

Abstract
Immune activation is a main driver of AIDS- and non-AIDS-linked morbidities in the course of HIV-1 infection. As CCR5, the main HIV-1 co-receptor, is not only a chemokine receptor but also a co-activation molecule expressed at the surface of T cells, it could be directly involved in this immune activation. To test this hypothesis, we measured by flow cytometry the mean number of CCR5 molecules at the surface of non-activated CD4+ T cells (CCR5 density), which determines the intensity of CCR5 signalling, and the percentage of CD8+ T cells over-expressing CD38 (CD38 expression), a major marker of immune activation, in the blood of 67 HIV-1-infected, non-treated individuals. CCR5 density was correlated with CD38 expression independently of viral load (P = 0·016). CCR5 density remained unchanged after highly active anti-retroviral therapy (HAART) introduction or cessation, whereas CD38 expression decreased and increased, respectively. Moreover, pre-therapeutic CCR5 density was highly predictive (r = 0·736, P < 10−4) of residual CD38 over-expression after 9 months of HAART. Hence, CCR5 might play an immunological role in HIV-1 infection as a driver of immune activation. This could explain why CCR5 antagonists may have an inhibitory effect on immune activation.
Keywords: activation marker, CD38, cell surface density, HIV co-receptor, T-cell activation
Introduction
Immune activation is a landmark of HIV-1 infection. It is even considered by some authors as a driver of CD4+ T-cell loss.1 In line with this hypothesis, biological parameters of T-cell activation, particularly the percentage of circulating CD8+ T-cells over-expressing CD38,2 have additive and stronger prognostic value in predicting progression to AIDS than CD4+ T-cell numbers or plasma HIV-1 RNA.3 Likewise, it has been observed that increased baseline levels of CD4+ and CD8+ T-cell activation before HIV-1 seroconversion were associated with a higher risk of developing AIDS after seroconversion.4 Furthermore, in virological responders to highly active anti-retroviral therapy (HAART), a non-immunological response is linked to residual immune activation.5 Finally, in aviraemic patients, residual immune activation is correlated with mortality,6 and with non-AIDS-linked morbidities such as atherosclerosis7 or hepatitis C virus-related cirrhosis.8 Altogether these observations show that immune activation is a key player in HIV pathogenesis. Therefore, it is of major importance to identify molecules involved in HIV-linked immune activation, biomarkers able to predict it, and therapeutic ways to combat it.
The C–C chemokine receptor CCR5 might be one of these molecules. CCR5 plays crucial virological roles in HIV-1 infection. As the major co-receptor, it is used by the virus to bind to target cells.9 This binding induces the activation of the target cell, increasing its infectibility and its capacity to produce virions.10 Moreover, CCR5 is involved in various cytopathogenic mechanisms including syncytia formation and viral envelope-induced apoptosis.11
It is becoming increasingly clear that, in addition to being a C–C chemokine receptor, CCR5 is a co-activation molecule. Previous studies in mice and humans have shown that the CCR5-binding chemokines MIP-1α (CCL3), MIP-1β (CCL4), and RANTES (CCL5) were able to induce a second in vitro activation signal in T-cell receptor-triggered T cells. This co-activation signal boosted the production of interleukin-2 (IL-2),12,13 interferon-γ (IFN-γ),14 IL-4,12 and CCL415; increased the cell-surface expression of the IL-12 receptor16 and of the activation markers CD25,13 CD28, CD40L;15 and increased T-lymphocyte proliferation.12,13 In mice, administration of CCL3, CCL4 or CCL5 together with an antigen increased the capacity of CD4+ T cells to express membrane activation markers, to produce IL-2, IFN-γ, IL-5 and IL-6 in vitro in response to the antigen, and the number of specific antibody-forming cells.16 Conversely, antigen-activated T cells from CCL5-deficient (CCL5−/−) mice displayed a decrease in IL-2 and IFN-γ production and in vitro proliferation, and a 50% reduction in cutaneous delayed-type hypersensitivity.17 In support of the notion that the C–C chemokine-CCR5 axis is a co-activation pathway, various groups have reported that blocking CCL3, CCL4, CCL512,13,18 or CCR519 with antibodies hinders T-cell activation. Recently, more direct arguments in favour of a role for CCR5 in T-cell activation have been provided. When a CD4+ T-cell recognizes a peptide at the surface of an antigen-presenting cell, CCR5 molecules at the surface of the lymphocyte are recruited into the immune synapse where they are stimulated, thereby providing co-activation signals as strong as those delivered through the co-activation molecule CD28.20,21 In addition to participating in T-lymphocyte activation at the single cell level, CCR5 also participates in the collaboration between T cells in the course of a response to an antigen. Hence, after immunization, there is CCR5 over-expression at the surface of naive CD8+ T cells in immunogen-draining lymph nodes that permits them to move chemotactically towards CD4+ T cells, recognizing the antigen presented by dendritic cells, where CCL3 and CCL4 are produced.22 The same phenomenon has been described for naive CD8+ T cells attracted in a CCR5-dependent way by specific CD8+ T cells interacting with an antigen-presenting cell.23 Altogether these data argue for the capacity of the CCR5 axis to amplify the immune response at T-cell and T-subpopulation levels. CCR5 also participates in macrophage24 as well as in mast cell activation.25 Moreover, CCR5 has been involved in the inflammatory gene expression induced by encephalomyocarditis virus infection.26
If CCR5 is involved in T-cell activation, it is tempting to speculate that it could participate in the polyclonal immune activation observed in HIV-infected subjects.
