

Abstract
Lysophosphatidic acid (LPA) is a potent bioactive phospholipid. As many other biological active lipids, LPA is an autacoid: it is formed locally on demand, and it acts locally near its site of synthesis. LPA has a plethora of biological activities on blood cells (platelets, monocytes) and cells of the vessel wall (endothelial cells, smooth muscle cells, macrophages) that are all key players in atherosclerotic and atherothrombotic processes. The specific cellular actions of LPA are determined by its multifaceted molecular structures, the expression of multiple G-protein coupled LPA receptors at the cell surface and their diverse coupling to intracellular signalling pathways. Numerous studies have now shown that LPA has thrombogenic and atherogenic actions. Here, we aim to provide a comprehensive, yet concise, thoughtful and critical review of this exciting research area and to pinpoint potential pharmacological targets for inhibiting thrombogenic and atherogenic activities of LPA. We hope that the review will serve to accelerate knowledge of basic and clinical science, and to foster drug development in the field of LPA and atherosclerotic/atherothrombotic diseases.
Keywords: Lysophosphatidic acid, LPA receptor, endothelial cells, smooth muscle cells, macrophage, platelet, neointima, hyperlipidaemia, chemokine, atherothrombosis
Introduction
Atherosclerosis is a slowly progressing, multifocal, chronic arterial disease that is characterized by inflammatory and regenerative processes, which lead to matrix remodelling and large lipid deposits. The retention of low-density lipoproteins (LDL) and activation of endothelial cells initiate atherosclerotic lesion formation in the inner layer (intima) of medium- and large-sized arteries, such as coronary arteries and cerebral arteries, predominantly at predilection sites, where the laminar blood flow is disturbed (Libby et al., 2011; Weber and Noels, 2011). In early lesions, monocytes are recruited to the arterial wall, where they engulf lipids and transform into foam cells. In the advanced stages of lesion formation, a fibroatheroma develops in the intima that is characterized by the extracellular accumulation of lipids and a fibrous cap consisting of vascular smooth muscle cells (VSMCs) and extracellular matrix proteins. Although patients with atherosclerosis may not develop clinical symptoms, the narrowing of the arterial lumen may compromise the oxygen supply and result in ischaemia of the tissues that are supplied by the affected artery, such as the myocardium or the brain. Furthermore, acute thrombotic occlusion of the arterial lumen can occur following erosion or rupture of atherosclerotic plaques, which leads to platelet activation and fibrin formation. These acute atherothrombotic events are potentially life-threatening sequelae of atherosclerosis as they may lead to myocardial infarction or stroke (Fuster et al., 2005; Thim et al., 2008). Percutaneous interventions, such as coronary stent implantation, can reopen the occluded or narrowed arteries and limit ischaemic symptoms, and are often life-saving. However, such interventions can result in a re-narrowing of the target vessel (restenosis) due to the accumulation of SMCs in the neointima.
Lysophosphatidic acid (LPA) is a phospholipid that mediates a plethora of activities in blood cells and cells of the vessel wall (Tigyi, 2001; Siess, 2002; Siess and Tigyi, 2004; Smyth et al., 2008). Similar to other lipid mediators such as prostaglandins and leukotrienes, LPA acts as an autacoid. After local formation in response to danger signals such as those initiated by vascular injury or inflammation LPA rapidly activates cells in the immediate vicinity (Tokumura, 1995). The action of LPA is mediated by its binding to and activation of surface GPCRs, including the Edg family of LPA receptors LPA1–3, LPA4 (GPR23), LPA5 (GPR92), and LPA6 (P2Y5), the latter three belonging to the purinoceptor cluster (Choi et al., 2010; Chun et al., 2010; Tigyi, 2010). Currently putative LPA GPCRs are GPR87, P2Y10 and GPR35; hence, there are in total nine LPA receptors (Choi et al., 2010; Chun et al., 2010; Tigyi, 2010). In addition to GPCRs, some of the effects of LPA appear to be mediated by the activation of the nuclear receptor PPARγ, specifically in macrophages and VSMCs (McIntyre et al., 2003; Zhang et al., 2004).
Our review is focused on the role of LPA and its receptors in atherosclerosis and cardiovascular diseases. Since the seminal observations that LPA is generated during mild oxidation of LDL and accumulates in the lipid core of human atherosclerotic plaques linking, for the first time, LPA to atherosclerosis (Siess et al., 1999), the area has evolved dramatically. We aim to cover this research field in the present review. General aspects of LPA receptor functions and their coupling to intracellular signalling pathways (Choi et al., 2010; Tigyi, 2010; Yanagida and Ishii, 2011), of intra- and extracellular LPA formation and metabolism (Nakanaga et al., 2010; Samadi et al., 2011; van Meeteren and Moolenaar, 2007), and of the role of LPA in cardiovascular physiology (Smyth et al., 2008) have been excellently reviewed previously.
LPA generation in vascular diseases
Although the absolute concentration of circulating LPA varies considerably in different assays (Smyth et al., 2008), it is clear that LPA is detectable in plasma in the low µM range. The generation of circulating LPA essentially requires the action of autotaxin (ATX), a secreted lysophospholipase D, which removes the polar head group from lysophospholipids (LPL), such as lysophosphatidylcholine (LPC) (Umezu-Goto et al., 2002; Tokumura et al., 2002c; Nakanaga et al., 2010). Accordingly, plasma LPA levels of healthy subjects strongly correlate with the serum ATX activity (Hosogaya et al., 2008) and are reduced by 50% in mice heterozygous for ATX (Tanaka et al., 2006; van Meeteren et al., 2006). Moreover, depletion of ATX completely prevents LPA production in serum (Tanaka et al., 2006; Tsuda et al., 2006), and ATX overexpression in transgenic mice increases LPA plasma levels (Pamuklar et al., 2009). Inhibition studies demonstrated that continual ATX activity is required to maintain a steady state concentration of LPA in the circulation (Albers et al., 2010; Gierse et al., 2010; Gupte et al., 2011). A boronic acid-based inhibitor in mice resulted in a rapid decline of LPA within minutes and the orally available ATX antagonist PF-8380 dose-dependently diminished the plasma LPA concentration by maximally 70% within 3 h (Albers et al., 2010; Gierse et al., 2010).
Adipocyte-derived ATX generates almost half of the LPA in plasma (Dusaulcy et al., 2011). In obesity, insulin resistance in adipocytes has been linked to increased expression of ATX, implicating a potential role of LPA in the sequelae of the metabolic syndrome (Boucher et al., 2005). Furthermore, hyperlipidaemia enhances the activity of circulating ATX in rabbits, which leads to enhanced generation of LPA from LPC in serum, and increases plasma LPA and adipose tissue expression of ATX in mice fed a high fat diet (Tokumura et al., 2002a; Dusaulcy et al., 2011).
Elevated circulating LPA levels may have pro-atherogenic effects, because systemic treatment with unsaturated LPAs has been shown to enhance atherosclerosis (Zhou et al., 2011). In humans, plasma LPA levels correlate with LPC, ATX and LDL levels, indicating that hyperlipidaemia may contribute to the generation of LPA in the circulation (Dohi et al., 2012). Moreover, circulating LPC is increased by hyperlipidaemia (Portman et al., 1970; McConnell and Hoefner, 2006; Matsumoto et al., 2007; Schmitz and Ruebsaamen, 2010). Plasma LPC, which is mainly bound to albumin is generated by endothelial lipase and by lecithin-cholesterol acyltransferase (LCAT) secreted from the liver (Wiesner et al., 2009; Schmitz and Ruebsaamen, 2010). Prolonged (10–50 h) incubation of plasma at 37°C leads to a large increase in LPA from 0.5 to 15 µM that is due to LPC generation by LCAT and not by lipoprotein-associated (Lp-) PLA2 activity (Aoki et al., 2002).
Progression of atherosclerosis is associated with conditions of chronic inflammation and oxidant stress. Under these conditions, circulating LPC can be generated by group IIA secretory PLA2 (sPLA2), which is an acute-phase reactant, and by Lp-PLA2 (identical to platelet-activating factor (PAF) acetylhydrolase) (Stafforini, 2009; Rosenson, 2010). sPLA2 acts on lipoproteins and microvesicles (Fourcade et al., 1995), whereas Lp-PLA2 specifically catalyses the removal of the acyl group at the sn-2 position of oxidatively truncated phospholipids present in oxidized LDL (ox-LDL) (Steinbrecher et al., 1984). LDL particles oxidized to different degrees have been detected at increased levels in the circulation of patients with acute coronary syndromes (Holvoet et al., 1998). Furthermore, electronegative LDL particles, which are pro-inflammatory by releasing chemokines from endothelial cells, are increased and physicochemically heterogeneous in familial hypercholesterolaemic patients (Sanchez-Quesada et al., 2002; 2003). Moreover, circulating levels and enzymatic activity of Lp-PLA2 and sPLA2 have been found to predict cardiovascular events (Packard et al., 2000; Koenig and Khuseyinova, 2009; Thompson et al., 2010). Lp-PLA2 is primarily bound to electronegative LDL in the circulation, and the levels of Lp-PLA2 positively correlate with the concentration of LDL (Packard et al., 2000; Benitez et al., 2003; Albert et al., 2005). Of note, the Lp-PLA2 inhibitor darapladib, which has currently been tested in two large cardiovascular outcome trials, limits atherosclerosis in mice (Charo and Taub, 2011; Wang et al., 2011). The sPLA2 inhibitor varespladib reduces atherosclerosis in mice and a phase III cardiovascular outcome trial on the effects of varespladib is ongoing (Charo and Taub, 2011).
Although it remains to be determined whether pro-atherogenic effects of Lp-PLA2 and sPLA2 are related to increased LPA production, a very recent study showing a correlation between levels of Lp-PLA2, LPCs, LPA and pro-inflammatory cytokines in human plaques supports such a link (Goncalves et al., 2012). Therefore, it can also be assumed that LPC levels elevated in hyperlipidaemia may lead to increased ATX-dependent LPA generation in the circulation. An interesting question in this regard is whether mice deficient in sPLA2 or Lp-PLA2 show decreased cellular LPA production or decreased LPA plasma levels. However, apart from one study that shows normal LPA generation of activated washed platelet suspensions prepared from mouse strains that are deficient in sPLA2 expression (such as the C57BL/6 and C3H strains) (le Balle et al., 1999), nothing is known about this topic.
LPA accumulates in human and mouse atherosclerotic lesions (Siess et al., 1999; Bot et al., 2010). LPA in atherosclerotic lesions is most likely to be derived from ATX-mediated hydrolysis of LPC. Although the expression of ATX in atherosclerotic lesions has not been studied, it has been shown that ATX can be secreted from arterial endothelial cells and that it is up-regulated in the arterial wall following vascular injury (Panchatcharam et al., 2008; Zhou et al., 2011). Structural analysis has suggested that ATX is capable of directing the LPA produced to the cognate GPCRs via binding of its somatomedin domains (SMD) to β3-integrins, thus contributing to localized LPA formation (Fulkerson et al., 2011; Hausmann et al., 2011; Nishimasu et al., 2011). However, it remains to be determined whether local LPA production and delivery by ATX via binding to β3-integrins, which are highly expressed in various cell types of atherosclerotic lesions, contributes to LPA formation in the lesion (Hoshiga et al., 1995).
In atherosclerotic lesions, the ATX substrate LPC is generated mainly by LDL oxidation. Polyunsaturated fatty acids in the sn-2 position of phosphatidylcholine (PC) in the outer layer of the LDL particle undergo oxidative fragmentation to oxidized short-chain fatty acids (Siess, 2006). The oxidized PC molecules are then specifically hydrolyzed by Lp-PLA2, which produces LPC (Steinbrecher et al., 1984). LPC in the vessel wall accumulates during hyperlipidaemia, and LPC accumulation in a mouse atherosclerotic model precedes the progressive accumulation of LPA in atherosclerotic tissue (Portman et al., 1970; Bot et al., 2010).
In addition, LPA in atherosclerotic lesions might derive indirectly from microparticles or microvesicles that are shed from activated and apoptotic cells (e.g. macrophages and VSMCs) and accumulate in inflamed atherosclerotic lesions. Membrane phospholipids of microvesicles, but not intact cells have been reported to be accessible to hydrolysis by sPLA2, and the concentration of lysophospholipids was found to be increased in microvesicles isolated from inflammatory fluids (Fourcade et al., 1995). Thus, this enzyme could also generate lysophospholipids from microparticles for ATX-dependent LPA formation in inflamed atherosclerotic lesions, although the role of microparticles in the production of lesional LPA remains to be determined.
Previous reports have described increased formation of LPA, preferentially of the alkyl-LPA species, during mild oxidation of LDL (Siess et al., 1999; Zhang et al., 2004), suggesting that a pathway of LPA formation during LDL oxidation may exist, which may be ATX-independent. However, the possibility that ATX, similar to other plasma proteins, might also be associated with LDL (Hoofnagle and Heinecke, 2009) cannot be excluded, although direct evidence is lacking.
Platelet activation importantly contributes to LPA formation in blood. Platelet depletion reduces the serum LPA levels by 50% in rats (Aoki et al., 2002). During blood clotting, the LPA concentration rises from low micromolar plasma concentrations (0.1–1 µM) up to 10 µM in serum predominantly by an increase of acyl-LPA species containing polyunsaturated fatty acids (mainly 18:2 and 20:4 fatty acids) (Siess, 2002; Tigyi, 2010). Activation of isolated platelets induces the formation of intracellular LPA via a pathway that involves the PLC-catalysed hydrolysis of phosphoinositides leading to the formation of diacylglycerol that is rapidly phosphorylated by diglyceride kinase to phosphatidic acid (PA) (Siess, 2002). Subsequently, degradation by platelet PLA2 and PLA1 enzymes specific for PA leads to the formation of 1-acyl-LPA and 2-acyl-LPA, respectively (Billah et al., 1981; Gaits et al., 1997). Intracellularly produced LPA is not released into the extracellular medium (Watson et al., 1985). LPA in the medium of activated platelets can only be detected in the presence of albumin (Eichholtz et al., 1993). Recent studies indicate that the small amounts of extracellular LPA that can be detected upon activation of isolated platelets are due to the activity of platelet-bound ATX (Pamuklar et al., 2009; Fulkerson et al., 2011). Thus, it seems likely that extracellular LPA detected upon activation of washed platelets is formed by platelet-bound ATX from LPC, and albumin by binding platelet-released LPC plays an essential role in this process.
