

Abstract
BACKGROUND AND PURPOSE
Ca2+ leak from the sarcoplasmic reticulum (SR) via ryanodine receptors (RyR2s) contributes to cardiomyocyte dysfunction. RyR2 Ca2+ leak has been related to RyR2 phosphorylation. In these conditions, JTV519 (K201), a 1,4-benzothiazepine derivative and multi-channel blocker, stabilizes RyR2s and decrease SR Ca2+ leak. We investigated whether JTV519 stabilizes RyR2s without increasing RyR2 phosphorylation in mice and in non-failing human myocardium and explored underlying mechanisms.
EXPERIMENTAL APPROACH
SR Ca2+ leak was induced by ouabain in murine cardiomyocytes. [Ca2+]-transients, SR Ca2+ load and RyR2-mediated Ca2+ leak (sparks/waves) were quantified, with or without JTV519 (1 µmol·L−1). Contribution of Ca2+-/calmodulin-dependent kinase II (CaMKII) was assessed by KN-93 and Western blot (RyR2-Ser2814 phosphorylation). Effects of JTV519 on contractile force were investigated in non-failing human ventricular trabeculae.
KEY RESULTS
Ouabain increased systolic and diastolic cytosolic [Ca2+]i, SR [Ca2+], and SR Ca2+ leak (Ca2+ spark (SparkF) and Ca2+ wave frequency), independently of CaMKII and RyR-Ser2814 phosphorylation. JTV519 decreased SparkF but also SR Ca2+ load. At matched SR [Ca2+], Ca2+ leak was significantly reduced by JTV519, but it had no effect on fractional Ca2+ release or Ca2+ wave propagation velocity. In human muscle, JTV519 was negatively inotropic at baseline but significantly enhanced ouabain-induced force and reduced its deleterious effects on diastolic function.
CONCLUSIONS AND IMPLICATIONS
JTV519 was effective in reducing SR Ca2+ leak by specifically regulating RyR2 opening at diastolic [Ca2+]i in the absence of increased RyR2 phosphorylation at Ser2814, extending the potential use of JTV519 to conditions of acute cellular Ca2+ overload.
Keywords: calcium leak, diastolic dysfunction, arrhythmia, non-failing human myocardium, JTV519, K201
Introduction
In cardiac muscle, excitation-contraction (EC) coupling relies on the precise regulation of intracellular Ca2+. Ca2+ also regulates a variety of intracellular processes, such as cellular metabolism, gene transcription and cell death. Dysregulation of cardiomyocyte Ca2+ homeostasis has been implicated in a variety of cardiac disease states (Bers, 2008). During EC coupling, Ca2+ influx through voltage-dependent L-type Ca2+ channels triggers Ca2+ release from the intracellular Ca2+ store, the sarcoplasmic reticulum (SR), via opening of ryanodine receptors (RyR2; receptor nomenclature follows Alexander et al., 2011) in the SR membrane (Ca2+-induced Ca2+ release). During diastole, RyR2s are closed and low cytosolic [Ca2+]i is restored mainly by the SR Ca2+ ATPase (SERCA) and the sarcolemmal Na+/Ca2+ exchanger (NCX). The contribution of NCX to cytosolic Ca2+ removal varies among species between 7% (rats), 28% (rabbits) and up to about 50% (failing human hearts) (Dipla et al., 1999; Bers, 2001).
Leak of Ca2+ from the SR through RyR2s during diastole has been implicated in arrhythmias and contractile dysfunction in heart failure (Marx et al., 2000; Lindner et al., 2002), myocardial ischaemia (Hirose et al., 2008), atrial fibrillation (Vest et al., 2005) and congenital arrhythmias (Mohamed et al., 2007). Several mechanisms have been proposed to explain RyR2 dysfunction, including altered RyR2 gating due to point mutations in the RyR2 molecule (Cerrone et al., 2005), alterations in RyR2 phosphorylation (Witcher et al., 1991; Wehrens et al., 2004b; Xiao et al., 2005) and the composition of the RyR2 macromolecular complex (Marx et al., 2000), and increased SR Ca2+ content (store-overload induced Ca2+ release) (Eisner et al., 2009). Increased RyR2 Ca2+ leak has been associated with PKA-dependent phosphorylation of the RyR2 at Ser2809 (Marx et al., 2000) or Ser2030 (Xiao et al., 2007), or Ca2+-calmodulin-dependent kinase II (CaMKII)-mediated phosphorylation at Ser2809 (Witcher et al., 1991) or Ser2815 (Wehrens et al., 2004b) (corresponding to Ser2808 and Ser2814 in mice) respectively. However, the exact role of site-specific phosphorylation of the RyR2 in promoting SR Ca2+ leak is still under debate (Blayney and Lai, 2009; Eisner et al., 2009). We have previously shown that cellular Na+- and Ca2+ overload induced by ouabain treatment leads to Ca2+ leak from the SR through the RyR2 in the absence of change in RyR2 phosphorylation at Ser2808 (Sedej et al., 2010). One aim of the present study was to investigate whether SR Ca2+ leak induced by intracellular Ca2+ overload can occur in the absence of CaMKII-mediated RyR2 phosphorylation at Ser2814.
JTV519 (K201) is a 1,4-benzothiazepine derivative and anti-arrhythmic drug currently under clinical investigation. While JTV519 also inhibits sarcolemmal currents, such as sodium (INa), L-type Ca2+ and potassium currents (IKr, IK1) (Kaneko et al., 2009), this multi-channel blocker has been investigated based on its potential to prevent arrhythmias related to increased Ca2+ leak from the SR. Heart failure and atrial fibrillation are associated with increased phosphorylation of the RyR2 (Marx et al., 2000; Greiser et al., 2009). In these conditions, JTV519 (K201) reduced RyR2-mediated SR Ca2+ leak and related cardiomyocyte dysfunction (Kohno et al., 2003; Wehrens et al., 2005; Toischer et al., 2010). However, whether JTV519 is effective in reducing arrhythmogenic RyR2 Ca2+ leak in the absence of CaMKII-mediated phosphorylation on Ser2814 has not yet been explored.
Ouabain, a cardiac glycoside, increases contractile force by increasing intracellular [Ca2+], and at higher doses induces arrhythmias, without changing phosphorylation at the PKA-dependent phosphorylation site Ser2808 (Sedej et al., 2010). In the present study, we investigated the mechanisms by which JTV519 reduced RyR2 Ca2+ leak resulting from ouabain-induced increase in intracellular [Ca2+]. We found that SR Ca2+ leak induced by ouabain occurred in the absence of increased RyR2 phosphorylation at the CaMKII-dependent site Ser2814. JTV519 reduced SR Ca2+ leak by stabilizing the RyR2 in its closed state during diastole without decreasing the gain of Ca2+-induced Ca2+ release.
Methods
The use of donor hearts and research protocols for human samples were approved by the local ethics committee (ref. nr. 20–277 ex 08/09). Investigations on human tissue conform with the principles outlined in the Declaration of Helsinki. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). Animals were treated according to the Guidelines for the Care and Use of Laboratory Animals (National Institute of Health, USA).
Cell isolation
Adult FVB/N mice (11–16 weeks old; total = 43) provided by the Abt. f. Labortierkunde, Medical University of Vienna, Austria, were used for all experiments. Ventricular cardiomyocytes were isolated as previously described (Heinzel et al., 2002; Sedej et al., 2010). Mice were anesthetized with isoflurane (Abbott, Wiesbaden, Germany) and killed by cervical dislocation. The heart was quickly removed and perfused on a Langendorff setup for 4 min using Ca2+-free Tyrode's solution (see below) with taurine (15 mmol·L−1), to which collagenase Type II (285 U·mg−1, Worthington, Lakewood, NJ, USA) was then added, and perfusion continued for 7 min. Following mechanical dispersion of the cells, extracellular [Ca2+] was increased stepwise to reach 1 mmol·L−1. Only quiescent, rod-shaped ventricular cardiomyocytes were used for experiments.