The level of expression of the CCR5 molecule at the surface of non-activated HLA-DR− (DR−) CD4+ T cells is variable among HIV-infected individuals, but constant over time in a given individual.27 In addition, CCR5 densities at the surface of CD4+ and CD8+ T cells are strongly correlated.28 This level of expression determines the intensity of the signalling triggered by CCR5 ligands. For instance, we have shown that over-expression of CCR5 at the surface of cells transduced with the CCR5 gene results in an increase in the intracellular calcium concentration upon CCL5 activation.29 In line with this, the capacity of primary CD8+ T cells to migrate towards CCL5 is linked to their level of surface CCR5 expression.30 In HIV-infected subjects, CD4+ T-cell surface CCR5 density, rather than the percentage of CD4+ T-cell-expressing CCR5, determines the intensity of virus production27 through gp120-triggered CCR5 signalling.29 For these reasons, we hypothesized that the level of T-cell surface CCR5 expression could influence the intensity of polyclonal immune activation in HIV-1-infected persons.
In the present study, we analysed the relationship between CCR5 expression on CD4+ T cells and CD8+ T-cell activation. We chose to measure CCR5 expression at the surface of non-activated, DR− CD4+ T cells, so that this factor would not be modified by T-cell activation. Our hypothesis is that an intrinsic level of CCR5 expression in T cells may predispose these cells to immune activation. To this purpose, we looked for a link between CCR5 density at the surface of DR− CD4+ T cells and the proportion of CD38hi CD8+ T cells in HIV-1-infected patients either untreated, or under treatment initiation or disruption to define the impact of the level of CCR5 expression on T-cell activation.
Methods
Patients
Peripheral blood samples for the routine follow-up of HIV-1-infected patients were used in this observational study. Patients were asymptomatic, non-treated adults over 18 years of age recruited at the Infectious Diseases Department of the University Hospital of Montpellier, France. No other eligibility criteria were used. Informed consent was obtained from the patients and human experimental guidelines of the local ethics committee were followed in the conduct of this research.
Determination of cell surface CCR5 density
DR− CD4+ T-cell surface CCR5 density was determined as previously described.27 Briefly, cells in 50 μl of blood were indirectly labelled with the anti-CCR5 monoclonal antibody (mAb) 2D7 (Pharmingen, San Diego, CA) and a fluorescein isothiocyanate (FITC) -conjugated anti-immunoglobulin probe (Beckman-Coulter, Margency, France), and directly labelled with phycoerythrin (PE) -conjugated anti-CD4 mAb and PE-cyanine 5 anti-DR mAb (Beckman-Coulter).
CCR5 expression was analysed after gating on DR− CD4+ T cells expressing CCR5 and quantified by converting FITC fluorescence intensity into antibody-binding capacity. This conversion was performed using a calibration curve obtained with commercial beads (QIFIKIT; Dako, Glostrup, Denmark) coated by the manufacturer with various densities of monoclonal antibody, and labelled with the FITC-conjugated anti-immunoglobulin probe.
Determination of the proportion of CD38hi CD8+ T cells
CD38 expression at the surface of CD8+ cells was determined using two-colour staining with FITC-conjugated anti-CD8 mAb and a PE-conjugated anti-CD38 mAb (Beckman-Coulter) as previously described.31 CD38 expression was analysed after gating on lymphocytes with high CD8 expression. The CD38hi threshold was established using the CELLQUANT CD38/CD8 kit (Biocytes, Marseille, France) in accordance with the manufacturer's instructions.
Plasma viraemia quantification
HIV-1 RNA levels in plasma were quantified with the COBAS AMPLICOR™ HIV-1 MONITOR v1.5 (Roche Diagnostics, Mannheim, Germany) in accordance with the manufacturer's instructions.
Statistical analysis
Pearson rank correlations were used to evaluate the link between age, HIV-1 RNA plasma levels, CD4 count, CCR5 density and percentage of CD38hi CD8+ T cells. A multivariate analysis by linear regression was performed on these parameters to test for a correlation between CCR5 density and immune activation independently of viral load.
The data were normally distributed excepting the viral loads. Therefore, for both analyses, a logarithmic transformation was made to normalize the distribution of viral loads. Variations in CCR5 density, and in the other parameters under treatment initiation or cessation, were analysed with the Student's t-test for paired data.
The effects of HAART initiation or interruption on the parameters measured were examined in eleven and six randomly selected patients, respectively. To determine the effects of HAART initiation, parameters before commencing and after 6 months of therapy were compared. To determine the effects of HAART interruption, parameters before and 6 months after stopping therapy were compared.
Results
The level of immune activation is linked to the level of CCR5 expression independently of viral load
Baseline characteristics of the 67 study subjects were as follows: 31% of patients were women and 69% were men, the mean age was 37 years (range 18–72), the mean HIV-1 RNA level was 101 293 copies/ml (range 22–776 658), and the mean CD4 count was 372 cells/μl (range 49–805).
