

Abstract
5,7-Dihydroxy-3′,4′,6′-trimethoxyflavone (eupatilin), the active pharmacological ingredient from Artemisia asiatica Nakai (Asteraceae), is reported to have a variety of anti-inflammatory properties in intestinal epithelial cells. However, little information is known about the molecular mechanism of eupatilin-induced attenuation of bronchial epithelial inflammation. This study investigates the role of eupatilin in the adhesion of inflammatory cells such as monocytes and eosinophils to bronchial epithelial cells. Stimulation of a human bronchial epithelial cell line (BEAS-2B) with tumour necrosis factor-α (TNF-α) increased the expression of surface adhesion molecules, including intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), in which eupatilin significantly inhibited the expression of those adhesion molecules in a dose-dependent manner. Eupatilin suppressed the TNF-α-induced activation of IκBα and nuclear factor-κB (NF-κB) signals in BEAS-2B cells. The IκB kinase (IKK) activation was also significantly reduced in eupatilin-pre-treated BEAS-2B and primary normal human bronchial epithelial (NHBE) cells. However, eupatilin did not influence AP-1 activity in TNF-α-stimulated cells. Suppression of NF-κB signalling induced by eupatilin resulted in the inhibition of the expression of adhesion molecules and the adhesion of monocytes and eosinophils to BEAS-2B cells. Furthermore, eupatilin suppressed the phosphorylation of Akt in TNF-α-stimulated BEAS-2B and NHBE cells, leading to down-regulation of NF-κB activation and adhesion molecule expression and finally to suppression of the inflammatory cell adhesion to epithelial cells. These results suggest that eupatilin can inhibit the adhesion of inflammatory cells to bronchial epithelial cells via a signalling pathway, including activation of Akt and NF-κB, as well as expression of adhesion molecules.
Keywords: adhesion, eupatilin, intercellular adhesion molecule -1, nuclear factor-κB, vascular cell adhesion molecule 1
Introduction
Asthma is a complex inflammatory disease involving the critical action of adhesion of inflammatory cells to airway epithelial cells. Therefore, the up-regulation of adhesion molecules on the surfaces of respiratory epithelial cells is an important cause in the development of asthma.1 The infiltration of inflammatory cells is known to be associated with chemokines in affected tissues. The infiltrating process of inflammatory cells is also largely the result of enhanced adhesion of leucocytes to epithelial cells via the expression of adhesion molecules.1 Surface adhesion molecules known to be involved in the process are intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1).2 It is possible that suppression of adhesion between respiratory epithelial cells and inflammatory cells could lead to attenuation of asthmatic symptoms.
Tumour necrosis factor-α (TNF-α) is a pleiotropic cytokine found in airways with strong pro-inflammatory and immunomodulatory properties.3 As TNF-α is increased in the blood of patients with severe asthma and can induce surface expression of ICAM-1 and VCAM-1 on epithelial cells,4,5 TNF-α is thought to play an important role in the pathogenesis of asthma. Therefore, strategies for developing therapies for asthma tend to be focused on suppressing TNF-α-induced biological responses.6
It is now recognized that respiratory epithelial cells play a central role in the regulation of airway inflammatory status, structure and function in normal and diseased airways.1 Nuclear factor-κB (NF-κB) is an important transcriptional factor that regulates inflammatory processes. It is present in the cytoplasm in its resting stage as a heterotrimer complex consisting of two subunits [i.e. RelA (p65), c-Rel, Rel B, NF-κB1 (p50), and NF-κB2 (p52)] and an additional inhibitory subunit, IκB (e.g. IκBα and IκBβ predominantly are associated with p65/p50 and p50/c-Rel heterodimers, IκBε preferentially associates with p65 and c-Rel homodimers, and Bcl-3 associates with nuclear p50 and p52 homodimers).7,8 During the activation process, activated IκB kinase (IKK) induces IκB phosphorylation, which is followed by degradation of the inhibitory subunit IκB and subsequently the activation of NF-κB. The released NF-κB dimers translocate to the nucleus.9 Therefore, TNF-α-induced transcription of ICAM-1 and VCAM-1 is critically dependent on the activation of NF-κB.10,11
Flavonoids are polyphenols found in a wide variety of edible plants and exert anti-inflammatory activities in several types of human cell lines.12,13 Extracts of Artemisia asiatica Nakai (Asteraceae) are known to have anti-inflammatory activities. One of the pharmacologically active ingredients from A. asiatica extracts is 5,7-dihydroxy-3′,4′,6′-trimethoxyflavone (eupatilin).14 Eupatilin has been shown to inhibit the gene expressions of TNF-α and interleukin-4 in rat basophilic leukaemia (RBL-2H3) cells stimulated by IgE–antigen complex, suggesting that eupatilin may offer protection from IgE-mediated allergic diseases.15 In addition, eupatilin can suppress the NF-κB signalling pathway in intestinal epithelial cells and attenuate Bacteroides fragilis enterotoxin-induced intestinal inflammation.16 Although eupatilin has anti-inflammatory actions in intestinal epithelial cells and is a potent inhibitor of NF-κB, little is known about the molecular mechanism of eupatilin-induced attenuation of respiratory epithelial cell inflammation.
Akt, also called protein kinase B, is a serine/threonine kinase that is implicated in a variety of cellular responses and in the pathogenesis of several diseases including metabolic diseases, cancers and asthma.17 Down-regulation of Akt activity in lipopolysaccharide-stimulated cells using a dominant-negative mutant resulted in the suppression of NF-κB activation.18 These results suggest a positive role of Akt on the NF-κB-dependent expression of adhesion molecules. However, the Akt signalling pathway has not yet been implicated in the expression of adhesion molecules during respiratory epithelial cell inflammation.
In this study, we asked whether eupatilin could affect the functions of human bronchial epithelial cells and inflammatory cell adhesion in response to TNF-α stimulation. Eupatilin was shown herein to inhibit an Akt-NF-κB signalling pathway, leading to the down-regulation of adhesion molecules such as ICAM-1 and VCAM-1 in bronchial epithelial BEAS-2B cells stimulated with TNF-α. In addition, eupatilin attenuated the adhesion of both monocytes and eosinophils to bronchial epithelial cells, suggesting the ability of eupatilin to modulate TNF-α-induced inflammation of airway epithelial cells, relevant to the pathogenesis of asthma, i.e. related to eosinophilic inflammation.
Materials and methods
Reagents
Lipopolysaccharide-free fetal bovine serum (FBS), antibiotics, l-glutamine, Trizol, and Ca2+ and Mg2+-free Hanks’ balanced salt solution (HBSS) were obtained from Gibco BRL (Gaithersburg, MD). Calcein AM, MG-132, LY294002, BSA, Histopaque-1077, and RPMI-1640 medium were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO). Monoclonal antibodies against phospho-IκBα, phospho-IKK-α/β, phospho-Akt, and actin were acquired from Cell Signaling Technology, Inc. (Beverly, MA). Human recombinant TNF-α was purchased from R&D Systems (Minneapolis, MN). Antibodies against ICAM-1, VCAM-1 and phospho-p65 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Goat anti-rabbit and anti-mouse secondary antibodies conjugated to horseradish peroxidase were purchased from Transduction Laboratories (Lexington, KY). Anti-mouse FITC-conjugated secondary goat antibody, anti-mouse tetramethylrhodamine isothiocyanate (TRITC)-conjugated secondary goat antibody, anti-rabbit FITC-conjugated secondary goat antibody and anti-rabbit TRITC-conjugated secondary goat antibody were obtained from Invitrogen (Carlsbad, CA). Vectashield mounting medium with DAPI was obtained from Vector Laboratories (Burlingame, CA).
Cell cultures
The human bronchial epithelial cell line BEAS-2B [CRL-9609; American Type Culture Collection (ATCC), Rockville, MD] was cultured in RPMI-1640 with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). Cells were seeded at 0·5–2 × 106 cells per well onto six-well plates and allowed to attach overnight. After 12 hr of serum starvation, cells were incubated with each stimulator for the indicated times. 5,7-Dihydroxy-3′,4′,6′-trimethoxy flavone (eupatilin, Dong-A Pharmaceutical, Yong-In, Korea) was dissolved in DMSO. In some experiments, specific inhibitors were used, including a specific inhibitor of NF-κB, MG-132, a specific inhibitor of Akt, LY294002, and a specific inhibitor of activator protein (AP)-1, curcumin.
The human monocytic THP-1 cell line (ATCC TIB-202) was cultured in RPMI-1640 medium, supplemented with 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml), l-glutamine (1·5 mm), sodium pyruvate (1 mm), and 2-mercaptoethanol (0·05 mm). Low passage-number THP-1 cells (< passage 20) were used for all experiments.19 The human eosinophilic EoL-1 cell line (RIKEN cell bank; Wako Pure Chemical Industries, Tokyo, Japan) was grown (106 cells/ml) in RPMI-1640 medium containing 10% FBS, penicillin (100 U/ml) and streptomycin (100 μg/ml).20,21 Cells were grown in a humidified atmosphere of 5% CO2 and 95% air at 37°.
