

Abstract
BACKGROUND AND PURPOSE
The analgesic action of 5-HT and noradrenaline reuptake inhibitors (SNRIs) on nociceptive synaptic transmission in the spinal cord is poorly understood. We investigated the effects of milnacipran, an SNRI, on C-fibre-evoked field potentials (FPs) in spinal long-term potentiation (LTP), a proposed synaptic mechanism of hypersensitivity, and on the FPs in a neuropathic pain model.
EXPERIMENTAL APPROACH
C-fibre-evoked FPs by electrical stimulation of the sciatic nerve fibres were recorded in the spinal dorsal horn of anaesthetized adult rats, and LTP was induced by high-frequency stimulation of the sciatic nerve fibres. A rat model of neuropathic pain was produced by L5 spinal nerve ligation and transection.
KEY RESULTS
Milnacipran produced prolonged inhibition of C-fibre-evoked FPs when applied spinally after the establishment of LTP of C-fibre-evoked FPs in naïve animals. In the neuropathic pain model, spinal administration of milnacipran clearly reduced the basal C-fibre-evoked FPs. These inhibitory effects of milnacipran were blocked by spinal administration of methysergide, a 5-HT1/2 receptor antagonist, and yohimbine or idazoxan, α2-adrenoceptor antagonists. However, spinal administration of milnacipran in naïve animals did not affect the basal C-fibre-evoked FPs and the induction of spinal LTP.
CONCLUSION AND IMPLICATIONS
Milnacipran inhibited C-fibre-mediated nociceptive synaptic transmission in the spinal dorsal horn after the establishment of spinal LTP and in the neuropathic pain model, by activating both spinal 5-hydroxytryptaminergic and noradrenergic systems. The condition-dependent inhibition of the C-fibre-mediated transmission by milnacipran could provide novel evidence regarding the analgesic mechanisms of SNRIs in chronic pain.
Keywords: 5-HT and noradrenaline reuptake inhibitor, spinal cord, long-term potentiation, neuropathic pain, C-fibre-evoked field potential, in vivo electrophysiology
Introduction
Long-lasting abhorrent pain can be induced by various factors such as tissue damage, inflammation and nerve injury. Although the mechanisms underlying the pathology of chronic pain syndromes remain largely unknown, it is widely accepted that enhanced responsiveness of central neurons to their normal afferent input is involved in the amplification of pain signalling (Sandkühler, 2009).
Long-term potentiation (LTP), which is proposed as a synaptic model of learning and memory in the hippocampus (Bliss and Collingridge, 1993), is an activity-dependent, long-lasting increase in the efficacy of synaptic transmission. Previous studies demonstrated the existence of LTP at excitatory synapses between primary sensory afferents and neurons in the spinal dorsal horn (Randićet al., 1993; Liu and Sandkühler, 1995; Ikeda et al., 2006). C-fibre afferents transfer nociceptive signals, and robust LTP of C-fibre-evoked, but not A-fibre-evoked, field potentials (FPs) is induced by high-intensity electrical stimulation of peripheral nerve, inflammation, and acute nerve injury, all of which also induce long-lasting hypersensitivity to peripheral stimuli (Sandkühler and Liu, 1998; Zhang et al., 2004; 2006; Ikeda et al., 2006). In addition, several clinically applied analgesic agents prevent the induction and/or maintenance of LTP of C-fibre-evoked FPs (Benrath et al., 2005; Ge et al., 2006; Tanabe et al., 2006). Therefore, LTP of C-fibre-evoked FPs in the spinal dorsal horn has been considered as a model of long-lasting abhorrent pain, underlying an increased efficacy of nociceptive transmission (Liu and Sandkühler, 1995; Sandkühler, 2009). However, the relationship between LTP and the induction and maintenance of chronic pain is not fully understood.