In contrast to activation of platelets in buffer, platelet activation in the presence of plasma leads to a large increase in extracellular LPA. This occurs by a multistep process involving the generation of LPL via intracellular PLAs and secreted PLA1 and/or PLA2 enzymes (Aoki et al., 2002; Sano et al., 2002). Very recent studies demonstrating the binding of ATX to β3-integrins of activated platelets (Fulkerson et al., 2011) and the identification of a new PLA enzyme secreted from platelets (Bolen et al., 2011) unravel how platelet activation may lead to the large increase in certain molecular LPA species (18:2-acyl-LPA and 20:4 acyl-LPA) in serum. The platelet PLA enzyme that may feed plasma ATX with LPLs upon platelet activation is acyl-protein thioesterase 1 (APT1), also known as lysophospholipase A-I (LYPLA-I) (Bolen et al., 2011). APT1 is released from activated platelets, acts like PLA1 and generates a pool of sn-2-esterified lysophospholipids containing mainly C18:2 and C20:4 (Bolen et al., 2011). The thermodynamically unstable sn-2-esterified lysophospholipids undergo acyl migration resulting in sn-1-esterified lysophospholipids, which are the preferred substrates of ATX (Bolen et al., 2011). Subsequently, ATX bound to activated β3-integrins on the activated platelet membrane via its two SMD produces LPA species containing mainly 18:2 and 20:4 fatty acids (Bolen et al., 2011; Fulkerson et al., 2011). This platelet-dependent LPA production may be crucial for vascular repair, for example, after erosion of atherosclerotic plaques or after stent implantation, where activated platelets rapidly adhere to the denuded vascular surface.
Extracellular LPA is degraded by the ecto-activities of lipid phosphate phosphatases (LPPs) (Morris et al., 2009; Samadi et al., 2011). LPPs hydrolyze a large variety of bioactive lipid phosphates and pyrophosphates; their active site resides on the outer surface of plasma membranes. Three different mammalian LPP isoforms are known. Whereas i.v. injected LPA has a half-life of only 3 min in mice, an extended half-life of circulating LPA up to 12 min and increased levels of plasma LPA have been found in LPP1-deficient mice (Tomsig et al., 2009). It is therefore likely that a balance between ATX and LPP activities controls the concentration of extracellular LPA and thus LPA receptor activation in a dynamic manner (Samadi et al., 2011).
In addition to LPA, cyclic phosphatidic acid (1-acyl-sn-glycerol-2,3-cyclic phosphate; cPA) probably formed by ATX, has been identified in human serum (Kobayashi et al., 1999; Tsuda et al., 2006; Fujiwara, 2008; Shan et al., 2008b). cPA is an LPA analogue in which the sn-2 hydroxyl group has formed a ring structure with the sn-3 phosphate. Although the function of extracellular cPA on vascular cells and blood cells is largely unknown (Fujiwara, 2008; Tigyi, 2010), intracellular cPA, formed by PLD activation, functions as inhibitor of PPARγ and might thereby modify atherosclerotic processes (Tsukahara et al., 2010).
Measurement of plasma LPA and circulating LPA levels in cardio- and cerebrovascular clinical studies
The two primary methods that are used to measure LPA in plasma are liquid chromatography-electrospray ionization tandem MS (LC-ESI-tandem MS) and enzymatic assays. Two enzymatic methods have been described: (i) a radioenzymatic assay uses recombinant LPA acid acyltransferase and a radioactive fatty acid to measure the formation of radiolabeled PA (Saulnier-Blache et al., 2000); and (b) an enzymatic cycling assay uses lysophospholipase, which generates glycerol 3-phosphate, followed by enzymatic cycling using glycerol 3-phosphate oxidase and glycerol 3-phosphate dehydrogenase. The amplified concentrations of hydrogen peroxide are then measured colorimetrically (Kishimoto et al., 2003). Both of these methods yielded LPA plasma levels (0.07–0.1 µM) that were 10-fold lower than the levels measured by the LC/MS/MS method (0.7–1 µM) (Baker et al., 2001; Liebisch and Scherer, 2012).
The determination of plasma LPA is not trivial, and the conditions of sample preparation are important for accurate measurement. The majority of LPA in the circulation is bound to albumin (Tigyi et al., 1991; Tigyi and Miledi, 1992), and acidic extraction methods are required. The addition of strong acids may lead to conversion of plasma LPC to LPA in plasma samples, leading to an artificial increase in LPA (Shan et al., 2008a; Liebisch and Scherer, 2012). This is important because plasma LPC levels are approximately 100–1000 times higher than that of LPA. To circumvent this problem, the butanolic extraction procedure (pH 4) described by Baker et al. is often the method of choice for LPA extraction (Baker et al., 2001; Liebisch and Scherer, 2012). Furthermore, direct mass spectrometric analysis of crude lipid extracts should be avoided when determining the levels of plasma LPA. In-source fragmentation of LPC and lysophosphatidylserine (LPS) to LPA has been reported after direct flow injection during plasma LPA analysis (Zhao and Xu, 2009; Liebisch and Scherer, 2012). LPC can lose its choline group at the ion source before the parent ions are detected, giving rise to signals that are indistinguishable from endogenous LPA.
Activated platelets also contribute significantly to LPA generation in the blood, which is an additional factor that makes it difficult to accurately determine the circulating LPA levels. Platelets are activated easily after venipuncture, and it is impossible to immediately inhibit platelet activation before the blood comes into contact with the anticoagulant and additional platelet inhibitors that are perhaps present in the blood collection cuvette. Similar problems have previously been encountered when determining the concentration of circulating thromboxane and other substances that are released from activated platelets (FitzGerald et al., 1987). However, platelet activation during the subsequent handling of the blood such as during the centrifugation of blood to obtain plasma can be controlled. The lowest plasma concentrations of LPA (approximately 0.1 µM, measured by the enzymatic cycling assay) (Kishimoto et al., 2003) and LPC (approximately 190 µM) were found when blood was drawn into 7.5 mM EDTA plus a mixture of 10% (v v−1) citrate, theophylline, adenosine and dipyridamole (Nakamura et al., 2007). The latter three substances are platelet inhibitors. In blood samples that were collected in this manner, the LPA concentration was significantly higher in women (0.1 µM) than in men (0.077 µM), and a positive correlation between the plasma LPA concentration and serum lysophospholipase D (lysoPLD) activity was found. However, the LPA concentration could be correlated with the plasma LPC concentration only in men (Hosogaya et al., 2008).
In most of the clinical studies that report LPA plasma levels, no precautions have been taken to control for the artificial contribution of LPA production from activated platelets in vitro during blood handling and processing. For future studies, the inclusion of a pharmacological ATX inhibitor would be ideal to circumvent this problem. After i.v. injection of a boronic acid-based inhibitor of ATX into mice, a rapid decrease in the total plasma LPA concentration was observed; this finding was interpreted to indicate that there is a dynamic turnover of LPA in the circulation (Albers et al., 2010). Another explanation for these results could be that the injected ATX inhibitor attenuated the in vitro formation of LPA during blood handling.
In a recent cross-sectional study of consecutive patients, it was reported that patients with acute coronary syndrome have significantly increased plasma LPA levels (0.54 µM) compared with patients with stable angina pectoris (0.36 µM) or angiographically normal coronary arteries (0.41 µM) (Dohi et al., 2012). In this study, LPA was measured enzymatically as previously described (Kishimoto et al., 2003); however, special precautions to inhibit LPA formation during blood handling in vitro (Nakamura et al., 2007) were not taken. The blood was collected into EDTA-containing tubes (Dohi et al., 2012), and the LPA plasma levels of the patients with angiographically normal coronary arteries (approximately 0.4 µM) were approximately four times higher than the LPA plasma levels that have been reported previously in healthy people. The interpretation of this clinical study is therefore difficult as patients with unstable angina pectoris might exhibit higher LPA plasma levels because their platelets are more easily activated in vitro.
Another clinical study investigated the influence of acetylsalicylate on plasma LPA levels in patients with ischaemic cerebral vascular diseases (Li et al., 2008). Elevated LPA levels were found in patients with ischaemic cerebrovascular disease (3.11 µM) compared with healthy controls (1.77 µM). Daily administration of aspirin (100 mg) for 1 month significantly lowered the LPA levels in the patients from 4.06 to 2.41 µM. The authors concluded that their findings support a close association between increased plasma LPA levels and platelet activation. Again, it is difficult to interpret whether the effect of aspirin on the plasma LPA levels occurred in vivo or in vitro.
A separate clinical study investigated, whether there is a relationship between LPA levels and the prevalence of silent brain infarction (SBI) in patients with non-valvular atrial fibrillation (NVAF) (Li et al., 2010). The plasma LPA levels in the NVAF patients with SBI were significantly higher than those in the control patients (P < 0.01) or the NVAF patients without SBI. The authors suggested that LPA might be a novel marker for estimating the status of platelet activation and the risk for SBI onset in NVAF patients.
The question when interpreting all of these studies is whether the plasma LPA that is measured truly reflects the circulating LPA concentration or whether it was formed in vitro from platelets that were activated during blood handling. In future studies, the addition of a pharmacological ATX inhibitor to the blood collection tube is recommended to minimize LPA formation during blood handling. Moreover, the measurement of total plasma LPA levels may not be sufficient to predict the cardiovascular risk, because the thrombogenicity and atherogenicity of LPA crucially depends on the type of bondage of the fatty acyl chain to the glycerol backbone (ester or ether) and the saturation of the fatty acid; alkyl-LPA species are more potent platelet activators than the corresponding acyl-LPA species (Simon et al., 1982; Tokumura et al., 2002d; Rother et al., 2003), and only unsaturated acyl-LPA species are atherogenic (Yoshida et al., 2003; Zhang et al., 2004; Zhou et al., 2011). In line with these findings, elevated levels of LPA during pregnancy, which is not associated with increased cardiovascular risk, is due to a rise predominantly of saturated LPA, such as LPA16:0 (Tokumura et al., 2002b). Furthermore, increased circulating LPA has been found in patients with chronic hepatitis C and liver fibrosis and in experimentally induced liver fibrosis (Watanabe et al., 2007a, b). In contrast to hepatitis C infection, which is associated with increased cardiovascular risk, liver cirrhosis does not lead to accelerated atherosclerosis (Petta et al., 2011; Purnak et al., 2011; Adinolfi et al., 2012). Unfortunately, it is currently unknown which LPA species are elevated in the different forms of liver diseases.
Further studies on the circulating levels of the various LPA species may help to clarify the role of elevated circulating LPA levels in atherogenesis. These could also give a hint for the origin of LPA, because circulating LPA might derive from activated platelets (which produce mainly 18:2-acyl-LPA and 20:4 acyl-LPA), microparticles originating from damaged or apoptotic cells (their LPA species are unknown) or other cell types (adipocytes). However, in general, the relevance of dosing LPA in plasma could be questioned, because LPA is mainly generated locally in the circulation and acts locally.
Effects of LPA on blood and vascular cells
LPA has multiple effects on blood cells and cells of the vessel wall (Siess, 2002; Smyth et al., 2008). In platelets, it induces directly shape change, and it stimulates platelet aggregation and secretion only in synergy with other platelet stimuli (Rother et al., 2003; Haseruck et al., 2004). In human monocytes, LPA increases cytosolic Ca2+ (Fueller et al., 2003), and in macrophages, it stimulates cell survival and ox-LDL uptake (Koh et al., 1998; McIntyre et al., 2003). In endothelial cells, LPA stimulates cell migration (Panetti et al., 2000; 2004; Ptaszynska et al., 2010), chemokine secretion (Lin et al., 2006; Zhou et al., 2011), adhesion molecule expression (Rizza et al., 1999; Zhou et al., 2011), actin stress fibre formation and cell contraction (Siess et al., 1999; Hirakawa et al., 2004). In confluent endothelial cells in vitro or in endothelium in vivo, LPA can either increase (van Nieuw Amerongen et al., 2000; Hirase et al., 2001; Sarker et al., 2010) or decrease (Alexander et al., 1998; Minnear et al., 2001) endothelial permeability. LPA also co-operates with VEGF to stimulate angiogenesis (Tanaka et al., 2006; van Meeteren et al., 2006; Ptaszynska et al., 2010). In VSMCs, LPA stimulates cell contraction, leading to increased vascular tone, cell migration, and proliferation [for ref. see (Siess, 2002; Smyth et al., 2008) ].
The specific cellular effects of LPA depend on the species of the LPA molecule, and the origin and the vascular environment of the target cells. There are multiple molecular species of LPA that have been described in biological fluids and that are present in plasma and the lipid-rich core of atherosclerotic plaques (Baker et al., 2001; Rother et al., 2003). The fatty acid is mainly attached at the sn-1 position of the glycerol backbone via an ester (1-acyl-LPA) or ether bond (1-alkyl-LPA, 1-alkenyl LPA). The fatty acids, which are of predominantly 16-, 18- and 20-carbon lengths, can be saturated, mono- or poly-unsaturated. Additionally, 2-acyl regio-isomers of LPA have been detected; however, sn-2-esterified lysophospholipids are unstable and undergo acyl migration to yield sn-1-esterified lysophospholipids (Bolen et al., 2011).
The types of LPA molecules that are present determine, qualitatively or quantitatively, the biological response of the cell. For example, acyl-LPA that contain unsaturated but not saturated fatty acid causes the phenotypic dedifferentiation of cultured VSMCs and elicits neointima formation in a non-injury model of the rat carotid artery (Hayashi et al., 2001; Yoshida et al., 2003; Zhang et al., 2004; Subramanian et al., 2010). Furthermore, LPA (20:4), but not LPA (18:0), triggers the adhesion of monocytes to the vessel wall and enhances the progression of atherosclerosis (Zhou et al., 2011). In platelets, alkyl-LPA (16:0) and acyl-LPA (20:4) are approximately 20- and 7-fold more potent than acyl-LPA (16:0), respectively (Tokumura et al., 2002d; Rother et al., 2003; Haseruck et al., 2004). In cells that express individual recombinant LPA receptors, most of the receptors show an agonist preference for mono- or poly-unsaturated acyl-LPA over saturated acyl-LPA (Tigyi, 2010). However, whether endogenous LPA receptors, which are expressed at much lower levels in mammalian cells, behave similarly is not known.
The type of LPA response can also vary between species. For example, human and cat platelets, but not rodent (rat and mouse) platelets, are activated by LPA (Schumacher et al., 1979; Tokumura et al., 1981). Interestingly, mouse platelets are inhibited by LPA (Pamuklar et al., 2009). Additionally, the type of vascular bed is important in considering the effect of LPA on endothelial cells because the endothelium shows a pronounced heterogeneity. There are varied biomechanical and biochemical inputs along the vasculature that alter the properties of the endothelium, and therefore its response to the same pathophysiological stimuli will be different (Aird, 2005). Thus, in vivo studies on the action of LPA on endothelial cells will be particularly important as these allow the study of endothelial cells in healthy and atherosclerotic arterial vessels in situ (Zhou et al., 2011). In contrast, in vitro culture of endothelial cells cannot preserve their vascular bed-specific phenotype, and cell culture also leads to a loss of glycocalyx on the cell surface, which is an important endothelial interface with circulating blood (Chappell et al., 2009).