The primary normal human bronchial epithelial (NHBE) cells were cultured as previously described.22,23 Briefly, commercially available NHBE cells (Lonza, Walkersville, MD) were seeded into T75 tissue-culture flasks at a density of 5 × 103 cells. Cells were expanded in growth medium (Lonza) at 5% CO2 at 37° to a confluence of ∼ 90%, dissociated from the flasks using 0·25% trypsin/EDTA, and frozen in liquid nitrogen as passage-2 cells (2 × 106 cells/ml). For experiments, NHBE cells were cultured with bronchial epithelial basal medium supplemented with growth factors and hormones (BEGM Bullet Kit; Lonza) in a humidified incubator at 37° with 5% CO2. Medium was replaced by fresh medium (bronchial epithelial basal medium without growth factors or hormones) 24 hr before treatment.
Peripheral blood mononuclear cells (PBMC) were isolated using Histopaque-1077 (Sigma-Aldrich). For the magnetic cell separation, PBMC were magnetically labelled with CD14 microbeads and separated on a column that was placed in the magnetic field of a MACS separator (Miltenyi Biotec, Bergisch Gladbach, Germany). The magnetically labelled CD14+ cells were retained in the column while the unlabelled CD14– cells ran through. The unlabelled cells were depleted of CD14+ cells. After removal of the column from the magnetic field, the magnetically retained CD14+ cells were eluted as positively selected cell fraction according to the manufacturer's instructions. Eosinophils were isolated from the peripheral blood of volunteers using a magnetic cell separation system (Miltenyi Biotec), as described previously.20,21 The Hanyang University College of Medicine Review Board approved the protocol used to obtain blood from volunteers. The purity of PBMC counted by flow cytometry using anti-CD14 antibody was > 95%. The purity of eosinophils counted using Randolph's stain was > 98%. Purified relatively pure mononuclear cells and eosinophils were used immediately for experiments using RPMI-1640 medium.
Quantitative reverse transcriptase (RT)-PCR
Total cellular RNA was extracted using Trizol. Reverse transcription and PCR amplification were performed as described previously.24 The primers and expected PCR product sizes were as follows:19 human ICAM-1, 5′-CTC AGC CTC GCT ATG GCT CCC-3′ (sense), 5′-GTA CAC GGT GAG GAA GGT TTT-3′ (anti-sense), 342 bp [NM_000201.2 Homo sapiens ICAM1, mRNA]; human VCAM-1, 5′-ATG ACA TGC TTG AGC CAG G-3′ (sense), 5′-GTG TCT CCT TCT TTG ACA CT-3′ (anti-sense), 260 bp [NM_001199834.1 Homo sapiens VCAM1, transcript variant 3, mRNA]; human β-actin, 5′-TGA CGG GGT CAC CCA CAC TGT GCC CAT CTA-3′ (sense) and 5′-CTA GAA GCA TTG CGG TGG ACG ATG GAG GG-3′ (anti-sense), 661 bp [NM_001101.3 Homo sapiens actin, β-actin, mRNA]. To quantify mRNA molecules, standard RNAs for human ICAM-1 and human VCAM-1 were generated by in vitro transcription using T7 RNA polymerase, as described previously.20 Standard RNA for human β-actin was kindly provided by Dr Martin F. Kagnoff of the University of California, San Diego. The sizes of PCR products generated from standard RNAs for human ICAM-1, human VCAM-1, and human β-actin were 480, 384 and 520 bp, respectively.
Flow cytometry analysis
After the indicated periods of incubation with eupatilin or TNF-α, cells were washed twice with cold Ca2+-free and Mg2+-free HBSS and trypsinized at 37° for 3 min. Resuspended cells were centrifuged at 200 g for 5 min at 4° and then washed with HBSS containing 0·5% BSA. The cells were transferred to flow cytometry tubes and centrifuged at 200 g for 5 min at 4°, and the supernatants were discarded. Thereafter, the cells were incubated with anti-human ICAM-1 or anti-human VCAM-1 mouse monoclonal antibodies with 0·5% BSA for 3 hr on ice. The cells were washed twice with cold HBSS containing 0·5% BSA and then incubated with anti-mouse FITC-conjugated secondary goat antibody on ice in the dark. After 1 hr, the cells were washed twice with cold HBSS with 0·5% BSA. The cells were analysed by flow cytometry (FACSCalibur cytometer; Becton Dickinson, San Jose, CA). Ten thousand cells were analysed per sample and the expression levels of ICAM-1 and VCAM-1 were expressed as mean fluorescence intensity (MFI).19,25
Electrophoretic mobility shift assay
Cells were harvested and nuclear extracts were prepared as previously described.22,26 Concentrations of protein in the extracts were determined using a Bradford assay (Bio-Rad, Hercules, CA). Electrophoretic mobility shift assay (EMSA) was performed using an assay kit (Promega, Madison, WI), as described previously.27 In brief, 5 μg of nuclear extract was incubated for 30 min at room temperature with a γ32P-labelled oligonucleotide probe (5′-AGT TGA GGG GAC TTT CCC AGG C-3′ for the NF-κB binding site; 5′-CGC TTG ATG ACT CAG CCG GAA-3′ for the AP-1 binding site). After incubation, both bound and free DNA was resolved on 5% polyacrylamide gels, as previously described.24 A TransAM AP-1 family kit (Catalog No. 44296; Active Motif, Carlsbad, CA) was used to measure the concentration of c-fos in the nuclear fractions of the cells via colorimetric DNA-binding ELISA.
Plasmids, transfection and luciferase assays
A wild-type over-expressing plasmid of Akt (constitutively active Akt plasmid) was provided by Dr Michael Weber (University of Virginia Health Sciences Center, Charlottesville, VA) via Dr Joo Young Lee (Gwangju Institute of Science and Technology, Gwangju, South Korea).28 Small interfering RNA (siRNA) against the Akt was obtained from Cell Signaling Technology, as previously described.28 Cells in six-well dishes were transfected with each plasmid, siRNA, or non-silencing siRNA using FuGene6 transfection reagent (Roche, Mannheim, Germany), as described previously.29 Briefly, 1 μg siRNA or 1·5 μg plasmid DNA was diluted in serum-free medium to produce a final volume of 100 μl, which was incubated with 3 μl Fugene 6 for 15 min at room temperature. The transfection mixture was added to the respective wells, each containing 300 μl medium (10% FBS content). Transfections were incubated for 48 hr before the assay.
Reporter plasmids containing pICAM-1, pVCAM-1, p2x NF-κB-, pβ-actin-, and pRSV-β-galactosidase-luciferase were kindly provided by Dr Martin F. Kagnoff of the University of California, San Diego.7 Cells in six-well dishes were transfected with 1·5 μg plasmid DNA using FuGene6 transfection reagent (Roche). The transfected cells were incubated for 24 hr at 37° in a 5% CO2 incubator and were then treated with eupatilin and/or TNF-α for the indicated times. Luciferase and β-galactosidase enzyme activities were determined using the Luciferase Assay System and β-galactosidase Enzyme System (Promega) according to the manufacturer's instructions. Light release was quantified using a luminometer (MicroLumat Plus; Berthold GmbH & Co. KG, Bad Wildbad, Germany) as described previously.30
Immunoblot analysis
Immunoblot analyses were performed as described previously.30 Briefly, cells were washed with ice-cold PBS and lysed in 0·5 ml/well lysis buffer [150 mm NaCl, 20 mm Tris–HCl at pH 7·5, 0·1% Triton X-100, 1 mm PMSF and 10 μg/ml aprotinin]. Fifteen to fifty micrograms of protein per lane was size-fractionated on a 6% polyacrylamide minigel and electrophoretically transferred to a nitrocellulose membrane (0·1-μm pore size). Specific proteins were detected using mouse anti-human phospho-IκBα, phospho-IKK-α/β, phospho-Akt, and actin as primary antibodies and peroxidase-conjugated anti-mouse IgG as a secondary antibody. Specifically bound peroxidase was detected by enhanced chemiluminescence and exposure to X-ray film.
In vitro kinase assay
IκB kinase activity on IκBα phosphorylation was determined using an immunocomplex kinase assay as described previously.31 Briefly, cells were lysed in Triton lysis buffer containing protease and phosphatase inhibitors and then cleared by centrifugation at 15 700 g for 10 min. Three hundred micrograms of whole cell extract was immunoprecipitated with anti-IKK-γ/protein-A beads. The kinase reaction was then performed by incubating 25 ml of kinase buffer containing 20 mm HEPES (pH 7·7), 10 mm MgCl2, 5 mm dithiothreitol, 50 mm ATP and 5 mCi [γ-32P] ATP with GST-IκBα substrate (Upstate Biotechnology Inc., Lake Placid, NY) for 30 min at 30°. The substrate protein was resolved by gel electrophoresis and phosphate incorporation was assessed by autoradiography. An HTScan IKK-β kinase assay kit was obtained from Cell Signaling Technology, Inc. This kit contains GST-IKK-β kinase protein, a biotinylated peptide substrate, and a phosphoserine antibody for detection of the phosphorylated form of the substrate peptide. The assay was performed according to the manufacturer's instruction.31 The activity of Akt was evaluated by PathScan® phospho-Akt (Thr308) sandwich ELISA kit (Cell Signaling Technology, Inc.), in accordance with the kit's protocols.