Confocal [Ca2+]i measurements
Cells were studied in a perfusion chamber mounted on the stage of an inverted microscope with a Plan Neofluar 40×/1.3 oil-immersion objective and equipped with a Zeiss LSM 510 Meta confocal laser point scanning system (Zeiss GmbH, Jena, Darmstadt, Germany). Fluo-4 AM (Invitrogen, Germany) was used as Ca2+ sensitive dye (excitation at 488 nm, emission >515 nm). Pinhole was set to 1 Airy unit, resulting in an optical slice thickness of 0.9 µm. Pixel width was between 0.17 and 0.33 µm. To record [Ca2+]i transients, a 512 pixel scan line was drawn along the longitudinal axis of the cell, avoiding scanning through the nuclei, and scanned every 1.54 ms. Consecutive scan lines were stacked over time and visualized as 2D image (line scan image).
Experimental protocol
Cells were superfused with Tyrode's solution, containing (in mmol·L−1) NaCl 136, KCl 5, CaCl2 3, MgCl2 1, HEPES 10, glucose 10; pH adjusted to 7.4 with NaOH at 37°C. Cells were electrically stimulated at 1 Hz for at least 10 min. Line scan images with [Ca2+]i transients were recorded at steady state of the [Ca2+]i transient amplitude (baseline). In order to quantify the effects of ouabain on [Ca2+]i transients and on SR Ca2+ leak (Ca2+ sparks frequency) in beating cardiomyocytes during steady-state electrical stimulation, ouabain (100 µmol·L−1, Sigma Aldrich, Vienna, Austria) was washed in for 7 min to induce a moderate increase in cytosolic [Na+]i (Sedej et al., 2010).
In a second set of experiments, SR Ca2+ leak was measured as a function of SR [Ca2+] at steady state of intracellular Ca2+ fluxes. For this, at steady-state baseline, stimulation was continued without (control) or with ouabain for 7 min. Then electrical stimulation was stopped, and external solution was rapidly switched to a Na+- and Ca2+-free solution to inhibit Na+- and Ca2+ transport via the sarcolemmal NCX [(Shannon et al., 2003); solution contained, in mmol·L−1, LiCl 91, LiOH 21, KCl 4, MgCl2 1, HEPES 20, EGTA 10, glucose 10; pH 7.4 with LiOH ± ouabain as above. 20 s were allowed for a steady state of intracellular Ca2+ fluxes, followed by 10 s to record Ca2+ spark frequency. Tyrode's solution with Na+ and Ca2+ (as above) was then re-introduced within <2 s, and SR Ca2+ content was assessed by rapid application of caffeine (30 mmol·L−1). In a subset of cells, the CaMKII-inhibitor KN-93 (1 µmol·L−1; Merck, Darmstadt, Germany) was added together with ouabain. Another group of cells was pre-incubated for at least 1 h with JTV519 (1 µmol·L−1) before running the protocols above. In these cells, JTV519 was also present throughout the entire protocol.
To allow quantitative spark analysis in all groups, we used supraphysiological [Ca2+] (3 mmol·L−1) in the external Tyrode's solution (see above), because in preceding experiments only a minority (32%) of control cardiomyocytes showed Ca2+ sparks during the scanning period at 1 mmol·L−1 external [Ca2+] (vs. 82% of all control cells in the present study at 3 mmol·L−1 external [Ca2+]).
Image analysis
For the steady-state [Ca2+]i transient analysis, line scan images were segmented by the onset of the whole-line averaged [Ca2+]i transients and four to five consecutive transients were averaged using custom made algorithms coded in IDL (IDL 7.0, ITT Visual Information Solutions, Paris, France) as previously described (Heinzel et al., 2008; Lenaerts et al., 2009). The amplitude of the cytosolic [Ca2+]i transient was calculated by normalizing the peak (Fpeak) of the [Ca2+]i-dependent fluorescence (F) to F averaged during 30 ms before the onset of the Ca2+ transient (F0). F50 was defined as the half-maximum of the [Ca2+]i transient amplitude. SR [Ca2+] was estimated by the amplitude of the caffeine-induced [Ca2+]i transient. The contribution of SERCA to the electrically stimulated cytosolic [Ca2+]i transient (TwitchSERCA%) was estimated from the decay rates (mono-exponential fit) of the caffeine-induced [Ca2+]i transient (λcaff) and the electrically stimulated [Ca2+]i transient (λstim) in the same cell by the formula: TwitchSERCA%= (λstim – λcaff) / λstim× 100 (Fowler et al., 2005). Relative changes in diastolic cytosolic [Ca2+]i were quantified in the same cell by dividing F0 at steady state in the treatment phase (see above) with F0 at steady-state baseline (F0,bsl). This F/F0 ratio could be slightly below 1.0 in control conditions, reflecting loss of fluorescence signal due to repetitive laser scanning. A pseudo-calibration of the Ca2+-dependent fluorescence signals was performed to estimate SR [Ca2+] in the presence of ouabain as outlined in the Appendix S1.
Ca2+ sparks were quantified during the decline of the [Ca2+]i transient at 1 Hz stimulation at baseline and during the treatment phase, as well as during the last 10 s of long diastole following stimulation (with the drugs present). Line scan images were analyzed offline, and Ca2+ sparks were quantified as singular Ca2+ release events with a maximum width smaller than 4 µm. Time to peak [Ca2+]i, peak amplitude, full width at half maximum (FWHM) and full duration at half maximum (FDHM) of Ca2+ sparks were calculated automatically from the local [Ca2+] transients. Ca2+ wave propagation speed was measured following linear fitting of the Ca2+ wave front.
Western blots
Suspensions of isolated cardiomyocytes (viability > 50%) were stimulated in an electrical field and exposed to extracellular solutions as in the treatment phase described above. A subset of cell suspensions was treated with isoprenaline (1 µmol·L−1) in combination with the serine/threonine phosphatase inhibitor calyculin A (1 µmol·L−1) to induce RyR2 phosphorylation at Ser2814 (positive control) (Huke and Bers, 2008). After treatment, the cells were pelleted and snap-frozen in liquid nitrogen. Samples were homogenized in lysis buffer composed of (in mmol·L−1) Tris–HCl 20, pH 7.4; NaCl 137, NaF 20, sodium orthovanadate 1, sodium pyrophosphate 1, β-glycerophosphate 50, EDTA 10, EGTA 1, PMSF 1, glycerol 10%, NP-40 1%, aprotinin 4 µg·mL−1, pepstatin A 4 µg·mL−1 and leupeptin 4 µg·mL−1. 40 µg of cell homogenates were run on 4–12% gradient SDS-polyacrylamide gels and transferred to nitrocellulose membranes overnight. Non-specific binding was blocked for 1 h at room temperature using 5% dried milk in Tris-buffered saline (pH 7.4) containing 0.1% Tween-20. Membranes were probed with anti-phospho-Ser2814-RyR2 antibody (Badrilla, Leeds, UK) overnight at 4°C. Anti-rabbit IgG linked with HRP (GE Healthcare, Berkshire, UK) was used as a secondary antibody. Signal was detected using the Pierce Supersignal West Pico Reagents (Thermo Fisher Scientific, Leicestershire, UK), and the optical density of the bands was determined by Quantity One software (Bio-Rad Laboratories, Hertfordshire, UK). After stripping, membranes were re-probed with the RyR-specific antibody MA3-916 (Thermo Scientific).
Human muscle strip experiments
Functional experiments were performed on 21 isolated endocardial muscle strips (trabeculae) from five non-failing hearts (ejection fraction 57 ± 4%) not suitable for transplantation (sepsis of the donor, n= 1; excessive myocardial hypertrophy, n= 1; advanced coronary artery disease, n= 1; old age, n= 2). Human endocardial trabeculae were prepared as previously described (von Lewinski et al., 2005). Donor hearts were kept in cardioplegic Tyrode's solution at 4°C containing (in mmol·L−1) Na+ 152, K+ 3.6, Cl- 135, HCO3− 25, Mg2+ 0.6, H2PO4− 1.3, SO42− 0.6, Ca2+ 0.2, glucose 11.2, insulin 10 I.U.·L−1 and 2,3-butanedione-monoxime (BDM) 30, equilibrated with carbogen (95% O2, 5% CO2) to a pH of 7.4, and transported to the laboratory. BDM was included in the cardioplegic solution to protect the myocardium during transportation from the operating room and from cutting injury. The cardioplegic effects of the solution are fully reversible upon wash-out (von Lewinski et al., 2008). Trabeculae (cross-sectional area < 0.6 mm2) were dissected under a stereo-microscope. Then the trabeculae were mounted between miniature hooks, connected to an isometric force transducer (Scientific Instruments GmbH, Gilching, Germany) and superfused with modified Tyrode's solution (no BDM, stepwise increase of [Ca2+]e to 2.5 mmol·L−1) at 37°C. Isometric twitches were evoked by electrical stimulation (1 Hz, pulse duration 5 ms) at the preload at which maximum steady-state twitch force was achieved (Lmax). JTV519 (1 µmol·L−1) was added to a subset of trabeculae. The trabeculae were stimulated for 1 h before the step-wise wash-in of ouabain at the increasing concentrations indicated.