CD8+ T cells were identified by gating on peripheral blood mononuclear cells with high CD8 expression as shown in Fig. 1(a,b). We have previously determined that activated CD8+ T cells express more than 8500 CD38 surface molecules per cell,31 and this threshold was used to define CD38hi expression (Fig. 1c). The proportion of CD38hi CD8+ T cells was not correlated with age (r = 0·121; P = 0·323), but was inversely correlated with the CD4 count (r = −0·491; P < 0·001; data not shown) and positively correlated with viraemia (r = 0·320; P = 0·008; Fig. 2a). DR− CD4+ T cells were identified by gating on peripheral blood mononuclear cells with high CD4 expression and no HLA-DR expression as shown in Fig. 1(a,d,e). The mean number of surface CCR5 molecules per cell labelled with an anti-CCR5 mAb (CCR5 density) was then quantified after the mean fluorescence intensity (Fig. 1f). We also measured CCR5 density on the cell subsets corresponding to naive (CCR7+ CD45RA+), central memory (CCR7+ CD45RA−), effector memory RA− (CCR7− CD45RA−), and effector memory RA+ (CCR7− CD45RA+) CD4+ T cells. The levels of cell surface CCR5 expression varied among these different subpopulations (Fig. 1g), but were linked. That is to say, low CCR5 expressors expressed low levels of CCR5 on all their CD4+ T-cell subsets, whereas high CCR5 expressors expressed high levels of CCR5 on all their CD4+ T-cell subsets, as illustrated in Fig. 1(h). Therefore, we chose CCR5 density at the surface of the whole DR− CD4+ T-cell population as a global marker of the level of CCR5 expression on all the CD4+ T cells.
Figure 1.

Analysis of CD38 over-expression (CD38hi) on CD8+ T cells and of CCR5 expression on DR− CD4+ T cells. Peripheral blood mononuclear cells were identified using side scatter versus forward scatter (a), and CD8+ T cells were identified using structure feature and CD8 over-expression (CD8hi, b). The percentage of CD8hi cells over-expression CD38 (more than 8500 CD38 molecules per CD8+ T cell) was then determined (c). DR− CD4+ T cells were identified in comparison with the labelling obtained with an isotype control within the peripheral blood mononuclear cells using side scatter versus CD4 expression (d), and then DR expression versus CD4 expression (e). CCR5 expression was then analysed on these DR− CD4+ T cells (black line, isotypic control; grey shaded histogram, anti-CCR5 antibody; f). CD4+ T cells of a donor were sorted into naive, central memory and effector memory subpopulations and the level of expression of CCR5 on each subpopulation was determined (g). CCR5 densities on CD4+ T-cell subpopulations of three different donors (h).
Figure 2.
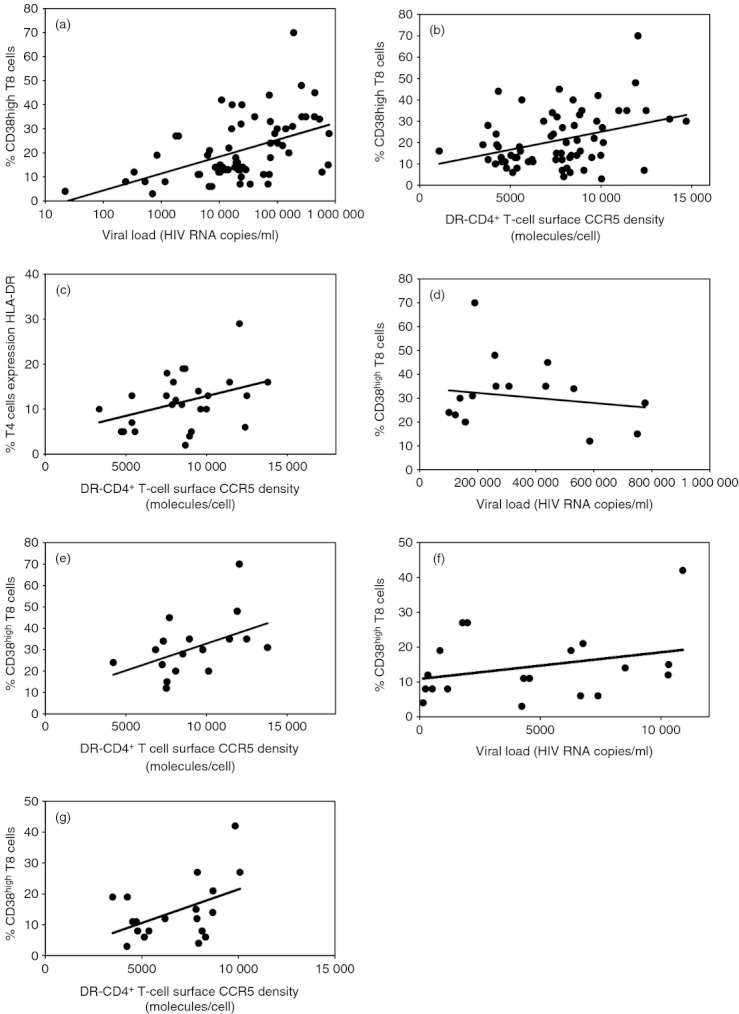
Correlation between the percentage of CD38hi CD8+ T cells and HIV-1 RNA plasma level (a), and the mean number of CCR5 molecules at the surface of DR− CD4+ T cells (b), and between the percentage of DR+ CD4+ T cells and the mean number of CCR5 molecules at the surface of DR− CD4+ T cells (c) in non-treated patients with HIV-1 infection. Correlation between the percentage of CD38hi CD8+ T cells and HIV-1 RNA plasma level (d, f), and the mean number of CCR5 molecules at the surface of DR− CD4+ T cells (e, g) in patients who presented with viraemia over 100 000 (d, e) or below 10 000 (e, f) copies/ml.