Confocal microscopy for adhesion molecules and phospho-p65
BEAS-2B were seeded (5 × 105 cells in 0·2 ml RPMI-1640/well) on four-well poly-d-lysine-coated culture microslides (Santa Cruz) overnight. After treatment with eupatilin and/or TNF-α for the indicated time period, cells were incubated with 100% methanol at −20° for 5 min. The cells were incubated with primary antibodies (rabbit anti-phospho-p65, mouse, mouse anti-ICAM-1 or mouse anti-VCAM-1) for 1 hr. Cells were then treated with secondary antibodies (FITC-conjugated goat anti-rabbit or TRITC-conjugated goat anti-mouse) for 40 min. Vectashield mounting medium with DAPI was applied to the cells and images were captured using a confocal microscope (Leica TCS SP5; Leica Microsystems CMS GmbH, Wetzlar, Germany).21
Adherence assay
To measure adhesion of monocytes or eosinophils to bronchial epithelial cells, THP-1, EoL-1, relatively pure mononuclear cells, or eosinophils isolated from blood were stained with Calcein AM (4 μm) for 20 min at 37°. Stained monocytes or eosinophils (1 × 106/ml) were added to eupatilin-exposed and TNF-α-exposed bronchial epithelial cells in four-well poly-d-lysine-coated culture microslides (Santa Cruz). After further incubation for 20 min at 37°, cells were fixed with 100% methanol at −20° for 5 min. To identify the morphology of each cell, primary antibody (rabbit anti-actin) was added for 1 hr. Cells were washed with PBS before the secondary antibody (goat TRITC-conjugated ant-rabbit antibody) was added for an additional 40 min. Vectashield mounting medium with DAPI was applied to the cells, and cells were then analysed under a fluorescence microscope (Leica DM 5000B).
Statistical analyses
Data of quantitative RT-PCR are presented as mean ± standard deviation (SD) and data of MFI, luciferase assays, and ELISA are presented as mean ± standard error of the mean (SEM). Wilcoxon's rank sum test was used for statistical analysis. P values < 0·05 were considered statistically significant.
Results
Eupatilin down-regulates the expression of ICAM-1 and VCAM-1 in BEAS-2B cells stimulated with TNF-α
Stimulation of BEAS-2B cells with TNF-α for 24 hr induced a significant increase in the expression of ICAM-1 surface molecules as assessed by flow cytometry. In this experimental system, eupatilin decreased ICAM-1 expression on TNF-α-stimulated cells (Fig. 1a). A similar decrease of VCAM-1 expression was observed following TNF-α-stimulated BEAS-2B cells when pre-treated with eupatilin. To quantify the expression of adhesion molecules, MFI using flow cytometric analysis was measured. As shown in Fig. 1(b,c), pre-treatment with eupatilin significantly down-regulated the increased MFI values of ICAM-1 and VCAM-1 induced by TNF-α stimulation. Inhibition of adhesion molecule expression was dependent on the concentration of eupatilin. To re-evaluate the expression of ICAM-1 molecules, quantitative RT-PCR analyses were performed using each standard RNA. Increased ICAM-1 mRNA expression in BEAS-2B cells was first noted 1 hr after stimulation with TNF-α and levels of ICAM-1 mRNA peaked at 6–12 hr post-stimulation (Fig. 2a). The pre-treatment with eupatilin significantly down-regulated the increased ICAM-1 mRNA expression induced by TNF-α stimulation. Similar to ICAM-1 mRNA expression, significant reduction of VCAM-1 mRNA expression by eupatilin was observed in TNF-α-stimulated cells compared with TNF-α alone. The β-actin mRNA levels in each experiment remained relatively constant throughout the same period.
Figure 1.
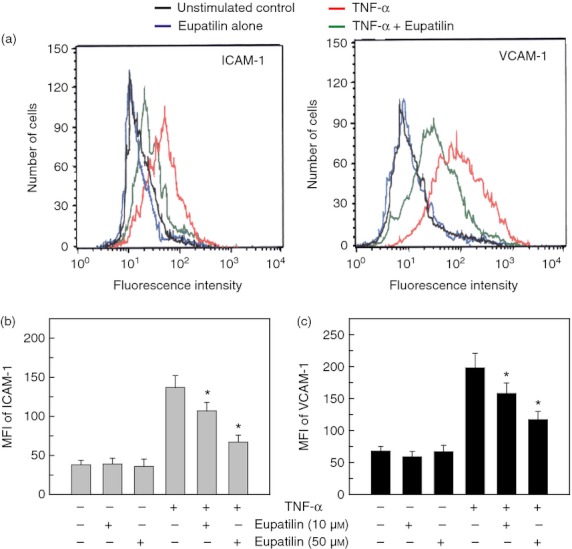
Effects of eupatilin on intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) surface molecules in a human bronchial epithelial cell line BEAS-2B stimulated with tumour necrosis factor-α (TNF-α). (a) BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr and then treated with TNF-α (20 ng/ml) for 24 hr. Cells were stained with a monoclonal antibody against ICAM-1 or VCAM-1 and were analysed using flow cytometry. Results are representative of more than five independent experiments. (b and c) BEAS-2B cells were pre-incubated with eupatilin for 18 hr and were then stimulated with TNF-α (20 ng/ml) for another 24 hr. Cells were stained with monoclonal antibody against ICAM-1 or VCAM-1 and were analysed using flow cytometry. The data represent the MFI ± SEM (n = 3). *P < 0·05 compared with TNF-α alone.
Figure 2.
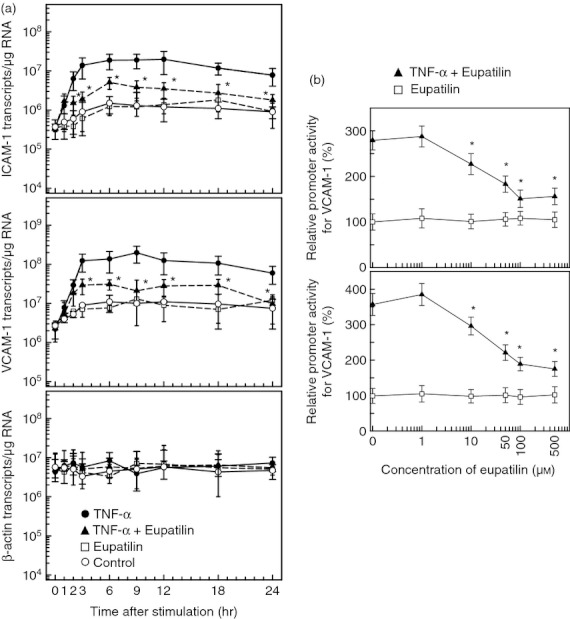
Effects of eupatilin on intercellular adhesion molecule 1 (ICAM-1) mRNA expression and reporter gene activation in BEAS-2B cells stimulated with tumour necrosis factor-α (TNF-α). (a) BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr and then treated with TNF-α (20 ng/ml) for the indicated periods. Levels of ICAM-1, vascular cell adhesion molecule 1 (VCAM-1), and β-actin mRNA were analysed by quantitative RT-PCR using each standard RNA. The values are expressed as mean ± SD (n = 3). Asterisks indicate a statistical significance in comparison with TNF-α alone (P < 0·05). (b) BEAS-2B cells were transfected with an ICAM-1- or VCAM-1-luciferase transcriptional reporter for 24 hr. Transfected cells were pre-treated with the indicated concentration of eupatilin for 18 hr and then combined with TNF-α (20 ng/ml) for another 9 hr, after which luciferase assays were performed. Data are expressed as the mean fold induction ± SEM in luciferase activity relative to that of the unstimulated controls (n = 5). The mean fold induction of the β-actin reporter gene relative to unstimulated controls remained relatively constant throughout each experiment. Asterisks indicate a statistical significance in comparison with cells stimulated with TNF-α alone (P < 0·05).
Inhibition of each adhesion molecule expression was dependent on the concentration of eupatilin. As shown in Fig. 2(b), the inhibition of the promoter activity in BEAS-2B cells transfected with a transcriptional reporter plasmid of ICAM-1 or VCAM-1 was correlated with the concentration of eupatilin. Significant decreases in promoter activities for ICAM-1 and VCAM-1 were noted at eupatilin concentrations exceeding 10 μm.
Eupatilin inhibits the expression of ICAM-1 and VCAM-1 through NF-κB signals in BEAS-2B human bronchial epithelial cells
Transcriptional expression of ICAM-1 and VCAM-1 is regulated primarily by the transcription factor NF-κB in TNF-α-stimulated bronchial epithelial cells. We previously demonstrated that eupatilin attenuated NF-κB DNA-binding activity in human intestinal epithelial cells;16 however, the effects of eupatilin on NF-κB signals in human bronchial epithelial cells have not yet been reported. For experiments assessing eupatilin-induced inhibition of NF-κB signalling, we determined the NF-κB DNA-binding activity in BEAS-2B cells using EMSA. Pre-treatment with eupatilin reduced the NF-κB DNA binding activity (Fig. 3a). Concurrently, phosphorylation of IκBα was observed in TNF-α-stimulated cells, and eupatilin reduced phosphorylated IκBα signals. In addition, pre-treatment with eupatilin reduced the levels of phosphorylated IKK-α/β signal (Fig. 3a). To confirm the eupatilin-induced suppression of NF-κB activity, a promoter assay was performed. As shown in Fig. 3(b), the inhibition of NF-κB promoter activity in BEAS-2B cells transfected with the NF-κB reporter plasmid was correlated with the concentration of eupatilin.
Figure 3.
