. The blocking of CCR3 in vivo did not reduce Treg-cell-mediated protection from GvHD, indicating that Treg-cell migration and function were not significantly affected. Previous studies had shown Treg-cell localization at sites of ongoing immune responses, which is linked to their suppressor function. Hence, Treg cells have been found in the synovial fluid from rheumatoid arthritis patients,21 inflamed lesions in mice infected with Pneumocystis carinii22 or Leishmania major,23 exogenously induced colitis24 and inflamed skin.25 These data indicate that a co-localization of Treg cells and inflammation supporting cells is crucial for their suppressive effect. This may also hold true for models that rely on Freund's adjuvant (CFA) which we had used in our allo-HCT model to recruit Treg cells. It was observed in collagen-induced arthritis enhanced by CFA that Treg cells accumulated at sites of inflammation.26 A comparable role for CFA on Treg-cell function was also evident in a model of non-obese diabetic (NOD) mice protected from the autoimmune rejection of transplanted islets.27 The CFA failed to protect interferon-γ receptor-deficient status, which was dependent on a change in the balance between Treg cells and pathogenic T cells.27 When discussing the observation that Treg cells accumulate at sites of inflammation it must be kept in mind that Treg cells are heterogeneous with regard to their migration behaviour based on their homing receptor expression profile. CD103 positivity discriminates a Treg-cell differentiation stage with the phenotype of effector/memory cells6 from CD103-negative Treg cells that appear more naive. Compatible with this concept, the CD103-negative naive-like Treg express molecules including CD62 ligand L and CC chemokine receptor 7 (CCR7), which is permissive for their recirculation through lymphoid tissues.6,28,29 Conversely, effector/memory-like Treg cells display a high chemokine and homing receptor versatility and efficiently migrate into peripheral tissues and inflamed sites.30,31 Besides these conventional homing receptors Treg cells may respond via other receptors including purinergic receptors or adenosine receptors.
It is known that nucleotides released from damaged cells work chemotactically in activating P2Y2,17,32 making it conceivable that ATP release may contribute to the strong migration of Treg cells towards inflamed tissues. Inflammation led to the recruitment of both Treg and Tconv cells, suggesting that they may respond to similar chemotaxis signals. The biological reason may be that both the pro-inflammatory effector T cells and the suppressive Treg cells which finally down-modulate the immune response need to localize in the inflamed region. Our observation that allo-HCT recipients receiving WT or P2Y2-deficient Treg cells were both protected from GvHD-related mortality indicates that the P2Y2 was not required for ATP-mediated Treg-cell recruitment. We found that the induction of acute GvHD altered the Treg trafficking pattern, leading to a pronounced accumulation in inflamed GvHD target organs including liver, intestines and spleen. This observation may explain why it is possible to attain the reported protection from GvHD while anti-tumour responses are still intact, as has been previously reported by several groups.2–5 Furthermore, this concept is compatible with clinical results indicating that acute33 and chronic34 GvHD correlate with lower relapse rates, possibly because of Treg-cell attraction towards inflamed tissues rather than towards the tumour microenvironment.
In conclusion, we observed that Treg cells are recruited towards tumour tissue after allo-HCT, that this process can be interrupted by strong local or systemic inflammation and P2Y2 deficiency in the Treg-cell compartment reduced their protective capacity. These data support the concept that a certain degree of inflammation in the situation where residual tumour burden is present could prevent Treg-cell accumulation in the tumour, and in turn enhance its rejection.
Acknowledgments
The authors are grateful to Klaus Geiger for excellent technical assistance. This study was supported by the Deutsche Forschungsgemeinschaft, Germany (ZE 872/1-1 to R.Z.).
Authors contribution
CD helped to design the experiments, performed experiments, analysed data and helped to write the manuscript; MF helped with experiments and WR helped to design experiments and to write the manuscript; RZ designed the studies, analysed data and wrote the manuscript. All authors have read and agreed to the final version of the manuscript.
Disclosure
The authors have no conflict of interest to disclose.