Voltage clamp experiments
A subset of cells was transferred to a recording bath chamber mounted on a confocal microscope (Zeiss LSM 700, Germany) equipped with a patch-clamp setup (Axopatch 200B, Molecular Devices, Sunnyvale, CA, USA). Cardiomyocytes were superfused at 37°C with Tyrode's solution and voltage clamped in whole-cell configuration. Pipette contained (in mmol·L−1) CsAsp 120, TEACl 20, MgATP 5, MgCl2 0.5, HEPES 10, pH 7.2 with CsOH. To measure L-type Ca2+ current, K+ was replaced by Cs+ in the external solution, and cells were clamped at −40 mV to inactivate INa. Ca2+ currents were then activated by 250 ms depolarizing steps to +10 mV, during 1 Hz stimulation. Simultaneously, confocal Ca2+ transients were recorded as described above. Current recordings were analysed using Clampfit 10.0 (Molecular Devices). L-type Ca2+ current was quantified as the difference between the negative peak and the current at the end of pulse. Ca2+ current density was determined by normalizing the current amplitude to cell capacitance.
Data analysis
Data are shown as mean ± SEM. As experiments were performed on mice of similar age and identical genetic and domestic background, data from individual experiments and observations (cardiomyocytes in Figures 1, 2C, 3 and 5; tissue homogenates from individual hearts in Figure 2A,B; solitary Ca2+ sparks in Figure 4) were pooled for statistical analysis. Data from experiments on individual human trabeculae were pooled as non-failing myocardium based on in vivo global left ventricular function before explantation, irrespective of co-morbidities (see section Human Muscle Strips Experiments). The number of experiments as a basis for statistical analysis is underlined in the figure legends. Data were compared using Student's t-test (Figures 1, 4, 5B), one-way (Figures 2, 3 and 5C) or two-way (Figure 6) anova followed by the Holm–Sidak post hoc tests for multiple comparisons when an overall significance was established. Chi-squared test was used to compare proportions (Figure 3C). P < 0.05 was considered significant.
Figure 1.
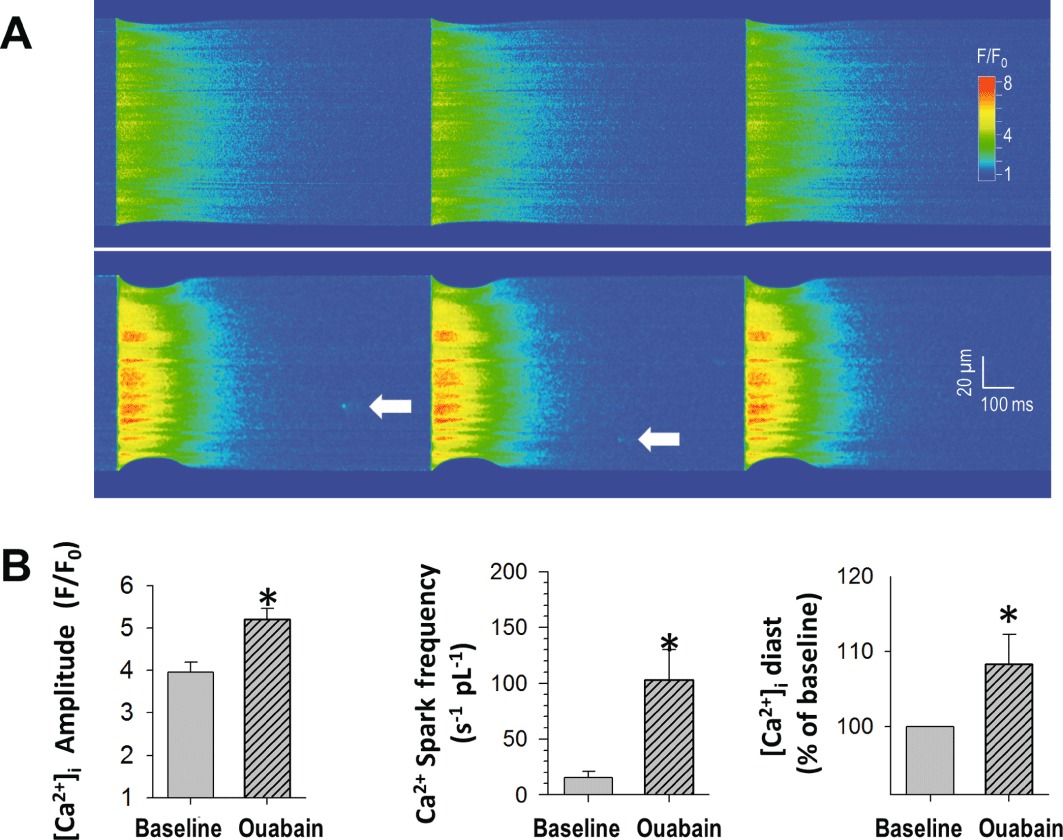
Increased cellular Ca2+ and SR Ca2+ leak with ouabain in beating ventricular myocytes. (A) Line scan image of a cardiomyocyte at baseline (above, 1 Hz stimulation) and following 7 min ouabain (100 µmol·L−1; below). During ouabain, the amplitude of the [Ca2+]i transients is larger, and Ca2+ sparks occur. (B) Peak [Ca2+]i, Ca2+ spark frequency and [Ca2+]i at diastole (30 ms before upstroke of [Ca2+]i, given as % of baseline) are significantly increased by application of ouabain (n= 18 cells from n= 7 mice, *P < 0.05 vs. baseline).
Figure 2.
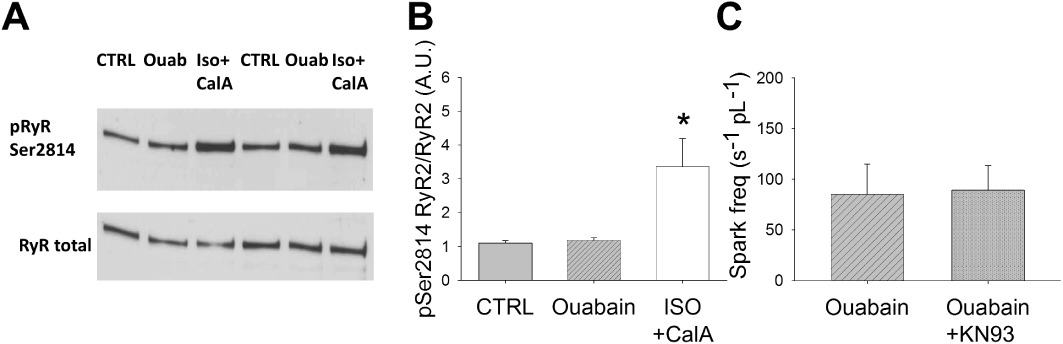
Ouabain-induced SR Ca2+ leak is independent of CaMKII-activation. (A) Western blot examples of RyR2 phosphorylated at Ser2814 and RyR2 total in mouse left ventricular cardiomyocyte suspensions. (B) Mean values of quantitative densitometry. While phosphorylation at the CaMKII-dependent RyR2 phosphorylation site Ser2814 was not altered with 7 min exposure to ouabain (100 µmol·L−1; n= 6 mice), isoprenaline (ISO) and calyculin A (CalA) induced a strong increase in phosphorylation (positive control; n= 3 mice). (C) In intact cardiomyocytes, spark frequency in the presence of ouabain was not altered by the CaMKII-inhibitor KN-93 (1 µmol·L−1; n= 8 cells from three mice). *P < 0.05 significantly different from control (CTRL) and ouabain (n= 18 cells from seven mice).
Figure 3.