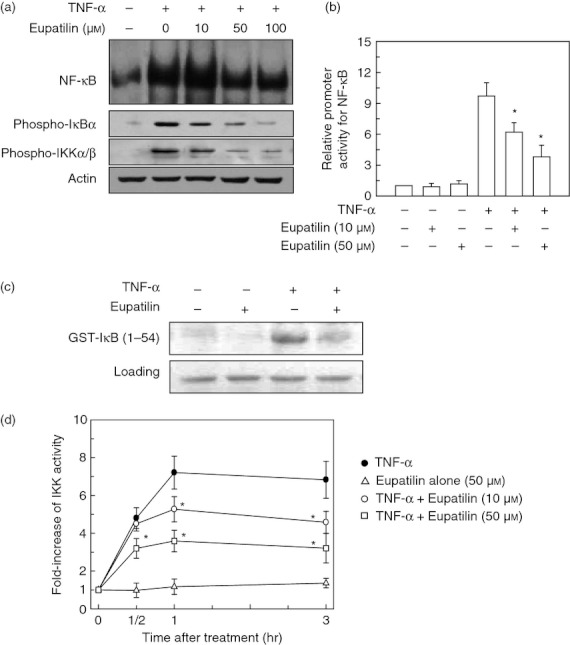
Eupatilin inhibits the nuclear factor-κB (NF-κB) signalling pathway in BEAS-2B cells stimulated with tumour necrosis factor-α (TNF-α). (a) BEAS-2B cells were pre-treated with the indicated concentration of eupatilin for 18 hr and then stimulated with TNF-α (20 ng/ml) for 1 hr. NF-κB DNA-binding activity in the nuclear extracts was assessed by EMSA. Expression of phospho-IκBα, phospho-IKK-α/β and actin was determined by immnoblot analysis. The data are representative of more than five independent experiments. (b) BEAS-2B cells were transfected with 2pNF-κB-luciferase transcriptional reporter. After 24 hr, transfected cells were pre-treated with the indicated concentration of eupatilin for 18 hr and were then combined with TNF-α (20 ng/ml) for 1 hr. The mean fold induction of the β-actin reporter gene activity relative to the unstimulated controls remained relatively constant throughout each experiment. Data are expressed as mean fold induction ± SEM relative to untreated controls (n = 5). *P < 0·05 compared with TNF-α alone. (c) BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr and then treated with TNF-α (20 ng/ml) for an additional 30 min. The whole cell extract was immunoprecipitated with anti-IKK-γ/protein-A beads and kinase reactions were performed using glutathione-S-transferase-IκB as a substrate. Results shown are representative of three independent experiments. (d) IKK kinase activity was measured using an HTS can IKK-β kinase assay kit. Data are expressed as the mean fold induction ± SEM of kinase activity relative to untreated controls (n = 5). *P < 0·05 compared with TNF-α alone.
To evaluate the effects of eupatilin on IKK activity, an in vitro kinase assay for IKK was performed. As shown in Fig. 3(c), stimulation of BEAS-2B cells with TNF-α resulted in a strong increase of IKK activity that was reduced by pre-treatment with eupatilin. To quantify the inhibition of IKK activity, an HTScan IKK-β kinase assay was performed. Stimulation of BEAS-2B cells with TNF-α resulted in an increase of IKK activity, whereas pre-treatment of cells with eupatilin significantly reduced the TNF-α-induced IKK activity (Fig. 3d). These results indicate that eupatilin can inhibit the NF-κB signalling pathway by blocking IKK activity in human bronchial epithelial cells.
We next evaluated whether NF-κB plays a role in regulating the expression of adhesion molecules in BEAS-2B cells treated with eupatilin. For the first experiment, immunohistochemistry and confocal microscopy were performed. As shown in Fig. 4, the phospho-p65 signals were mainly observed in the nuclei of TNF-α-treated cells. Concurrently, increased signals of ICAM-1 expression were shown on TNF-α-treated cells. In these experimental conditions, pre-treatment with eupatilin clearly reduced the translocation of phospho-p65 signals and the expression of ICAM-1 molecules. Cells with phospho-p65 and ICAM-1 signals were observed in ∼ 65% of the TNF-α-treated group and ∼ 30% of the eupatilin and TNF-α-treated group.
Figure 4.
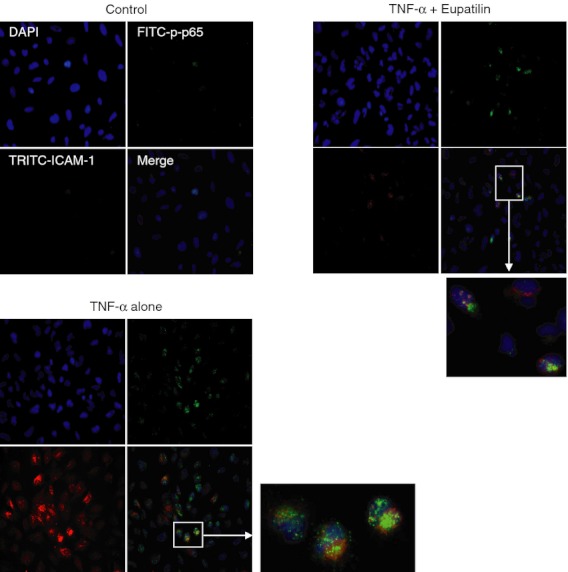
Expression of surface intercellular adhesion molecule 1 (ICAM-1) molecules and translocation of phospho-p65 nuclear factor-κB (NF-κB) signal in BEAS-2B cells stimulated with eupatilin and tumour necrosis factor-α (TNF-α). BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr and then stimulated with TNF-α (20 ng/ml) for 24 hr. For immunofluorescence, cells were stained with FITC-conjugated anti-p65 antibody (green), TRITC-conjugated anti-ICAM-1 antibody (red), and DAPI (blue, nucleus). The data are representative of at least five experiments.
For physiological relevance, primary cultures of NHBE cells were treated with eupatilin and IKK activity was measured. As shown in Fig. 5(a), pre-treatment of cells with eupatilin significantly decreased the TNF-α-induced phospho-IKK activity. In this experimental system, pre-treatment with eupatilin significantly down-regulated the increased MFI values of ICAM-1 and VCAM-1 induced by TNF-α stimulation (Fig. 5b,c). These results suggest that the eupatilin inhibits the NF-κB signalling pathway by blocking IKK activity, leading to suppression of the adhesion molecules ICAM-1 and VCAM-1 in bronchial epithelial cells.
Figure 5.
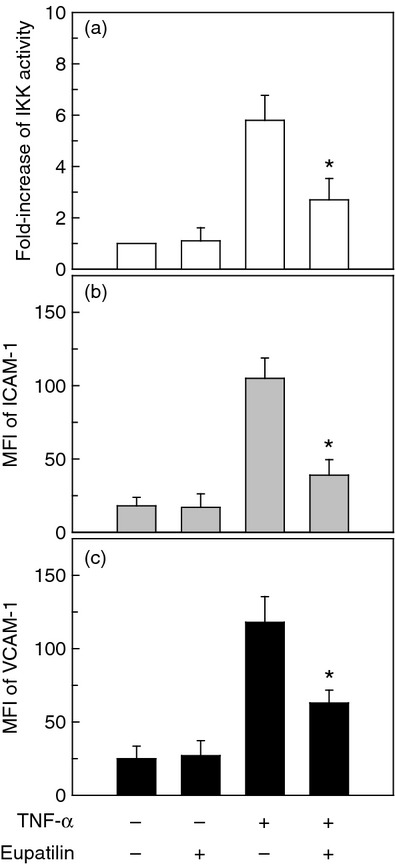
Eupatilin inhibits the IKK signalling and adhesion surface molecules in primary normal human bronchial epithelial (NHBE) cells stimulated with tumour necrosis factor-α (TNF-α). (a) NHBE cells were pre-treated with eupatilin (50 μm) for 18 hr and then stimulated with TNF-α (20 ng/ml) for 1 hr. IKK kinase activity was measured using an HTScan IKK-β kinase assay kit. Data are expressed as the mean fold induction ± SEM of kinase activity relative to untreated controls (n = 3). (b and c) NHBE cells were pre-incubated with eupatilin (50 μm) for 18 hr and were then stimulated with TNF-α (20 ng/ml) for another 24 hr. Cells were stained with monoclonal antibody against intercellular adhesion moelcule 1 (ICAM-1) or vascular cell adhesion molecule 1 (VCAM-1) and were analysed using flow cytometry. The data represent the MFI ± SEM (n = 3). * P < 0·05 compared with TNF-α alone.
Eupatilin does not inhibit activation of AP-1 in BEAS-2B human bronchial epithelial cells stimulated with TNF-α
As the promoter for ICAM-1 or VCAM-1 gene induction contains binding sites for both AP-1 and NF-κB, we investigated the possibility that activated AP-1 can affect the expression of adhesion molecules in bronchial epithelial cells treated with eupatilin. To validate the hypothesis, we first performed EMSA. Stimulation of BEAS-2B cells with TNF-α increased AP-1-DNA binding activity, and pre-treatment with eupatilin did not suppress AP-1 activation in BEAS-2B cells stimulated with TNF-α (Fig. 6a). To confirm these results, activity of c-fos in the nuclear fraction of cells was measured using the TransAM AP-1 family kit. As shown in Fig. 6(b), stimulation of BEAS-2B cells with TNF-α resulted in an increase of c-fos activity that was significantly reduced by pre-treatment with the AP-1 inhibitor curcumin. However, treatment with both eupatilin and TNF-α did not significantly change the levels of c-fos activity compared with TNF-α alone.