The DR− CD4+ T-cell surface CCR5 density correlated positively with the proportion of CD38hi CD8+ T cells (r = 0·356; P = 0·003; Fig. 2b). The percentage of CD4+ T cells expressing another activation marker, HLA-DR, was also determined in a subgroup of 26 patients. Here again, we observed a correlation between DR− CD4+ T-cell surface CCR5 density and the frequency of activated CD4+ T cells (r = 0·382; P = 0·054; Fig. 2c).
To determine whether the link between CCR5 expression and T8 activation depended on the level of viraemia or not, we first performed a multivariate analysis. This analysis established that the proportion of CD38hi CD8+ T cells was linked to DR− CD4+ T-cell surface CCR5 density independently of HIV-1 RNA level (P = 0·016). Second, we stratified the patients according to their level of viraemia. This stratification made clear that CCR5 expression and CD8+ T-cell activation are linked independently of the viraemia. For instance, in the 17 patients harbouring > 100 000 HIV-1 RNA copies/ml, the proportion of CD38hi CD8+ T cells did not appear to be linked to viraemia (r = −0·214; P = 0·426; Fig. 2d), but was linked to the level of CCR5 expression (r = 0·481; P = 0·050; Fig. 2e). Likewise, in the 19 patients harbouring < 10 000 HIV-1 RNA copies/ml, the proportion of CD38hi CD8+ T cells did not appear to be linked to viraemia (r = 0·348; P = 0·144; Fig. 2f), but was linked to the level of CCR5 expression (r = 0·466; P = 0·044; Fig. 2g).
The decrease in immune activation under therapy does not lower DR− CD4+ T-cell surface CCR5 density
To test the hypothesis that the level of CD8+ T-cell activation might determine the level of CCR5 expression on CD4+ T cells, we followed 11 patients who started an anti-retroviral therapy. Six months after initiation of the treatment, HIV-1 viraemias and the percentages of CD38hi CD8+ T cells dropped drastically, and CD4 counts increased, in all patients (Fig. 3a–c). Overall, the reductions in the viral loads and percentages of CD38hi CD8+ T cells were statistically significant, as were the increases in CD4 counts (Table 1). Yet, DR− CD4+ T-cell surface CCR5 densities remained remarkably stable (Fig. 3d and Table 1). Hence, HAART-mediated reduction in the percentage of CD38hi CD8+ T cells was not followed by a decrease in CCR5 expression at the surface of non-activated CD4+ T cells.
Figure 3.
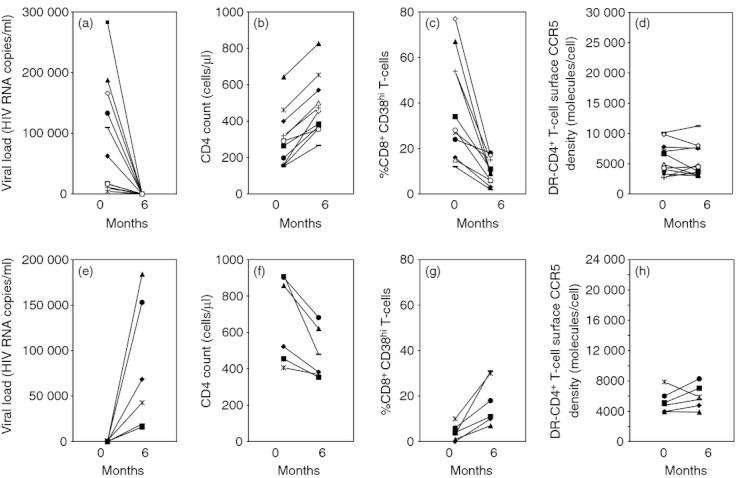
Evolution of HIV-1 RNA plasma level (a), CD4 count (b), percentage of CD38hi CD8+ T cells (c), and CCR5 density at the surface of DR− CD4+ T cells (d) in HIV-1-infected patients before and after treatment with highly active anti-retroviral therapy (HAART). Months represent time from initiation of therapy. Evolution of HIV-1 RNA plasma level (e), CD4 count (f), percentage of CD38hi CD8+ T cells (g), and CCR5 density at the surface of DR− CD4+ T cells (h) in HIV-1-infected patients before and after interruption of highly active anti-retroviral therapy. Months represent time from cessation of therapy.
Table 1.