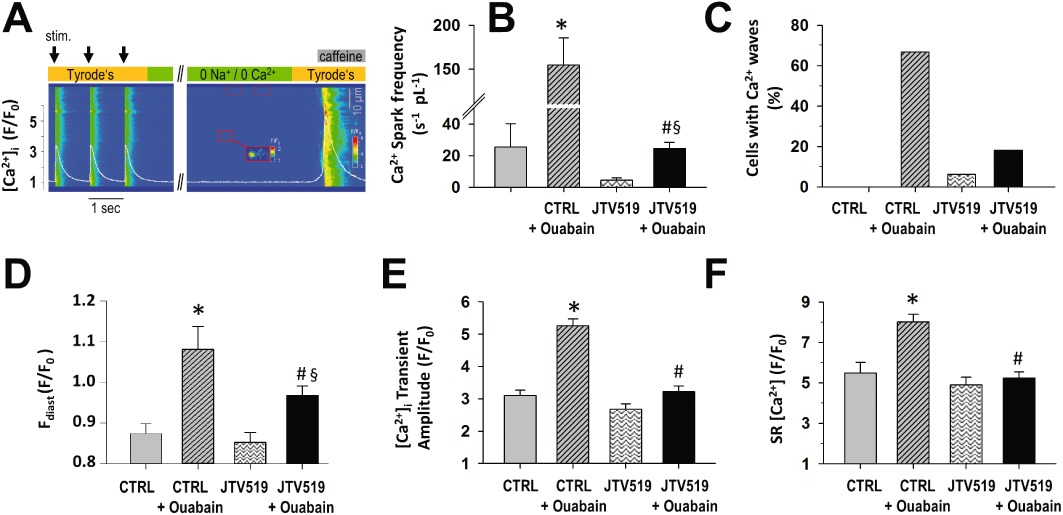
JTV519 decreases SR Ca2+ leak and SR Ca2+ content. (A) Confocal line scan image of [Ca2+]i transients during electrical stimulation, Ca2+ sparks during prolonged diastole and caffeine-induced release of SR Ca2+ content (right side) in a mouse cardiomyocyte (see Methods for details). White line represents average [Ca2+]i across the scan line. With intracellular Ca2+ fluxes at equilibrium during a prolonged diastole, Ca2+ spark frequency (B), the incidence of Ca2+ waves (C), diastolic cytosolic [Ca2+]i (D) and [Ca2+]I transient amplitude (E) were increased with ouabain (control (CTRL) + Ouabain), related to an increased SR [Ca2+] (F). In the presence of JTV519 (1 µM), Ca2+ spark frequency and diastolic cytosolic [Ca2+]i with ouabain were significantly lower than in CTRL + Ouabain. JTV519 decreased the amplitude of the [Ca2+]i transient and SR [Ca2+]. n mice or cells per group: CTRL: 7/11; ouabain: 7/18; JTV: 8/16; JTV + ouabain: 6/33. *P < 0.05 versus CTRL; #P < 0.05 significantly different from CTRL + Ouabain; §P < 0.05 significantly different from JTV519 P < 0.01 for group differences in Figure 3C.
Figure 5.
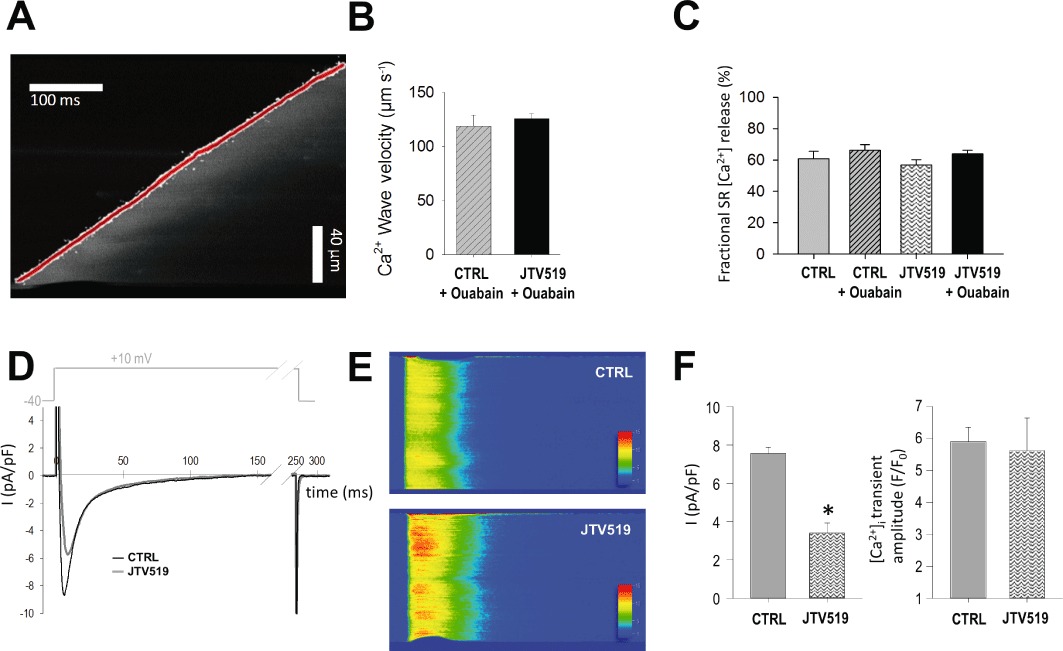
JTV519 does not affect wave propagation or fractional SR Ca2+ release despite inhibition of L-type Ca2+ influx. (A) Example of a line scan image with the slope of the Ca2+ wave front marked red. (B) Ca2+ wave propagation velocity in ouabain-treated cells was unchanged in the presence of JTV519 (ouabain: n= 12 cells from three mice; JTV519 + ouabain: n= 7 cells from three mice; one wave/cell). (C) Fractional Ca2+ release ([Ca2+]i transient amplitude/SR [Ca2+]) at 1 Hz stimulation was not influenced by JTV519 (n cells as in Figure 3). Representative recording of a L-type Ca2+ current (D) and simultaneously recorded line scan image of the cytosolic Ca2+ transient (E) in a control (CTRL) and JTV519-treated cardiomyocyte (1 Hz). (F) Mean values for L-type Ca2+ current density and Ca2+ transient amplitude (CTRL: n= 7 cells from three mice; JTV519: n= 6 cells from three mice). *P < 0.05 significantly different from control.
Figure 4.
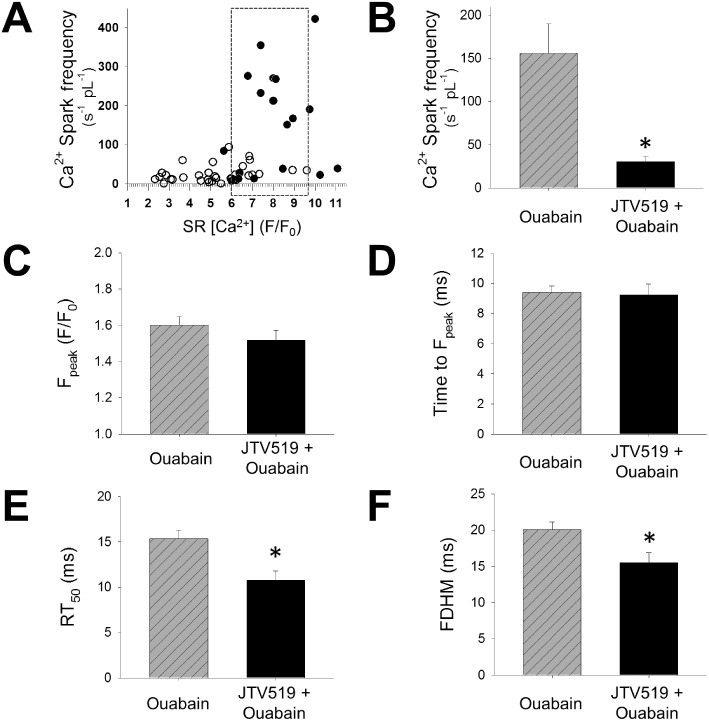
JTV519 has SR [Ca2+]i– independent effects on SR Ca2+ leak. (A) Ca2+ spark frequency with ouabain as a function of SR [Ca2+] in the absence and presence of JTV519 (each symbol represents one cell; n cells or mice as in Figure 3). (B) In cardiomyocytes matched for similar SR [Ca2+] (box in A), Ca2+ spark frequency was reduced in the presence of JTV519 (n= 13 cells from seven mice for ouabain and n= 12 cells from five mice for JTV + ouabain). *P < 0.05 significantly different from ouabain without JTV519. Quantitative analysis of sparks from cardiomyocytes with matched SR [Ca2+] revealed similar Ca2+ spark amplitude (C) and time to peak (D), but faster Ca2+ decay (time to 50% decay, RT50) (E) and thus shorter spark duration (full duration at half maximum, FDHM) (F) in the presence of JTV519 (n= 98 sparks with ouabain, n= 66 sparks with JTV + ouabain). *P < 0.05 significantly different from ouabain without JTV519.