Figure 6.
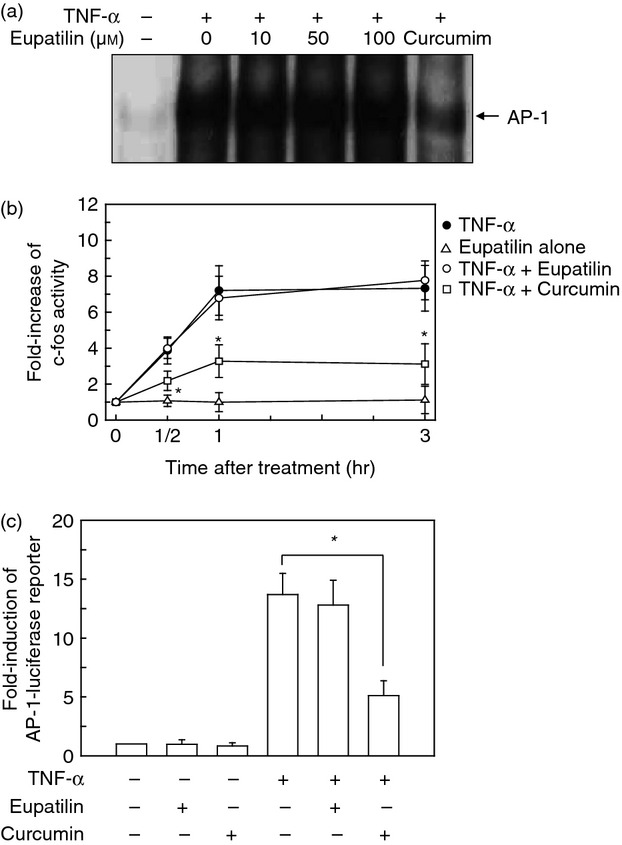
Effect of eupatilin on AP-1 activation in BEAS-2B cells stimulated with tumour necrosis factor-α (TNF-α). (a) BEAS-2B cells were pre-treated with the indicated concentration of eupatilin for 18 hr and then stimulated with TNF-α (20 ng/ml) for 1 hr. AP-1 DNA-binding activity in the nuclear extracts was assessed by EMSA. The data are representative of more than five independent experiments. (b) BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr or curcumin (20 μm) for 30 min and then stimulated with TNF-α (20 ng/ml) for the indicated period of time. Nuclear proteins were extracted and 10 μg of each sample was subjected to assay. Data are expressed as mean ± SEM (n = 5). *P < 0·05 compared with TNF-α alone. (c) BEAS-2B cells were transfected with pAP-1-luciferase transcriptional reporter. After 24 hr, transfected cells were pre-treated with eupatilin (50 μm) for 18 hr or curcumin (20 μm) for 30 min and were then stimulated with TNF-α (20 ng/ml) for 1 hr. The mean fold induction of the β-actin reporter gene activity relative to the unstimulated controls remained relatively constant throughout each experiment. Data are expressed as the mean fold induction ± SEM relative to untreated controls (n = 5). *P < 0·05 compared with TNF-α alone.
We next performed an AP-1 reporter gene luciferase assay. Results showed that TNF-α increased the levels of the AP-1 reporter gene in BEAS-2B cells; however, pre-treatment with eupatilin did not significantly reduce the levels of AP-1 reporter gene activity in TNF-α-stimulated cells (Fig. 6c). In contrast, curcumin, as the control, significantly reduced the AP-1 activation induced by TNF-α. These results suggest that eupatilin does not influence AP-1 signalling in TNF-α-stimulated BEAS-2 B cells.
Suppression of NF-κB activation induced by eupatilin inhibits monocyte adhesion to bronchial epithelial cells
To evaluate whether pre-treatment with eupatilin could affect monocyte adhesion to the bronchial epithelial cells, monocytic THP-1 cells were incubated with TNF-α-treated BEAS-2B cells. Stimulation of BEAS-2B cells with TNF-α resulted in a significant increase in THP-1 cell adherence (Fig. 7a). In this experimental system, pre-treatment of BEAS-2B cells with eupatilin or an NF-κB inhibitor MG-132 before TNF-α stimulation resulted in a significant decrease in monocyte adhesion to BEAS-2B cells (Fig. 7b). In addition, pre-treatment with the Akt inhibitor LY294002 significantly reduced THP-1 cell adhesion to BEAS-2B cells. For physiological relevance, relatively pure mononuclear cells were incubated with TNF-α-treated primary NHBE cells. The ratio of adherence in treated compared with control cultures was similar with each of the experimental groups, although absolute numbers differed (Fig. 7c).
Figure 7.
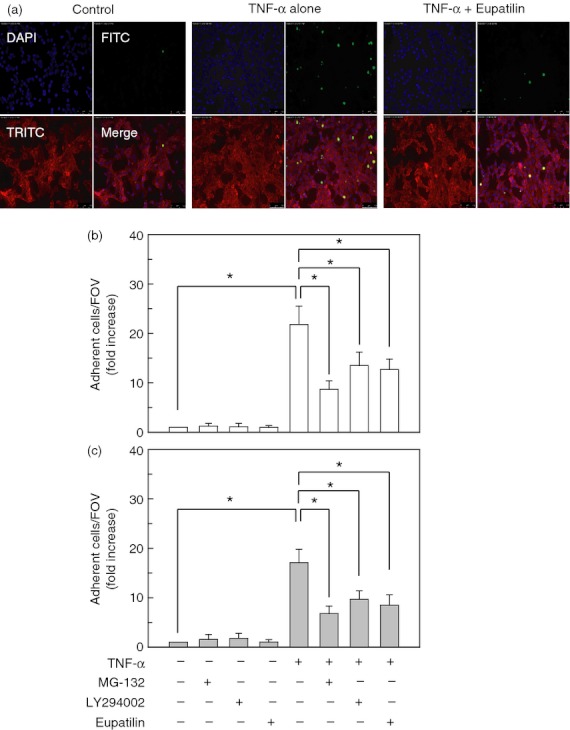
Relationship between suppression of nuclear factor-κB (NF-κB) activity and monocytic adhesion to BEAS-2B cells pre-treated with eupatilin. (a) BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr and then stimulated with tumour necrosis factor-α (TNF-α) (20 ng/ml) for 24 hr. Cells were further incubated with Calcein AM-stained human monocytes (THP-1, green) for 30 min. Cells were stained with the primary antibody (rabbit anti-actin) and the secondary antibody (anti-rabbit, red). Monocyte adhesion to BEAS-2B cells was analysed by fluorescence microscopy. Original magnification, × 200. (b) BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr or NF-κB inhibitor MG-132 (50 μm) for 30 min, after which the cells were stimulated with TNF-α (20 ng/ml) for another 24 hr. The staining procedure was identical to that in (a). Data represent the mean fold increases ± SD relative to that of the unstimulated controls (n = 5). (c) The primary normal human bronchial epithelial (NHBE) cells were pre-treated with eupatilin (50 μm) for 18 hr and then stimulated with TNF-α (20 ng/ml) for 24 hr. Cells were further incubated with Calcein AM-stained relatively pure mononuclear cells for 30 min. The staining procedure was identical to that in (a). Mononuclear cell adhesion to NHBE cells was analysed by fluorescence microscopy. Data represent the mean fold increases ± SD relative to that of the unstimulated controls (n = 5). *P < 0·05; FOV, field of observation.
Effect of eupatilin on TNF-α-induced phosphorylation of Akt in BEAS-2B cells
The phosphatidylinositol 3-kinase (PI3K)/Akt pathway has been known to be associated with NF-κB activation in lipopolysaccharide-stimulated cells.18 To investigate whether the inhibition of ICAM-1 and VCAM-1 expression by eupatilin may be mediated through the modulation of the Akt signal, we examined the effect of eupatilin on the TNF-α-induced phosphorylation of Akt in BEAS-2B cells using an immunoblot analysis. As shown in Fig. 8(a), stimulation of BEAS-2B cells with TNF-α increased the phosphorylated signals of Akt that were definitely inhibited by pre-treatment with eupatilin. These results were confirmed by experiments using the PathScan® phospho-Akt ELISA kit (Fig. 8b). In addition, transfection with a wild-type over-expressing plasmid of Akt recovered the eupatilin-induced down-regulation of phosphorylated activities of Akt. In contrast, the control plasmid-transfected cells combined with eupatilin and TNF-α did not show any significant change in phosphorylation activity of Akt compared with that of untransfected cells treated with eupatilin and TNF-α. Furthermore, pre-treatment of primary NHBE cells with eupatilin significantly decreased the TNF-α-induced phospho-Akt activity (Fig. 8c).
Figure 8.

