

Abstract
BACKGROUND AND PURPOSE
Excess morbidity/mortality in rheumatoid arthritis (RA) is associated with increased incidence of cardiovascular disease. In this ‘proof-of-concept’ study, vascular function was characterized in the murine collagen-induced arthritis (mCIA) model, the benchmark choice for evaluation of the pathological processes and assessment of new therapies.
EXPERIMENTAL APPROACH
Mice in the very early stages of arthritis development [and appropriate naïve (non-immunized) age-matched controls] were used in the study. Blood pressure was measured using tail cuff plethysmography. Vascular function in rings of isolated aorta was studied with isometric tension myography. Levels of NO metabolites (NOx), MMP-9 protein and IL-1β in plasma and MMP-9 protein in aortic homogenates were quantified.
KEY RESULTS
Impaired vascular contractile responses in arthritis were unaffected by ex vivo inhibition of NOS (endothelial/neuronal and inducible) or COX activities. Endothelium-dependent and -independent relaxation, plasma NOx and blood pressure were unaffected by arthritis. Plasma and aortic homogenate MMP-9 protein levels were increased significantly in arthritis. Incubation of aortic tissues from naïve control animals with exogenous MMP-9 impaired subsequent contractile responses, mirroring that observed in arthritis. A role for IL-1β in perpetuating contractile dysfunction and increasing aortic MMP-9 was excluded.
CONCLUSIONS AND IMPLICATIONS
These data identify for the first time a relationship between early arthritis and contractile dysfunction and a possible role for MMP-9 therein, in the absence of overt endothelial dysfunction or increased NO production. As such, MMP-9 may constitute a significant target for early intervention in RA patients with a view to decreasing risk of cardiovascular disease.
Keywords: arthritis, cardiovascular disease, aorta: contractile dysfunction, matrix metalloproteinase 9
Introduction
It has long been established that excess morbidity and mortality is associated with rheumatoid arthritis (RA). Furthermore, significant evidence now suggests that an increased incidence of cardiovascular disease (CVD) is responsible for this outcome (Roman and Salmon, 2007; Meune et al., 2009; Gabriel, 2010). More specifically, coronary artery disease has been identified as one of the most common causes of death in RA (Solomon et al., 2003; Maradit-Kremers et al., 2005) and seems to occur at a younger age than in the general population.
In a recent study of RA patients, whilst traditional cardiovascular risk (CVR) factors were shown to predict a new cardiovascular event, the latter was precipitated by high disease activity (Innala et al., 2011). Whether traditional risk factors for CVD are more common, or simply more deleterious, in RA patients remains open to investigation and debate. What seems clear is that the increased CVR in RA is caused by a combination of such factors and the persistently high levels of inflammation that characterizes these individuals. Interestingly a recent study suggests that RA now equals type 2 diabetes, a disease associated with low-grade systemic inflammation (Kolb and Mandrup-Poulsen, 2010), as an independent risk factor for CVD (Peters et al., 2009). However, although the parallels between inflammatory/autoimmune diseases and other ‘atherosclerotic vascular diseases’ are clear, the mechanisms underlying increased CVD in RA patients are ill-defined.
To investigate such mechanisms in humans is very difficult, if not impossible, and under such circumstances, appropriate animal models are useful for surrogate studies. With regard to the evaluation of immunological/pathological processes in RA, and importantly the assessment of new therapeutic strategies, the murine collagen-induced arthritis (mCIA) model is the benchmark choice (Hegen et al., 2008; Bevaart et al., 2010). Indeed, this is a highly relevant model due largely to its histopathology, inflammatory, immune and macroscopic features (Cho et al., 2007). Significantly, mCIA represents a first presentation autoinflammatory insult to the joint and not the relapsing/remitting condition seen in well-established human RA. Since increased CVR appears to precede RA onset (Young et al., 2007), this model offers an ideal opportunity to identify the very earliest pathological changes within the vasculature that could subsequently contribute to full-blown CVD.
Given the high CVD burden associated with RA, it is surprising that little is known regarding the vascular phenotype associated with mCIA. Indeed, the assessment of changes in vascular function during the time course of such a relevant inflammatory arthritis model is essential if we are to learn more about these processes in RA. In this ‘proof-of-concept’ study, we investigated joint pathology, systemic inflammation and vascular function in mice with mild early-onset disease to identify possible pathological mechanisms, which underlie CVD risk in RA. For the first time, the data identify a relationship between arthritic disease and contractile dysfunction that appears in the absence of overt endothelial dysfunction or increased NO production. Moreover, a possible association with increased MMP-9 at this early phase of arthritis development is observed. As such, these observations have significant potential to influence clinical practice and to affect the prevention and treatment of CVD in RA patients.
Methods
Animals and induction of mCIA
All animal care and experimental procedures complied with the United Kingdom Animals (Scientific Procedures) Act 1986 and were under the authority of Home Office Personal and Project Licences. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). A total of 64 animals were used in these experiments. Inflammatory arthritis was induced in male DBA/1 mice (aged 6–8 weeks) as previously described (Bull et al., 2008; Nowell et al., 2009). Briefly, on day 0, mice were immunized by intradermal injection with 100 µL of chick collagen (2 mg·mL−1) dissolved in 2.5 mL of 0.02% (v/v) acetic acid and emulsified with an equal volume of complete Freund's adjuvant. Each animal received a second identical injection on day 21, after which arthritis became progressively evident. Arthritis severity was scored in each paw using a scale ranging from 0 to 4, as follows: 0, normal; 1, mild/moderate erythema and swelling; 2, severe erythema and swelling affecting the entire paw joint; 3, up to three joints affected by arthritis; 4, greater than three joints affected by arthritis. Every day after mCIA induction, individual paw scores were summed for each mouse and the combined paw score (CPS) for each animal gave the clinical score. Animals were also weighed daily throughout this process.
The severity of arthritis at a defined time point was very variable and precluded daily comparative analysis. In previous studies with this model, animals were allowed to progress to the maximum severity or time allowed by the Home Office Project Licence (up to CPS = 15 or day 35 respectively) (Bull et al., 2008; Nowell et al., 2009; Evans et al., 2011). Therefore, to investigate vascular changes in the very early stages of arthritis development, mice were killed (by inhalation of CO2) between days 24 and 27 when mild disease (CPS = 1–5) was evident. A second group of naïve (non-immunized) age-matched mice were used as controls throughout.
Blood pressure measurement
At time points coincident with isometric tension analysis (see below), systolic blood pressure (BP) measurements were obtained in control and mildly arthritic mice using a Harvard mouse tail photoplethysmography pressure monitor (Harvard Apparatus, Edenbridge, UK).