Figure 6.
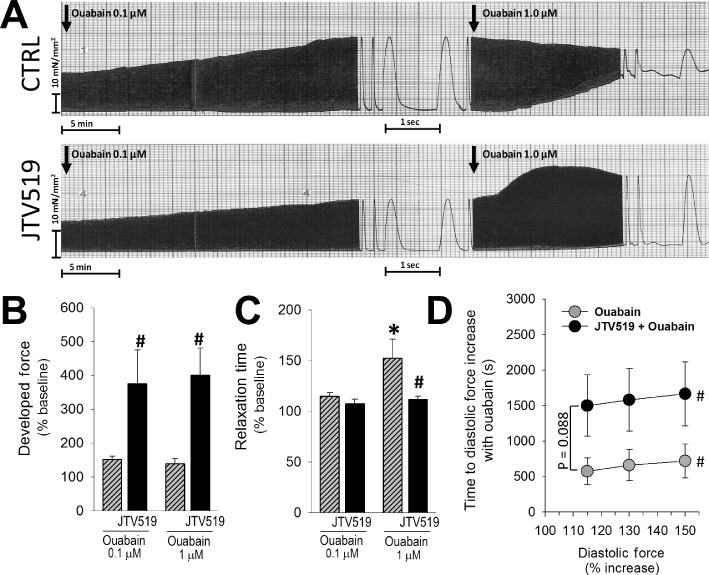
JTV519 effects in human non-failing muscle strips. (A) Original recordings of force development over time in control (CTRL: upper panel) and JTV519-treated muscle strips stimulated at 1 Hz in response to ouabain. Force development at baseline tended to be lower with JTV519 (see text). (B) Ouabain-induced increase in systolic force development was significantly higher in the presence of JTV519. (C) Time to half-maximal relaxation was measured at an ouabain-induced 25% increase of systolic force. Inotropy at higher ouabain concentration (1 µmol·L−1) was associated with impaired relaxation (longer relaxation time) in CTRL but not in JTV519-treated muscle strips. (D) Increase in diastolic force indicating diastolic deterioration with prolonged ouabain exposure tended to occur later in JTV519-treated cells. n muscle strips/hearts: 10/5 for ouabain, 11/5 for JTV + ouabain. *P < 0.05 significantly different from control, #P < 0.05 significant differences between 115%, 130% and 150% diastolic force in both groups.
Materials
All chemicals were purchased from Sigma Aldrich, if not otherwise stated.
Results
Ouabain increases intracellular [Ca2+]i and diastolic SR Ca2+ release
Treatment with ouabain (100 µmol·L−1) for 7 min during 1 Hz stimulation significantly increased systolic [Ca2+]i (Figure 1). Time to peak [Ca2+]i was increased with ouabain (46 ± 4 ms vs. 33 ± 3 ms at baseline, P < 0.01, n= 18), whereas half-time of decay of the [Ca2+]i transient was unchanged (185 ± 10 vs. 193 ± 10 ms). The frequency of Ca2+ sparks during the decay of the [Ca2+]i transient was significantly increased in the presence of ouabain, indicating increased RyR2-mediated Ca2+ leak (Figure 1). Additionally, ouabain treatment significantly increased diastolic cytosolic [Ca2+]i (Figure 1).
Acute treatment with ouabain is not associated with CaMKII-mediated RyR2 phosphorylation
We have previously shown that ouabain treatment does not alter phosphorylation of RyR2 at Ser2808 (Sedej et al., 2010). We now quantified RyR2 phosphorylation at the CaMKII-dependent site Ser2814. The ouabain-induced increase in cellular Ca2+ load was not associated with altered phosphorylation of RyR2 at Ser2814 (Figure 2A/B, n= 4). Isoprenaline (100 nmol·L−1) and calyculin A (1 µmol·L−1), however, induced a significant increase in Ser2814 phosphorylation (positive control). To confirm that CaMKII activation was not involved in increased Ca2+ spark frequency observed after acute ouabain treatment, Ca2+ spark frequency was measured in isolated cardiomyocytes during prolonged diastole in the presence of the CaMKII-inhibitor KN-93 (1 µmol·L−1, n= 8) and was found to be unchanged, compared with ouabain alone (Figure 2C).
Effects of JTV519 on SR Ca2+ leak and [Ca2+]i transient
The effects of JTV519 (1 µmol·L−1) on ouabain-induced SR Ca2+ leak were investigated during prolonged diastole to allow for a steady state of intracellular Ca2+ fluxes (Figure 3A-F: control: n= 11; ouabain: n= 18; JTV: n= 16; JTV + ouabain: n= 33). With sarcoplasmic Ca2+ reuptake and RyR2-mediated Ca2+ flux at balance, net SR Ca2+ leak remained significantly elevated with ouabain as reflected by a significant increase in Ca2+ spark frequency (Figure 3B) and Ca2+ waves (Figure 3C). SR Ca2+ content as assessed by caffeine-induced [Ca2+]i transient was increased with ouabain (Figure 3D). In JTV519-treated cardiomyocytes (1 µmol·L−1, pre-incubation for 1 h), Ca2+ spark frequency in response to ouabain was significantly reduced as compared with ouabain alone (Figure 3B), indicating a reduced SR Ca2+ leak. Similarly, in the presence of JTV519 the number of cardiomyocytes developing Ca2+ waves was lower (Figure 3C, 12/19 = 67% for ouabain, 6/33 = 18% for JTV519 + ouabain, P < 0.05).Ouabain induced a significant increase in diastolic cytosolic [Ca2+] also in the presence of JTV519. However, in the presence of JTV519, diastolic cytosolic [Ca2+] with ouabain was significantly lower than in the absence of JTV519 (Figure 3D).
JTV519 did not affect the [Ca2+]i transient amplitude in control conditions (Figure 3E) but reduced the effect of ouabain on the systolic peak [Ca2+]i transient (Figure 3E) and on SR Ca2+ load (Figure 3F). JTV519 did not affect the decay of the [Ca2+]i transient at 1 Hz (τ: 153 ± 15 ms in control vs. 140 ± 10 ms in JTV519). The calculated contribution of SERCA to the decay tended to be reduced with JTV519 (87.9 ± 1.1% vs. 90.6 ± 1.0% in control; P= 0.089; n= 11 cells from five mice for control and n= 13 cells from four mice for JTV519).
JTV519 has SR Ca2+ load-independent effects on SR Ca2+ leak
As SR Ca2+ leak from the RyR2s is a function of the SR Ca2+ content (Shannon et al., 2002), reduced Ca2+ spark frequency in the presence of JTV519 and ouabain may in part be explained by a lower SR Ca2+ load. Figure 4A shows Ca2+ spark frequency as a function of SR Ca2+ load in ouabain-treated cardiomyocytes without and with JTV519. While JTV519-treated cardiomyocytes (open circles) were found to be more on the left side of the x axis, reflecting reduced SR Ca2+ load, there was considerable overlap allowing matching cardiomyocytes of both groups with comparable SR Ca2+ load (marked by box in Figure 4A). At similar SR Ca2+ load, JTV519-treated cells showed a significantly lower Ca2+ spark frequency as compared with cardiomyocytes treated with ouabain alone (P < 0.01; Figure 4B).