Effect of eupatilin on the Akt signalling of BEAS-2B cells stimulated with tumour necrosis factor-α (TNF-α). (a) BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr and then stimulated with TNF-α (20 ng/ml) for the indicated period. Protein expression of phosphorylated Akt and actin were assessed by immunoblot analysis. Results are representative of three independent experiments. (b) Inhibition of Akt phosphorylation in TNF-α-stimulated BEAS-2B cells by treatment with eupatilin. BEAS-2B cells were transfected with a wild-type over-expressing Akt (WT Akt) or control plasmid for 48 hr. Untransfected or transfected cells were incubated with eupatilin (50 μm) for 18 hr and then stimulated with TNF-α (20 ng/ml) for the indicated period of time. Akt phosphorylation was measured using an Akt ELISA kit. Data are expressed as the mean fold induction ± SEM of phosphorylated Akt activity relative to untreated controls (n = 5). *P < 0·05 compared with TNF-α alone. (c) The primary normal human bronchial epithelial (NHBE) cells were pre-treated with eupatilin (50 μm) for 18 hr and then stimulated with TNF-α (20 ng/ml) for 1 hr. Akt phosphorylation was measured using an Akt ELISA kit. Data are expressed as the mean fold induction ± SEM of phosphorylated Akt activity relative to untreated controls (n = 3). *P < 0·05 compared with TNF-α alone.
We next investigated whether the Akt signal could be involved in the NF-κB-mediated expression of adhesion molecules in BEAS-2B cells. For this experiment, siRNA against Akt was used. The phosphorylation of Akt was almost completely suppressed in cells transfected with Akt siRNA (Fig. 9a). In this system, BEAS-2B cells transfected with Akt siRNA exhibited a significantly lower level of IκBα phosphorylation in TNF-α-stimulated cells (Fig. 9b). In this experimental system, TNF-α strongly increased MFI values of adhesion molecules such as ICAM-1 and VCAM-1 in untransfected cells, but Akt siRNA transfection significantly reduced the MFI values of ICAM-1 and VCAM-1. Moreover, transfection with Akt over-expressing plasmid augmented TNF-α-induced up-regulation of IκBα phosphorylation as well as both MFI values of ICAM-1 and VCAM-1 (Fig. 9c). However, there was no significant difference in MFI values between the TNF-α alone and eupatilin + TNF-α group in the Akt over-expressing plasmid-transfected cells. In the control plasmid-transfected cells, combined treatment with eupatilin and TNF-α significantly decreased IκBα phosphorylation as well as MFI values of both ICAM-1 and VCAM-1 compared with TNF-α alone. These results suggest that a signalling pathway including Akt and NF-κB is associated with the eupatilin-mediated inhibition of adhesion molecule expression in bronchial epithelial cells.
Figure 9.

Akt signal is involved in the nuclear factor-κB (NF-κB) -mediated expression of adhesion molecules in BEAS-2B cells treated with eupatilin and tumour necrosis factor-α (TNF-α). BEAS-2B cells were transfected with small interfering RNA (siRNA) against Akt or non-silencing control RNA (NS RNA) for 48 hr. (a) The siRNA-transfected cells were stimulated with TNF-α (20 ng/ml) for 1 hr. Cell lysates were analysed by immunoblotting with the indicated antibodies. Results shown are representative of three independent experiments. (b) Transfected or untransfected cells were incubated with eupatilin (50 μm) for 18 hr and then stimulated with TNF-α (20 ng/ml) for 1 hr (phosphorylated IκBα) or 24 hr [MFI values for intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1)]. IκBα phosphorylation was measured using an IκBα ELISA kit. Data are expressed as the mean fold induction ± SEM of phosphorylated activity relative to untreated controls (n = 5). Values of MFI were analysed by flow cytometry using cells stained with monoclonal antibody against ICAM-1 or VCAM-1. The data represent the MFI ± SEM (n = 3). (c) BEAS-2B cells were transfected with a wild-type over-expressing Akt (WT Akt) or control plasmid for 48 hr. Culture conditions for the stimulation with eupatilin and TNF-α were identical to those in (a). *P < 0·05; NS, not significant.
Eupatilin inhibits the adhesion of eosinophils to bronchial epithelial cells
Allergic diseases such as asthma are characterized by the infiltration of eosinophils;32 as a consequence, we determined whether pre-treatment with eupatilin could affect eosinophil adhesion to the bronchial epithelial cells. For this experiment, the eosinophilic EoL-1 cell line was used. As shown in Fig. 10(a), TNF-α-treated BEAS-2B cells promoted a significant increase in EoL-1 cell adherence. In this experimental system, pre-treatment of BEAS-2B cells with eupatilin before TNF-α stimulation resulted in a significant decrease of eosinophil adhesion to BEAS-2B cells. In addition, pre-treatment with the Akt inhibitor LY294002 or the NF-κB inhibitor MG-132 significantly reduced eosinophil adhesion to BEAS-2B cells (Fig. 10b). Similar results were observed in the adhesion between NHBE cells and isolated primary human eosinophils (Fig. 10c).
Figure 10.
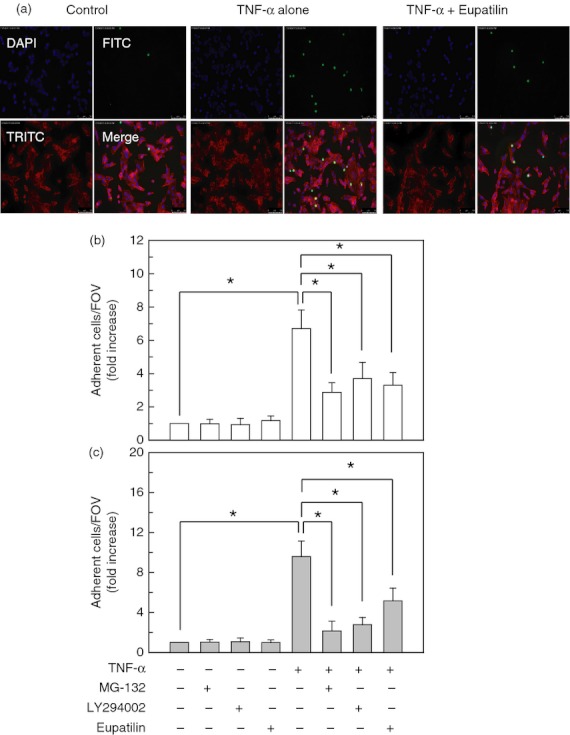
Eupatilin inhibits the adhesion of eosinophils to bronchial epithelial cells. (a) BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr and then stimulated with tumour necrosis factor-α (TNF-α) (20 ng/ml) for 24 hr. Cells were further incubated with Calcein AM-stained human eosinophils (EoL-1, green) for 30 min. Cells were stained with the primary antibody (rabbit anti-actin) and the secondary antibody (anti-rabbit, red). Eosinophil adhesion to BEAS-2B cells was analysed by fluorescence microscopy. Original magnification, × 200. (b) BEAS-2B cells were pre-treated with eupatilin (50 μm) for 18 hr, nuclear factor-κB (NF-κB) inhibitor MG-132 (50 μm) for 30 min, or Akt inhibitor LY294002 (20 μm) for 30 min. Cells were then stimulated with TNF-α (20 ng/ml) for another 24 hr. The staining procedure was identical to that in (a). Data represent the mean fold increases ± SD relative to that of the unstimulated controls (n = 5). (c) The primary normal human bronchial epithelial (NHBE) cells were pre-treated with eupatilin (50 μm) for 18 hr and then stimulated with TNF-α (20 ng/ml) for 24 hr. Cells were further incubated with Calcein AM-stained primary human eosinophils for 30 min. The staining procedure was identical to that in (a). Eosinophil adhesion to NHBE cells was analysed by fluorescence microscopy. Data represent the mean fold increases ± SD relative to that of the unstimulated controls (n = 5). * P < 0·05; FOV, field of observation.
Discussion
In the present study, eupatilin significantly inhibited both phosphorylation of Akt and IKK as well as the activation of NF-κB, which in turn resulted in the down-regulated expression of adhesion molecules such as ICAM-1 and VCAM-1 in TNF-α-stimulated bronchial epithelial cells. This effect finally led to the decreased adhesion of inflammatory cells such as monocytes and eosinophils to bronchial epithelial cells. These findings regarding the suppression of a signalling pathway, including Akt-NF-κB-induced expression of adhesion molecules and cell adhesion, support a novel mechanism of eupatilin function in anti-inflammatory responses.
Pro-inflammatory cytokine TNF-α induces activation of a central transcription factor known as NF-κB, which is a key regulator of gene expression. The activated NF-κB signal translocates to the nucleus and mediates transcription of inflammatory genes;9 for this reason, inhibitors of NF-κB activation may have therapeutic potential and are actively being researched.6,33 The present study showed that TNF-α strongly induced IκBα phosphorylation, NF-κB DNA binding activity, and translocation of the p65 NF-κB subunit in BEAS-2B cells, whereas these responses were suppressed in eupatilin-treated cells.
In this study, treatment with eupatilin blocked TNF-α-induced IKK activity and decreased the expression of both ICAM-1 and VCAM-1 surface molecules. These results suggest that eupatilin affects an IKK site in the inflammatory signalling pathway induced by TNF-α stimulation. Although we previously demonstrated that eupatilin induces the dissociation of the heatshock protein 90–IKK-γ complex in Bacteroides fragilis enterotoxin-stimulated intestinal epithelial cells, eupatilin did not completely inhibit Bacteroides fragilis enterotoxin-induced activation of NF-κB.16 Consistent with this, the present study revealed that eupatilin did not completely inhibit the increased signals of IKK and NF-κB or the up-regulation of adhesion molecules in TNF-α-stimulated cells. These results indicate that there may be other pathways that allow TNF-α to induce the expression of adhesion molecules.