Collection of experimental samples
Blood (approximately 0.2–1 mL) was collected via cardiac puncture and transferred to EDTA-coated vacutainers (BD, Oxford, UK) on ice. Samples were centrifuged for 20 min at 16 464×g at 4°C, and the resulting plasma was stored at −80°C for further analysis.
Subsequently, the thoracic aorta was exposed, vented just above the renal arteries and the left ventricle slowly perfused with ∼1 mL of gassed (95% O2/5% CO2) Krebs buffer (see below) to remove blood. The vessel was then carefully dissected, placed in fresh Krebs buffer and loose fat and connective tissue removed. Aortae were then used either for myography or snap-frozen in liquid N2 before subsequent homogenization for protein analysis (see below).
Hind paws were removed and fixed in neutral buffered formal saline prior to processing for histological evaluation.
Histological assessment of arthritis
All joints were decalcified with 10% (v/v) formic acid for 2 weeks at 4°C before embedding in paraffin wax as previously described (Bull et al., 2008; Nowell et al., 2009). Serial 7 µm thick sections were taken and stained routinely with H&E. Two independent observers, unaware of the treatments, scored the sections for synovial hyperplasia (0–3), cellular infiltration (0–5), cellular exudate (0–3), and cartilage and bone degradation (0–3), with 0 representing a normal joint.
Vascular function
Isometric tension studies using a myograph are commonly used to assess vascular function ex vivo in isolated blood vessels. Such experiments were carried out as described previously (Cai et al., 2005). Briefly, the thoracic aorta, isolated from control and mildly arthritic mice, was cut into rings of 2 mm length and mounted in separate wells of a Mulvany myograph (Danish Myo-Technology, Aarhus, Denmark) and bathed in Krebs buffer (composition (mM): NaCl 118, KCl 4.7, KH2PO4 1.2, MgSO4.7H2O 1.2, NaHCO3 25, glucose 11, CaCl2·7H2O 1.5) at 37°C gassed with 95% O2/5% CO2. Myograph output was recorded and analysed using Myodaq and Myodata (Aarhus, Denmark) software respectively.
Following establishment of a 5 mN baseline tension and subsequent equilibration, tissues were exposed to 60 mM K+ for 5 min. After washing and re-equilibration, tissues were sequentially exposed to single concentrations of 5-HT (the most effective constrictor agonist in preliminary experiments; data not shown) and ACh (both 1 µM) to establish viable agonist-induced contraction and relaxation responses respectively. Again following washing and re-equilibration, tissues were initially constricted to their maximum by adding increasing concentrations of 5-HT (1 nM to 10 µM). Subsequently, tissues were washed, and then those from arthritic animals were re-constricted to 70–80% of the previous maximum with appropriate concentrations of 5-HT (0.3–1 µM). At the same time, control tissues were constricted (with 5-HT, 0.1–0.3 µM) to match these levels of agonist-induced tension. At the contraction plateau, vessels were subjected to increasing concentrations of ACh (1 nM to 10 µM) to assess endothelium-dependent relaxation. Following washing and appropriate re-constriction (as above), endothelium-independent relaxation was assessed by exposing the tissues to increasing concentrations of the NO donor sodium nitroprusside (SNP; 1 nM to 10 µM). Responses to ACh and SNP were then calculated as a percentage of the 5-HT-induced tone.
In other experiments, the above responses to K+, 5-HT and ACh were repeated in the presence of either the inhibitor of inducible NOS (iNOS), N-(3-(aminomethyl)benzyl)acetamidine (1400W, 10 µM), the non- selective NOS inhibitor NG-nitro-l-arginine methyl ester (l -NAME, 300 µM) or the non-selective COX inhibitor indomethacin (1 µM). Each inhibitor was added 30 min before the first exposure to K+ and then re-added after each subsequent washing step.
NO metabolites in blood
It is possible that a global systemic change in NO production, from iNOS or eNOS subsequent to increased production of inflammatory mediators, could contribute to changes in vascular function. A common method of assessing in vivo NO production is the determination of plasma concentrations of the more stable oxidative products of NO metabolism, namely nitrite and nitrate, described as total NO metabolites (NOx). As such, in appropriate plasma samples, NOx (µM) was measured by ozone-based chemiluminescence as previously described (Pinder et al., 2009) and using the Sievers® 280i NO Analyzer (NOA, Analytix, Boldon, UK).
Homogenization of aortae
The frozen aorta was placed in 100 µL of ice cold RIPA buffer in a small glass homogenization tube (VWR International, Lutterworth, UK) and homogenized on ice for 6 × 10 s bursts with a close-fitting glass rod. The homogenate was then centrifuged for 30 min at 16 464×g at 4°C, and the resulting supernatant was frozen at −80°C for further use.
MMP-9/IL-1β by elisa
Levels of the gelatinase MMP-9 in plasma samples and aortic homogenates were detected by commercial elisa (mouse MMP-9, total: pro-MMP-9, active-MMP-9 and MMP-9 bound to TIMPs). Levels of the potent endogenous pyrogen IL-1β were also measured in plasma samples by commercial elisa (Quantikine elisa Kit, R&D System, Abingdon, UK). Concentrations in plasma were expressed as ng·mL−1 (MMP-9) and pg·mL−1 (IL-1β). Levels in aortic homogenates were normalized to sample protein content (see below) and expressed as pg·mg−1 protein.
Gelatin zymography
Gelatine zymography was used in the specific quantitation of the levels of pro- and active MMP-9 in aortic tissues (George et al., 1997). Briefly, frozen aortae were homogenized in ice-cold lysis buffer (50 mmol·L−1 Tris–HCl, pH 6.8, 10% glycerol and 1% SDS) then centrifuged at 10 000×g for 5 min, and the supernatant was collected for analysis. Subsequently, equal amounts of tissue extract protein (15 µg per sample) were loaded onto 10% SDS-PAGE gels containing 1% gelatin. After Triton-X100 exchange, incubation overnight in developing buffer and staining with Coomassie Blue, bands of lysis were quantified by densitometry and expressed as arbitrary optical units.
Protein detection
For most samples, protein content was measured using the commercially available Coomassie Plus assay kit (Thermoscientific, Epsom, UK). For the zymography, the Micro BCA assay kit (Thermoscientific) was used.