Differential effects of JTV519 on RyR2-mediated SR Ca2+ release
In order to evaluate the effects of JTV519 on RyR2-mediated Ca2+ leak in more detail and independent on its effect on SR [Ca2+], we analysed Ca2+ spark morphology in cardiomyocytes matched for SR Ca2+ content (Figure 4C-F; n= 78 sparks in 13 cells with ouabain and 30 sparks in 12 cells for ouabain + JTV519). Using pseudo-calibration as detailed in the Appendix S1, we calculated an amplitude of the caffeine-induced [Ca2+] transient of 694 ± 81 nM in control, 1550 ± 199 nM with ouabain and 681 ± 62 nM in ouabain + JTV519 based on the data in Fig 3. When comparing cardiomyocytes with similar average SR Ca2+ load (1241 ± 39 nM in ouabain vs. 1140 ± 131 nM in ouabain + JTV519), spontaneous Ca2+ sparks in the presence of JTV519 had a similar amplitude (Figure 4C), time to peak [Ca2+]i (Figure 4D) and width (full width at half maximum, 1.84 ± 0.08 vs. 1.77 ± 0.06 µm), but significantly faster decay (Figure 4E) and thus reduced duration (Figure 4F), suggesting a shorter open duration of RyR2 in the presence of JTV519. Ca2+ waves represent SR Ca2+ release propagated (triggered) by cytosolic Ca2+ released from neighbouring RyR2 clusters. We quantified the velocity of Ca2+ waves in cardiomyocytes as a measure of RyR2 opening properties during Ca2+ release triggered by cytosolic [Ca2+]i increase. Ca2+ wave velocity was not different in cardiomyocytes treated with JTV519+ouabain as compared to ouabain alone (Figure 5A,B), despite a reduced SR Ca2+ load with JTV519 (F/F0: 5.7 ± 0.4 in control + ouabain vs. 7.4 ± 0.6 in JTV519 + ouabain, n= 6 and five cells with waves respectively; P < 0.05). This suggests that JTV519 did not lower the opening probability of RyR2s in response to an increase in cytosolic [Ca2+]i. During steady-state electrical stimulation, the ratio between the amplitude of the cytosolic [Ca2+]i transient and SR Ca2+ content (i.e. fractional Ca2+ release) was not altered by JTV519 (Figure 5C), again supporting the notion that JTV519 reduced the rate of spontaneous (diastolic) RyR2 opening, but not its sensitivity to cytosolic [Ca2+]i.
In order to investigate the effects of JTV519 on the gain of EC coupling, we recorded whole-cell L-type Ca2+ influx and the triggered cytosolic [Ca2+] transients in control and JTV519-treated cardiomyocytes (Figure 5D-F). In the absence of ouabain, JTV519 did not affect SR Ca2+ content (Figure 3D). However, L-type Ca2+ current amplitude was significantly reduced with JTV519 (Figure 5F). On the other hand, cytosolic [Ca2+] transients were unchanged, confirming our observations in intact cells (Figure 3E). Together, these results suggest that the gain of EC coupling is increased with JTV519.
Effects of JTV519 on ouabain-induced contractile dysfunction in human myocardium
We evaluated the effects of JTV519 (1 µmol·L−1) in conditions of ouabain-induced Ca2+ overload in human, non-failing, ventricular myocardium. In the contracting muscle strips, ouabain time-dependently increased diastolic force (Figure 6A), indicating an increase in diastolic [Ca2+]. Increasing ouabain from 0.1 to 1.0 µmol·L−1 did not further increase developed (systolic) force, but instead led to impaired relaxation (Figure 6A, upper panel and Figure 6C), reflecting cytosolic Ca2+ overload. In JTV519-treated muscle strips, average systolic force development at baseline tended to be lower as compared with control (8.0 ± 2.0 vs. 16.3 ± 3.3 mN·mm−2, P= 0.06; n= 11 and 10 respectively). However, the positive inotropic effect of ouabain on developed force at 0.1 and at 1.0 µmol·L−1 was significantly greater in the presence of JTV519 (Figure 6B). The ouabain-induced impairment of relaxation at comparable systolic force response was prevented in the presence of JTV519 (Figure 6A lower panel, Figure 6C). Similarly, the time to deterioration of diastolic function upon prolonged exposure to ouabain tended to be longer in the presence of JTV519 (Figure 6D). Comparable to our findings in murine cardiomyocytes, these results indicated that, JTV519 could reduce Ca2+ overload-induced diastolic dysfunction in non-failing human myocardium.
Discussion
In the present study, we showed that ouabain-induced acute SR Ca2+ leak was not attenuated by CaMKII inhibition and occurred without change in RyR2 phosphorylation at Ser2814. In light of our previous findings (Sedej et al., 2010), we conclude that JTV519 has an SR Ca2+ load-independent, RyR2-stabilizing effect that does not depend on increased RyR2 phosphorylation at PKA- and CaMKII-dependent sites. Furthermore, our data suggest that, while reducing the RyR2 open probability with regard to spontaneous openings during diastole, JTV519 does not decrease the efficacy of Ca2+-induced Ca2+ release that triggers contraction. In human ventricular muscle strips, JTV519 acts negative inotropic but improves diastolic function and preserves the inotropic response in conditions of ouabain-induced Ca2+ overload.
Several studies have associated an increased phosphorylation of the RyR2 at its PKA-(Marx et al., 2000; Xiao et al., 2005) or CaMKII-dependent (Ai et al., 2005; Neef et al., 2010) sites with an increased open probability of the channel resulting in SR Ca2+ leak. CaMKII is activated by increased cytosolic Ca2+ turnover and could promote SR Ca2+ leak also by its phospholamban-dependent effect to increase SERCA activity and thus SR [Ca2+] (Picht et al., 2007). In contrast, in rat cardiomyocytes, overexpression of constitutively active CaMKII even reduced SR Ca2+ leak (Yang et al., 2007) despite increased RyR2 phosphorylation. As a result, it has been proposed that changes in RyR2 phosphorylation may not be an accurate reflection of altered RyR2 activity (Yamaguchi and Meissner, 2007). We have shown previously that in conditions of ouabain-induced SR Ca2+ overload, SR Ca2+ leak occurs in the absence of RyR2 phosphorylation at the PKA-dependent site (Sedej et al., 2010). Our present results extend these findings, indicating that RyR2 phosphorylation at Ser2814 was also not a prerequisite for increased SR Ca2+ leak, implying that therapeutic approaches targeted at upstream kinases modulating RyR2 phosphorylation such as CaMKII may not be protective in similar conditions of acute Ca2+ overload. Increased SR Ca2+ leak with ouabain was observed in the decay phase of the [Ca2+] transient in steadily beating cardiomyocytes (Figure 1B), as well as in prolonged diastole following stimulation when intracellular Ca2+ fluxes are at steady state (Figure 3D).
JTV519 has been shown to reduce SR Ca2+ leak and related arrhythmias in some (Kohno et al., 2003; Wehrens et al., 2005; Toischer et al., 2010), but not all (Liu et al., 2006) cardiac disease models. Despite recent advances in the understanding of the molecular interactions of JTV519 and the RyR2 (Tateishi et al., 2009), the molecular mechanisms by which JTV519 modifies RyR2 gating to attenuate acquired gain-of-function defects of the RyR2 are not completely understood. Dissociation of the RyR2-associated protein FKBP12.6 was thought to induce instability of the RyR2 in the closed state resulting in increased SR Ca2+ leak (Marx et al., 2000). JTV519 increases the association of FKBP12.6 with the RyR2 (Wehrens et al., 2005) and was reportedly ineffective in FKBP12.6 knock-out mice (Wehrens et al., 2004a). However, Hunt et al. (2007) showed that K201 (JTV519) suppressed spontaneous SR Ca2+ release in heterologous expression systems, irrespective of FKBP12.6 association, suggesting that JTV519 may alter RyR2 gating by inducing conformational changes of the channel protein itself. In line with these findings, Tateishi et al. (2009) found that JTV519 was able to correct domain unzipping between the central and N-terminal domain of the RyR2 to stabilize the channel.