It seems reasonable to assume that the eupatilin may affect the expression levels or activation state of lymphocyte function-associated antigen-1 (LFA-1) and the integrin α4β1 (very late antigen-4, VLA-4) on leucocytes. The LFA-1 on leucocytes is involved in recruitment to the site of infection. It binds to ICAM-1 on antigen-presenting cells and functions as an adhesion molecule. In addition, VCAM-1 as an integrin receptor binds to VLA-4, which is normally expressed on leucocyte plasma membranes. Further studies are necessary to clearly establish the role of eupatilin on the expression levels or activation state of LFA-1 and VLA-4 on leucocytes.
In addition to NF-κB signalling, the expression of adhesion molecules is also regulated by transcriptional factor AP-1.34 Mitogen-activated protein kinase (MAPK) signalling cascades have been shown to regulate AP-1 activity and are implicated in controlling gene transcription such as interleukin-8 and adhesion molecules.35–38 As eupatilin decreased the hydrogen peroxide-induced activation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in human gastric AGS cells,39 it may affect the MAPK and AP-1 signalling pathway. However, eupatilin did not influence AP-1 signalling in bronchial epithelial cells stimulated with TNF-α. Nevertheless, it is possible that an association between MAPK and IKK activation may activate NF-κB signalling in TNF-α-stimulated bronchial epithelial cells because MAPK signalling can induce NF-κB activation.18,29 Further studies are required to clarify other factors that contribute to the eupatilin-induced decrease of adhesion molecule expression in bronchial epithelial cells.
Activation of Akt, a serine/threonine kinase, is initiated by binding of its N-terminal pleckstrin homology (PH) domain to phosphatidylinositol-3,4,5-triphosphates produced by PI3K and translocation of Akt to the plasma membrane.40,41 Akt is then phosphorylated at Thr308 in the activation loop by phosphatidyl inositol-dependent kinase-1 and at Ser473 in the hydrophobic motif of the C-terminus by various kinases including the rictor–mammalian target of rapamycin complex.42 However, the Akt signalling pathway has not yet been implicated in the regulation of adhesion molecules in bronchial epithelial cells. In the present study, transfection with Akt siRNA significantly suppressed the NF-κB signals and reduced MFI values of ICAM-1 and VCAM-1 in TNF-α-stimulated BEAS-2B cells. These results indicate that a signalling pathway including Akt and NF-κB may be associated with the expression of adhesion molecules in bronchial epithelial cells. In this experimental model, the phosphorylated Akt signal in TNF-α-stimulated cells was significantly reduced after treatment with eupatilin. In addition, transfection with a constitutively active Akt plasmid recovered the eupatilin-induced down-regulation of phosphorylation activities of Akt. Moreover, pre-treatment of BEAS-2B cells with eupatilin, the NF-κB inhibitor MG-132, or the Akt inhibitor LY294002 resulted in a significant decrease of eosinophil adhesion to bronchial epithelial cells. These results suggest that eupatilin-induced suppression of eosinophil adhesion via the NF-κB signal seems to be largely the result of the inhibition of Akt.
Recently, Song et al.43 reported that a relatively high concentration of eupatilin (150 μm) induced the phosphorylation of Akt in cultured feline oesophageal epithelial cells. In contrast, we found that eupatilin (50 μm) did not activate the phosphorylation of Akt in BESA-2B human bronchial epithelial cells. Moreover, the up-regulated phosphorylated Akt signals by TNF-α stimulation was significantly suppressed when treated with eupatilin. Therefore, the mechanism for eupatilin-associated Akt signalling in human bronchial epithelial cells may be cell-specific and different from those in feline oesophageal epithelial cells.
It is possible that the inhibitory effect seen on adhesion molecules and NF-κB activation may be a result of apoptosis or cell toxicity. Our preliminary experiment showed that the cell viability was significantly decreased over 500 μm of eupatilin. Therefore, the concentration of eupatilin (50 μm) used in the present study seems not to be associated with cell toxicity. However, some studies demonstrated that eupatilin induced apoptosis at ranges between 20 and 150 μm in several cell types such as HL-60 promyelocytic leukaemia cells,14 Jurkat T cells,44 and AGS gastric epithelial cells.45 In contrast to HL-60 and Jurkat T cells, apoptotic features of AGS cells were apparent from 48 to 96 hr after treatment with eupatilin (more than 100 μm). These results suggest that 50 μm of eupatilin may not induce apparent apoptosis of BESE-2B bronchial epithelial cells within 24 hr. Nevertheless, further studies are needed to clarify the potential role of eupatilin-induced apoptosis in the expression of adhesion molecules via the Akt/NF-κB signalling pathway.
In summary, we have demonstrated that eupatilin has anti-inflammatory activity in bronchial epithelial cells through the suppression of a signalling pathway such as Akt-NF-κB-induced adhesion of inflammatory cells. This novel action of eupatilin suggests that it may play a role in developing a therapeutic strategy to counter the infiltration of inflammatory cells in asthma.
Acknowledgments
Jireh Jung and Su Hyuk Ko contributed equally to this work. We thank Dr Martin F. Kagnoff for providing several luciferase plasmids and standard β-actin RNA, Dr Michael Weber and Dr Joo Young Lee for assistance with the Akt-over-expressing plasmid, and Hyunsuk Frank Roh for the excellent technical help. This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (MEST) (No. 2010-0008594) and a grant from the NRF of Korea Grant funded by the Korean Government (MEST) (MRC Program No. 2010-0029507).
References
- 1.Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev. 2011;242:186–204. doi: 10.1111/j.1600-065X.2011.01033.x. [DOI] [PubMed] [Google Scholar]
- 2.Cook-Mills JM, Deem TL. Active participation of endothelial cells in inflammation. J Leukoc Biol. 2005;77:487–95. doi: 10.1189/jlb.0904554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holtmann MH, Neurath MF. Differential TNF-signaling in chronic inflammatory disorders. Curr Mol Med. 2004;4:439–44. doi: 10.2174/1566524043360636. [DOI] [PubMed] [Google Scholar]
- 4.Ciebiada M, Gorska-Ciebiada M, Gorski P. sICAM-1 and TNF-α in asthma and rhinitis: relationship with the presence of atopy. J Asthma. 2011;48:660–6. doi: 10.3109/02770903.2011.604886. [DOI] [PubMed] [Google Scholar]
- 5.Silvestri M, Bontempelli M, Giacomelli M, Malerba M, Rossi GA, Di Stefano A, Rossi A, Ricciardolo FL. High serum levels of tumour necrosis factor-α and interleukin-8 in severe asthma: markers of systemic inflammation? Clin Exp Allergy. 2006;36:1373–81. doi: 10.1111/j.1365-2222.2006.02502.x. [DOI] [PubMed] [Google Scholar]
- 6.Chen S. Natural products triggering biological targets – a review of the anti-inflammatory phytochemicals targeting the arachidonic acid pathway in allergy asthma and rheumatoid arthritis. Curr Drug Targets. 2011;12:288–301. doi: 10.2174/138945011794815347. [DOI] [PubMed] [Google Scholar]
- 7.Elewaut D, DiDonato JA, Kim JM, Truong F, Eckmann L, Kagnoff MF. NF-κB is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J Immunol. 1999;163:1457–66. [PubMed] [Google Scholar]
- 8.Kim JM, Cho SJ, Oh YK, Jung HY, Kim YJ, Kim N. Nuclear factor-κB activation pathway in intestinal epithelial cells is a major regulator of chemokine gene expression and neutrophil migration induced by Bacteroides fragilis enterotoxin. Clin Exp Immunol. 2002;130:59–66. doi: 10.1046/j.1365-2249.2002.01921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuk JM, Jo EK. Toll-like receptors and innate immunity. J Bacteriol Virol. 2011;41:225–35. [Google Scholar]
- 10.Mackesy DZ, Goalstone ML. Insulin augments tumor necrosis factor-α stimulated expression of vascular cell adhesion molecule-1 in vascular endothelial cells. J Inflamm. 2011;8:34. doi: 10.1186/1476-9255-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mankan AK, Lawless MW, Gray SG, Kelleher D, McManus R. NF-κB regulation: the nuclear response. J Cell Mol Med. 2009;13:631–43. doi: 10.1111/j.1582-4934.2009.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biesalski HK. Polyphenols and inflammation: basic interactions. Curr Opin Clin Nutr Metab Care. 2007;10:724–8. doi: 10.1097/MCO.0b013e3282f0cef2. [DOI] [PubMed] [Google Scholar]