MMP-9, IL-1β and vascular contractility
In order to investigate the direct effect of MMP-9 or IL-1β on aortic contractility, tissues from control non-immunized mice were incubated ex vivo for 24 h (at 37°C in an atmosphere of 5% CO2 in air) in DMEM (Life Technologies, Paisley, UK) (supplemented with fetal calf serum 1% v/v, benzylpenicillin 25 U·mL−1, streptomycin 25 µg·mL−1 and glutamine 200 mM) in the absence or presence of either exogenous MMP-9 (75 ng·mL−1, approximately 3-fold excess of that observed in mCIA-derived plasma, but similar to that found in mCIA aortic homogenates; see Figure 3A and B) or IL-1β (R&D Systems; 70 or 700 pg·mL−1 approximately equal and 10-fold excess of that observed in mCIA-derived plasma respectively; see Figure 6A). Subsequently, tissues were washed in fresh warmed Krebs, removing the MMP-9 or IL-1β, and mounted on the myograph for vascular function assessment as described above.
Figure 3.
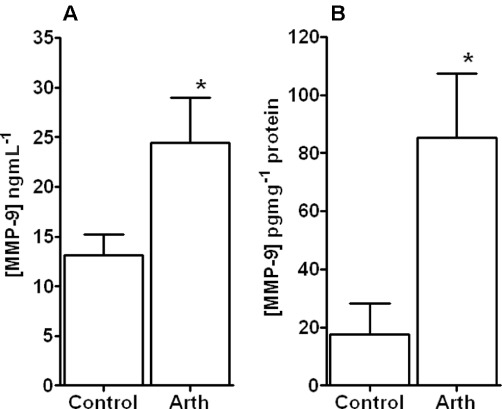
MMP-9 protein levels in plasma and aorta. Graph showing MMP-9 levels in plasma samples (A) and homogenates of aorta (B) from control animals and those with mild inflammatory arthritis. A significant increase in plasma and aortic MMP-9 protein was observed following the development of arthritis. *P < 0.05 significantly different from control, n≥ 4 for all groups.
Figure 6.
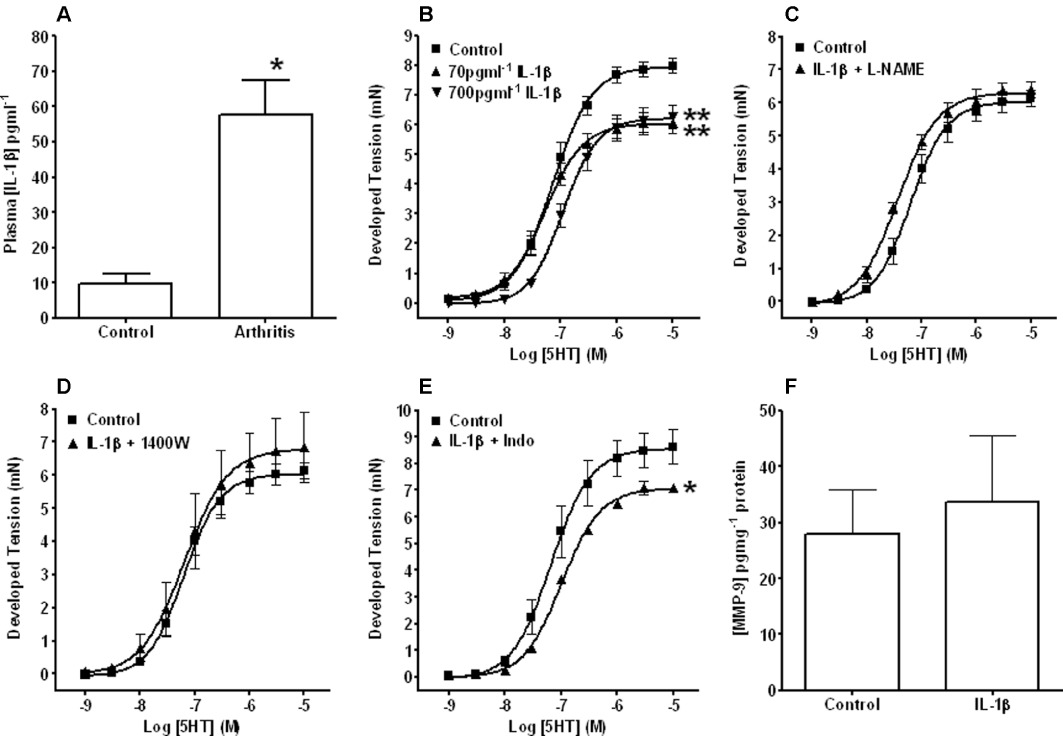
Potential role of IL-1β in mediating contractile dysfunction. Graphs showing plasma levels of IL-1β from control and arthritic animals (A) and vasoconstriction concentration–response curves to 5-HT in aortic tissues taken from naïve non-immunized animals and incubated for 24 h in the absence (Control) and presence of either 70 or 700 pg·mL−1 IL-1β alone (B) or IL-1β (700 pg·mL−1) in the presence of L-NAME (C), 1400W (D) or indomethacin (Indo) (E). Also shown are elisa-derived levels of MMP-9 in aortic tissues taken from naïve non-immunized animals and incubated for 24 h in the absence (control) and presence of 700 pg·mL−1 IL-1β (F). Plasma levels of IL-1β were elevated in mCIA animals and ex vivo incubation with IL-1β impaired contractile responses to 5-HT. However, the latter was prevented by inhibitors of NO/H2O2 and (to a lesser extent) vasodilatory prostanoids. Moreover, the observed ex vivo contractile dysfunction was not associated with a change in MMP-9 protein. *P < 0.05, **P < 0.01 significantly different from the appropriate control, n≥ 4 for all groups.
In the case of IL-1β (700 pg·mL−1), experiments were repeated in the presence of either 1400W, l -NAME or indomethacin as described above.
IL-1β and vascular MMP-9 production
To investigate the direct affect of IL-1β on aortic MMP-9 production, tissues from control non-immunized mice were incubated ex vivo for 24 h (as described above) in the absence or presence of IL-1β (700 pg·mL−1). Subsequently, tissues were washed in fresh warmed Krebs, snap-frozen in liquid N2, homogenized and MMP-9 concentrations were measured by elisa as described above.
Statistical analysis
All data are expressed as means ± SEM, n≥ 3 for all groups (not all animals were used for each experiment). Constriction responses to K+ and 5-HT are expressed as developed tension in mN. Relaxation responses to ACh are expressed as a percentage of the appropriate 5-HT-induced constriction. All concentration–response data were fitted to sigmoid curves using GraphPad Prism software. Rmax (maximum response) and EC50 (concentration to produce 50% of maximum response) values for responses to 5-HT and ACh and all other data were compared by t-test or one-way anova followed by Student–Newman–Keul's multiple range test as appropriate. Differences were considered significant where P < 0.05.
Materials
All chemicals were supplied by Sigma-Aldrich, (Gillingham, UK) unless otherwise stated.