References
- 1.Curiel T, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 2.Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, Negrin RS. CD4+ CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-v-host disease after bone marrow transplantation. Nat Med. 2003;9:1144–9. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 3.Trenado A, Charlotte F, Fisson S, et al. Recipient-type specific CD4+ CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest. 2003;112:1688–96. doi: 10.1172/JCI17702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeiser R, Nguyen VH, Beilhack A, Buess M, Schulz S, Baker J, Contag CH, Negrin RS. Inhibition of CD4+ CD25+ regulatory T cell function by calcineurin dependent interleukin-2 production. Blood. 2006;108:390–9. doi: 10.1182/blood-2006-01-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones SC, Murphy GF, Korngold R. Post-hematopoietic cell transplantation control of graft-versus-host disease by donor CD425 T cells to allow an effective graft-versus-leukemia response. Biol Blood Marrow Transplant. 2003;9:243–56. doi: 10.1053/bbmt.2003.50027. [DOI] [PubMed] [Google Scholar]
- 6.Huehn J, Siegmund K, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199:303–13. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegmund K, Feuerer M, Siewert C, et al. Migration matters: regulatory T-cell compartmentalization determines suppressive activity in vivo. Blood. 2005;106:3097–104. doi: 10.1182/blood-2005-05-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dürr C, Pfeifer D, Claus R, et al. CXCL12 mediates immunosuppression in the lymphoma microenvironment after allogeneic transplantation of hematopoietic cells. Cancer Res. 2010;70:10170–81. doi: 10.1158/0008-5472.CAN-10-1943. [DOI] [PubMed] [Google Scholar]
- 9.Wilhelm K, Ganesan J, Müller T, et al. Graft-versus-host disease enhanced by extracellular adenosine triphosphate activating P2X7R. Nat Med. 2010;12:1434–8. doi: 10.1038/nm.2242. [DOI] [PubMed] [Google Scholar]
- 10.McDonald B, Pittman K, Menezes GB, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–6. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 11.Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, Hou JZ, Negrin RS. Differential impact of mTOR inhibition on CD4+ CD25+Foxp3+ regulatory T cells as compared to conventional CD4+ T cells. Blood. 2008;111:453–62. doi: 10.1182/blood-2007-06-094482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeiser R, Nguyen VH, Hou JZ, Beilhack A, Zambricki EA, Buess M, Contag CH, Negrin RS. Early CD30 signaling is critical for adoptively transferred CD4+ CD25+ regulatory T cells in prevention of acute graft versus host disease. Blood. 2007;109:2225–33. doi: 10.1182/blood-2006-07-038455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplan DH, Anderson BE, McNiff JM, Jain D, Shlomchik MJ, Shlomchik WD. Target antigens determine graft-versus-host disease phenotype. J Immunol. 2004;173:5467–75. doi: 10.4049/jimmunol.173.9.5467. [DOI] [PubMed] [Google Scholar]
- 14.Mori A, et al. Selective suppression of Th2-mediated airway eosinophil infiltration by low-molecular weight CCR3 antagonists. Int Immunol. 2007;19:913–21. doi: 10.1093/intimm/dxm049. [DOI] [PubMed] [Google Scholar]
- 15.Wysocki C, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, Su L, Blazar BR, Serody JS. Critical role for CCR5 in the function of donor CD4+ CD25+ regulatory T cells during acute graft-versus-host disease. Blood. 2005;106:3300–7. doi: 10.1182/blood-2005-04-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS ONE. 2008;3:e2599. doi: 10.1371/journal.pone.0002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller T, Robaye B, Vieira RP, et al. The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy. 2010;65:1545–53. doi: 10.1111/j.1398-9995.2010.02426.x. [DOI] [PubMed] [Google Scholar]
- 18.Kronlage M, Song J, Sorokin L, Isfort K, Schwerdtle T, Leipziger J, et al. Autocrine purinergic receptor signaling is essential for macrophage chemotaxis. Sci Signal. 2010;3:55. doi: 10.1126/scisignal.2000588. [DOI] [PubMed] [Google Scholar]