Effects of highly active anti retroviral therapy (HAART) initiation or interruption on viral load, CD4 counts, CD38hi CD8+ T cells and CCR5 density on the surface of DR− CD4+ T cells in eleven and six HIV-1-infected patients, respectively. For HAART initiation and interruption, months represent time from initiation of therapy and time from cessation of therapy, respectively
| Months | |||
|---|---|---|---|
| 0 | 6 | P-value | |
| HAART initiation, mean (95% CI) | |||
| Viral load, HIV-1 RNA copies/ml | 91 042 (28 599–153 484) | 53 (38–68) | 0·009 |
| CD4 counts, cells/μl | 281 (168–394) | 472 (364–581) | < 10−4 |
| CD38hi CD8+ T cells,% | 38 (21–55) | 10 (6–15) | 0·003 |
| CCR5 density, molecules/cell | 5762 (3956–7567) | 5476 (3645–7308) | 0·541 |
| HAART interruption, mean (95% CI) | |||
| Viral load, HIV-1 RNA copies/ml | 238 (0–713) | 80 583 (5720–155 447) | 0·040 |
| CD4 counts, cells/μl | 677 (425–928) | 473 (317–629) | 0·015 |
| CD38hi CD8+ T cells, % | 4 (0–8) | 18 (7–29) | 0·011 |
| CCR5 density, molecules/cell | 5281 (3723–6839) | 5903 (4246–7540) | 0·366 |
The increase in immune activation secondary to treatment interruption does not result in a rise in DR− CD4+ T-cell surface CCR5 density
Conversely, we followed six other patients who interrupted their anti-retroviral therapy. Six months after interruption of HAART, augmentation of viral load and an increase in the percentage of CD38hi CD8+ T cells was observed in all patients as well as a reduction in CD4 counts (Fig. 3e–g). Overall, increases in the viral loads and percentages of CD38hi CD8+ T cells were statistically significant, as were the decreases in CD4 counts (Table 1). By contrast, CCR5 expression at the surface of DR− CD4+ T cells remained stable (Fig. 3h and Table 1). Hence, when immune activation is modified either by introduction or by interruption of HAART, CCR5 density is not modified. This proves that it is not the level of immune activation that determines the level of CCR5 expression, but rather argues for the opposite.
The baseline level of CCR5 expression on CD4+ T cells is predictive of the residual immune activation under HAART
Even after the disappearance of measurable viraemia under treatment, immune activation may persist in some patients. We wanted to check whether the baseline level of CCR5 expression might determine the intensity of this residual immune activation. To test this hypothesis, we compared DR− CD4+ T-cell surface CCR5 density in 30 patients who received HAART, measured before the treatment, with the percentage of CD38hi CD8+ T cells persisting after the initiation of therapy. All the patients had become aviraemic at month 6. Figure 4 shows that the percentage of CD38hi CD8+ T cells at month 3 (r = 0·565; P = 0·001), month 6 (r = 0·584; P < 0·001), and month 9 (r = 0·736; P < 10−4) was strongly correlated with the pre-therapeutic CCR5 density on DR− CD4+ T cells.
Figure 4.
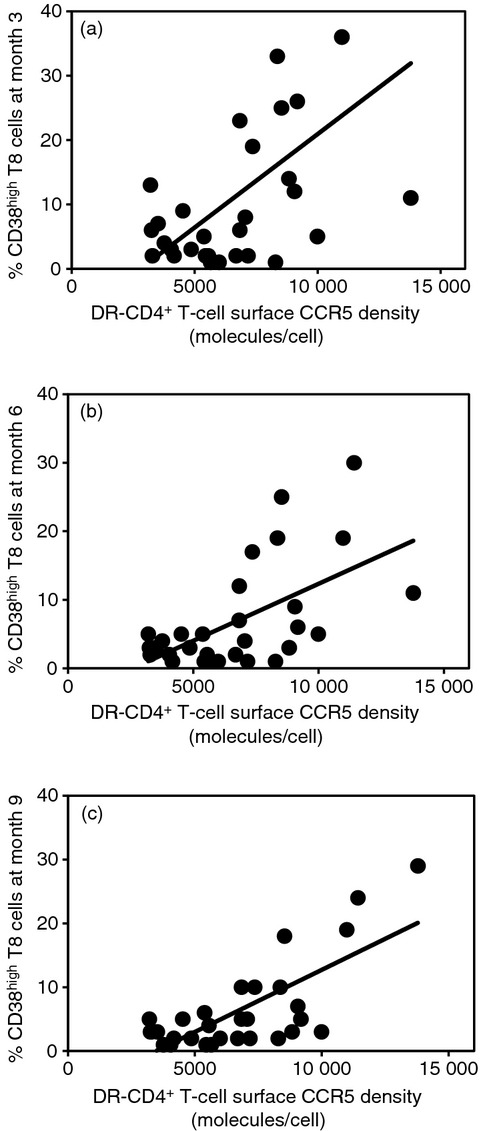
Correlation between the baseline pre-therapeutic mean number of CCR5 molecules at the surface of the DR− CD4+ T cells and the percentage of CD38hi CD8+ T cells after 3 (a), 6 (b), and 9 (c) months of HAART in 30 virological responders.
Discussion
This study shows that the level of CCR5 expression is linked to and predictive of the level of immune activation in patients infected with HIV-1, independently of the viral load.
As T-cell activation may result in CCR5 over-expression27 it could be argued that, although we measured CCR5 density at the surface of non-activated CD4+ T cells, the link observed between immune activation and CCR5 density was a consequence of the level of T-cell activation determining the level of CCR5 expression. If immune activation drives CCR5 expression, viral rebound observed in patients following HAART interruption would result in an increase in the percentage of CD38hi CD+ T cells and thereby in CCR5 density. However, the changes in immune activation induced by either the introduction or the cessation of HAART did not result in any change in CCR5 density; thereby appearing to indicate that the baseline level of CCR5 expression impacts on the intensity of lymphocyte activation.
Hence, T-cell surface CCR5 density may not only be a driver of viral production, as suggested by Reynes et al.,27 but may also be a driver of the polyclonal T-cell activation (Fig. 4). That is to say, CCR5 could play an immunological role in HIV-1 infection in addition to its roles in viral replication and cytopathogenicity.