Results
Clinical and histological assessment of arthritis
To assess disease progression of mCIA, paws were clinically scored with regard to the criteria outlined above. The average CPS was 2.3 ± 0.3 (CPS range = 1–4, n= 17).
The clinical progression of arthritis was reflected in the mild histological changes within the inflamed paws. Slight thickening of the synovial membrane (0.5 ± 0.1, range = 0–1, n= 17), a small influx of inflammatory cells (1.3 ± 0.2, range = 0–3, n= 15), a minor increase in the level of joint space exudate (0.2 ± 0.1, range = 0–1, n= 17) and minimal cartilage and bone damage (0.2 ± 0.1, range = 0–1, n= 17) were observed.
In age-matched control animals, all the above parameters were scored as zero by definition.
Body weight
No significant differences in body weight were observed within or between groups of animals (mCIA; weight at day 21, 21.1 ± 0.5 g, weight at termination, 20.3 ± 0.6 g, both n= 17: age-matched controls; weight at day 21, 20.9 ± 0.4 g, weight at termination, 21.2 ± 0.4 g, both n= 17).
Blood pressure
No change in BP was observed between control (112.5 ± 4.38 mmHg) and mildly arthritic animals (115.8 ± 4.25 mmHg) (both n= 6).
Vascular contractile function
Following exposure to 60 mM K+ for 5 min, Rmax values for tissues from arthritic animals (2.21 ± 0.16 mN) were significantly (P < 0.01) depressed compared with those from non-immunized controls (3.32 ± 0.15 mN) (both n= 5). Subsequent Rmax values for contraction responses to 5-HT were also significantly lower in tissues from arthritic mice when compared with controls (Figure 1A). No differences in EC50 values were observed.
Figure 1.
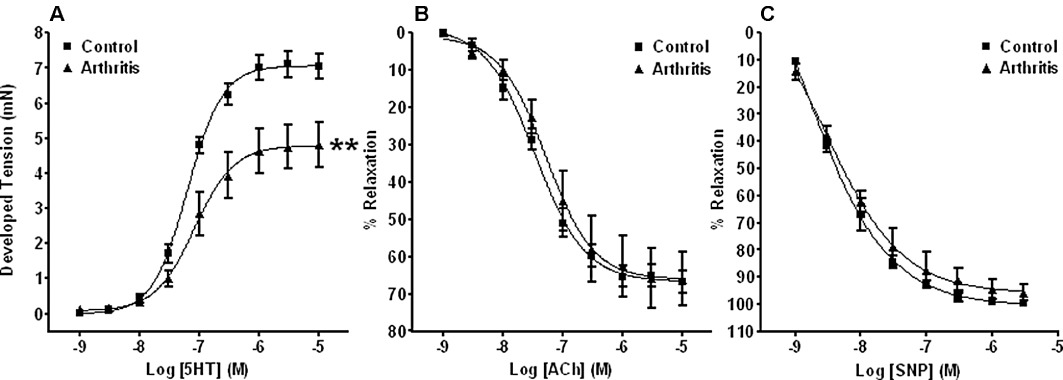
Assessment of vascular function in rings of isolated aorta. Graphs showing vasoconstriction concentration–response curves to 5-HT (A) and relaxation concentration–response curves to ACh (B) or SNP (C) (following pre-constriction to 70–80% of their appropriate Rmax response to 5-HT) in aortic rings from control animals and those with mild inflammatory arthritis. Impaired contraction responses to 5-HT were observed in the presence of intact endothelium-dependent and -independent relaxation responses to ACh and SNP respectively. **P < 0.01 significantly different from control, n≥ 5 for all groups.
Endothelium-dependent relaxation
Because the extent to which tissues are constricted can have a significant effect on their subsequent ability to relax (more constriction/less relaxation and vice versa), tissues from mildly arthritic animals were again constricted to 70–80% of their appropriate Rmax response to 5-HT. Importantly, control tissues were constricted to corresponding levels by altering the concentration of 5-HT applied (as described above). Subsequently, no differences in relaxation responses to ACh were observed between tissues from control and mildly arthritic mice (Figure 1B). In other experiments where tissues were exposed to the same concentration of 5-HT (1 µM), and thus constricted to different levels, no differences in relaxation responses were observed (data not shown).
Endothelium-independent relaxation
Relaxation responses to increasing concentrations (1 nM to 10 µM) of the NO donor SNP were similar in tissues from control and mildly arthritic animals, and no significant differences were identified (Figure 1C).
Constriction following NOS or COX inhibition
Excess production of NO from either iNOS or endothelial (eNOS) or neuronal (nNOS) NOS, vasodilatory prostanoids from COX or indeed hydrogen peroxide (H2O2) from nNOS (Capettini et al., 2008), in the face of acute inflammation may affect vascular smooth muscle (VSM) contraction. Therefore, in separate experiments, contractile responses to 5-HT were evaluated in the presence of their respective inhibitors, namely 1400W (iNOS), l -NAME (both eNOS and nNOS) and indomethacin (COX). In control tissues, contractile responses to 5-HT were not affected by the presence of any of these agents (data not shown). Figure 2 shows that none of the inhibitors were able to reverse the impaired contractile responses to 5-HT in tissues from mildly arthritic animals.
Figure 2.
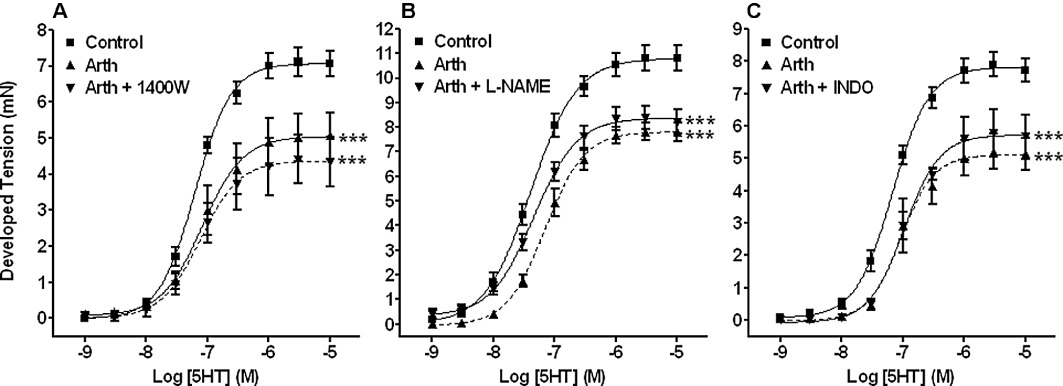
Assessment of contractile function in rings of isolated aorta following iNOS, eNOS and COX inhibition. Graphs showing vasoconstriction concentration–response curves to 5-HT in aortic tissues from control animals and those with mild inflammatory arthritis, the latter in the absence or presence of the iNOS inhibitor 1400W, the eNOS inhibitor L-NAME or the non-selective COX inhibitor indomethacin (INDO). The impaired responses to 5-HT were still evident in the presence of the various inhibitors suggesting that excess NO, H2O2 and vasodilatory prostanoids do not mediate this contractile dysfunction. ***P < 0.001 significantly different from control, n≥ 3 for all groups.