An important consideration when investigating the activity of circulating LPA is that plasma albumin might limit the activity of LPA. Albumin binds LPA with a stoichiometry of 3 mol of LPA to 1 mol of albumin (Tigyi and Miledi, 1992; Thumser et al., 1994), and albumin dose-dependently inhibits LPA-induced platelet shape change and aggregation (Tokumura et al., 2002d; Haseruck et al., 2004; Khandoga et al., 2008). This finding explains the observation that approximately 1000-fold higher LPA concentrations (0.5–20 µM) are required to stimulate platelets in plasma and blood, as compared with the stimulation of washed platelet suspensions. It is therefore unlikely that the low LPA concentrations found in plasma (total LPA: 0.1–1 µM) affect circulating platelets, and these low levels may also not affect other blood cells or endothelial cells as well.
For many of the effects of LPA on blood cells and cells of the vessel wall, the LPA receptors that are responsible have not been identified. The availability of mice that are deficient for individual LPA receptors (Choi et al., 2010) has advanced our knowledge of the involvement of specific LPA receptors in atherosclerotic processes (Panchatcharam et al., 2008). Often, in vitro studies of LPA receptor expression in individual cells have to rely on quantitative PCR analysis of mRNA transcripts because the possibly low endogenous expression of LPA receptor proteins is difficult to determine by immunoblot with specific LPA-receptor antibodies. Furthermore, the fact that one cell type often expresses many different LPA receptors, and that a single LPA receptor can couple to different G-proteins in the same cell (Choi et al., 2010), thereby stimulating a complex LPA signalling network, makes it difficult to dissect the function of individual LPA receptors. However, studies using the siRNA technology to down-regulate individual LPA receptors in vascular cells, blood cells or isolated arteries are helpful to assign a specific LPA receptor to its cellular or vascular effect (Subramanian et al., 2010; Khandoga et al., 2011; Zhou et al., 2011).
LPA in acute atherothrombosis
Myocardial infarction and ischaemic stroke are leading causes of morbidity and mortality in humans. The trigger for approximately 70% of myocardial infarctions is plaque rupture, and the erosion of vulnerable atherosclerotic plaques accounts for the other 30%. These events lead to the exposure of thrombogenic plaque material to circulating blood (Fernandez-Ortiz et al., 1994; van Zanten et al., 1994; Toschi et al., 1997; Kolodgie et al., 2004). Subsequent platelet activation and fibrin formation can lead to the development of an occluding thrombus with possibly fatal consequences for the patient. LPA may play different roles in thrombus formation after erosion and plaque rupture. After plaque erosion, the main thrombogenic stimulus is possibly subendothelial versican-hyaluronan matrix (Kolodgie et al., 2004), whereas after plaque rupture, collagen of the disrupted cap and possibly LPA of the plaque core are the platelet stimuli (van Zanten et al., 1994; Siess et al., 1999; Corti and Badimon, 2002; Rother et al., 2003; Penz et al., 2005; Nakanaga et al., 2010). The lipid core contains alkyl-LPA and acyl-LPA species with high platelet-activating potency (Rother et al., 2003) (Figure 1). On the other hand, LPA locally formed by ATX bound to activated platelets that cover eroded plaques might, in concert with other platelet-derived mediators such as sphingosine 1-phosphate (S1P) and VEGF, activate neighbouring endothelial cells to migrate and proliferate, and thereby help in healing the endothelial defect (Panetti et al., 2000; 2004; Ptaszynska et al., 2010).
Figure 1.
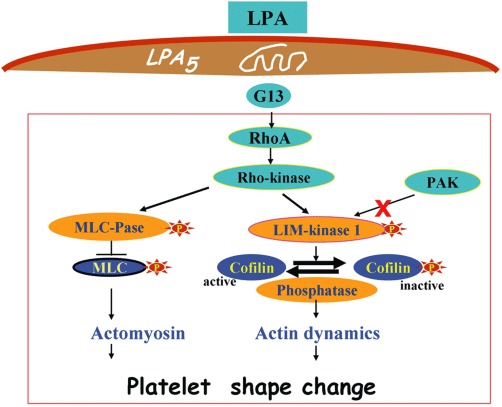
LPA-induced platelet signalling during platelet shape change. Activation of the LPA5 receptor coupled to the heterotrimeric G13 protein stimulates Rho and Rho-kinase. The subsequent bifurcating pathway directed to either the myosin-binding subunit of MLC phosphatase or the LIM-kinase 1 leads to enhanced phosphorylation of MLC and stimulation of phospho-cofilin turnover, respectively. Phosphorylated myosin develops actin-activated ATPase activity, interacts with F-actin, and assembles into filaments, whereas cofilin regulates actin dynamics by enhancing both actin-polymerization and actin filament severing. These cytoskeleton changes underlie the folding of the surface membrane, the formation of pseudopods and the contractile wave centralizing the secretory granules during platelet shape change.
LPA that is produced by activated platelets after plaque erosion or rupture may play a role as a positive feedback mediator of platelet activation. However, in a previous study, no inhibitory effects of LPA receptor antagonists were found on aggregation of washed platelets stimulated by collagen or thrombin, indicating that the small amounts of LPA that were generated by stimulated, washed platelets do not mediate or support stimulus-induced platelet aggregation (Haseruck et al., 2004). Whether LPA plays a role as a positive feedback mediator of platelet activation in stimulated blood where ATX-dependent LPA formation rises drastically is not known.
Thrombin activation of isolated platelets leads to the activation of the integrin αIIbβ3 and binding of extracellular ATX to the β3 integrin through its tandem somatomedin B (SMB) domains (Fulkerson et al., 2011; Hausmann et al., 2011). Echistatin, an arginine–glycine–aspartic acid (RGD)-containing peptide, and a monoclonal antibody against integrin αIIbβ3 (10E5) reduced platelet adhesion to ATX and inhibited ATX-mediated LPA formation by thrombin-activated, isolated platelets (Fulkerson et al., 2011). However, the RGD sequence of the tandem SMB domains is not involved in the binding of ATX to the β3 integrin of activated platelets, as demonstrated by an ATX–RGE mutant that showed a similar binding to activated platelets as that of wild-type ATX (Hausmann et al., 2011). Although it has not been tested directly, it is likely that LPA formation in blood also depends on binding of ATX to the β3 integrins of activated platelets. In blood, ATX, which is present at a concentration of approximately 100 nM in plasma (Nakamura et al., 2008), has to compete with the much higher plasma concentration of fibrinogen (5–8 µM) for binding to the β3 integrin on activated platelets.
Healthy people display individual heterogeneity of the LPA-induced platelet aggregation response when measured in washed platelets, PRP or blood (Haseruck et al., 2004; Pamuklar et al., 2008). However, all of the blood donors showed a similar shape change response when tested in blood (Haseruck et al., 2004). LPA-induced platelet aggregation was completely dependent on ADP-mediated activation of P2Y1 and P2Y12 receptors in whole blood (Haseruck et al., 2004). Thus, LPA does not directly induce human platelet aggregation in blood, as LPA-induced platelet aggregation requires the presence of extracellular ADP. In stirred blood, ADP may be either secreted from platelet-dense granules or released from red cells. In a recent study using isolated platelets, the individual heterogeneity of LPA-induced platelet aggregation was hypothesized to be due to LPA-induced activation of an inhibitory pathway in the non-responders (Pamuklar et al., 2008).
Low nM concentrations of LPA directly induce shape change in washed platelets (Siess et al., 1999; Rother et al., 2003). LPA binds to GPCRs on the platelet surface, and the signal that is emitted by the activated platelet receptor is transduced by the heterotrimeric G13 protein to activate Rho and Rho-kinase (Bauer et al., 1999; Klages et al., 1999; Gratacap et al., 2001; Moers et al., 2003). Rho-kinase phosphorylates the 130-kD myosin-binding subunit of myosin phosphatase, thereby decreasing the activity of myosin phosphatase and increasing myosin light chain (MLC) phosphorylation (Kimura et al., 1996; Bauer et al., 1999; Retzer and Essler, 2000). Rho-kinase also phosphorylates and activates LIM-kinase 1, which increases the turnover of phospho-cofilin (Pandey et al., 2006; 2007). These biochemical pathways converge in the remodelling of actin–myosin structures that underlie platelet shape change (Figure 1). Cytosolic Ca2+ increase, Rac activation and p21-activated kinase (PAK) are not involved in LPA-induced shape change (Maschberger et al., 2000; Pandey et al., 2007). High concentrations of LPA (10 µM) induce an increase in cytosolic Ca2+ in washed platelets that is primarily due to the stimulation of Ca2+ entry. This leads to cofilin dephosphorylation and secretion; the latter response requires, in addition, integrin αIIβ3 outside-in signalling (Maschberger et al., 2000; Pandey et al., 2007). LPA does not activate the heterotrimeric G-protein Gi in platelets, yet it shows a strong synergism in the induction of platelet aggregation with platelet stimuli such as adrenaline and ADP that activate Gi (Rother et al., 2003; Haseruck et al., 2004).
Until recently, the identity of the LPA receptor that mediates platelet shape change remained obscure. Human platelets express mRNA for the Edg receptors LPA1–3, and the purinergic cluster LPA4 (GPR23), LPA5 (GPR92), LPA6 (P2Y5), and the putative receptors GPR87 and P2Y10 (Amisten et al., 2008; Khandoga et al., 2008; Pamuklar et al., 2008). The receptors that are most abundantly expressed at the mRNA level are LPA4 and LPA5 (Amisten et al., 2008; Khandoga et al., 2008). A previous study suggested that LPA1 and LPA3 play a role in inducing platelet shape change based on the ability of the LPA receptor subtype-specific antagonist diacylglycerol pyrophosphate to inhibit LPA-induced platelet shape change. However, the inhibitory effect of this compound increased with the time of pre-incubation (Rother et al., 2003), which is not typical for a receptor antagonist. Two recent studies described the involvement of LPA4 and LPA5 in mediating LPA-induced platelet shape change (Khandoga et al., 2008; Williams et al., 2009). However, firm evidence that these receptors were the functional platelet LPA receptors was lacking. The LPA response of platelets did not match the pharmacological properties of the LPA4 and LPA5 receptors in heterologous expression systems (Khandoga et al., 2008), and the pharmacological receptor agonists and antagonists that were used were not selective for LPA5 (Williams et al., 2009).
More recently, it was demonstrated that LPA-induced shape change in two human megakaryocytic cell lines was inhibited by siRNA against LPA5, but not by knock-down of each of the other receptors LPA1–4,6,7 (Khandoga et al., 2011). The rank order of activation by LPA species in these cells, with alkyl-LPA 18:1 and alkyl-LPA 16:0 being the most potent, was similar to that of human platelets, supporting the hypothesis that LPA5 is the functional LPA receptor-mediating platelet shape change (Figure 1). Importantly, siRNA against LPA5 also inhibited shape change of the megakaryocytic cells that was induced by the lipid-rich core of human atherosclerotic plaques (Khandoga et al., 2011). Thus, LPA5 may be a novel target for anti-thrombotic therapy for patients with ischaemic cardio- and cerebrovascular disease.
Upon plaque rupture, LPA exposed by the lipid core might, by binding to LPA5, induce platelet shape change and stimulate, in synergy with ADP, platelet aggregation and thrombus formation (Haseruck et al., 2004; Rother et al., 2003) (Figure 2). ADP can derive from damaged red cells (Born and Wehmeier, 1979) and from activated platelets adhering to the collagenous matrix of the ruptured cap and secreting their granule contents (Penz et al., 2005; Reininger et al., 2010) (Figure 2). Thus, plaque LPA may act as a cofactor in acute atherothrombosis. However, the importance of LPA in the lipid-rich core for platelet activation in whole blood remains to be demonstrated. Specific LPA5 receptor antagonists will be helpful to answer this question. Indirect evidence supports a role for plaque LPA in plaque-induced platelet activation and thrombus formation. Low (sub-µM) concentrations of alkyl-LPA species, that may be reached locally after plaque rupture, activate platelets in blood (Haseruck et al., 2004), and aspirin is equally ineffective at inhibiting LPA-triggered platelet aggregation in blood (Haseruck et al., 2004) and plaque-induced platelet thrombus formation under arterial flow conditions (Penz et al., 2007).
Figure 2.
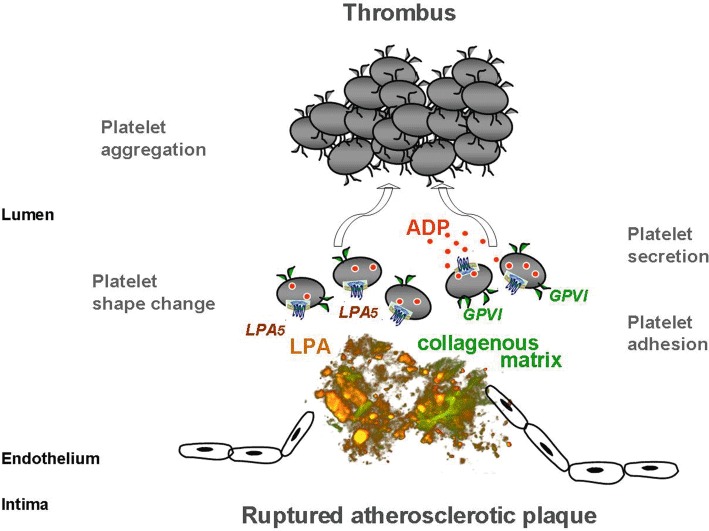
Hypothetical role of plaque LPA and the platelet LPA5 receptor in acute atherothrombosis after plaque rupture. LPA in the lipid core of atherosclerotic plaques may act as a cofactor with platelet-adhesive matrix proteins such as collagen type I and III in platelet activation (van Zanten et al., 1994; Penz et al., 2005; Schulz et al., 2008). These matrix proteins are over-expressed in plaques as compared with healthy arterial intima. LPA induces, through binding to the LPA5 receptor, shape change of circulating platelets and has a synergistic effect with ADP at inducing platelet aggregation and thrombus formation. ADP is secreted from dense granules of platelets adhering to the collagenous matrix of the ruptured cap. GPVI, glycoprotein VI.
LPA in atherosclerosis
Hypercholesterolaemia promotes the continuous recruitment of circulating monocytes to the arterial wall, which drives the progression of atherosclerotic plaques (Swirski, 2006). LDL enters the vessel wall and is retained in the subendothelial space, where LDL is oxidatively modified. However, the type and degree of LDL modifications are diverse, resulting in distinct biological activities. Oxidized LDL is generally divided into two main categories: minimally modified or mildly oxidized LDL (MM–LDL or mox-LDL) and extensively oxidized LDL (ox-LDL) (Stocker and Keaney, 2004; Levitan et al., 2010). Although both are chemically modified, MM–LDL differs from oxLDL, because it still binds to the LDL receptor and is not recognized by most scavenger receptors (Stocker and Keaney, 2004; Levitan et al., 2010). In contrast to native LDL, MM-LDL can induce the adhesion of monocytes to endothelial cells by up-regulating the CXC chemokine, CXCL1, on the endothelial surface (Berliner et al., 1990; Schwartz et al., 1994). Oxidized LDL or components of LDL that are released during the oxidation, like oxidized phospholipids, trigger the inflammatory response by activating endothelial cells (Hansson and Hermansson, 2011; Weber and Noels, 2011). Activated endothelial cells express adhesion molecules and chemokines that direct the adhesion and transmigration of circulating monocytes. These monocytes transform into macrophages in the vessel wall, where they engulf ox-LDL and propagate the inflammatory reaction in concert with T-cells. During the progression of atherosclerosis, SMCs accumulate and form a fibrous cap that encloses a highly thrombogenic core comprising extracellular lipids.