This hypothesis is in line with a recent study, which reported that the level of CCR5 expression at the surface of a lymphocyte determines the level of activation of the transcription factor nuclear factor of activated T cells (NFAT) and thereby the level of IL-2 production.18 It is also in keeping with a study by Pandrea et al.,32 which demonstrated that the level of CCR5 expression at the surface of CD4+ T cells and immune activation, but not the level of viral replication, were predictive of progression in primates with simian immunodeficiency virus (SIV) infection. This suggests that in high CCR5 expressors, SIV infection may induce high immune activation responsible for a high CD4+ T-cell loss, whereas in low CCR5 expressors a low level of immune activation, in spite of high viral loads, might hinder disease progression. Moreover, this model might explain why Ahuja et al.33 observed that patients with different CCR5 genotypes and doses of gene encoding the CCR5-binding chemokine CCL3L1 have different rates of CD4+ T-cell recovery under HAART. CCR5–CCL3L1 genotypes resulting in a strong efficiency of the CCR5 axis might favour residual T-cell activation and thereby limit immune recovery.
Assuming that CCR5 plays both virological and immunological roles in the course of HIV-1 infection, CCR5 antagonists would have a double effect. It is possible that currently developed CCR5 antagonists, inhibiting C–C chemokines–CCR5 interactions, could inhibit the co-activation function of CCR5 in addition to blocking HIV-1 replication and cytopathogenicity. Specifically, HAART intensification with a CCR5 antagonist has resulted, in some34,35 but not all36 studies on virological responders, in a decrease in the percentage of activated peripheral CD4+ and CD8+ T cells.
This may account for the observation that CCR5 blockers appear to have a higher than expected effect on CD4+ T-cell counts.37 For example, the anti-CCR5 antibody HGS004 induced increases in CD4 cell counts independently of antiviral response in a study in 63 patients infected with CCR5-tropic HIV-1.38 In another study, 167 patients harbouring non-pure R5 strains were treated with maraviroc. After 24 weeks, a significant rise in CD4 counts was observed, whereas the HIV-1 RNA plasma levels were not significantly reduced.39 Moreover, clinical trials testing regimens including a CCR5 antagonist appear to have a relatively better immune recovery than regimens without a CCR5 antagonist.40
This might be particularly important for patients at risk of a non-immunological response. About 15% of patients aviraemic under HAART have an incomplete immune response with CD4 counts remaining below 350 CD4+ T cells/ml,41 caused by the persistence of polyclonal immune activation in some patients.5 The possibility that blocking the CCR5 axis could boost CD4+ T-cell recovery in non-immunological responders has been tested by various groups. In some34,35 but not all36 trials, HAART intensification with a CCR5 antagonist resulted in an increase in CD4 slope.
The putative effect of CCR5 blockers on immune activation may also be beneficial in HIV-associated inflammatory disorders. Immune reconstitution inflammatory syndrome (IRIS) may be considered as immune hyperactivation triggered by host, tumour or infective antigens consecutive to HAART-induced CD4+ T-cell recovery. It results in pathological inflammation with a heterogeneous range of clinical manifestations. The introduction of a CCR5 antagonist in the anti-retroviral regimen of persons at risk of IRIS seems worth testing. Also, CCR5 antagonists may help to prevent non-AIDS linked morbidities in which chronic inflammation is believed to play a causative role.
In conclusion, the link between T4 cell surface CCR5 density and the proportion of activated T8 cells in HIV-1-infected adults evidenced in this study raises the interesting hypothesis that CCR5, as a co-activation molecule, could participate in polyclonal immune activation. By blocking both the virological roles and this newly described immunological role of CCR5 in HIV-1 infection, CCR5 antagonists could have a double therapeutic effect.
Acknowledgments
We thank Andrea Bothwell of Science Communications who provided copy-editing and journal styling before submission. PP, ET and TM performed assays, acquired, analysed and interpreted data, and revised and approved the article; JPV and JR designed assays, and revised and approved the article; PC conceived and designed the study, and drafted and approved the article. This study was supported by the French National Agency for Research on AIDS and Viral Hepatitis (ANRS, grant number 09397), Pfizer, and ViiV.
Disclosures
JR has received honoraria for lectures or advisory boards and/or research support from Abbott, BMS, Boehringer Ingelheim, Gilead, GSK, Merck, Pfizer, Roche, Schering-Plough, Tibotec, ViiV; PC has received grants from Pfizer, Merck Sharp and Dohme-Chibret, Gilead, ViiV and GlaxoSmithKline. No conflicts are declared for PP, ET, TM and J-PV.