A gain-of-function defect of the RyR2 can also result from point mutations in the channel protein, as found in patients presenting with catecholaminergic polymorphic ventricular tachycardia (CPVT). In knock-in mice carrying the RyR2R4496C mutation, JTV519 (1 µmol·L−1) was not able to prevent arrhythmias induced by the β-adrenoceptor agonist isoprenaline, either in vitro or in vivo (Liu et al., 2006). We have recently shown in the same model of CPVT, that delayed afterdepolarizations and spontaneous action potentials can also be induced by ouabain. Interestingly, ouabain-induced cellular arrhythmias could be prevented by JTV519 treatment (Sedej et al., 2010), suggesting that SR Ca2+ leak triggered by isoprenaline and store-overload induced SR Ca2+ leak induced by ouabain result from different pathophysiological states of the RyR2 complex. Based on these findings, we chose to further explore the effects of JTV519 underlying ouabain-induced arrhythmias in the mouse model. JTV519 significantly attenuated the increase in Ca2+ spark frequency. In JTV519-treated cells ouabain still induced an increase in diastolic cytosolic [Ca2+] which may be attributed to NCX-mediated Ca2+ influx upon ouabain-induced Na+/K+-ATPase inhibition. However, diastolic cytosolic [Ca2+] with ouabain was significantly lower in JTV519 treated cells as compared with control cells, reflecting less contribution of SR Ca2+ leak to diastolic cytosolic [Ca2+] in the presence of JTV519. On the other hand, JTV519 also reduced SR Ca2+ content. In rat cardiomyocytes, JTV519 dose-dependently reduced the rate of Ca2+ uptake by SERCA (Loughrey et al., 2007); however, at 1 µmol·L−1 JTV519, SR Ca2+ content remained unchanged in rats as well as in dogs (Kohno et al., 2003; Loughrey et al., 2007). Similarly, the authors reported unchanged amplitude of the L-type Ca2+ current at 1 µmol·L−1. In contrast, in our study, L-type Ca2+ currents were significantly reduced (Figure 5F), and we found a trend towards a small decrease in SERCA activity already at 1 µmol·L−1 JTV519. Our results suggest that in murine cardiomyocytes, L-type Ca2+ channels and maybe also SERCA are more susceptible to inhibition by JTV519.
Given that SR Ca2+ leak is a function of SR [Ca2+] (Shannon et al., 2002), reduced SR Ca2+ load with JTV519 may contribute to its anti-arrhythmic effect in conditions of Ca2+ overload. However, in cardiomyocytes matched for SR Ca2+ load in the presence of ouabain, Ca2+ spark frequency was still significantly lower in JTV519-treated cardiomyocytes, confirming that JTV519 reduced RyR2-mediated Ca2+ leak, independently of SR Ca2+ content. In line with a specific effect on RyR2 open time, at similar SR Ca2+ load, the duration of individual Ca2+ sparks was significantly shorter with JTV519 (Figure 4E/F).
Altered gating properties of the RyR2 may be related to an altered sensitivity to SR luminal [Ca2+] (Kubalova et al., 2005) or altered sensitivity to triggering [Ca2+]i at the cytosolic site of the receptor (Marx et al., 2000). While Ca2+ spark frequency at similar SR Ca2+ was reduced with JTV519, we found that the propagation velocity of cytosolic Ca2+ waves was unchanged despite lower SR Ca2+ load in the presence of JTV519. Thus, at an average amplitude of the caffeine-induced [Ca2+]i transients of ∼1200 nM, reflecting elevated SR [Ca2+] (Howlett et al., 2006; Ozdemir et al., 2008; Dybkova et al., 2011), cytosolic Ca2+ release events of similar amplitude (Figure 4C) were at least as effective in triggering Ca2+ release from neighbouring RyR2 in the absence and presence of JTV519. Our results suggest that luminal rather than cytosolic Ca2+ sensitivity of the RyR2 was reduced with JTV519. In line with this assumption, fractional Ca2+ release was unchanged by JTV519 (Figure 5C). Simultaneous measurement of L-type Ca2+ currents and cytosolic [Ca2+] transients revealed that [Ca2+]; transients of similar amplitude were triggered by significantly lower Ca2+ influx in the presence of JTV519 (Figure 5), suggesting that the gain of Ca2+-induced Ca2+ release was increased during treatment with JTV519. Inhibition of L-type Ca2+ channels by JTV519 has been reported earlier (Inagaki et al., 2000; Loughrey et al., 2007). In contrast, however, Loughrey et al. (2007) reported that, in rat cardiomyocytes, acute (90 s) application of JTV519 (1 µmol·L−1) induced a reduction in Ca2+ wave propagation velocity (permeabilized cells) and fractional Ca2+ release (whole-cell patch clamp). Inhibition of L-type Ca2+ currents was only observed at higher concentrations of JTV519 (3 µmol·L−1). The reason for these differences are unclear but may be related to species differences, time dependent effects (1 h pre-incubation with JTV519 in the present study) and recording conditions (intact vs. permeabilized/dialysed cells, 3 mmol·L−1 external [Ca2+] in the present study).
In a recent study, Toischer et al. (2010) investigated the acute effects of JTV519 on diastolic function in terminally failing human myocardium. In failing trabeculae, 1 µmol·L−1 JTV519 decreased systolic (developed) tension, whereas at a lower dose (0.3 µmol·L−1) developed tension was increased. JTV519 decreased diastolic tension with higher external [Ca2+] or higher stimulation frequency, indicating that diastolic dysfunction, was attenuated by JTV519. In the present study, we characterized for the first time the effects of JTV519 in non-failing human ventricular muscle strips. As in failing human myocardium (Toischer et al., 2010) and in accordance with what we observed in murine cardiomyocytes, 1 µmol·L−1 JTV519 was negatively inotropic. The positive inotropic effect of ouabain, however, was significantly enhanced by JTV519, suggesting that in the presence of JTV519, more of the ouabain-induced additional intracellular Ca2+ was retained in the SR for release with each beat. Accordingly, JTV519-treated cardiomyocytes were relatively protected from diastolic dysfunction (prolonged relaxation time, Figure 6), reflecting cytosolic Ca2+ accumulation at higher doses of ouabain. Thus, in non-failing human ventricular myocardium, JTV519 protects from diastolic dysfunction in vitro induced by cardiomyocyte Ca2+ overload.
In summary, increased PKA- or CaMKII-dependent phosphorylation at Ser2808/Ser2814 of mouse RyR2 was not required for increased SR Ca2+ leak during acute ouabain exposure. JTV519 decreased SR Ca2+ leak induced by ouabain through its specific effects on RyR2 opening properties, and protected the ventricular myocardium from Ca2+-overload-induced diastolic dysfunction in vitro and from arrhythmogenic events. Our results extend the potential use of JTV519 in conditions of acute cellular Ca2+ overload.
Acknowledgments
The authors thank Eva-Maria Gutschi for excellent technical support. This work was supported by the Molecular Medicine PhD program of the Medical University of Graz (BP) and the European Union 6th Framework Programme for Research and Technological Development (EU-FP6) grant LSHM-CT-2005–018802 (CONTICA).