LPA accumulates during atherogenesis induced by perivascular collar placement in Apoe−/− mice and is increased in the lipid core region of human atherosclerotic plaques (Rother et al., 2003; Bot et al., 2010). In vitro, LPA has been shown to activate NF-κB and increase the adhesion of monocytes to endothelial cells under static conditions by up-regulating adhesion molecules and chemokines, such as intracellular adhesion molecule-1 (ICAM-1), E-selectin, and vascular cell adhesion molecule-1 (VCAM-1) (Palmetshofer et al., 1999; Rizza et al., 1999; Lee et al., 2004; Lin et al., 2007). Whereas LPA-induced endothelial ICAM-1 expression is mediated by the LPA1 receptor, both LPA1 and LPA3 are involved in the chemotactic activity generated by the secretion of IL-8 and chemokine ligand 2 (CCL2), also known as monocyte chemotactic protein-1 (MCP-1) from LPA-stimulated endothelial cells (Lee et al., 2004; Gustin et al., 2008b). Additionally, the endothelial release of pentraxin-3 is induced by LPA and enhances monocyte migration (Gustin et al., 2008a). However, the role of LPA-induced endothelial pentraxin-3 secretion in atherogenesis is unclear.
In murine carotid arteries, atherogenic monocyte adhesion under flow conditions is primarily mediated by the CXC chemokine CXCL1 (human GRO-α/murine KC), which is in contrast to CCL2 immobilized on the endothelial surface (Weber et al., 1999; Huo et al., 2001). Whereas most chemokines are transcriptionally up-regulated, CXCL1 is stored in intracellular vesicles of endothelial cells (Oynebraten, 2004; Zhou et al., 2011). The modified LDL-induced secretion of endothelial CXCL1 is mediated by an unsaturated LPA species and requires the activity of ATX (Zhou et al., 2011). This secretagogue effect of LPA is mediated by the LPA1/3-induced activation of Rho-associated coiled-coil containing protein kinase. However, activation of NF-κB by unsaturated LPA induces transcriptional up-regulation of CXCL1 in endothelial cells, suggesting a biphasic response (Zhou et al., 2011). This mechanism of LPA on CXCL1 is crucial in promoting atherogenic monocyte recruitment and atherosclerosis in vivo (Figure 3). Hyperlipidaemia-induced monocyte adhesion to carotid arteries is almost completely abolished by pharmacological inhibition of LPA1/3, indicating that LPA is an important mediator of the pro-atherogenic effects of ox-LDL (Zhou et al., 2011). This reduced monocyte recruitment may explain the inhibition of atherogenesis observed by blocking LPA1/3 receptors (Zhou et al., 2011).
Figure 3.
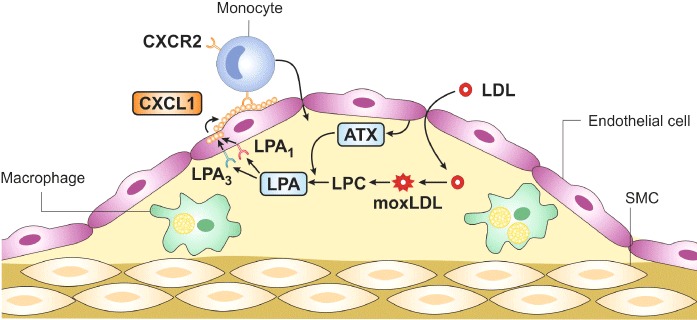
LPA promotes the accumulation of macrophages in atherosclerotic lesions. The moxLDL leads to increased formation of LPC, which is converted by endothelial-derived ATX into LPA. LPA triggers the release of the chemokine CXCL1 from endothelial cells through the activation of LPA1 and LPA3. CXCL1 is immobilized on the endothelial surface and induces the adhesion of monocytes to the vessel wall via its receptor CXCR2 on monocytes. These monocytes migrate into the subendothelial space and transform into macrophages, which are the primary cells in early atherosclerotic plaques.
Apart from the recruitment of monocytes, several cellular functions of monocytes and macrophages, including the activation status, uptake of LDL and survival, play an important role in atherogenesis (Moore and Tabas, 2011). Human monocytes and macrophages primarily express LPA1 and LPA2, and LPA has been shown to mediate the effects of MM-LDL on monocyte activation via LPA1 (Fueller et al., 2003; D'Aquilio et al., 2007). In agreement with this finding, LPA stimulates the expression of the pro-atherogenic cytokine IL-1β in a GPCR-dependent manner in a murine macrophage cell line (Chang et al., 2008b). Moreover, LPA increases the uptake of ox-LDL in monocytes/macrophages (Llodra et al., 2004; Chang et al., 2008b). In J774A murine macrophages, which express LPA2 and LPA3, LPA-induced lipid accumulation is mediated by the up-regulation of the scavenger receptor A and can be inhibited by the LPA1/LPA3 receptor antagonist Ki16425 (Chang et al., 2008a). In addition, LPA binds to the nuclear receptor PPARγ and induces PPARγ-dependent CD36 expression in murine macrophages (McIntyre et al., 2003). PPARγ activation in macrophages has anti-inflammatory effects, and conditional deletion of PPARγ in macrophages enhances atherosclerosis (Babaev et al., 2005; Bouhlel et al., 2007). Therefore, an atheroprotective role of LPA by activation of PPARγ could be envisioned, if this mechanism is relevant in vivo. Although LPA is known to increase monocyte migration (Zhou et al., 1995; Gustin et al., 2008b), inhibition of reverse transmigration of monocytes by LPA has been found, which may play a role in the regression of atherosclerosis (Llodra et al., 2004). Moreover, LPA protects macrophages from apoptosis by activating phosphoinositide 3-kinase (PI3K), indicating that LPA impairs the removal of plaque macrophages (Koh et al., 1998).
SMCs accumulate in advanced atherosclerotic plaques, and apoptosis of SMCs is associated with accelerated atherosclerosis. However, exacerbated proliferation of SMCs and the conversion to a pro-inflammatory SMC phenotype may promote the progression of atherosclerosis by enhanced monocyte recruitment (Zeiffer et al., 2004). LPA potently stimulates the proliferation of cultured SMCs and most probably contributes to ox-LDL-induced SMC proliferation (Tokumura et al., 1994; Natarajan et al., 1995; Kim et al., 2006; Damirin et al., 2007; Komachi et al., 2009). LPA mediates the MM–LDL-induced expression of CCL20 in SMCs via LPA receptors, which is secreted from atherosclerotic plaques and elevated in the circulation of patients with hyperlipidaemia (Calvayrac et al., 2011). However, the functional role of LPA-induced CCL20 in atherogenesis needs to be determined. Tissue factor (TF) is a critical determinant of atherosclerotic plaque thrombogenicity. LPA up-regulates TF expression in SMCs via Gi-protein–mediated activation of ERK1/2 (Cui et al., 2003). Furthermore, LPA induces the pro-atherogenic factors CCL2 (MCP-1) and IL-6 in human vascular SMCs in vitro (Kaneyuki et al., 2007; Hao et al., 2010).
Taken together, lipoprotein-derived LPA plays a crucial role in atherogenesis by promoting the recruitment of monocytes. Therapeutic targeting of LPA1 or LPA3 on endothelial cells may be a promising approach against atherosclerosis.
LPA in vascular remodelling
The arterial vessel wall can adapt to variety of environmental cues, such as changes in pressure, flow, or oxygen supply, by a process called vascular remodelling that mainly includes a response of the SMCs and results in structural alterations of the tunica media (Gibbons and Dzau, 1994). The media may enlarge, for example in arterial or pulmonary hypertension, or diminish, as in the formation of aneurysms. Furthermore, neointimal accummulation of SMCs is characteristic of vascular repair after injury, for example following stent implantation into atherosclerotic arteries (called restenosis). In addition, endothelial cells and inflammatory cells like monocytes/macrophages affect SMC function and the production of extracellular matrix proteins, thereby playing an important role in vascular remodelling (Schober, 2008). The phenotypic switch of SMCs from a contractile, quiescent state towards a synthetic, proliferative state is a common feature during vascular remodelling and may be causally related to neointima formation (Owens et al., 2004).
Both migration and proliferation of medial SMCs following vascular injury have been implicated in neointima formation (Schwartz et al., 1995). In vitro, LPA has been shown to induce the proliferation of SMCs via activation of LPA1 and Gi/q proteins and involves PKC, ERK1/2, the PI3K/PKB (Akt) pathway and MAPK cascades (Tokumura et al., 1994; Seewald et al., 1997; 1999; Gennero et al., 1999; Schmitz et al., 2002; Xu et al., 2003; Gouni-Berthold et al., 2004; Baldini et al., 2005; Kim et al., 2006; Komachi et al., 2009). In contrast, combined genetic deletion of LPA1 and LPA2 is required to inhibit serum-induced growth of SMCs (Panchatcharam et al., 2008). Furthermore, LPA enhances migratory activity of vascular SMCs by Gi/q protein-coupled LPA1 receptor-mediated activation of the p38MAPK pathway (Kim et al., 2006; Damirin et al., 2007; Komachi et al., 2009; Zhou et al., 2009). However, SMCs from LPA1−/− mice showed increased migration due to compensatory up-regulation of LPA3 (Panchatcharam et al., 2008). Whereas the migration of LPA2-deficient SMCs is not impaired, combined deletion of LPA1 and LPA2 inhibits SMC migration (Panchatcharam et al., 2008). These results indicate that LPA3 also promotes SMC migration in the presence of LPA2. Neointimal SMCs display a pro-inflammatory phenotype driven by activated NF-κB signalling, which may promote leucocyte recuitment to the injured artery (Zeiffer et al., 2004). LPA increases the expression of IL-6 and CCL2 in vascular SMCs via LPA1 and PKC-mediated p38 MAPK and NADPH oxidase-dependent generation of reactive oxygen species, respectively (Kaneyuki et al., 2007; Hao et al., 2010). The early growth response gene 1 (Egr1) is crucial for SMC proliferation and neointima formation (Khachigian, 2006). Egr1 is induced by LPA-mediated activation of the transcription factors cAMP response element-binding (CREB) and serum response factor (SRF) in SMCs (Cui et al., 2006). However, the role of LPA-induced Egr1 in vascular remodelling remains to be defined. The reduced expression of contractile proteins is characteristic for synthetic SMCs, and this dedifferentiation process has been implicated in neointima formation (Owens et al., 2004). LPA species with an unsaturated fatty acyl chain selectively down-regulate contractile protein expression in SMCs via sustained activation of ERK1/2 and p38 MAPK (Hayashi et al., 2001). This effect of LPA on SMC dedifferentiation has been attributed to the activation of LPA3 using LPA receptor-specific antagonists and agonists (Zhou et al., 2010). In agreement with this finding, no differences in LPA-induced SMC dedifferentiation have been observed between wild-type, LPA1−/−, LPA2−/−, LPA1−/−/LPA2−/− and PPARγ−/− SMCs (Guo et al., 2008; Panchatcharam et al., 2008). However, the PPARγ agonist rosiglitazone reduced the expression of SMC markers, like unsaturated LPA, indicating different pathways for PPARγ- and LPA-induced SMC differentiation (Zhang et al., 2004). Taken together, LPA acts in multiple ways on SMCs in vitro, which suggests that LPA promotes vascular remodelling and neointima formation.
To study the role of LPA in vascular remodelling in vivo, short-term incubation of the carotid arteries with LPA has been performed. Intriguingly, transient treatment only with unsaturated LPAs, like LPA18:1, LPA20:4 or the LPA analogue 1-AGP18:1 (1-O-octadecenyl glycerophosphate), triggers the formation within weeks of a neointima that primarily consists of SMCs (Yoshida et al., 2003; Zhang et al., 2004; Cheng et al., 2009; Subramanian et al., 2010). Blocking LPA18:1-induced ERK and p38MAPK activation prevented neointima formation (Yoshida et al., 2003). Furthermore, LPA20:4 and 1-AGP18:1-induced neointima formation was reduced by treatment with a PPARγ antagonist and in mice with PPARγ-deficiency in vascular wall cells (Zhang et al., 2004; Cheng et al., 2009). Although LPA20:4-induced neointima formation was partially inhibited by pertussis toxin and the LPA1/LPA3 antagonist dioctylglycerol pyrophosphate in rats, which do not express LPA3 in the carotid wall, neointimal growth following 1-AGP18:1 incubation was not impaired in LPA1−/−, LPA2−/− and LPA1−/−/LPA2−/− mice (Zhang et al., 2004; Cheng et al., 2009). Neointima formation by carotid ligation is increased in LPA1−/− mice, whereas a combined deficiency of LPA1 and LPA2 diminishes neointimal growth (Panchatcharam et al., 2008). The compensatory overexpression of LPA3 in SMCs from LPA1−/− mice, which is absent in LPA1−/−/LPA2−/− SMCs, is associated with enhanced neointima formation (Panchatcharam et al., 2008). PPARγ activation in SMCs has been shown to inhibit proliferation and migration and attenuates neointimal hyperplasia after vascular injury (Lim et al., 2006; Lee et al., 2009; Zhang et al., 2011). Accordingly, 1-AGP18:1, but not rosiglitazone, increases injury-induced neointima formation, indicating that LPA-mediated PPARγ activation does not play a role in vascular remodelling following vascular injury (Cheng et al., 2009). Taken together, these results show that molecular mechanisms of LPA-dependent neointima formation may differ between local treatment with LPA and in established animal models of vascular remodelling.