- 13.González-Gallego J, Sánchez-Campos S, Tuñón MJ. Anti-inflammatory properties of dietary flavonoids. Nutr Hosp. 2007;22:287–93. [PubMed] [Google Scholar]
- 14.Seo HJ, Surh YJ. Eupatilin, a pharmacologically active flavone derived from Artemisia plants, induces apoptosis in human promyelocytic leukemia cells. Mutat Res. 2001;496:191–8. doi: 10.1016/s1383-5718(01)00234-0. [DOI] [PubMed] [Google Scholar]
- 15.Lee SH, Bae EA, Park EK, Shin YW, Baek NI, Han EJ, Chung HG, Kim DH. Inhibitory effect of eupatilin and jaceosidin isolated from Artemisia princeps in IgE-induced hypersensitivity. Int Immunopharmacol. 2007;7:1678–84. doi: 10.1016/j.intimp.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 16.Kim JM, Lee DH, Kim JS, et al. 5,7-dihydroxy-3,4,6-trimethoxyflavone inhibits the inflammatory effects induced by Bacteroides fragilis enterotoxin via dissociating the complex of heat shock protein 90 and IκBα and IκB kinase-γ in intestinal epithelial cell culture. Clin Exp Immunol. 2009;155:541–51. doi: 10.1111/j.1365-2249.2008.03849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim MS, Rådinger M, Gilfillan AM. The multiple roles of phosphoinositide 3-kinase in mast cell biology. Trends Immunol. 2008;29:493–501. doi: 10.1016/j.it.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–51. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- 19.Roh HC, Yoo do Y, Ko SH, Kim YJ, Kim JM. Bacteroides fragilis enterotoxin upregulates intercellular adhesion molecule-1 in endothelial cells via an aldose reductase-, MAPK-, and NF-κB-dependent pathway, leading to monocyte adhesion to endothelial cells. J Immunol. 2011;187:1931–41. doi: 10.4049/jimmunol.1101226. [DOI] [PubMed] [Google Scholar]
- 20.Kim JM, Kim JS, Lee JY, et al. Vacuolating cytotoxin in Helicobacter pylori water-soluble proteins upregulates chemokine expression in human eosinophils via Ca2+ influx, mitochondrial reactive oxygen intermediates, and NF-κB activation. Infect Immun. 2007;75:3373–81. doi: 10.1128/IAI.01940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JM, Kim JS, Lee JY, et al. Dual effects of Helicobacter pylori vacuolating cytotoxin on human eosinophil apoptosis in early and late periods of stimulation. Eur J Immunol. 2010;40:1651–62. doi: 10.1002/eji.200939882. [DOI] [PubMed] [Google Scholar]
- 22.Raiford KL, Park J, Lin KW, Fang S, Crews AL, Adler KB. Mucin granule-associated proteins in human bronchial epithelial cells: the airway goblet cell “granulome”. Respir Res. 2011;12:118. doi: 10.1186/1465-9921-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krunkosky TM, Fischer BM, Martin LD, Jones N, Akley NJ, Adler KB. Effects of TNF-α on expression of ICAM-1 in human airway epithelial cells in vitro. Signaling pathways controlling surface and gene expression. Am J Respir Cell Mol Biol. 2000;22:685–92. doi: 10.1165/ajrcmb.22.6.3925. [DOI] [PubMed] [Google Scholar]
- 24.Kim JM, Oh YK, Kim YJ, Oh HB, Cho YJ. Polarized secretion of CXC chemokines by human intestinal epithelial cells in response to Bacteroides fragilis enterotoxin: NF-κB plays a major role in the regulation of IL-8 expression. Clin Exp Immunol. 2001;123:421–7. doi: 10.1046/j.1365-2249.2001.01462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JY, Kim H, Cha MY, Park HG, Kim YJ, Kim IY, Kim JM. Clostridium difficile toxin A promotes dendritic cell maturation and chemokine CXCL2 expression through p38, IKK, and the NF-κB signaling pathway. J Mol Med. 2009;87:169–80. doi: 10.1007/s00109-008-0415-2. [DOI] [PubMed] [Google Scholar]
- 26.Park H, Kim NI, Kim JM, et al. Expression of eotaxin in gastric epithelial cells stimulated with Helicobacter pylori vacuolating cytotoxin. J Bacteriol Virol. 2006;36:11–20. [Google Scholar]
- 27.Kim JM, Lee JY, Yoon YM, Oh YK, Kang JS, Kim YJ, Kim KH. Bacteroides fragilis enterotoxin induces cyclooxygenase-2 and fluid secretion in intestinal epithelial cells through NF-κB activation. Eur J Immunol. 2006;36:2446–56. doi: 10.1002/eji.200535808. [DOI] [PubMed] [Google Scholar]
- 28.Joung SM, Park ZY, Rani S, Takeuchi O, Akira S, Lee JY. Akt contributes to activation of the TRIF-dependent signaling pathways of TLRs by interacting with TANK-binding kinase 1. J Immunol. 2011;186:499–507. doi: 10.4049/jimmunol.0903534. [DOI] [PubMed] [Google Scholar]
- 29.Yoon YM, Lee JY, Yoo D, et al. Bacteroides fragilis enterotoxin induces human β-defensin-2 expression in intestinal epithelial cells via a mitogen-activated protein kinase/I κB kinase/NF-κB-dependent pathway. Infect Immun. 2010;78:2024–33. doi: 10.1128/IAI.00118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JM, Kim JS, Yoo DY, Ko SH, Kim N, Kim H, Kim YJ. Stimulation of dendritic cells with Helicobacter pylori vacuolating cytotoxin negatively regulates their maturation via the restoration of E2F1. Clin Exp Immunol. 2011;166:34–45. doi: 10.1111/j.1365-2249.2011.04447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JM, Kim JS, Kim YJ, Oh YK, Kim IY, Chee YJ, Han JS, Jung HC. Conjugated linoleic acids produced by Lactobacillus dissociates IKK-γ and Hsp90 complex in Helicobacter pylori-infected gastric epithelial cells. Lab Invest. 2008;88:541–52. doi: 10.1038/labinvest.2008.16. [DOI] [PubMed] [Google Scholar]
- 32.Rådinger M, Lötvall J. Eosinophil progenitors in allergy and asthma – do they matter? Pharmacol Ther. 2009;121:174–84. doi: 10.1016/j.pharmthera.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki J, Ogawa M, Muto S, Itai A, Isobe M, Hirata Y, Nagai R. Novel IκB kinase inhibitors for treatment of nuclear factor-κB-related diseases. Expert Opin Investig Drugs. 2011;20:395–405. doi: 10.1517/13543784.2011.559162. [DOI] [PubMed] [Google Scholar]
- 34.Serafini M, Peluso I, Raguzzini A. Flavonoids as anti-inflammatory agents. Proc Nutr Soc. 2010;69:273–8. doi: 10.1017/S002966511000162X. [DOI] [PubMed] [Google Scholar]
- 35.Kim JM, Jung HY, Lee JY, Youn J, Lee CH, Kim KH. Mitogen-activated protein kinase and activator protein-1 dependent signals are essential for Bacteroides fragilis enterotoxin-induced enteritis. Eur J Immunol. 2005;35:2648–57. doi: 10.1002/eji.200526321. [DOI] [PubMed] [Google Scholar]
- 36.Lee JY, Park HR, Oh YK, Kim YJ, Youn J, Han JS, Kim JM. Effects of transcription factor activator protein-1 on interleukin-8 expression and enteritis in response to Clostridium difficile toxin A. J Mol Med. 2007;85:1393–404. doi: 10.1007/s00109-007-0237-7. [DOI] [PubMed] [Google Scholar]
- 37.Lin WN, Luo SF, Lin CC, Hsiao LD, Yang CM. Differential involvement of PKC-dependent MAPKs activation in lipopolysaccharide-induced AP-1 expression in human tracheal smooth muscle cells. Cell Signal. 2009;21:1385–95. doi: 10.1016/j.cellsig.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Ksiazek K, Mikuła-Pietrasik J, Catar R, et al. Oxidative stress-dependent increase in ICAM-1 expression promotes adhesion of colorectal and pancreatic cancers to the senescent peritoneal mesothelium. Int J Cancer. 2010;127:293–303. doi: 10.1002/ijc.25036. [DOI] [PubMed] [Google Scholar]
- 39.Lee S, Lee M, Kim SH. Eupatilin inhibits H2O2-induced apoptotic cell death through inhibition of mitogen-activated protein kinases and nuclear factor-κB. Food Chem Toxicol. 2008;46:2865–70. doi: 10.1016/j.fct.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 40.Bellacosa A, Chan TO, Ahmed NN, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–25. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 41.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 42.Du K, Tsichlis PN. Regulation of the Akt kinase by interacting proteins. Oncogene. 2005;24:7401–9. doi: 10.1038/sj.onc.1209099. [DOI] [PubMed] [Google Scholar]
- 43.Song HJ, Shin CY, Oh TY, Min YS, Park ES, Sohn UD. Eupatilin with heme oxygenase-1-inducing ability protects cultured feline esophageal epithelial cells from cell damage caused by indomethacin. Biol Pharm Bull. 2009;32:589–96. doi: 10.1248/bpb.32.589. [DOI] [PubMed] [Google Scholar]
- 44.Kim YD, Choi SC, Oh TY, Chun JS, Jun CD. Eupatilin inhibits T-cell activation by modulation of intracellular calcium flux and NF-κB and NF-AT activity. J Cell Biochem. 2009;108:225–36. doi: 10.1002/jcb.22244. [DOI] [PubMed] [Google Scholar]
- 45.Choi EJ, Oh HM, Wee H, et al. Eupatilin exhibits a novel anti-tumor activity through the induction of cell cycle arrest and differentiation of gastric carcinoma AGS cells. Differentiation. 2009;77:412–23. doi: 10.1016/j.diff.2008.12.004. [DOI] [PubMed] [Google Scholar]