NO metabolites
No differences were observed between NOx levels in plasma samples from control (1.16 ± 0.34 µM, n= 7) and mildly arthritic animals (1.23 ± 0.23 µM, n= 9).
Plasma and aorta levels of MMP-9
In plasma samples and aorta homogenates, concentrations of total MMP-9 protein, as measured by elisa, were seen to increase significantly (P < 0.01) following the onset of arthritis (Figure 3A and B respectively). In the arthritic group, aortic MMP-9 levels increased by almost fivefold above control compared with the approximately twofold increase seen in plasma samples. Moreover, the high level of MMP-9 protein in the aortic homogenate was reflected in a significant level of the active form of MMP-9 as measured by zymography (Figure 4).
Figure 4.
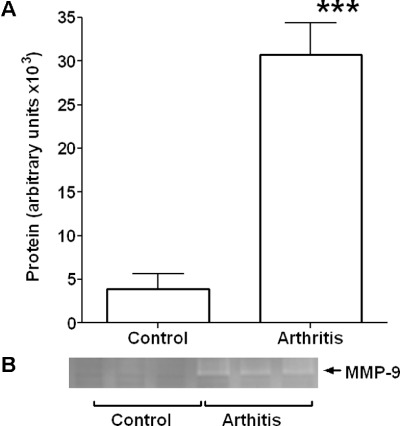
Aortic MMP-9 activity. Graph showing densitometry data (A) and representative zymograph (B) for MMP-9 activity in homogenates of aorta from control animals and those with mild inflammatory arthritis. A significant increase was observed following the development of arthritis. ***P < 0.001 significantly different from control, n= 6 for all groups.
MMP-9 and ex vivo vascular contractility
A significant decrease in the Rmax responses to both 60 mM K+ and 5-HT (Figure 5A and B respectively) was noted in aortic tissue taken from non-immunized mice following a 24 h pre-incubation period with exogenous MMP-9 (75 ng·mL−1) compared with timed controls. No changes in EC50 values for 5-HT responses were observed. The higher contraction responses observed in this particular series of experiments has been attributed to batch to batch variability in the DBA/1 mice.
Figure 5.
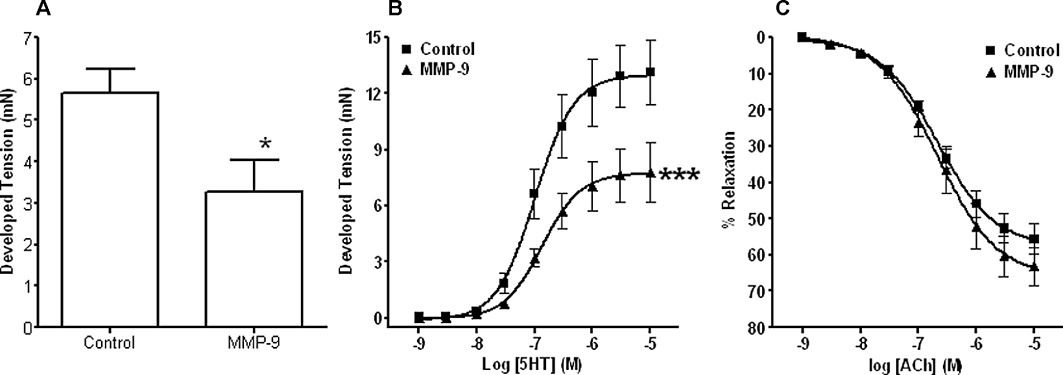
In vitro effect of MMP-9 on vascular function. Graphs showing Rmax responses to high K+ (A), vasoconstriction concentration–response curves to 5-HT (B) and relaxation concentration–response curves to ACh (C) (following pre-constriction to 70–80% of their appropriate Rmax response to 5-HT) in aortic tissues taken from naïve non-immunized animals and incubated in the absence (Control) and presence of exogenous MMP-9 (24 h, 75 ng·mL−1). Impaired contraction responses to K+ and 5-HT were observed in the presence of intact endothelium-dependent relaxation responses to ACh following this incubation. *P < 0.05, ***P < 0.001 significantly different from control, n≥ 3 for all groups.
MMP-9 incubation was without effect on the ACh-induced relaxation responses (Figure 5C).
Plasma levels of IL-1β
Plasma concentrations of IL-1β were seen to increase significantly following the onset of arthritis (Figure 6A).
IL-1β and ex vivo vascular contractility
K+-induced contraction responses (2.88 ± 0.26 mN) in control tissues were not different to those incubated with IL-1β for 24 h (2.98 ± 0.29 mN for 70 pg·mL−1 and 2.40 ± 0.48 mN for 700 pg·mL−1). Conversely, significant (P < 0.01) decreases in Rmax responses to 5-HT were observed with both IL-1β concentrations (Figure 6B). l -NAME and 1400W restored the IL-1β/5HT constriction response to control levels (Figure 6C and D respectively). Indomethacin did not normalise the impaired IL-1β/5HT-induced contractile responses (Figure 6E).
IL-1β and vascular MMP-9 production
In isolated aortic tissues from non-immunized mice, incubation with IL-1β (700 pg·mL−1 for 24 h) had no effect on control MMP-9 levels (Figure 6F).
Discussion
The data presented in this study showed that the impaired contractile responses to both depolarisation with high K+ and the receptor-dependent agonist 5-HT were early vascular pathological changes in mCIA animals. This contractile dysfunction was not associated with a change in BP or affected by ex vivo iNOS and eNOS/nNOS inhibition. Furthermore, plasma NOx levels were similar in both groups, which suggests that excess NO production did not mediate the aberrant vascular contractility observed in this model of inflammatory arthritis. The fact that this vascular dysfunction was unaffected by ex vivo nNOS and COX inhibition suggests that H2O2 (Capettini et al., 2008) and vasodilatory prostanoids were not involved.
Interestingly, contractile dysfunction was not accompanied by overt endothelial dysfunction given that endothelium-dependent relaxation responses to ACh were similar to those seen in tissues from non-immunized control mice. Moreover, relaxation responses to exogenously donated NO were unaffected. Together, these observations suggest that there is ample endothelium-derived NO bioavailability during mCIA, and that the VSM functionally responds to both endogenous and exogenous NO to produce vasodilation. As such, this finding is contrary to what is expected from data already published, given the widely described endothelial dysfunction in patients with established RA (Khan et al., 2010).