In atherosclerosis-prone Apoe−/− mice, treatment with the LPA1/LPA3 antagonist Ki16425 diminished neointimal hyperplasia after carotid wire injury by reducing the SMC and macrophage content in the lesions, indicating that LPA promotes injury-induced neointima formation via LPA receptors (Subramanian et al., 2010). Interestingly, unsaturated but not saturated LPA induces sustained up-regulation of the chemokine CXCL12 (stromal cell-derived factor 1) in the vessel wall, which mobilizes and recruits smooth muscle progenitor cells (SPCs) from the bone marrow into the injured artery (Schober, 2008; Subramanian et al., 2010) (Figure 4). Silencing of either LPA1 or LPA3 in the vessel wall impaired the LPA20:4-induced neointima formation and SPC mobilization, suggesting that, similar to atherogenic monocyte recruitment, these two LPA receptors independently trigger the vascular response (Subramanian et al., 2010). It remains to be determined whether this functional independency might be due to the formation of heterodimers between LPA1 and LPA3, and whether LPA1/LPA3 heterodimers have an increased binding affinity to unsaturated LPAs (Zaslavsky et al., 2006). The different mechanisms that were observed in LPA-induced neointimal hyperplasia might be dose-dependent. Whereas 40 µM of LPA20:4 has been found to trigger CXCL12 expression, unsaturated LPAs in a dosage range between 1 and 10 µM induce ERK1/2/p38MAPK- or PPARγ–dependent neointima formation (Yoshida et al., 2003; Zhang et al., 2004; Cheng et al., 2009; Subramanian et al., 2010). However, the fact that pharmacological inhibition of LPA1 and LPA3 after carotid injury inhibits CXCL12 expression and impairs CXCL12-dependent SPC mobilization indicates that the 40 µM LPA20:4 dose used for local carotid treatment closely mirrors the role of LPA after vascular injury (Subramanian et al., 2010). Microvesicles released from apoptotic SMCs following injury have been found to up-regulate CXCL12 expression in non-injured SMCs, and blocking the apoptosis of SMCs following vascular injury greatly inhibits CXCL12 expression (Zernecke et al., 2005). Taking into account the role of microvesicles in LPA generation (Fourcade et al., 1995), increased LPA production due to the release of microvesicles from apoptotic SMCs may contribute to the up-regulation of CXCL12 in the injured arteries. In addition, activated platelets adhering to the denudated vascular surface may not only present CXCL12 during the recruitment of SPCs, but also increase CXCL12 expression by enhancing LPA formation (Zernecke et al., 2005).
Figure 4.
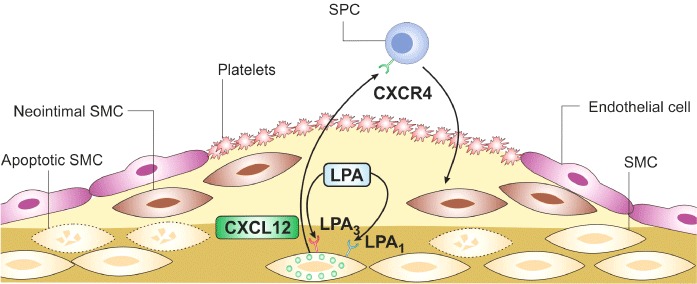
LPA induces neointima formation after vascular injury. Following vascular injury, activated platelets adhere to the denuded surface of the vessel wall and medial SMCs undergo apoptosis. These early events after vascular injury may induce the production of LPA, which increases CXCL12 in the vessel wall through its receptors LPA1 and LPA3. CXCL12 is released into the circulation and recruits SPC via its receptor CXCR4 to the injury site. These SPCs differentiate into neointimal SMCs, which form the neointimal lesion.
Remodelling of the pulmonary vasculature occurs during the hypoxia-driven development of pulmonary hypertension and is characterized by the thickening of the media and adventitia in pulmonary arteries and muscularization of non-muscular alveolar wall vessels (Stenmark et al., 2006). Interestingly, hypoxia-induced pulmonary hypertension and remodelling is accelerated in both ATX+/− mice and LPA1−/−/LPA2−/− mice (Cheng et al., 2012). In LPA1−/−/LPA2−/− mice, thickening of the arteriolar vessel wall is already evident during normoxia, and aging results in severe pulmonary hypertension (Cheng et al., 2012). This increased remodelling of pulmonary vessels was associated with increased expression of genes involved in endothelin-1 signalling (Cheng et al., 2012). In contrast, the deficiency of either LPA1 or LPA2 is not associated with pulmonary hypertension. This indicates that LPA signalling via LPA1 and LPA2 in the pulmonary vasculature has an important homeostatic role and appears to have protective effects in hypoxia-induced pulmonary hypertension.
Conclusions and future directions
LPA has atherogenic actions, and specific LPA receptors have been identified that may mediate some of these harmful LPA activities. The experimental evidence is based not only on numerous in vitro studies, but also on several in vivo studies. LPA does not only enhance atherosclerosis, but also vice versa, cardiovascular risk factors affect the ATX/LPA axis thereby further aggravating the progression of atherosclerotic diseases. Obesity and hyperlipidaemia enhance ATX expression and activity, and the generation of LPA in the circulation. Increased ATX-dependent LPA formation from LDL-derived LPC promotes endothelial chemokine (CXCL1) secretion leading to the recruitment of monocytes to the atherosclerotic lesion, and thereby enhancing atherosclerosis (Figure 3). Endothelial LPA1/3 receptors and Rho-kinase are critical here. LPA also increases the uptake of ox-LDL in monocytes/macrophages by up-regulation of the scavenger receptor A, and inhibits monocyte egression from plaques. LPA may also induce these effects by binding to LPA1/3 and to the nuclear receptor PPARγ. LPA also stimulates VSMC dedifferentiation, proliferation and neointima formation. Also here, LPA1 and LPA3 receptors as well as PPARγ seem to be involved; the situation is, however, complex, as the mechanism of LPA-dependent neointima formation seems to differ between local treatment with LPA and established animal models of vascular remodelling. Lastly but not least, LPA accumulating in the lipid-rich core may contribute to acute thrombus formation after plaque rupture by stimulation of the platelet LPA5 receptor and Rho-kinase (Figures 1, 2). Future studies might be directed to explore the role of the non-Edg receptor family of LPA receptors in various animal models of atherosclerosis, to determine the expression of individual LPA receptors in human atherosclerotic lesions in situ and to evaluate the effects of novel LPA receptor antagonists on the development, progression and regression of atherosclerosis in animal models, as well as on thrombosis induced by atherosclerotic plaques in human models. Clinical studies could determine whether plasma levels of defined LPA molecular species could be a novel biomarker of cardiovascular risk. In such studies, the addition of pharmacological ATX inhibitors to the blood collection tube might be important to minimize artificial in vitro LPA formation during blood handling.
Acknowledgments
This publication was made possible by grants to AS and WS from the Deutsche Forschungsgemeinschaft (Scho 1056/2, Scho 1056/3, Si 274/9; Si 274/11), the August-Lenz-Stiftung and the Bayern University.
Glossary
- APT1
acyl-protein thioesterase 1
- ATX
autotaxin
- CCL2
chemokine ligand 2
- cPA
cyclic phosphatidic acid
- CREB
cAMP response element-binding
- Edg
endothelial differentiation gene
- Egr
early growth response gene
- ESI
electrospray ionization
- ICAM-1
intracellular adhesion molecule-1
- LC
liquid chromatography
- LCAT
lecithin-cholesterol acyltransferase
- LDL
low-density lipoprotein
- LPA
lysophosphatidic acid
- LPC
lysophosphatidylcholine
- LPL
lysophospholipids
- LPP
lipid phosphate phosphatase
- Lp-PLA2
lipoprotein-associated PLA2
- LPS
lysophosphatidylserine
- LYPLA-I
lysophospholipase A-I
- lysoPLD
lysophospholipase D
- MCP-1
monocyte chemotactic protein-1
- MLC
myosin light chain
- MM–LDL
minimally modified LDL
- mox-LDL
mildly oxidized LDL
- MS
mass spectrometry
- NVAF
nonvalvular atrial fibrillation
- PA
phosphatidic acid
- PAF
platelet-activating factor
- PAK
p21-activated kinase
- PC
phosphatidylcholine
- PI3K
phosphoinositide 3-kinase
- PLA
phospholipase A
- ox-LDL
oxidized LDL
- SBI
silent brain infarction
- S1P
sphingosine 1-phosphate
- SMD
somatomedin domain
- SPCs
smooth muscle progenitor cells
- sPLA2
secretory PLA2
- SRF
serum response factor
- TF
tissue factor
- VCAM-1
vascular cell adhesion molecule-1
- VSMC
vascular smooth muscle cells
Statement of conflict of interest
The authors have no conflict of interest.
References
- Adinolfi LE, Restivo L, Zampino R, Guerrera B, Lonardo A, Ruggiero L, et al. Chronic HCV infection is a risk of atherosclerosis. Role of HCV and HCV-related steatosis. Atherosclerosis. 2012;221:496–502. doi: 10.1016/j.atherosclerosis.2012.01.051. [DOI] [PubMed] [Google Scholar]
- Aird WC. Spatial and temporal dynamics of the endothelium. J Thromb Haemost. 2005;3:1392–1406. doi: 10.1111/j.1538-7836.2005.01328.x. [DOI] [PubMed] [Google Scholar]
- Albers HM, Dong A, van Meeteren LA, Egan DA, Sunkara M, van Tilburg EW, et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc Natl Acad Sci U S A. 2010;107:7257–7262. doi: 10.1073/pnas.1001529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MA, Glynn RJ, Wolfert RL, Ridker PM. The effect of statin therapy on lipoprotein associated phospholipase A2 levels. Atherosclerosis. 2005;182:193–198. doi: 10.1016/j.atherosclerosis.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Alexander JS, Patton WF, Christman BW, Cuiper LL, Haselton FR. Platelet-derived lysophosphatidic acid decreases endothelial permeability in vitro. Am J Physiol. 1998;274(1 Pt 2):H115–H122. doi: 10.1152/ajpheart.1998.274.1.H115. [DOI] [PubMed] [Google Scholar]
- Amisten S, Braun OO, Bengtsson A, Erlinge D. Gene expression profiling for the identification of G-protein coupled receptors in human platelets. Thromb Res. 2008;122:47–57. doi: 10.1016/j.thromres.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, et al. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J Biol Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, et al. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1647–1653. doi: 10.1161/01.ATV.0000173413.31789.1a. [DOI] [PubMed] [Google Scholar]
- Baker DL, Desiderio DM, Miller DD, Tolley B, Tigyi GJ. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal Biochem. 2001;292:287–295. doi: 10.1006/abio.2001.5063. [DOI] [PubMed] [Google Scholar]
- Baldini PM, De Vito P, D'Aquilio F, Vismara D, Zalfa F, Bagni C, et al. Role of atrial natriuretic peptide in the suppression of lysophosphatydic acid-induced rat aortic smooth muscle (RASM) cell growth. Mol Cell Biochem. 2005;272:19–28. doi: 10.1007/s11010-005-4779-0. [DOI] [PubMed] [Google Scholar]
- le Balle F, Simon MF, Meijer S, Fourcade O, Chap H. Membrane sidedness of biosynthetic pathways involved in the production of lysophosphatidic acid. Adv Enzyme Regul. 1999;39:275–284. doi: 10.1016/s0065-2571(98)00024-7. [DOI] [PubMed] [Google Scholar]
- Bauer M, Retzer M, Wilde JI, Maschberger P, Essler M, Aepfelbacher M, et al. Dichotomous regulation of myosin phosphorylation and shape change by Rho-kinase and calcium in intact human platelets. Blood. 1999;94:1665–1672. [PubMed] [Google Scholar]
- Benitez S, Sanchez-Quesada JL, Ribas V, Jorba O, Blanco-Vaca F, Gonzalez-Sastre F, et al. Platelet-activating factor acetylhydrolase is mainly associated with electronegative low-density lipoprotein subfraction. Circulation. 2003;108:92–96. doi: 10.1161/01.CIR.0000072791.40232.8F. [DOI] [PubMed] [Google Scholar]
- Berliner JA, Territo MC, Sevanian A, Ramin S, Kim JA, Bamshad B, et al. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clin Invest. 1990;85:1260–1266. doi: 10.1172/JCI114562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billah MM, Lapetina EG, Cuatrecasas P. Phospholipase A2 activity specific for phosphatidic acid. A possible mechanism for the production of arachidonic acid in platelets. J Biol Chem. 1981;256:5399–5403. [PubMed] [Google Scholar]
- Bolen AL, Naren AP, Yarlagadda S, Beranova-Giorgianni S, Chen L, Norman D, et al. The phospholipase A1 activity of lysophospholipase A-I links platelet activation to LPA production during blood coagulation. J Lipid Res. 2011;52:958–970. doi: 10.1194/jlr.M013326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born GV, Wehmeier A. Inhibition of platelet thrombus formation by chlorpromazine acting to diminish haemolysis. Nature. 1979;282:212–213. doi: 10.1038/282212a0. [DOI] [PubMed] [Google Scholar]
- Bot M, Bot I, Lopez-Vales R, van de Lest CHA, Saulnier-Blache JS, Helms JB, et al. Atherosclerotic lesion progression changes lysophosphatidic acid homeostasis to favor its accumulation. Am J Pathol. 2010;176:3073–3084. doi: 10.2353/ajpath.2010.090009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher J, Quilliot D, Praderes JP, Simon MF, Gres S, Guigne C, et al. Potential involvement of adipocyte insulin resistance in obesity-associated up-regulation of adipocyte lysophospholipase D/autotaxin expression. Diabetologia. 2005;48:569–577. doi: 10.1007/s00125-004-1660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Calvayrac O, Rodriguez-Calvo R, Alonso J, Orbe J, Martin-Ventura JL, Guadall A, et al. CCL20 is increased in hypercholesterolemic subjects and is upregulated by LDL in vascular smooth muscle cells: role of NF-kappaB. Arterioscler Thromb Vasc Biol. 2011;31:2733–2741. doi: 10.1161/ATVBAHA.111.235721. [DOI] [PubMed] [Google Scholar]
- Chang CL, Hsu HY, Lin HY, Chiang W, Lee H. Lysophosphatidic acid-induced oxidized low-density lipoprotein uptake is class A scavenger receptor-dependent in macrophages. Prostaglandins Other Lipid Mediat. 2008a;87:20–25. doi: 10.1016/j.prostaglandins.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Chang CL, Lin ME, Hsu HY, Yao CL, Hwang SM, Pan CY, et al. Lysophosphatidic acid-induced interleukin-1 beta expression is mediated through Gi/Rho and the generation of reactive oxygen species in macrophages. J Biomed Sci. 2008b;15:357–363. doi: 10.1007/s11373-007-9223-x. [DOI] [PubMed] [Google Scholar]