References
- 1.Levacher M, Hulstaert F, Tallet S, Ullery S, Pocidalo JJ, Bach BA. The significance of activation markers on CD8 lymphocytes in human immunodeficiency syndrome: staging and pronostic value. Clin Exp Immunol. 1992;90:376–82. doi: 10.1111/j.1365-2249.1992.tb05854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coetzee LM, Tay SS, Lawrie D, Janossy G, Glencross DK. From research tool to routine test: CD38 monitoring in HIV patients. Cytometry B Clin Cytom. 2009;76:375–84. doi: 10.1002/cyto.b.20478. [DOI] [PubMed] [Google Scholar]
- 3.Froebel KS, Raab GM, D'Alessandro C, Armitage MP, MacKenzie KM, Struthers M, Whitelaw JM, Yang S. A single measurement of CD38CD8 cells in HIV+, long-term surviving injecting drug users distinguishes those who will progress to AIDS from those who will remain stable. Clin Exp Immunol. 2000;122:72–8. doi: 10.1046/j.1365-2249.2000.01348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koning FA, Otto SA, Hazenberg MD, Dekker L, Prins M, Miedema F, Schuitemaker H. Low-level CD4+ T cell activation is associated with low susceptibility to HIV-1 infection. J Immunol. 2005;175:6117–22. doi: 10.4049/jimmunol.175.9.6117. [DOI] [PubMed] [Google Scholar]
- 5.Goicoechea M, Smith DM, Liu L, May S, Tenorio AR, Ignacio CC, Landay A, Haubrich R. Determinants of CD4+ T cell recovery during suppressive antiretroviral therapy: association of immune activation, T cell maturation markers, and cellular HIV-1 DNA. J Infect Dis. 2006;194:29–37. doi: 10.1086/504718. [DOI] [PubMed] [Google Scholar]
- 6.Kuller LH, Tracy R, Belloso W, et al. Inflammatory and coagulation biomarkers in patients with HIV infection. PLoS Med. 2008;5:e203. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsue PY, Hunt PW, Schnell A, Kalapus SC, Hoh R, Ganz P, Martin JN, Deeks SG. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS. 2009;23:1059–67. doi: 10.1097/QAD.0b013e32832b514b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balagopal A, Philp FH, Astemborski J, et al. Human immunodeficiency virus-related microbial translocation and progression of hepatitis C. Gastroenterology. 2008;135:226–33. doi: 10.1053/j.gastro.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dragic T, Litwin V, Allaway GP, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–73. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 10.Wu Y, Yoder A. Chemokine coreceptor signaling in HIV-1 infection and pathogenesis. PLoS Pathog. 2009;5:e1000520. doi: 10.1371/journal.ppat.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahr B, Robert-Hebmann V, Devaux C, Biard-Piechaczyk M. Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirology. 2004;23:12. doi: 10.1186/1742-4690-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taub DD, Ortaldo JR, Turcovski-Correlates SM, Key ML, Longo DL, Murphy WJ. Beta chemokines costimulate lymphocyte cytolysis, proliferation, and lymphokine production. J Leukoc Biol. 1996;59:81–6. doi: 10.1002/jlb.59.1.81. [DOI] [PubMed] [Google Scholar]
- 13.Taub DD, Turcovski-Correlates SM, Key ML, Longo DL, Murphy WJ. Chemokines and T lymphocyte activation: I. Beta chemokines costimulate human T lymphocyte activation in vitro. J Immunol. 1996;156:2095–103. [PubMed] [Google Scholar]
- 14.Karpus WJ, Lukacs NW, Kennedy KJ, Smith WS, Hurst SD, Barrett TA. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J Immunol. 1997;158:4129–36. [PubMed] [Google Scholar]
- 15.Appay V, Dunbar PR, Cerundolo V, McMichael A, Czaplewski L, Rowland-Jones SL. RANTES activates antigen-specific cytotoxic T lymphocytes in a mitogen-like manner through cell surface aggregation. Int Immunol. 2000;12:1173–82. doi: 10.1093/intimm/12.8.1173. [DOI] [PubMed] [Google Scholar]
- 16.Lillard JJW, Boyaka PN, Taub DD, McGhee JR. RANTES potentiates antigen-specific mucosal immune responses. J Immunol. 2001;166:162–9. doi: 10.4049/jimmunol.166.1.162. [DOI] [PubMed] [Google Scholar]
- 17.Makino Y, Cook DN, Smithies O, et al. Impaired T cell function in RANTES-deficient mice. Clin Immunol. 2002;102:302–9. doi: 10.1006/clim.2001.5178. [DOI] [PubMed] [Google Scholar]
- 18.Camargo JF, Quinones MP, Mummidi S, et al. CCR5 expression levels influence NFAT translocation, IL-2 production, and subsequent signaling events during T lymphocyte activation. J Immunol. 2009;182:171–82. doi: 10.4049/jimmunol.182.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou W, Borvak J, Marches F, Wei S, Galanaud P, Emilie D, Curiel TJ. Macrophage-derived dendritic cells have strong Th1-polarizing potential mediated by beta-chemokines rather than IL-12. J Immunol. 2000;165:4388–96. doi: 10.4049/jimmunol.165.8.4388. [DOI] [PubMed] [Google Scholar]
- 20.Contento RL, Molon B, Boularan C, Pozzan T, Manes S, Marullo S, Viola A. CXCR4-CCR5: a couple modulating T cell functions. Proc Natl Acad Sci USA. 2008;105:10101–6. doi: 10.1073/pnas.0804286105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molon B, Gri G, Bettella M, Gomez-Mouton C, Lanzavecchia A, Martinez-A C, Viola A. T cell costimulation by chemokine receptors. Nat Immunol. 2005;6:465–71. doi: 10.1038/ni1191. [DOI] [PubMed] [Google Scholar]
- 22.Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell–dendritic cell interaction. Nature. 2006;440:890–5. doi: 10.1038/nature04651. [DOI] [PubMed] [Google Scholar]