Endothelial dysfunction is characterized by reduced NO bioavailability and impaired endothelium-dependent relaxation responses. With regard to causative mechanisms in CVD, such changes constitute well-described primary events and the presence of endothelial dysfunction predicts the risk of future adverse cardiovascular events in older high-risk and younger populations alike (Martin and Anderson, 2009). However, it would seem that, in the mCIA model, pathological changes at the VSM rather than the endothelial level are the first to present, at least functionally.
It is highly likely that the observed departure from the traditional endothelial dysfunction model of CVD development is related to the fact that mCIA represents a first presentation autoinflammatory insult and not the relapsing/remitting condition seen in well-established human RA. Interestingly, recent independent studies also describe the absence of measurable endothelial dysfunction in both new onset inflammatory arthritis (Foster et al., 2012) and newly diagnosed RA (Sodergren et al., 2010; van Eijk et al., 2011) patients. Moreover, in a sub-group of RA patients 18 months after the initial evaluation, a significant increase in common carotid artery intima-media thickening (IMT) was observed in the absence of any change in endothelial function (Sodergren et al., 2010). Given that increased IMT is a result of phenotypical/structural changes at a VSM level, these findings tend to complement those described in the present study. The question remains as to the underlying mechanism(s) responsible for the VSM dysfunction and impaired constrictor responses.
Our data rule out a role for increased NO production or prostanoids in the contractile dysfunction observed. This finding is surprising given the major role of the inflammatory cytokine TNF-α in both RA and iNOS induction. In fact, previous studies using the same model have reported increases in iNOS mRNA (Juarranz et al., 2005) and plasma NOx (Sakaguchi et al., 2004). However, in those studies, animals were killed 14 days (Juarranz et al., 2005) and 12 weeks (Sakaguchi et al., 2004) after the onset of arthritis, and the increases observed were relatively minor. Moreover, the animals in the present study were used well within the times of those described above. Again, this indicates that early non-endothelium/non-NO-dependent events may underlie the observed contractile dysfunction. Any possible role for inflammation-associated excess production of NO may be involved in much later pathological changes in RA.
Whilst it is possible that the systemic inflammatory milieu associated with arthritis could have a negative effect on receptor-mediated VSM contraction, the fact that both depolarization and agonist-induced responses are impaired would argue against this concept. Consequently, the fact that similar contractile dysfunction in an unrelated disease model was accompanied by increases in MMP-9 (Chung et al., 2007) leads us to hypothesise that this gelatinase may play a role in the vascular impairment associated with the mCIA model (see below).
As indicated, the present study demonstrates that levels of MMP-9 protein in plasma samples and aorta homogenates are elevated following the development of arthritis. The much greater change in aortic levels, combined with the significant increase in the active form of MMP-9 in the aortae, strongly suggests a possible role for tissue-derived MMP-9 in the impaired contractile responses described above. Furthermore, the finding that incubation of aortic tissues from naïve control animals with exogenous MMP-9 (at a concentration similar to that seen in mCIA-derived aortic homogenates) produced a detrimental effect on vessel contractility also implies a possible role for circulating MMP-9 in this process. Interestingly, such incubations were without negative effects on subsequent endothelium-dependent relaxation responses, a fact that emphasises the significance of the earliest changes in contractile function with regard to subsequent vascular responsiveness.
The observation of increased levels of aortic MMP-9 is a novel finding in this model and perhaps represents the earliest pathological change in the vasculature in response to systemic inflammation. A role for MMP-9 in the inhibition of vasoconstriction has been described previously (Chew et al., 2004; Chung et al., 2007), with possible mechanisms related to the inhibition of extracellular calcium entry into the VSM cells having been identified (Raffetto and Khalil, 2008). Importantly, it is suggested that this effect is both concentration and time-dependent and is reversible (Chew et al., 2004). Although the role of concentration and time cannot be refuted, data from the present study would argue against the reversibility. In the previous study (Chew et al., 2004), only short 1 h incubations with MMP-9 were used, and such short incubation periods might allow the consequent acute effects to be reversed following removal of MMP-9. However, in our experiments, following 24 h incubations with MMP-9, a significant contractile dysfunction remained, even though the enzyme was completely washed away. While dysfunctional mechanisms of extracellular calcium entry into the VSM cells cannot be ruled out as an underlying cause, no such evidence has been published. Therefore, given the irreversible nature of the MMP-9-induced effect, it is likely that more permanent damage is responsible. Interestingly such a hypothesis is supported by preliminary experiments in which aortic tissues from mCIA animals were incubated ex vivo with a MMP-9 inhibitor and contractile dysfunction was still observed (unpubl. obs.).
Despite the fact that IL-1β has been intrinsically linked to both RA and MMP-9 secretion (Goldbach-Mansky, 2009), it would seem that this pyrogen does not play a role in the contractile dysfunction observed in this study. Whilst plasma levels of IL-1β were significantly elevated in mCIA over the course of disease progression, the contractile dysfunction produced following ex vivo incubations of aortic tissues from naïve animals with IL-1β was not associated with increased MMP-9 production. Indeed given that l -NAME and 1400W, and to a certain extent indomethacin, prevented the IL-1β-induced impairment of contraction suggests the well-described up-regulation of NO production, and to a lesser extent COX products, in these tissues in response to this inflammatory mediator. Such an action in the mCIA model has already been ruled out by experiments described above.
MMP-9 would seem to be a common denominator in the pathogenesis of RA and cardiovascular-related diseases (Ram et al., 2006). Importantly, this gelatinase is associated with destructive roles resulting in compromised blood vessel function. For instance, in an animal model of Kawasaki disease, a disease characterized by vasculitis or inflammation of middle-sized arteries, such arterial inflammation was associated with MMP-9 up-regulation and the breakdown of elastin (Lau et al., 2008). Similarly in a mouse model of Marfan syndrome, a disease characterized by increased arterial stiffness and life-threatening aortic aneurysm formation (Kingwell and Boutouyrie, 2007), the degeneration of elastic fibre integrity and the deterioration in vascular compliance is associated with an increased presence of gelatinases (Chung et al., 2007). Because MMP-9 has been implicated in the development of arterial stiffness in human studies (Yasmin et al., 2005; Vlachopoulos et al., 2007), an important question with regard to RA is what consequences could such MMP-9-associated effects have on the subsequent vascular health of affected individuals?