- Chappell D, Jacob M, Paul O, Rehm M, Welsch U, Stoeckelhuber M, et al. The glycocalyx of the human umbilical vein endothelial cell: an impressive structure ex vivo but not in culture. Circ Res. 2009;104:1313–1317. doi: 10.1161/CIRCRESAHA.108.187831. [DOI] [PubMed] [Google Scholar]
- Charo IF, Taub R. Anti-inflammatory therapeutics for the treatment of atherosclerosis. Nat Rev Drug Discov. 2011;10:365–376. doi: 10.1038/nrd3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HY, Dong A, Panchatcharam M, Mueller P, Yang F, Li Z, et al. Lysophosphatidic Acid signaling protects pulmonary vasculature from hypoxia-induced remodeling. Arterioscler Thromb Vasc Biol. 2012;32:24–32. doi: 10.1161/ATVBAHA.111.234708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Makarova N, Tsukahara R, Guo H, Shuyu E, Farrar P, et al. Lysophosphatidic acid-induced arterial wall remodeling: requirement of PPARgamma but not LPA1 or LPA2 GPCR. Cell Signal. 2009;21:1874–1884. doi: 10.1016/j.cellsig.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, et al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 2010;50:157–186. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol Rev. 2010;62:579–587. doi: 10.1124/pr.110.003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti R, Badimon JJ. Biologic aspects of vulnerable plaque. Curr Opin Cardiol. 2002;17:616–625. doi: 10.1097/00001573-200211000-00007. [DOI] [PubMed] [Google Scholar]
- Cui MZ, Zhao G, Winokur AL, Laag E, Bydash JR, Penn MS, et al. Lysophosphatidic acid induction of tissue factor expression in aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:224–230. doi: 10.1161/01.atv.0000054660.61191.7d. [DOI] [PubMed] [Google Scholar]
- Cui MZ, Laag E, Sun L, Tan M, Zhao G, Xu X. Lysophosphatidic acid induces early growth response gene 1 expression in vascular smooth muscle cells: CRE and SRE mediate the transcription. Arterioscler Thromb Vasc Biol. 2006;26:1029–1035. doi: 10.1161/01.ATV.0000214980.90567.b5. [DOI] [PubMed] [Google Scholar]
- D'Aquilio F, Procaccini M, Izzi V, Chiurchiu V, Giambra V, Carotenuto F, et al. Activatory properties of lysophosphatidic acid on human THP-1 cells. Inflammation. 2007;30:167–177. doi: 10.1007/s10753-007-9034-2. [DOI] [PubMed] [Google Scholar]
- Damirin A, Tomura H, Komachi M, Liu JP, Mogi C, Tobo M, et al. Role of lipoprotein-associated lysophospholipids in migratory activity of coronary artery smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;292:H2513–H2522. doi: 10.1152/ajpheart.00865.2006. [DOI] [PubMed] [Google Scholar]
- Dohi T, Miyauchi K, Ohkawa R, Nakamura K, Kishimoto T, Miyazaki T, et al. Increased circulating plasma lysophosphatidic acid in patients with acute coronary syndrome. Clin Chim Acta. 2012;413:207–212. doi: 10.1016/j.cca.2011.09.027. [DOI] [PubMed] [Google Scholar]
- Dusaulcy R, Rancoule C, Gres S, Wanecq E, Colom A, Guigne C, et al. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J Lipid Res. 2011;52:1247–1255. doi: 10.1194/jlr.M014985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichholtz T, Jalink K, Fahrenfort I, Moolenaar WH. The bioactive phospholipid lysophosphatidic acid is released from activated platelets. Biochem J. 1993;291((Pt 3)):677–680. doi: 10.1042/bj2910677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Ortiz A, Badimon JJ, Falk E, Fuster V, Meyer B, Mailhac A, et al. Characterization of the relative thrombogenicity of atherosclerotic plaque components: implications for consequences of plaque rupture. J Am Coll Cardiol. 1994;23:1562–1569. doi: 10.1016/0735-1097(94)90657-2. [DOI] [PubMed] [Google Scholar]
- FitzGerald GA, Healy C, Daugherty J. Thromboxane A2 biosynthesis in human disease. Fed Proc. 1987;46:154–158. [PubMed] [Google Scholar]
- Fourcade O, Simon MF, Viode C, Rugani N, Leballe F, Ragab A, et al. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell. 1995;80:919–927. doi: 10.1016/0092-8674(95)90295-3. [DOI] [PubMed] [Google Scholar]
- Fueller M, Wang DA, Tigyi G, Siess W. Activation of human monocytic cells by lysophosphatidic acid and sphingosine-1-phosphate. Cell Signal. 2003;15:367–375. doi: 10.1016/s0898-6568(02)00117-1. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y. Cyclic phosphatidic acid – a unique bioactive phospholipid. Biochim Biophys Acta. 2008;1781:519–524. doi: 10.1016/j.bbalip.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulkerson Z, Wu T, Sunkara M, Kooi CV, Morris AJ, Smyth SS. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J Biol Chem. 2011;286:34654–34663. doi: 10.1074/jbc.M111.276725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol. 2005;46:937–954. doi: 10.1016/j.jacc.2005.03.074. [DOI] [PubMed] [Google Scholar]
- Gaits F, Fourcade O, Le Balle F, Gueguen G, Gaige B, Gassama-Diagne A, et al. Lysophosphatidic acid as a phospholipid mediator: pathways of synthesis. FEBS Lett. 1997;410:54–58. doi: 10.1016/s0014-5793(97)00411-0. [DOI] [PubMed] [Google Scholar]
- Gennero I, Xuereb JM, Simon MF, Girolami JP, Bascands JL, Chap H, et al. Effects of lysophosphatidic acid on proliferation and cytosolic Ca++ of human adult vascular smooth muscle cells in culture. Thromb Res. 1999;94:317–326. doi: 10.1016/s0049-3848(99)00004-3. [DOI] [PubMed] [Google Scholar]
- Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330:1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- Gierse J, Thorarensen A, Beltey K, Bradshaw-Pierce E, Cortes-Burgos L, Hall T, et al. A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J Pharmacol Exp Ther. 2010;334:310–317. doi: 10.1124/jpet.110.165845. [DOI] [PubMed] [Google Scholar]
- Goncalves I, Edsfeldt A, Ko NY, Grufman H, Berg K, Bjorkbacka H, et al. Evidence supporting a key role of Lp-PLA2-generated lysophosphatidylcholine in human atherosclerotic plaque inflammation. Arterioscler Thromb Vasc Biol. 2012;32:1505–1512. doi: 10.1161/ATVBAHA.112.249854. [DOI] [PubMed] [Google Scholar]
- Gouni-Berthold I, Seewald S, Hescheler J, Sachinidis A. Regulation of mitogen-activated protein kinase cascades by low density lipoprotein and lysophosphatidic acid. Cell Physiol Biochem. 2004;14:167–176. doi: 10.1159/000078108. [DOI] [PubMed] [Google Scholar]
- Gratacap MP, Payrastre B, Nieswandt B, Offermanns S. Differential regulation of Rho and Rac through heterotrimeric G-proteins and cyclic nucleotides. J Biol Chem. 2001;276:47906–47913. doi: 10.1074/jbc.M104442200. [DOI] [PubMed] [Google Scholar]
- Guo H, Makarova N, Cheng Y, E S, Ji RR, Zhang C, et al. The early- and late stages in phenotypic modulation of vascular smooth muscle cells: differential roles for lysophosphatidic acid. Biochim Biophys Acta. 2008;1781:571–581. doi: 10.1016/j.bbalip.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte R, Patil R, Liu J, Wang Y, Lee SC, Fujiwara Y, et al. Benzyl and naphthalene methylphosphonic acid inhibitors of autotaxin with anti-invasive and anti-metastatic activity. ChemMedChem. 2011;6:922–935. doi: 10.1002/cmdc.201000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin C, Delaive E, Dieu M, Calay D, Raes M. Upregulation of pentraxin-3 in human endothelial cells after lysophosphatidic acid exposure. Arterioscler Thromb Vasc Biol. 2008a;28:491–497. doi: 10.1161/ATVBAHA.107.158642. [DOI] [PubMed] [Google Scholar]
- Gustin C, Van Steenbrugge M, Raes M. LPA modulates monocyte migration directly and via LPA-stimulated endothelial cells. Am J Physiol Cell Physiol. 2008b;295:C905–C914. doi: 10.1152/ajpcell.00544.2007. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- Hao F, Tan M, Wu DD, Xu X, Cui MZ. LPA induces IL-6 secretion from aortic smooth muscle cells via an LPA1-regulated, PKC-dependent, and p38{alpha}-mediated pathway. Am J Physiol Heart Circ Physiol. 2010;298:H974–H983. doi: 10.1152/ajpheart.00895.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseruck N, Erl W, Pandey D, Tigyi G, Ohlmann P, Ravanat C, et al. The plaque lipid lysophosphatidic acid stimulates platelet activation and platelet-monocyte aggregate formation in whole blood: involvement of P2Y1 and P2Y12 receptors. Blood. 2004;103:2585–2592. doi: 10.1182/blood-2003-04-1127. [DOI] [PubMed] [Google Scholar]
- Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat Struct Mol Biol. 2011;18:198–204. doi: 10.1038/nsmb.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Takahashi M, Nishida W, Yoshida K, Ohkawa Y, Kitabatake A, et al. Phenotypic modulation of vascular smooth muscle cells induced by unsaturated lysophosphatidic acids. Circ Res. 2001;89:251–258. doi: 10.1161/hh1501.094265. [DOI] [PubMed] [Google Scholar]
- Hirakawa M, Oike M, Karashima Y, Ito Y. Sequential activation of RhoA and FAK/paxillin leads to ATP release and actin reorganization in human endothelium. J Physiol. 2004;558((Pt 2)):479–488. doi: 10.1113/jphysiol.2004.065334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase T, Kawashima S, Wong EY, Ueyama T, Rikitake Y, Tsukita S, et al. Regulation of tight junction permeability and occludin phosphorylation by Rhoa-p160ROCK-dependent and -independent mechanisms. J Biol Chem. 2001;276:10423–10431. doi: 10.1074/jbc.M007136200. [DOI] [PubMed] [Google Scholar]
- Holvoet P, Vanhaecke J, Janssens S, Van de Werf F, Collen D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 1998;98:1487–1494. doi: 10.1161/01.cir.98.15.1487. [DOI] [PubMed] [Google Scholar]
- Hoofnagle AN, Heinecke JW. Lipoproteomics: using mass spectrometry-based proteomics to explore the assembly, structure, and function of lipoproteins. J Lipid Res. 2009;50:1967–1975. doi: 10.1194/jlr.R900015-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiga M, Alpers CE, Smith LL, Giachelli CM, Schwartz SM. Alpha-v beta-3 integrin expression in normal and atherosclerotic artery. Circ Res. 1995;77:1129–1135. doi: 10.1161/01.res.77.6.1129. [DOI] [PubMed] [Google Scholar]
- Hosogaya S, Yatomi Y, Nakamura K, Ohkawa R, Okubo S, Yokota H, et al. Measurement of plasma lysophosphatidic acid concentration in healthy subjects: strong correlation with lysophospholipase D activity. Ann Clin Biochem. 2008;45:364–368. doi: 10.1258/acb.2008.007242. [DOI] [PubMed] [Google Scholar]
- Huo Y, Weber C, Forlow SB, Sperandio M, Thatte J, Mack M, et al. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest. 2001;108:1307–1314. doi: 10.1172/JCI12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneyuki U, Ueda S, Yamagishi S, Kato S, Fujimura T, Shibata R, et al. Pitavastatin inhibits lysophosphatidic acid-induced proliferation and monocyte chemoattractant protein-1 expression in aortic smooth muscle cells by suppressing Rac-1-mediated reactive oxygen species generation. Vascul Pharmacol. 2007;46:286–292. doi: 10.1016/j.vph.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Khachigian LM. Early growth response-1 in cardiovascular pathobiology. Circ Res. 2006;98:186–191. doi: 10.1161/01.RES.0000200177.53882.c3. [DOI] [PubMed] [Google Scholar]
- Khandoga AL, Fujiwara Y, Goyal P, Pandey D, Tsukahara R, Bolen A, et al. Lysophosphatidic acid-induced platelet shape change revealed through LPA(1-5) receptor-selective probes and albumin. Platelets. 2008;19:415–427. doi: 10.1080/09537100802220468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandoga AL, Pandey D, Welsch U, Brandl R, Siess W. GPR92/LPA lysophosphatidate receptor mediates megakaryocytic cell shape change induced by human atherosclerotic plaques. Cardiovasc Res. 2011;90:157–164. doi: 10.1093/cvr/cvq369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Keys JR, Eckhart AD. Vascular smooth muscle migration and proliferation in response to lysophosphatidic acid (LPA) is mediated by LPA receptors coupling to Gq. Cell Signal. 2006;18:1695–1701. doi: 10.1016/j.cellsig.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Matsuoka T, Imamura S, Mizuno K. A novel colorimetric assay for the determination of lysophosphatidic acid in plasma using an enzymatic cycling method. Clin Chim Acta. 2003;333:59–67. doi: 10.1016/s0009-8981(03)00165-7. [DOI] [PubMed] [Google Scholar]
- Klages B, Brandt U, Simon MI, Schultz G, Offermanns S. Activation of G12/G13 results in shape change and Rho/Rho-kinase-mediated myosin light chain phosphorylation in mouse platelets. J Cell Biol. 1999;144:745–754. doi: 10.1083/jcb.144.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Tanaka-Ishii R, Taguchi R, Ikezawa H, Murakami-Murofushi K. Existence of a bioactive lipid, cyclic phosphatidic acid, bound to human serum albumin. Life Sci. 1999;65:2185–2191. doi: 10.1016/s0024-3205(99)00483-x. [DOI] [PubMed] [Google Scholar]
- Koenig W, Khuseyinova N. Lipoprotein-associated and secretory phospholipase A2 in cardiovascular disease: the epidemiological evidence. Cardiovasc Drugs Ther. 2009;23:85–92. doi: 10.1007/s10557-008-6135-6. [DOI] [PubMed] [Google Scholar]
- Koh JS, Lieberthal W, Heydrick S, Levine JS. Lysophosphatidic acid is a major serum noncytokine survival factor for murine macrophages which acts via the phosphatidylinositol 3-kinase signaling pathway. J Clin Invest. 1998;102:716–727. doi: 10.1172/JCI1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodgie FD, Burke AP, Wight TN, Virmani R. The accumulation of specific types of proteoglycans in eroded plaques: a role in coronary thrombosis in the absence of rupture. Curr Opin Lipidol. 2004;15:575–582. doi: 10.1097/00041433-200410000-00012. [DOI] [PubMed] [Google Scholar]
- Komachi M, Damirin A, Malchinkhuu E, Mogi C, Tobo M, Ohta H, et al. Signaling pathways involved in DNA synthesis and migration in response to lysophosphatidic acid and low-density lipoprotein in coronary artery smooth muscle cells. Vascul Pharmacol. 2009;50:178–184. doi: 10.1016/j.vph.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Lee CS, Kwon YW, Yang HM, Kim SH, Kim TY, Hur J, et al. New mechanism of rosiglitazone to reduce neointimal hyperplasia: activation of glycogen synthase kinase-3beta followed by inhibition of MMP-9. Arterioscler Thromb Vasc Biol. 2009;29:472–479. doi: 10.1161/ATVBAHA.108.176230. [DOI] [PubMed] [Google Scholar]