Neuropathic pain is defined as long-lasting pain resulting from injury or dysfunction in the periphery and/or CNS. It is not often relieved adequately by conventional treatments such as non-steroidal anti-inflammatory drugs or morphine (Mao and Chen, 2000; Minami et al., 2009). Several lines of evidence suggest that 5-HT and noradrenaline reuptake inhibitors (SNRIs), which inhibit both 5-HT and noradrenaline reuptake transporters and thus elevate the extracellular concentrations of these monoamines in the synaptic cleft, exert potent analgesic effects in neuropathic conditions (Sindrup et al., 2005; Lee and Chen, 2010). The drugs are thought to suppress pain transmission by activating the bulbospinal descending 5-hydroxytryptaminergic and noradrenergic pathways and induce more potent analgesic efficacy than selective 5-HT reuptake inhibitors (SSRIs) or noradrenaline reuptake inhibitors (Coluzzi and Mattia, 2005; Lee and Chen, 2010). SNRIs have limited affinities for other receptors and ion channels tested and exhibit acceptable safety profiles (Moret et al., 1985; Bymaster et al., 2001; Lee and Chen, 2010), supporting the notion that SNRIs should be a first-line treatment for neuropathic pain (Sindrup et al., 2005). Although SNRIs exert analgesic activity when administered by various routes in neuropathic pain model animals (Iyengar et al. 2004; King et al., 2006; Suzuki et al., 2008), the spinal cord may be an important site for SNRI-induced analgesia, given that the intrathecal administration of monoamine reuptake inhibitors effectively ameliorates abnormal pain behaviours in the animal models (Shin and Eisenach, 2004; Obata et al., 2005; Honda et al., 2006; Ikeda et al., 2009). Therefore, it could be important to clarify the analgesic mechanism(s) of spinally administered SNRIs on spinal nociceptive transmission without affecting the supraspinal and peripheral regions.
Using in vivo electrophysiological recordings from the spinal dorsal horn, we can observe pain-related changes and LTP in the spinal nociceptive transmission and evaluate the efficacy of drugs on the transmission at a synaptic level in the intact nervous system. Employing this approach, we investigated the effects of local spinal administration of milnacipran, a representative SNRI, on C-fibre-evoked FPs in naïve and neuropathic pain model animals and on the induction and maintenance of LTP of C-fibre-evoked FPs in the rat spinal dorsal horn. Our results demonstrated potent inhibitory effects of milnacipran on spinal nociceptive synaptic transmission in the limited conditions where spinal LTP or neuropathic pain was established.
Methods
All animal care and experimental procedures complied with the guidelines of the National Institutes of Health (Tokyo, Japan) and the Japanese Pharmacological Society (Tokyo, Japan) and were approved by the Animal Care and Use Committee of Nagoya City University (Nagoya, Japan) and the Animal Care and Use Committee of Shionogi Research Laboratories (Osaka, Japan). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). A total of 103 animals were used in the experiments reported here.
Preparation of animals
Male Wistar/ST rats (6–9 weeks old; SLC, Shizuoka, Japan) were anaesthetized with urethane (1.5 g·kg−1; i.p.). Surgery was initiated after confirming the loss of righting, and corneal and cutaneous reflexes, and additional doses of urethane (∼0.3 g·kg−1) were given if needed. The dose range of urethane has been described elsewhere (e.g. Liu and Sandkühler, 1995; 1997; Ge et al., 2006; Tanabe et al., 2006) and shown to produce a steady level of anaesthesia lasting for more than 6 h in experiments without muscle paralysis (Svendsen et al., 1997). The trachea was cannulated for mechanical ventilation with air. The rectal temperature was kept constant at 36–37°C using a feedback-controlled heating blanket and radiant heat. In each rat, a laminectomy was performed at vertebrae T13-L1, and the dura mater was incised longitudinally. A plastic chamber (handmade; 10 × 14 × 10 mm; wall thickness, 0.3 mm) was placed on the cord dorsum at the recording site and stabilized with 4% agar in distilled water. Artificial cerebrospinal fluid (in mM: 113 NaCl, 3 KCl, 1 NaHPO4, 25 NaHCO3, 11 glucose, 2 CaCl2, 1 MgCl2) was applied to the chamber (0.6–0.8 mL/5–10 s) using a 1 mL syringe with a 23 G needle (Terumo, Tokyo, Japan). The left sciatic nerve was exposed, and the nerve fibres were placed on bipolar Ag–AgCl electrodes for stimulation. At the end of the surgery, the animals were paralysed with pancuronium bromide (0.5 mg·kg−1 for 90 min; i.v.) administered through a cannula inserted into the femoral vein to prevent the muscle contraction evoked by electrical stimulation. After the electrophysiological recordings were completed, the animals were allowed to recover from muscle relaxation and the deepness of anaesthesia was confirmed by observing the loss of the reflexes as above. The recovery was confirmed by observing the muscle constriction evoked by the electrical stimulation to the sciatic nerve fibres and the spontaneous respiration. For early recovery, the dose of pancuronium was decreased near the end of a recording period.