A change in VSM phenotype from contractile to synthetic is important in the development and progression of diseases such as hypertension and atherosclerosis. The consequence of such ‘phenotypic switching’ is the promotion of VSM proliferation and extracellular matrix remodelling leading to a thickened arterial wall and narrowing of the vessel lumen. It has been suggested that VSM cell-derived MMP-9 activity mediates this process (Cho and Reidy, 2002) via its basement-membrane and elastin degrading capabilities (Senior et al., 1991). Such a notion is substantiated by the observation that MMP-9 inhibition or gene deletion prevents VSM cell proliferation and migration (Galis et al., 2002). Importantly, IMT (Tanasescu et al., 2009) and arterial stiffness (loss of vascular elasticity and compliance) (Yildiz, 2010) are commonplace in patients with RA and correlate directly with cumulative inflammatory burden and disease severity independently of established cardiovascular risk factors (Crilly et al., 2009). As such, it would seem that a MMP-9/IMT/arterial stiffness pathological triangle may exist in RA.
In conclusion, the data presented here demonstrate an early impairment of vascular contractile responses associated with disease activity in the mCIA model of inflammatory arthritis in the absence of overt endothelial dysfunction. Because the progression of RA is not linear, the health of sufferers deteriorates quickly following its development. As such, there is now significant interest in an ‘early window of opportunity’ with regard to treatment of the disease. Nonetheless, despite changes in the course of RA in recent years, the increased risk of CVD has not altered. Perhaps the window of opportunity should also be concentrated on the development of vascular disease. Structural dysregulation in the vasculature associated with increased MMP-9 would seem a potential target for novel early intervention.
Acknowledgments
We wish to thank the British Heart Foundation for supporting this study (grant number FS/05/109/19952).
Glossary
- CPS
combined paw score
- CVD
cardiovascular disease
- CVR
cardiovascular risk
- eNOS
endothelial NOS
- IMT
intima-media thickening
- iNOS
inducible NOS
- mCIA
murine collagen-induced arthritis
- NOx
NO metabolites
- RA
rheumatoid arthritis
- SNP
sodium nitroprusside
- l -NAME
NG-nitro-l-arginine methyl ester
- VSM
vascular smooth muscle
- 1400W
N-(3-(aminomethyl)benzyl)acetamidine
Conflicts of interest
None.
References
- Bevaart L, Vervoordeldonk MJ, Tak PP. Evaluation of therapeutic targets in animal models of arthritis: how does it relate to rheumatoid arthritis? Arthritis Rheum. 2010;62:2192–2205. doi: 10.1002/art.27503. [DOI] [PubMed] [Google Scholar]
- Bull MJ, Williams AS, Mecklenburgh Z, Calder CJ, Twohig JP, Elford C, et al. The Death Receptor 3-TNF-like protein 1A pathway drives adverse bone pathology in inflammatory arthritis. J Exp Med. 2008;205:2457–2464. doi: 10.1084/jem.20072378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S, Khoo J, Mussa S, Alp NJ, Channon KM. Endothelial nitric oxide synthase dysfunction in diabetic mice: importance of tetrahydrobiopterin in eNOS dimerisation. Diabetologia. 2005;48:1933–1940. doi: 10.1007/s00125-005-1857-5. [DOI] [PubMed] [Google Scholar]
- Capettini LS, Cortes SF, Gomes MA, Silva GA, Pesquero JL, Lopes MJ, et al. Neuronal nitric oxide synthase-derived hydrogen peroxide is a major endothelium-dependent relaxing factor. Am J Physiol Heart Circ Physiol. 2008;295:H2503–H2511. doi: 10.1152/ajpheart.00731.2008. [DOI] [PubMed] [Google Scholar]
- Chew DK, Conte MS, Khalil RA. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J Vasc Surg. 2004;40:1001–1010. doi: 10.1016/j.jvs.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Cho A, Reidy MA. Matrix metalloproteinase-9 is necessary for the regulation of smooth muscle cell replication and migration after arterial injury. Circ Res. 2002;91:845–851. doi: 10.1161/01.res.0000040420.17366.2e. [DOI] [PubMed] [Google Scholar]
- Cho YG, Cho ML, Min SY, Kim HY. Type II collagen autoimmunity in a mouse model of human rheumatoid arthritis. Autoimmun Rev. 2007;7:65–70. doi: 10.1016/j.autrev.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Chung AW, Au Yeung K, Sandor GG, Judge DP, Dietz HC, van Breemen C. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007;101:512–522. doi: 10.1161/CIRCRESAHA.107.157776. [DOI] [PubMed] [Google Scholar]
- Crilly MA, Kumar V, Clark HJ, Scott NW, Macdonald AG, Williams DJ. Arterial stiffness and cumulative inflammatory burden in rheumatoid arthritis: a dose-response relationship independent of established cardiovascular risk factors. Rheumatology (Oxford) 2009;48:1606–1612. doi: 10.1093/rheumatology/kep305. [DOI] [PubMed] [Google Scholar]
- Evans L, Williams AS, Hayes AJ, Jones SA, Nowell M. Suppression of leukocyte infiltration and cartilage degradation by selective inhibition of pre-B cell colony-enhancing factor/visfatin/nicotinamide phosphoribosyltransferase: Apo866-mediated therapy in human fibroblasts and murine collagen-induced arthritis. Arthritis Rheum. 2011;63:1866–1877. doi: 10.1002/art.30338. [DOI] [PubMed] [Google Scholar]
- Foster W, Lip GY, Raza K, Carruthers D, Blann AD. An observational study of endothelial function in early arthritis. Eur J Clin Invest. 2012;42:510–516. doi: 10.1111/j.1365-2362.2011.02607.x. [DOI] [PubMed] [Google Scholar]
- Gabriel SE. Heart disease and rheumatoid arthritis: understanding the risks. Ann Rheum Dis. 2010;69(Suppl. 1):i61–i64. doi: 10.1136/ard.2009.119404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galis ZS, Johnson C, Godin D, Magid R, Shipley JM, Senior RM, et al. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ Res. 2002;91:852–859. doi: 10.1161/01.res.0000041036.86977.14. [DOI] [PubMed] [Google Scholar]
- George SJ, Zaltsman AB, Newby AC. Surgical preparative injury and neointima formation increase MMP-9 expression and MMP-2 activation in human saphenous vein. Cardiovasc Res. 1997;33:447–459. doi: 10.1016/s0008-6363(96)00211-8. [DOI] [PubMed] [Google Scholar]