- 23.Kohlmeier JE, Miller SC, Smith J, Lu B, Gerard C, Cookenham T, Roberts AD, Woodland DL. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity. 2008;29:101–13. doi: 10.1016/j.immuni.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zamilpa R, Kanakia R, Cigarroa JI, et al. CC chemokine receptor 5 deletion impairs macrophage activation and induces adverse remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol. 2011;300:H1418–26. doi: 10.1152/ajpheart.01002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Yang H, Ma W, He S. Induction of IL-13 production and upregulated expression of protease activated receptor-1 by RANTES in a mast cell line. Cytokine. 2011;523:231–8. doi: 10.1016/j.cyto.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Chritmann BS, Moran JM, McGraw JA, Buller RM, Cornbett JA. CCR5 regulates inflammatory gene expression in response to encephalomyocarditis virus infection. Am J Pathol. 2011;179:2941–51. doi: 10.1016/j.ajpath.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynes J, Portales P, Segondy M, et al. CD4+ T cell surface CCR5 density as a determining factor of viral load in HIV-1-infected individuals. J Infect Dis. 2000;181:927–32. doi: 10.1086/315315. [DOI] [PubMed] [Google Scholar]
- 28.Vincent T, Portales P, Baillat V, et al. T-cell surface CCR5 density is not correlated with hepatitis severity in hepatitis C virus/HIV-coinfected individuals: implications for the therapeutic use of CCR5 antagonists. J Acquir Immune Defic Syndr. 2005;38:305–9. [PubMed] [Google Scholar]
- 29.Lin Y-L, Mettling C, Portalès P, Réant B, Robert-Hebmann V, Reynes J, Clot J, Corbeau P. The efficiency of R5 infection is determined by CD4 T cell surface CCR5 density through Gai-protein signaling. AIDS. 2006;20:1369–77. doi: 10.1097/01.aids.0000233570.51899.e2. [DOI] [PubMed] [Google Scholar]
- 30.Desmetz C, Lin Y-L, Mettling C, Portalès P, Rabesandratana H, Clot J, Corbeau P. The strength of the chemotactic response to a CCR5 binding chemokine is determined by the level of cell surface CCR5 density. Immunology. 2006;119:551–61. doi: 10.1111/j.1365-2567.2006.02470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tuaillon E, Al Tabaa Y, Baillat V, Segondy M, Picot M-C, Reynes J, Vendrell J-P. Close association of CD8+/CD38bright with HIV-1 reolication and complex relationship with CD4+ T-cell count. Cytometry B Clin Cytom. 2009;76B:249–60. doi: 10.1002/cyto.b.20467. [DOI] [PubMed] [Google Scholar]
- 32.Pandrea I, Apetrei C, Gordon S, et al. Paucity of CD4+CCR5+ T cells is a typical feature of natural SIV hosts. Blood. 2007;109:1069–76. doi: 10.1182/blood-2006-05-024364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahuja SK, Kulkarni H, Catano G, et al. CCL3L1-CCR5 genotype influences durability of immune recovery during antiretroviral therapy of HIV-1-infected individuals. Nat Med. 2008;14:413–20. doi: 10.1038/nm1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilkin T, Lalama C, Tenorio AR, et al. Maraviroc intensification for suboptimal CD4+ cell response despite sustained virologic suppression: ATCG 5256. 17th Conference on Retroviruses and Opportunistic Infections 2010; Abstract 285.
- 35.Gutiérrez C, Diaz L, Hernandez-Novoa B, et al. Effect of the intensification with a CCR5 antagonist on the decay of the HIV-1 latent reservoir and residual viremia17th Conference on Retroviruses and Opportunistic Infections 2010; Abstract 284.
- 36.Hunt P, Shulman N, Hayes T, et al. Boston: 2011. Immunomodulatory effects of MVC intensification in HIV-infected individuals with incomplete CD4+ T cell recovery during suppressive ART. 18th Conference on Retroviruses and Opportunistiv Infections, 27 February–2 March 2011. [Google Scholar]
- 37.Trottier B, Cooper DA, Wood R, Carosi G, Van der Ryst E, Heera J, et al. MERIT: Week 96 virologic response and tropism at virological failure by baseline CD4+ cell count. 49th ICAAC, San Francisco, CA, USA, September 12–15, Poster H-921 2009; Poster H-921.
- 38.Lalezari J, Yadavalli GK, Para M, Richmond G, DeJesus E, Brown SJ, et al. Safety, pharmacokinetics, and antiviral activity of HGS004, a novel fully human IgG4 monoclonal antibody against CCR5, in HIV-1-infected patients. J Infect Dis. 2008;197:721–7. doi: 10.1086/527327. [DOI] [PubMed] [Google Scholar]
- 39.Saag M, Goodrich J, Fätkenheuer G, et al. A double-blind, placebo-controlled trial of maraviroc in treatment-experienced patients infected with non-R5 HIV-1. J Infect Dis. 2009;199:1638–47. doi: 10.1086/598965. [DOI] [PubMed] [Google Scholar]
- 40.Wilkin T, Ribaudo H, Gulick R. The relationship of CCR5 inhibitors to CD4 count changes: a meta-analysis of recent clinical trials in treatment-experienced subjects. 15th Conference on Retroviruses and Opportunistic Infections, Boston 2008 Feb 2–6 2008; Abstract 800.
- 41.Gazzola L, Tincati C, Bellistri GM, d'Arminio Monforte A, Marchetti G. The absence of CD4+ T cell count recovery despite receipt of virologically suppressive highly active antiretroviral therapy: clinical risk, immunological gaps, and therapeutic options. Clin Infect Dis. 2009;48:328–37. doi: 10.1086/595851. [DOI] [PubMed] [Google Scholar]