- Lee H, Lin CI, Liao J-J LY-W, Yang HY, Lee C-Y, et al. Lysophospholipids increase ICAM-1 expression in HUVEC through a Gi- and NF-{kappa}B-dependent mechanism. Am J Physiol Cell Physiol. 2004;287:C1657–C1666. doi: 10.1152/ajpcell.00172.2004. [DOI] [PubMed] [Google Scholar]
- Levitan I, Volkov S, Subbaiah PV. Oxidized LDL: diversity, patterns of recognition, and pathophysiology. Antioxid Redox Signal. 2010;13:39–75. doi: 10.1089/ars.2009.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZG, Yu ZC, Wang DZ, Ju WP, Zhan X, Wu QZ, et al. Influence of acetylsalicylate on plasma lysophosphatidic acid level in patients with ischemic cerebral vascular diseases. Neurol Res. 2008;30:366–369. doi: 10.1179/174313208X300369. [DOI] [PubMed] [Google Scholar]
- Li ZG, Yu ZC, Yu YP, Ju WP, Wang DZ, Zhan X, et al. Lysophosphatidic acid level and the incidence of silent brain infarction in patients with nonvalvular atrial fibrillation. Int J Mol Sci. 2010;11:3988–3998. doi: 10.3390/ijms11103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- Liebisch G, Scherer M. Quantification of bioactive sphingo- and glycerophospholipid species by electrospray ionization tandem mass spectrometry in blood. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;883–884:141–146. doi: 10.1016/j.jchromb.2011.10.037. [DOI] [PubMed] [Google Scholar]
- Lim S, Jin CJ, Kim M, Chung SS, Park HS, Lee IK, et al. PPARgamma gene transfer sustains apoptosis, inhibits vascular smooth muscle cell proliferation, and reduces neointima formation after balloon injury in rats. Arterioscler Thromb Vasc Biol. 2006;26:808–813. doi: 10.1161/01.ATV.0000204634.26163.a7. [DOI] [PubMed] [Google Scholar]
- Lin CI, Chen CN, Chen JH, Lee H. Lysophospholipids increase IL-8 and MCP-1 expressions in human umbilical cord vein endothelial cells through an IL-1-dependent mechanism. J Cell Biochem. 2006;99:1216–1232. doi: 10.1002/jcb.20963. [DOI] [PubMed] [Google Scholar]
- Lin CI, Chen CN, Lin PW, Chang KJ, Hsieh FJ, Lee H. Lysophosphatidic acid regulates inflammation-related genes in human endothelial cells through LPA1 and LPA3. Biochem Biophys Res Commun. 2007;363:1001–1008. doi: 10.1016/j.bbrc.2007.09.081. [DOI] [PubMed] [Google Scholar]
- Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779–11784. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell JP, Hoefner DM. Lipoprotein-associated phospholipase A2. Clin Lab Med. 2006;26:679–697. doi: 10.1016/j.cll.2006.06.003. vii. [DOI] [PubMed] [Google Scholar]
- McIntyre TM, Pontsler AV, Silva AR, St Hilaire A, Xu Y, Hinshaw JC, et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc Natl Acad Sci U S A. 2003;100:131–136. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschberger P, Bauer M, Baumann-Siemons J, Zangl KJ, Negrescu EV, Reininger AJ, et al. Mildly oxidized low density lipoprotein rapidly stimulates via activation of the lysophosphatidic acid receptor Src family and Syk tyrosine kinases and Ca2+ influx in human platelets. J Biol Chem. 2000;275:19159–19166. doi: 10.1074/jbc.M910257199. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Kobayashi T, Kamata K. Role of lysophosphatidylcholine (LPC) in atherosclerosis. Curr Med Chem. 2007;14:3209–3220. doi: 10.2174/092986707782793899. [DOI] [PubMed] [Google Scholar]
- van Meeteren LA, Moolenaar WH. Regulation and biological activities of the autotaxin-LPA axis. Prog Lipid Res. 2007;46:145–160. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradere JP, et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol. 2006;26:5015–5022. doi: 10.1128/MCB.02419-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnear FL, Patil S, Bell D, Gainor JP, Morton CA. Platelet lipid(s) bound to albumin increases endothelial electrical resistance: mimicked by LPA. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1337–L1344. doi: 10.1152/ajplung.2001.281.6.L1337. [DOI] [PubMed] [Google Scholar]
- Moers A, Nieswandt B, Massberg S, Wettschureck N, Gruner S, Konrad I, et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9:1418–1422. doi: 10.1038/nm943. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AJ, Panchatcharam M, Cheng HY, Federico L, Fulkerson Z, Selim S, et al. Regulation of blood and vascular cell function by bioactive lysophospholipids. J Thromb Haemost. 2009;7(Suppl 1):38–43. doi: 10.1111/j.1538-7836.2009.03405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kishimoto T, Ohkawa R, Okubo S, Tozuka M, Yokota H, et al. Suppression of lysophosphatidic acid and lysophosphatidylcholine formation in the plasma in vitro: proposal of a plasma sample preparation method for laboratory testing of these lipids. Anal Biochem. 2007;367:20–27. doi: 10.1016/j.ab.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Igarashi K, Ide K, Ohkawa R, Okubo S, Yokota H, et al. Validation of an autotaxin enzyme immunoassay in human serum samples and its application to hypoalbuminemia differentiation. Clin Chim Acta. 2008;388:51–58. doi: 10.1016/j.cca.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Nakanaga K, Hama K, Aoki J. Autotaxin – an LPA producing enzyme with diverse functions. J Biochem. 2010;148:13–24. doi: 10.1093/jb/mvq052. [DOI] [PubMed] [Google Scholar]
- Natarajan V, Scribner WM, Hart CM, Parthasarathy S. Oxidized low density lipoprotein-mediated activation of phospholipase D in smooth muscle cells: a possible role in cell proliferation and atherogenesis. J Lipid Res. 1995;36:2005–2016. [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, Vermeer MA, van Hinsbergh VW. Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol. 2000;20:E127–E133. doi: 10.1161/01.atv.20.12.e127. [DOI] [PubMed] [Google Scholar]
- Nishimasu H, Okudaira S, Hama K, Mihara E, Dohmae N, Inoue A, et al. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat Struct Mol Biol. 2011;18:205–212. doi: 10.1038/nsmb.1998. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Oynebraten I. Rapid chemokine secretion from endothelial cells originates from 2 distinct compartments. Blood. 2004;104:314–320. doi: 10.1182/blood-2003-08-2891. [DOI] [PubMed] [Google Scholar]
- Packard CJ, O'Reilly DS, Caslake MJ, McMahon AD, Ford I, Cooney J, et al. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. West of Scotland Coronary Prevention Study Group. N Engl J Med. 2000;343:1148–1155. doi: 10.1056/NEJM200010193431603. [DOI] [PubMed] [Google Scholar]
- Palmetshofer A, Robson SC, Nehls V. Lysophosphatidic acid activates nuclear factor kappa B and induces proinflammatory gene expression in endothelial cells. Thromb Haemost. 1999;82:1532–1537. [PubMed] [Google Scholar]
- Pamuklar Z, Lee JS, Cheng HY, Panchatcharam M, Steinhubl S, Morris AJ, et al. Individual heterogeneity in platelet response to lysophosphatidic acid: evidence for a novel inhibitory pathway. Arterioscler Thromb Vasc Biol. 2008;28:555–561. doi: 10.1161/ATVBAHA.107.151837. [DOI] [PubMed] [Google Scholar]
- Pamuklar Z, Federico L, Liu S, Umezu-Goto M, Dong A, Panchatcharam M, et al. Autotaxin/lysopholipase D and lysophosphatidic acid regulate murine hemostasis and thrombosis. J Biol Chem. 2009;284:7385–7394. doi: 10.1074/jbc.M807820200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchatcharam M, Miriyala S, Yang F, Rojas M, End C, Vallant C, et al. Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ Res. 2008;103:662–670. doi: 10.1161/CIRCRESAHA.108.180778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey D, Goyal P, Bamburg JR, Siess W. Regulation of LIM-kinase 1 and cofilin in thrombin-stimulated platelets. Blood. 2006;107:575–583. doi: 10.1182/blood-2004-11-4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey D, Goyal P, Siess W. Lysophosphatidic acid stimulation of platelets rapidly induces Ca2+-dependent dephosphorylation of cofilin that is independent of dense granule secretion and aggregation. Blood Cells Mol Dis. 2007;38:269–279. doi: 10.1016/j.bcmd.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Panetti TS, Nowlen J, Mosher DF. Sphingosine-1-phosphate and lysophosphatidic acid stimulate endothelial cell migration. Arterioscler Thromb Vasc Biol. 2000;20:1013–1019. doi: 10.1161/01.atv.20.4.1013. [DOI] [PubMed] [Google Scholar]
- Panetti TS, Hannah DF, Avraamides C, Gaughan JP, Marcinkiewicz C, Huttenlocher A, et al. Extracellular matrix molecules regulate endothelial cell migration stimulated by lysophosphatidic acid. J Thromb Haemost. 2004;2:1645–1656. doi: 10.1111/j.1538-7836.2004.00902.x. [DOI] [PubMed] [Google Scholar]
- Penz S, Reininger AJ, Brandl R, Goyal P, Rabie T, Bernlochner I, et al. Human atheromatous plaques stimulate thrombus formation by activating platelet glycoprotein VI. FASEB J. 2005;19:898–909. doi: 10.1096/fj.04-2748com. [DOI] [PubMed] [Google Scholar]
- Penz SM, Reininger AJ, Toth O, Deckmyn H, Brandl R, Siess W. Glycoprotein Ibalpha inhibition and ADP receptor antagonists, but not aspirin, reduce platelet thrombus formation in flowing blood exposed to atherosclerotic plaques. Thromb Haemost. 2007;97:435–443. [PubMed] [Google Scholar]
- Petta S, Torres D, Fazio G, Camma C, Cabibi D, Di Marco V, et al. Carotid atherosclerosis and chronic hepatitis C: a prospective study of risk associations. Hepatology. 2011;55:1317–1323. doi: 10.1002/hep.25508. [DOI] [PubMed] [Google Scholar]
- Portman OW, Soltys P, Alexander M, Osuga T. Metabolism of lysolecithin in vivo: effects of hyperlipemia and atherosclerosis in squirrel monkeys. J Lipid Res. 1970;11:596–604. [PubMed] [Google Scholar]
- Ptaszynska MM, Pendrak ML, Stracke ML, Roberts DD. Autotaxin signaling via lysophosphatidic acid receptors contributes to vascular endothelial growth factor-induced endothelial cell migration. Mol Cancer Res. 2010;8:309–321. doi: 10.1158/1541-7786.MCR-09-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purnak T, Efe C, Beyazit Y, Ozaslan E, Astan R, Milanloglu A, et al. Recent insights into the relationship between inflammatory liver diseases and atherosclerosis. J Investig Med. 2011;59:904–911. doi: 10.2310/JIM.0b013e318217f3a0. [DOI] [PubMed] [Google Scholar]
- Reininger AJ, Bernlochner I, Penz SM, Ravanat C, Smethurst P, Farndale RW, et al. A 2-step mechanism of arterial thrombus formation induced by human atherosclerotic plaques. J Am Coll Cardiol. 2010;55:1147–1158. doi: 10.1016/j.jacc.2009.11.051. [DOI] [PubMed] [Google Scholar]
- Retzer M, Essler M. Lysophosphatidic acid-induced platelet shape change proceeds via Rho/Rho kinase-mediated myosin light-chain and moesin phosphorylation. Cell Signal. 2000;12:645–648. doi: 10.1016/s0898-6568(00)00108-x. [DOI] [PubMed] [Google Scholar]
- Rizza C, Leitinger N, Yue J, Fischer DJ, Wang DA, Shih PT, et al. Lysophosphatidic acid as a regulator of endothelial/leukocyte interaction. Lab Invest. 1999;79:1227–1235. [PubMed] [Google Scholar]
- Rosenson RS. Phospholipase A2 inhibition and atherosclerotic vascular disease: prospects for targeting secretory and lipoprotein-associated phospholipase A2 enzymes. Curr Opin Lipidol. 2010;21:473–480. doi: 10.1097/MOL.0b013e32833eb581. [DOI] [PubMed] [Google Scholar]
- Rother E, Brandl R, Baker DL, Goyal P, Gebhard H, Tigyi G, et al. Subtype-selective antagonists of lysophosphatidic acid receptors inhibit platelet activation triggered by the lipid core of atherosclerotic plaques. Circulation. 2003;108:741–747. doi: 10.1161/01.CIR.0000083715.37658.C4. [DOI] [PubMed] [Google Scholar]
- Samadi N, Bekele R, Capatos D, Venkatraman G, Sariahmetoglu M, Brindley DN. Regulation of lysophosphatidate signaling by autotaxin and lipid phosphate phosphatases with respect to tumor progression, angiogenesis, metastasis and chemo-resistance. Biochimie. 2011;93:61–70. doi: 10.1016/j.biochi.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Sanchez-Quesada JL, Benitez S, Otal C, Franco M, Blanco-Vaca F, Ordonez-Llanos J. Density distribution of electronegative LDL in normolipemic and hyperlipemic subjects. J Lipid Res. 2002;43:699–705. [PubMed] [Google Scholar]
- Sanchez-Quesada JL, Camacho M, Anton R, Benitez S, Vila L, Ordonez-Llanos J. Electronegative LDL of FH subjects: chemical characterization and induction of chemokine release from human endothelial cells. Atherosclerosis. 2003;166:261–270. doi: 10.1016/s0021-9150(02)00374-x. [DOI] [PubMed] [Google Scholar]
- Sano T, Baker D, Virag T, Wada A, Yatomi Y, Kobayashi T, et al. Multiple mechanisms linked to platelet activation result in lysophosphatidic acid and sphingosine 1-phosphate generation in blood. J Biol Chem. 2002;277:21197–21206. doi: 10.1074/jbc.M201289200. [DOI] [PubMed] [Google Scholar]
- Sarker MH, Hu DE, Fraser PA. Regulation of cerebromicrovascular permeability by lysophosphatidic acid. Microcirculation. 2010;17:39–46. doi: 10.1111/j.1549-8719.2010.00001.x. [DOI] [PubMed] [Google Scholar]
- Saulnier-Blache JS, Girard A, Simon MF, Lafontan M, Valet P. A simple and highly sensitive radioenzymatic assay for lysophosphatidic acid quantification. J Lipid Res. 2000;41:1947–1951. [PMC free article] [PubMed] [Google Scholar]
- Schmitz G, Ruebsaamen K. Metabolism and atherogenic disease association of lysophosphatidylcholine. Atherosclerosis. 2010;208:10–18. doi: 10.1016/j.atherosclerosis.2009.05.029. [DOI] [PubMed] [Google Scholar]
- Schmitz U, Thommes K, Beier I, Vetter H. Lysophosphatidic acid stimulates p21-activated kinase in vascular smooth muscle cells. Biochem Biophys Res Commun. 2002;291:687–691. doi: 10.1006/bbrc.2002.6493. [DOI] [PubMed] [Google Scholar]
- Schober A. Chemokines in vascular dysfunction and remodeling. Arterioscler Thromb Vasc Biol. 2008;28:1950–1959. doi: 10.1161/ATVBAHA.107.161224. [DOI] [PubMed] [Google Scholar]
- Schulz C, Penz S, Hoffmann C, Langer H, Gillitzer A, Schneider S, et al. Platelet GPVI binds to collagenous structures in the core region of human atheromatous plaque and is critical for atheroprogression in vivo. Basic Res Cardiol. 2008;103:356–367. doi: 10.1007/s00395-008-0722-3. [DOI] [PubMed] [Google Scholar]