Electrophysiological recordings
The electrophysiological set-up was essentially as described previously (Ohnami et al., 2011). A tungsten microelectrode was inserted at a depth of 100–500 µm from the spinal dorsal surface. As a test stimulus, single, monophasic square-wave pulses of 0.5 ms duration were delivered to the sciatic nerve fibres through the bipolar electrodes. The evoked spinal FPs were amplified, band-pass filtered between 3 kHz and 0.1 Hz (DAM80, World Precision Instruments, Sarasota, FL, USA), and digitized for computer analysis at a sampling rate of 2 kHz using a PowerLab A/D converter (AD Instruments, Mountain View, CA, USA). The C-fibre-evoked component of an FP was distinguished as a response that peaked with 100–200 ms latency following stimulation and that had a threshold greater than 5 V stimulus intensity (Liu and Sandkühler, 1995; Tanabe et al., 2006). Following the determination of this threshold, the test stimuli (at a constant voltage intensity 1.2–2 times the threshold) were applied at 1 min intervals to the sciatic nerve fibres. High-frequency stimulation (HFS; 0.5 ms duration, 40–45 V, 100 Hz, given in two trains of 1 s duration at 20 s intervals) was used to induce LTP. Recordings were performed for a minimum of 180 min from the HFS.
Animal models of neuropathic pain and behavioural tests
The surgical procedure was performed with some modifications to the original L5 and L6 spinal nerve ligation (SNL) model (Kim and Chung, 1992) described by Fukuoka et al. (1998). Briefly, male Wistar/ST rats (4–5 weeks old; SLC, Shizuoka, Japan) were anaesthetized with pentobarbital sodium (60 mg·kg−1; i.p.). The left paraspinal muscles were separated from the spinous processes at the L5–6 level, and the L6 transverse process was removed. The left L5 spinal nerve was isolated, tightly ligated with 3-0 silk thread, and cut just distal to the ligation (∼1 mm apart). The distance from the dorsal root ganglion to the site of nerve injury was approximately 8–12 mm.
About 14–17 days after the operation, the threshold of the paw withdrawal response was determined by the up-down method (Chaplan et al., 1994) using a series of calibrated von Frey filaments (Semmes-Weinstein Monofilaments; North Coast Medical, Morgan Hill, CA, USA). Each rat was placed on a metal mesh floor covered with a clear plastic cage, and the filaments were applied from underneath the floor to the plantar surface of the paw. Only those animals that show a lowered threshold at the injured paw (left; ≤4 g, right; ≥10 g) were used for the electrophysiological studies described.