- Goldbach-Mansky R. Blocking interleukin-1 in rheumatic diseases. Ann N Y Acad Sci. 2009;1182:111–123. doi: 10.1111/j.1749-6632.2009.05159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegen M, Keith JC, Jr, Collins M, Nickerson-Nutter CL. Utility of animal models for identification of potential therapeutics for rheumatoid arthritis. Ann Rheum Dis. 2008;67:1505–1515. doi: 10.1136/ard.2007.076430. [DOI] [PubMed] [Google Scholar]
- Innala L, Moller B, Ljung L, Magnusson S, Smedby T, Sodergren A, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther. 2011;13:R131. doi: 10.1186/ar3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarranz Y, Abad C, Martinez C, Arranz A, Gutierrez-Canas I, Rosignoli F, et al. Protective effect of vasoactive intestinal peptide on bone destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1034–R1045. doi: 10.1186/ar1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan F, Galarraga B, Belch JJ. The role of endothelial function and its assessment in rheumatoid arthritis. Nat Rev Rheumatol. 2010;6:253–261. doi: 10.1038/nrrheum.2010.44. [DOI] [PubMed] [Google Scholar]
- Kingwell B, Boutouyrie P. Genetic influences on the arterial wall. Clin Exp Pharmacol Physiol. 2007;34:652–657. doi: 10.1111/j.1440-1681.2007.04655.x. [DOI] [PubMed] [Google Scholar]
- Kolb H, Mandrup-Poulsen T. The global diabetes epidemic as a consequence of lifestyle-induced low-grade inflammation. Diabetologia. 2010;53:10–20. doi: 10.1007/s00125-009-1573-7. [DOI] [PubMed] [Google Scholar]
- Lau AC, Duong TT, Ito S, Yeung RS. Matrix metalloproteinase 9 activity leads to elastin breakdown in an animal model of Kawasaki disease. Arthritis Rheum. 2008;58:854–863. doi: 10.1002/art.23225. [DOI] [PubMed] [Google Scholar]
- Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE. Cardiovascular death in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2005;52:722–732. doi: 10.1002/art.20878. [DOI] [PubMed] [Google Scholar]
- Martin BJ, Anderson TJ. Risk prediction in cardiovascular disease: the prognostic significance of endothelial dysfunction. Can J Cardiol. 2009;25(Suppl. A):15A–20A. doi: 10.1016/s0828-282x(09)71049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meune C, Touze E, Trinquart L, Allanore Y. Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford) 2009;48:1309–1313. doi: 10.1093/rheumatology/kep252. [DOI] [PubMed] [Google Scholar]
- Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, Jones GW, et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol. 2009;182:613–622. doi: 10.4049/jimmunol.182.1.613. [DOI] [PubMed] [Google Scholar]
- Peters MJ, van Halm VP, Voskuyl AE, Smulders YM, Boers M, Lems WF, et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 2009;61:1571–1579. doi: 10.1002/art.24836. [DOI] [PubMed] [Google Scholar]
- Pinder AG, Rogers SC, Khalatbari A, Ingram TE, James PE. The measurement of nitric oxide and its metabolites in biological samples by ozone-based chemiluminescence. Methods Mol Biol. 2009;476:10–27. doi: 10.1007/978-1-59745-129-1_2. [DOI] [PubMed] [Google Scholar]
- Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75:346–359. doi: 10.1016/j.bcp.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram M, Sherer Y, Shoenfeld Y. Matrix metalloproteinase-9 and autoimmune diseases. J Clin Immunol. 2006;26:299–307. doi: 10.1007/s10875-006-9022-6. [DOI] [PubMed] [Google Scholar]
- Roman MJ, Salmon JE. Cardiovascular manifestations of rheumatologic diseases. Circulation. 2007;116:2346–2355. doi: 10.1161/CIRCULATIONAHA.106.678334. [DOI] [PubMed] [Google Scholar]
- Sakaguchi Y, Shirahase H, Ichikawa A, Kanda M, Nozaki Y, Uehara Y. Effects of selective iNOS inhibition on type II collagen-induced arthritis in mice. Life Sci. 2004;75:2257–2267. doi: 10.1016/j.lfs.2004.02.037. [DOI] [PubMed] [Google Scholar]
- Senior RM, Griffin GL, Fliszar CJ, Shapiro SD, Goldberg GI, Welgus HG. Human 92- and 72-kilodalton type IV collagenases are elastases. J Biol Chem. 1991;266:7870–7875. [PubMed] [Google Scholar]
- Sodergren A, Karp K, Boman K, Eriksson C, Lundstrom E, Smedby T, et al. Atherosclerosis in early rheumatoid arthritis: very early endothelial activation and rapid progression of intima media thickness. Arthritis Res Ther. 2010;12:R158. doi: 10.1186/ar3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DH, Karlson EW, Rimm EB, Cannuscio CC, Mandl LA, Manson JE, et al. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation. 2003;107:1303–1307. doi: 10.1161/01.cir.0000054612.26458.b2. [DOI] [PubMed] [Google Scholar]
- Tanasescu C, Jurcut C, Jurcut R, Ginghina C. Vascular disease in rheumatoid arthritis: from subclinical lesions to cardiovascular risk. Eur J Intern Med. 2009;20:348–354. doi: 10.1016/j.ejim.2008.09.005. [DOI] [PubMed] [Google Scholar]
- van Eijk IC, Serne EH, Dijkmans BA, Smulders Y, Nurmohamed M. Microvascular function is preserved in newly diagnosed rheumatoid arthritis and low systemic inflammatory activity. Clin Rheumatol. 2011;30:1113–1118. doi: 10.1007/s10067-011-1750-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachopoulos C, Aznaouridis K, Dima I, Ioakeimidis N, Vasiliadou C, Zervoudaki A, et al. Negative association between serum levels of matrix metalloproteinases-2 and -9 and aortic stiffness in healthy adults. Int J Cardiol. 2007;122:232–238. doi: 10.1016/j.ijcard.2006.11.099. [DOI] [PubMed] [Google Scholar]
- Yasmin, McEniery CM, Wallace S, Dakham Z, Pulsalkar P, Maki-Petaja K, et al. Matrix metalloproteinase-9 (MMP-9), MMP-2, and serum elastase activity are associated with systolic hypertension and arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25:372–378. doi: 10.1161/01.ATV.0000151373.33830.41. [DOI] [PubMed] [Google Scholar]
- Yildiz M. Arterial distensibility in chronic inflammatory rheumatic disorders. Open Cardiovasc Med J. 2010;4:83–88. doi: 10.2174/1874192401004020083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A, Koduri G, Batley M, Kulinskaya E, Gough A, Norton S, et al. Mortality in rheumatoid arthritis. Increased in the early course of disease, in ischaemic heart disease and in pulmonary fibrosis. Rheumatology (Oxford) 2007;46:350–357. doi: 10.1093/rheumatology/kel253. [DOI] [PubMed] [Google Scholar]