- Schumacher KA, Classen HG, Spath M. Platelet aggregation evoked in vitro and in vivo by phosphatidic acids and lysoderivatives: identity with substances in aged serum (DAS) Thromb Haemost. 1979;42:631–640. [PubMed] [Google Scholar]
- Schwartz D, Andalibi A, Chaverri-Almada L, Berliner JA, Kirchgessner T, Fang ZT, et al. Role of the GRO family of chemokines in monocyte adhesion to MM-LDL-stimulated endothelium. J Clin Invest. 1994;94:1968–1973. doi: 10.1172/JCI117548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz SM, deBlois D, O'Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–465. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- Seewald S, Sachinidis A, Dusing R, Ko Y, Seul C, Epping P, et al. Lysophosphatidic acid and intracellular signalling in vascular smooth muscle cells. Atherosclerosis. 1997;130:121–131. doi: 10.1016/s0021-9150(96)06055-8. [DOI] [PubMed] [Google Scholar]
- Seewald S, Schmitz U, Seul C, Ko Y, Sachinidis A, Vetter H. Lysophosphatidic acid stimulates protein kinase C isoforms alpha, beta, epsilon, and zeta in a pertussis toxin sensitive pathway in vascular smooth muscle cells. Am J Hypertens. 1999;12:532–537. doi: 10.1016/s0895-7061(98)00269-6. [DOI] [PubMed] [Google Scholar]
- Shan L, Jaffe K, Li S, Davis L. Quantitative determination of lysophosphatidic acid by LC/ESI/MS/MS employing a reversed phase HPLC column. J Chromatogr B Analyt Technol Biomed Life Sci. 2008a;864:22–28. doi: 10.1016/j.jchromb.2008.01.031. [DOI] [PubMed] [Google Scholar]
- Shan L, Li S, Jaffe K, Davis L. Quantitative determination of cyclic phosphatidic acid in human serum by LC/ESI/MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2008b;862:161–167. doi: 10.1016/j.jchromb.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Siess W. Athero- and thrombogenic actions of lysophosphatidic acid and sphingosine-1-phosphate. Biochim Biophys Acta. 2002;1582:204–215. doi: 10.1016/s1388-1981(02)00173-7. [DOI] [PubMed] [Google Scholar]
- Siess W. Platelet interaction with bioactive lipids formed by mild oxidation of low-density lipoprotein. Pathophysiol Haemost Thromb. 2006;35:292–304. doi: 10.1159/000093222. [DOI] [PubMed] [Google Scholar]
- Siess W, Tigyi G. Thrombogenic and atherogenic activities of lysophosphatidic acid. J Cell Biochem. 2004;92:1086–1094. doi: 10.1002/jcb.20108. [DOI] [PubMed] [Google Scholar]
- Siess W, Zangl KJ, Essler M, Bauer M, Brandl R, Corrinth C, et al. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc Natl Acad Sci U S A. 1999;96:6931–6936. doi: 10.1073/pnas.96.12.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MF, Chap H, Douste-Blazy L. Human platelet aggregation induced by 1-alkyl-lysophosphatidic acid and its analogs: a new group of phospholipid mediators? Biochem Biophys Res Commun. 1982;108:1743–1750. doi: 10.1016/s0006-291x(82)80113-7. [DOI] [PubMed] [Google Scholar]
- Smyth SS, Cheng HY, Miriyala S, Panchatcharam M, Morris AJ. Roles of lysophosphatidic acid in cardiovascular physiology and disease. Biochim Biophys Acta. 2008;1781:563–570. doi: 10.1016/j.bbalip.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafforini DM. Biology of platelet-activating factor acetylhydrolase (PAF-AH, lipoprotein associated phospholipase A2) Cardiovasc Drugs Ther. 2009;23:73–83. doi: 10.1007/s10557-008-6133-8. [DOI] [PubMed] [Google Scholar]
- Steinbrecher UP, Parthasarathy S, Leake DS, Witztum JL, Steinberg D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc Natl Acad Sci U S A. 1984;81:3883–3887. doi: 10.1073/pnas.81.12.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- Subramanian P, Karshovska E, Reinhard P, Megens RTA, Zhou Z, Akhtar S, et al. Lysophosphatidic acid receptors LPA1 and LPA3 promote CXCL12-mediated smooth muscle progenitor cell recruitment in neointima formation. Circ Res. 2010;107:96–105. doi: 10.1161/CIRCRESAHA.109.212647. [DOI] [PubMed] [Google Scholar]
- Swirski FK. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103:10340–10345. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M, et al. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem. 2006;281:25822–25830. doi: 10.1074/jbc.M605142200. [DOI] [PubMed] [Google Scholar]
- Thim T, Hagensen MK, Bentzon JF, Falk E. From vulnerable plaque to atherothrombosis. J Intern Med. 2008;263:506–516. doi: 10.1111/j.1365-2796.2008.01947.x. [DOI] [PubMed] [Google Scholar]
- Thompson A, Gao P, Orfei L, Watson S, Di Angelantonio E, Kaptoge S, et al. Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 2010;375:1536–1544. doi: 10.1016/S0140-6736(10)60319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumser AE, Voysey JE, Wilton DC. The binding of lysophospholipids to rat liver fatty acid-binding protein and albumin. Biochem J. 1994;301((Pt 3)):801–806. doi: 10.1042/bj3010801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigyi G. Physiological responses to lysophosphatidic acid and related glycero-phospholipids. Prostaglandins Other Lipid Mediat. 2001;64:47–62. doi: 10.1016/s0090-6980(01)00107-1. [DOI] [PubMed] [Google Scholar]
- Tigyi G. Aiming drug discovery at lysophosphatidic acid targets. Br J Pharmacol. 2010;161:241–270. doi: 10.1111/j.1476-5381.2010.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigyi G, Miledi R. Lysophosphatidates bound to serum albumin activate membrane currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J Biol Chem. 1992;267:21360–21367. [PubMed] [Google Scholar]
- Tigyi G, Henschen A, Miledi R. A factor that activates oscillatory chloride currents in Xenopus oocytes copurifies with a subfraction of serum albumin. J Biol Chem. 1991;266:20602–20609. [PubMed] [Google Scholar]
- Tokumura A. A family of phospholipid autacoids: occurrence, metabolism and bioactions. Prog Lipid Res. 1995;34:151–184. doi: 10.1016/0163-7827(95)00001-g. [DOI] [PubMed] [Google Scholar]
- Tokumura A, Fukuzawa K, Isobe J, Tsukatani H. Lysophosphatidic acid-induced aggregation of human and feline platelets: structure-activity relationship. Biochem Biophys Res Commun. 1981;99:391–398. doi: 10.1016/0006-291x(81)91758-7. [DOI] [PubMed] [Google Scholar]
- Tokumura A, Iimori M, Nishioka Y, Kitahara M, Sakashita M, Tanaka S. Lysophosphatidic acids induce proliferation of cultured vascular smooth muscle cells from rat aorta. Am J Physiol. 1994;267(1 Pt 1):C204–C210. doi: 10.1152/ajpcell.1994.267.1.C204. [DOI] [PubMed] [Google Scholar]
- Tokumura A, Kanaya Y, Kitahara M, Miyake M, Yoshioka Y, Fukuzawa K. Increased formation of lysophosphatidic acids by lysophospholipase D in serum of hypercholesterolemic rabbits. J Lipid Res. 2002a;43:307–315. [PubMed] [Google Scholar]
- Tokumura A, Kanaya Y, Miyake M, Yamano S, Irahara M, Fukuzawa K. Increased production of bioactive lysophosphatidic acid by serum lysophospholipase D in human pregnancy. Biol Reprod. 2002b;67:1386–1392. doi: 10.1095/biolreprod.102.004051. [DOI] [PubMed] [Google Scholar]
- Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, et al. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem. 2002c;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- Tokumura A, Sinomiya J, Kishimoto S, Tanaka T, Kogure K, Sugiura T, et al. Human platelets respond differentially to lysophosphatidic acids having a highly unsaturated fatty acyl group and alkyl ether-linked lysophosphatidic acids. Biochem J. 2002d;365((Pt 3)):617–628. doi: 10.1042/BJ20020348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomsig JL, Snyder AH, Berdyshev EV, Skobeleva A, Mataya C, Natarajan V, et al. Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem J. 2009;419:611–618. doi: 10.1042/BJ20081888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toschi V, Gallo R, Lettino M, Fallon JT, Gertz SD, Fernandez-Ortiz A, et al. Tissue factor modulates the thrombogenicity of human atherosclerotic plaques. Circulation. 1997;95:594–599. doi: 10.1161/01.cir.95.3.594. [DOI] [PubMed] [Google Scholar]
- Tsuda S, Okudaira S, Moriya-Ito K, Shimamoto C, Tanaka M, Aoki J, et al. Cyclic phosphatidic acid is produced by autotaxin in blood. J Biol Chem. 2006;281:26081–26088. doi: 10.1074/jbc.M602925200. [DOI] [PubMed] [Google Scholar]
- Tsukahara T, Tsukahara R, Fujiwara Y, Yue J, Cheng Y, Guo H, et al. Phospholipase D2-dependent inhibition of the nuclear hormone receptor PPARgamma by cyclic phosphatidic acid. Mol Cell. 2010;39:421–432. doi: 10.1016/j.molcel.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Zhang J, Wu WY, Li J, Ma YL, Chen WH, et al. Inhibition of lipoprotein-associated phospholipase A2 ameliorates inflammation and decreases atherosclerotic plaque formation in ApoE-deficient mice. PLoS ONE. 2011;6:e23425. doi: 10.1371/journal.pone.0023425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Ikeda H, Nakamura K, Ohkawa R, Kume Y, Aoki J, et al. Both plasma lysophosphatidic acid and serum autotaxin levels are increased in chronic hepatitis C. J Clin Gastroenterol. 2007a;41:616–623. doi: 10.1097/01.mcg.0000225642.90898.0e. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Ikeda H, Nakamura K, Ohkawa R, Kume Y, Tomiya T, et al. Plasma lysophosphatidic acid level and serum autotaxin activity are increased in liver injury in rats in relation to its severity. Life Sci. 2007b;81:1009–1015. doi: 10.1016/j.lfs.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Watson SP, McConnell RT, Lapetina EG. Decanoyl lysophosphatidic acid induces platelet aggregation through an extracellular action. Evidence against a second messenger role for lysophosphatidic acid. Biochem J. 1985;232:61–66. doi: 10.1042/bj2320061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- Weber KS, von Hundelshausen P, Clark-Lewis I, Weber PC, Weber C. Differential immobilization and hierarchical involvement of chemokines in monocyte arrest and transmigration on inflamed endothelium in shear flow. Eur J Immunol. 1999;29:700–712. doi: 10.1002/(SICI)1521-4141(199902)29:02<700::AID-IMMU700>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Wiesner P, Leidl K, Boettcher A, Schmitz G, Liebisch G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J Lipid Res. 2009;50:574–585. doi: 10.1194/jlr.D800028-JLR200. [DOI] [PubMed] [Google Scholar]
- Williams JR, Khandoga AL, Goyal P, Fells JI, Perygin DH, Siess W, et al. Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate receptor indicates role in human platelet activation. J Biol Chem. 2009;284:17304–17319. doi: 10.1074/jbc.M109.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YJ, Rathi SS, Chapman DC, Arneja AS, Dhalla NS. Mechanisms of lysophosphatidic acid-induced DNA synthesis in vascular smooth muscle cells. J Cardiovasc Pharmacol. 2003;41:381–387. doi: 10.1097/00005344-200303000-00006. [DOI] [PubMed] [Google Scholar]
- Yanagida K, Ishii S. Non-Edg family LPA receptors: the cutting edge of LPA research. J Biochem. 2011;150:223–232. doi: 10.1093/jb/mvr087. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Nishida W, Hayashi K, Ohkawa Y, Ogawa A, Aoki J, et al. Vascular remodeling induced by naturally occurring unsaturated lysophosphatidic acid in vivo. Circulation. 2003;108:1746–1752. doi: 10.1161/01.CIR.0000089374.35455.F3. [DOI] [PubMed] [Google Scholar]
- van Zanten GH, de Graaf S, Slootweg PJ, Heijnen HF, Connolly TM, de Groot PG, et al. Increased platelet deposition on atherosclerotic coronary arteries. J Clin Invest. 1994;93:615–632. doi: 10.1172/JCI117014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslavsky A, Singh LS, Tan H, Ding H, Liang Z, Xu Y. Homo- and hetero-dimerization of LPA/S1P receptors, OGR1 and GPR4. Biochim Biophys Acta. 2006;1761:1200–1212. doi: 10.1016/j.bbalip.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Zeiffer U, Schober A, Lietz M, Liehn EA, Erl W, Emans N, et al. Neointimal smooth muscle cells display a proinflammatory phenotype resulting in increased leukocyte recruitment mediated by P-selectin and chemokines. Circ Res. 2004;94:776–784. doi: 10.1161/01.RES.0000121105.72718.5C. [DOI] [PubMed] [Google Scholar]
- Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Mopps B, et al. SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res. 2005;96:784–791. doi: 10.1161/01.RES.0000162100.52009.38. [DOI] [PubMed] [Google Scholar]
- Zhang C, Baker DL, Yasuda S, Makarova N, Balazs L, Johnson LR, et al. Lysophosphatidic acid induces neointima formation through PPARgamma activation. J Exp Med. 2004;199:763–774. doi: 10.1084/jem.20031619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LL, Gao CY, Fang CQ, Wang YJ, Gao D, Yao GE, et al. PPARgamma attenuates intimal hyperplasia by inhibiting TLR4-mediated inflammation in vascular smooth muscle cells. Cardiovasc Res. 2011;92:484–493. doi: 10.1093/cvr/cvr238. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Xu Y. Measurement of endogenous lysophosphatidic acid by ESI-MS/MS in plasma samples requires pre-separation of lysophosphatidylcholine. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3739–3742. doi: 10.1016/j.jchromb.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Luini W, Bernasconi S, Diomede L, Salmona M, Mantovani A, et al. Phosphatidic acid and lysophosphatidic acid induce haptotactic migration of human monocytes. J Biol Chem. 1995;270:25549–25556. doi: 10.1074/jbc.270.43.25549. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Niu J, Zhang Z. The role of lysophosphatidic acid receptors in phenotypic modulation of vascular smooth muscle cells. Mol Biol Rep. 2010;37:2675–2686. doi: 10.1007/s11033-009-9798-6. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Subramanian P, Sevilmis G, Globke B, Soehnlein O, Karshovska E, et al. Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 2011;13:592–600. doi: 10.1016/j.cmet.2011.02.016. [DOI] [PubMed] [Google Scholar]
- Zhou ZB, Niu JP, Zhang ZJ. Receptor-mediated vascular smooth muscle migration induced by LPA involves p38 mitogen-activated protein kinase pathway activation. Int J Mol Sci. 2009;10:3194–3208. doi: 10.3390/ijms10073194. [DOI] [PMC free article] [PubMed] [Google Scholar]