Drugs and pharmacological treatment
Milnacipran hydrochloride was purchased from Janssen Pharmaceutical K.K. (Tokyo, Japan) and was purified at Shionogi & Co. (Osaka, Japan). Urethane, yohimbine hydrochloride, idazoxan hydrochloride and methysergide maleate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Pancuronium bromide was purchased from Schering-Plough K.K. (Osaka, Japan), and pentobarbital sodium was purchased from Tokyo Chemical Industry (Tokyo, Japan). Milnacipran, yohimbine, idazoxan and methysergide were dissolved in the artificial cerebrospinal fluid. For spinal administration (Ohnami et al., 2011), approximately > 90% of the vehicle (with or without drugs) was removed from the chamber by suction, and the chamber was refilled with the next solution to be tested by manual injection, using a 1 mL syringe with a 23 G needle (Terumo, Tokyo, Japan). It took roughly 15–20 s for each wash. The procedure was repeated twice to completely exchange the solution in the chamber. The volume of solution used for administration and washing was approximately 0.8 mL. As the temperature of the solution in the chamber on the spinal column was approximately 27–30°C during the recording period, the vehicle with or without drugs was warmed to roughly 30°C just prior to the administration. The effects of the antagonists and each dose of milnacipran were monitored for more than 180 min. The penetration of spinally administered drugs into the spinal cord tissue was described by Beck et al. (1995).
Data analysis
The area of C-fibre-evoked FPs was determined offline, as described previously (Figure 1, inset) (Ohnami et al., 2011). The mean areas of the basal C-fibre-evoked FPs were 1.4 ± 0.1 µV ms (range, 0.3–4.0 µV ms; n= 56) and 1.9 ± 0.2 µV ms (range, 0.4–6.9 µV ms; n= 38) in naïve and the modified SNL-model animals, respectively, at 0–60 min prior to HFS or the first drug administration. As demonstrated previously (Matthews and Dickenson, 2001; Buesa et al., 2008; Ohnami et al., 2011), the actual value of basal C-fibre-evoked responses in the nerve-injured animals was similar to that in naïve animals despite the loss and/or damage of spinal nerves, suggesting that nociceptive transmission was already enhanced in the spinal cord after the modified SNL. The mean area of C-fibre-evoked FPs in the maintenance phase of LTP in naïve animals was 3.1 ± 0.2 µV ms (range, 0.7–8.2 µV ms; n= 51) at 30–60 min after HFS. In the studies evaluating the effect of milnacipran on the induction of spinal LTP, FPs were normalized to the average of 30 consecutive FPs obtained prior to HFS. In the other studies, 60 consecutive FPs recorded prior to HFS or the administration of vehicle or milnacipran were averaged for normalization. The maximal inhibition of C-fibre-evoked FPs was calculated using the mean area of 30 consecutive FPs during 90–120 min following vehicle or milnacipran administration. Student's t-test and a two-tailed t-test with Bonferroni correction following one-way anova were used for the two-group and multiple comparisons (Wallenstein et al., 1980) respectively. Differences with P < 0.05 were considered significant. All data are presented as means ± SEM.
Figure 1.
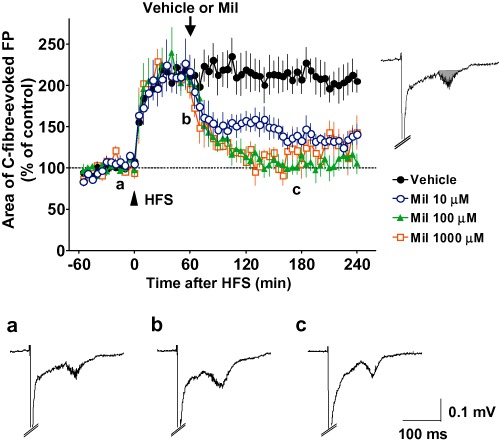
Effects of spinally administered milnacipran (Mil) on C-fibre-evoked field potentials (FPs) following the establishment of long-term potentiation (LTP) in naïve animals. FPs in the spinal dorsal horn were elicited by electrical stimulation of the sciatic nerve fibres at 1 min intervals in anaesthetized naïve adult rats. LTP of C-fibre-evoked FPs was induced by high-frequency stimulation (HFS; 0.5 ms duration, 40–45 V, 100 Hz, given in two trains of 1 s duration at 20 s intervals; arrowhead) of the sciatic nerve fibres. Each area of C-fibre-evoked FPs was normalized to the mean of 60 consecutive responses obtained prior to HFS (–60 to 0 min in the graph) and five consecutive responses were averaged. The vehicle (n= 6) or 10, 100 and 1000 µM milnacipran (n= 6, n= 5 and n= 3, respectively) was administered spinally 60 min after HFS (arrow), when the enhancement of C-fibre-evoked FPs by HFS was fully established. Example-averaged traces of five consecutive FPs from the 10 µM milnacipran group recorded during the times indicated on the graph (a, b and c) are shown. Inset represents the calculated area of C-fibre-evoked FPs. Data shown are means ± SEM.