- 19.Viguier M, Lemaitre F, Verola O, et al. Foxp3 expressing CD4+ CD25high regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol. 2004;172:1444–53. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- 20.Yang Z, Novak AJ, Stenson MJ, Witzig ET, Ansell SM. Intratumoral CD4+ CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood. 2006;107:3639–46. doi: 10.1182/blood-2005-08-3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao D, Malmstrom V, Baecher-Allan C, Hafler D, Klareskog L, Trollmo C. Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol. 2003;33:215–23. doi: 10.1002/immu.200390024. [DOI] [PubMed] [Google Scholar]
- 22.Hori S, Carvalho TL, Demengeot J. CD25+ CD4+ regulatory T cells suppress CD4+ T cell-mediated pulmonary hyperinflammation driven by Pneumocystis carinii in immunodeficient mice. Eur J Immunol. 2002;32:1282–91. doi: 10.1002/1521-4141(200205)32:5<1282::AID-IMMU1282>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 23.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+ CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–7. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 24.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+ CD25+ regulatory T cells. J Immunol. 2003;170:3939–43. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 25.Park SY, Gupta D, Kim CH, Dziarski R. Differential effects of peptidoglycan recognition proteins on experimental atopic and contact dermatitis mediated by Treg and Th17 cells. PLoS ONE. 2011;6:e24961. doi: 10.1371/journal.pone.0024961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelchtermans H, De Klerck B, Mitera T, Van Balen M, Bullens D, Billiau A, Leclercq G, Matthys P. Defective CD4+CD25+ regulatory T cell functioning in collagen-induced arthritis: an important factor in pathogenesis, counter-regulated by endogenous IFN-gamma. Arthritis Res Ther. 2005;7:402–15. doi: 10.1186/ar1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mori Y, Kodaka T, Kato T, Kanagawa EM, Kanagawa O. Critical role of IFN-gamma in CFA-mediated protection of NOD mice from diabetes development. Int Immunol. 2009;21:1291–9. doi: 10.1093/intimm/dxp097. [DOI] [PubMed] [Google Scholar]
- 28.Szanya V, Ermann J, Taylor C, Holness C, Fathman CG. The subpopulation of CD4+ CD25+ splenocytes that delays adoptive transfer of diabetes expresses l-selectin and high levels of CCR7. J Immunol. 2002;169:246102465. doi: 10.4049/jimmunol.169.5.2461. [DOI] [PubMed] [Google Scholar]
- 29.Valmori D, Merlo A, Souleimanian NE, Hesdorffer CS, Ayyoub M. A peripheral circulating compartment of natural naive CD4 Tregs. J Clin Invest. 2005;115:1953–62. doi: 10.1172/JCI23963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, D'Ambrosio D. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4+ CD25+ regulatory T cells. J Exp Med. 2001;194:847–53. doi: 10.1084/jem.194.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleinewietfeld M, Puentes F, Borsellino G, Battistini L, Rotzschke O, Falk K. CCR6 expression defines regulatory effector/memory-like cells within the CD25+ CD4+ T-cell subset. Blood. 2005;105:2877–86. doi: 10.1182/blood-2004-07-2505. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Corriden R, Inoue Y, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–5. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 33.Kim HJ, Min WS, Eom KS, et al. Anti-leukaemic role of acute GvHD after unrelated haematopoietic stem cell transplantation in intermediate- to high-risk acute myelogenous leukaemia. Bone Marrow Transplant. 2007;40:1069–74. doi: 10.1038/sj.bmt.1705873. [DOI] [PubMed] [Google Scholar]
- 34.Valcárcel D, Martino R, Caballero D, Martin J, Ferra C, Nieto JB, et al. Sustained remissions of high-risk acute myeloid leukemia and myelodysplastic syndrome after reduced-intensity conditioning allogeneic hematopoietic transplantation: chronic graft-versus-host disease is the strongest factor improving survival. J Clin Oncol. 2008;26:577–84. doi: 10.1200/JCO.2007.11.1641. [DOI] [PubMed] [Google Scholar]