Glossary
- λcaff
decay rate of the caffeine-induced cytosolic [Ca2+]i transient
- λstim
decay rate of the electrically stimulated cytosolic [Ca2+]i transient
- CalA
calyculin A
- CaMKII
Ca2+-calmodulin-dependent kinase II
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- EC
excitation-contraction
- F0
minimal fluorescence signal during end-diastole (treatment phase)
- F0
bsl, minimal fluorescence signal during end-diastole (baseline)
- FDHM
full duration at half maximum
- Fdiast
normalized diastolic Ca2+-dependent fluorescence signal
- Fpeak
systolic peak amplitude of the Ca2+-dependent fluorescence signal
- FWHM
full width at half maximum
- Lmax
length at maximum steady-state force
- pRyR
phosphorylated RyR
- RyR2
ryanodine receptor 2
- SERCA
sarco-endoplasmic reticulum Ca2+ ATPase
- SR
sarcoplasmic reticulum
- TwitchSERCA%
percentage contribution of SERCA to cytosolic [Ca2+]i transient decay
Conflict of interest
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Appendix S1 Extended methods section.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. The Netherlands: Kluver Academic Publishers; 2001. [Google Scholar]
- Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- Blayney LM, Lai FA. Ryanodine receptor-mediated arrhythmias and sudden cardiac death. Pharmacol Ther. 2009;123:151–177. doi: 10.1016/j.pharmthera.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, et al. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–e82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- Dipla K, Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ Res. 1999;84:435–444. doi: 10.1161/01.res.84.4.435. [DOI] [PubMed] [Google Scholar]
- Dybkova N, Sedej S, Napolitano C, Neef S, Rokita AG, Hunlich M, et al. Overexpression of CaMKIIdeltac in RyR2R4496C+/- knock-in mice leads to altered intracellular Ca2+ handling and increased mortality. J Am Coll Cardiol. 2011;57:469–479. doi: 10.1016/j.jacc.2010.08.639. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Kashimura T, O'Neill SC, Venetucci LA, Trafford AW. What role does modulation of the ryanodine receptor play in cardiac inotropy and arrhythmogenesis? J Mol Cell Cardiol. 2009;46:474–481. doi: 10.1016/j.yjmcc.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Fowler MR, Naz JR, Graham MD, Bru-Mercier G, Harrison SM, Orchard CH. Decreased Ca2+ extrusion via Na+/Ca2+ exchange in epicardial left ventricular myocytes during compensated hypertrophy. Am J Physiol Heart Circ Physiol. 2005;288:H2431–H2438. doi: 10.1152/ajpheart.01069.2004. [DOI] [PubMed] [Google Scholar]
- Greiser M, Neuberger HR, Harks E, El-Armouche A, Boknik P, de Haan S, et al. Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J Mol Cell Cardiol. 2009;46:385–394. doi: 10.1016/j.yjmcc.2008.11.012. [DOI] [PubMed] [Google Scholar]
- Heinzel FR, Bito V, Volders PG, Antoons G, Mubagwa K, Sipido KR. Spatial and temporal inhomogeneities during Ca2+ release from the sarcoplasmic reticulum in pig ventricular myocytes. Circ Res. 2002;91:1023–1030. doi: 10.1161/01.res.0000045940.67060.dd. [DOI] [PubMed] [Google Scholar]
- Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, et al. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338–346. doi: 10.1161/CIRCRESAHA.107.160085. [DOI] [PubMed] [Google Scholar]
- Hirose M, Stuyvers BD, Dun W, Ter Keurs HE, Boyden PA. Function of Ca2+) release channels in Purkinje cells that survive in the infarcted canine heart: a mechanism for triggered Purkinje ectopy. Circ Arrhythm Electrophysiol. 2008;1:387–395. doi: 10.1161/CIRCEP.107.758110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett SE, Grandy SA, Ferrier GR. Calcium spark properties in ventricular myocytes are altered in aged mice. Am J Physiol Heart Circ Physiol. 2006;290:H1566–H1574. doi: 10.1152/ajpheart.00686.2005. [DOI] [PubMed] [Google Scholar]
- Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DJ, Jones PP, Wang R, Chen W, Bolstad J, Chen K, et al. K201 (JTV519) suppresses spontaneous Ca2+ release and [3H]ryanodine binding to RyR2 irrespective of FKBP12.6 association. Biochem J. 2007;404:431–438. doi: 10.1042/BJ20070135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K, Kihara Y, Izumi T, Sasayama S. The cardioprotective effects of a new 1,4-benzothiazepine derivative, JTV519, on ischemia/reperfusion-induced Ca2+ overload in isolated rat hearts. Cardiovasc Drugs Ther. 2000;14:489–495. doi: 10.1023/a:1007884905461. [DOI] [PubMed] [Google Scholar]
- Kaneko N, Matsuda R, Hata Y, Shimamoto K. Pharmacological characteristics and clinical applications of K201. Curr Clin Pharmacol. 2009;4:126–131. doi: 10.2174/157488409788184972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno M, Yano M, Kobayashi S, Doi M, Oda T, Tokuhisa T, et al. A new cardioprotective agent, JTV519, improves defective channel gating of ryanodine receptor in heart failure. Am J Physiol Heart Circ Physiol. 2003;284:H1035–H1042. doi: 10.1152/ajpheart.00722.2002. [DOI] [PubMed] [Google Scholar]
- Kubalova Z, Terentyev D, Viatchenko-Karpinski S, Nishijima Y, Gyorke I, Terentyeva R, et al. Abnormal intrastore calcium signaling in chronic heart failure. Proc Natl Acad Sci U S A. 2005;102:14104–14109. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D'hooge J, et al. Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. 2009;105:876–885. doi: 10.1161/CIRCRESAHA.109.206276. [DOI] [PubMed] [Google Scholar]
- von Lewinski D, Bruns S, Walther S, Kogler H, Pieske B. Insulin causes [Ca2+]i-dependent and [Ca2+]i-independent positive inotropic effects in failing human myocardium. Circulation. 2005;111:2588–2595. doi: 10.1161/CIRCULATIONAHA.104.497461. [DOI] [PubMed] [Google Scholar]
- von Lewinski D, Zhu D, Khafaga M, Kockskamper J, Maier LS, Hasenfuss G, et al. Frequency-dependence of the slow force response. Front Biosci. 2008;13:7202–7209. doi: 10.2741/3222. [DOI] [PubMed] [Google Scholar]
- Lindner M, Brandt MC, Sauer H, Hescheler J, Bohle T, Beuckelmann DJ. Calcium sparks in human ventricular cardiomyocytes from patients with terminal heart failure. Cell Calcium. 2002;31:175–182. doi: 10.1054/ceca.2002.0272. [DOI] [PubMed] [Google Scholar]
- Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2R4496C knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- Loughrey CM, Otani N, Seidler T, Craig MA, Matsuda R, Kaneko N, et al. K201 modulates excitation-contraction coupling and spontaneous Ca2+ release in normal adult rabbit ventricular cardiomyocytes. Cardiovasc Res. 2007;76:236–246. doi: 10.1016/j.cardiores.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed U, Napolitano C, Priori SG. Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2007;18:791–797. doi: 10.1111/j.1540-8167.2007.00766.x. [DOI] [PubMed] [Google Scholar]
- Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, et al. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. 2010;106:1134–1144. doi: 10.1161/CIRCRESAHA.109.203836. [DOI] [PubMed] [Google Scholar]
- Ozdemir S, Bito V, Holemans P, Vinet L, Mercadier JJ, Varro A, et al. Pharmacological inhibition of na/ca exchange results in increased cellular Ca2+ load attributable to the predominance of forward mode block. Circ Res. 2008;102:1398–1405. doi: 10.1161/CIRCRESAHA.108.173922. [DOI] [PubMed] [Google Scholar]
- Picht E, DeSantiago J, Huke S, Kaetzel MA, Dedman JR, Bers DM. CaMKII inhibition targeted to the sarcoplasmic reticulum inhibits frequency-dependent acceleration of relaxation and Ca2+ current facilitation. J Mol Cell Cardiol. 2007;42:196–205. doi: 10.1016/j.yjmcc.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedej S, Heinzel FR, Walther S, Dybkova N, Wakula P, Groborz J, et al. Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc Res. 2010;87:50–59. doi: 10.1093/cvr/cvq007. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- Tateishi H, Yano M, Mochizuki M, Suetomi T, Ono M, Xu X, et al. Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovasc Res. 2009;81:536–545. doi: 10.1093/cvr/cvn303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toischer K, Lehnart SE, Tenderich G, Milting H, Korfer R, Schmitto JD, et al. K201 improves aspects of the contractile performance of human failing myocardium via reduction in Ca2+ leak from the sarcoplasmic reticulum. Basic Res Cardiol. 2010;105:279–287. doi: 10.1007/s00395-009-0057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, et al. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005;111:2025–2032. doi: 10.1161/01.CIR.0000162461.67140.4C. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004a;304:292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004b;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, et al. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci U S A. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266:11144–11152. [PubMed] [Google Scholar]
- Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, et al. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ Res. 2005;96:847–855. doi: 10.1161/01.RES.0000163276.26083.e8. [DOI] [PubMed] [Google Scholar]
- Xiao B, Tian X, Xie W, Jones PP, Cai S, Wang X, et al. Functional consequence of protein kinase A-dependent phosphorylation of the cardiac ryanodine receptor: sensitization of store overload-induced Ca2+ release. J Biol Chem. 2007;282:30256–30264. doi: 10.1074/jbc.M703510200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi N, Meissner G. Does Ca2+/calmodulin-dependent protein kinase deltac activate or inhibit the cardiac ryanodine receptor ion channel? Circ Res. 2007;100:293–295. doi: 10.1161/01.RES.0000259327.56377.55. [DOI] [PubMed] [Google Scholar]
- Yang D, Zhu WZ, Xiao B, Brochet DX, Chen SR, Lakatta EG, et al. Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ Res. 2007;100:399–407. doi: 10.1161/01.RES.0000258022.13090.55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.