Results
Effects of milnacipran on C-fibre-evoked FPs following the establishment of LTP in naïve rats
C-fibre-evoked FPs were continually observed in the spinal dorsal horn of anaesthetized naïve adult rats by electrical stimulation of the sciatic nerve fibres (Liu and Sandkühler, 1995). The area of C-fibre-evoked FPs was stable for more than 60 min with 1 min interval stimulation of the sciatic nerve fibres (Figure 1). In naïve animals, HFS (see Methods) of the sciatic nerve fibres after ≥60 min of stable baseline recordings induced a large increase in the area of C-fibre-evoked FPs to 214 ± 14% (n= 6, averaged at 30–60 min following HFS) of the pre-HFS values (basal responses, averaged at 0–60 min prior to HFS; Figure 1). Enhancement of the C-fibre-evoked FPs continued until termination of the experimental protocol (≥240 min), despite the spinal administration of the vehicle alone at 60 min after HFS, accomplished by exchanging the entire volume of the vehicle solution in the chamber placed on the recording segments, indicating the development and maintenance of LTP of C-fibre-evoked FPs (Liu and Sandkühler, 1995; Ohnami et al., 2011).
We applied milnacipran (0.8 mL of 10, 100 and 1000 µM, corresponding to about 2.26, 22.6 and 226 µg, respectively) directly onto the spinal dorsal surface at the recording segments 60 min after HFS. As shown in Figure 1, all concentrations of milnacipran induced a long-lasting depression of C-fibre-evoked FPs and at 100 µM, milnacipran depressed FPs to the pre-HFS level. When the concentration of milnacipran was increased to 1000 µM, the inhibition of C-fibre-evoked FPs did not increase. In summary, 90–120 min following the spinal administration (150–180 min following HFS), the mean C-fibre-evoked FPs of 10, 100, and 1000 µM milnacipran groups were 137 ± 10% (n = 6), 104 ± 7% (n = 5), and 110 ± 16% (n = 3), respectively, all of which were significantly smaller (P < 0.01) than that of the vehicle group (211 ± 18%, n = 6). When compared with the pre-drug values (30–60 min following HFS), the C-fibre-evoked FPs were maximally decreased by 35 ± 4% (n= 6), 50 ± 6% (n= 5) and 47 ± 9% (n= 3) from 90–120 min following administration of 10, 100 and 1000 µM milnacipran (150–180 min following HFS) respectively. We have to take into consideration that up to 5% of drugs can be absorbed into the blood after the spinal administration (Beck et al., 1995), in particular when the drug dose is increased. To reduce any possible extraspinal or nonspecific effects, therefore, the lowest dose of milnacipran in this study (10 µM) was used in the subsequent experiments to assess spinal receptors involved in the milnacipran-induced inhibition of C-fibre-evoked FPs.
Previous behavioural studies suggested that the analgesic effect of intrathecally administered milnacipran in animal models of chronic pain is blocked by yohimbine (Obata et al., 2005) and idazoxan (Obata et al., 2010), α2-adrenoceptor antagonists, and methysergide (Obata et al., 2005; 2010), a nonselective 5-HT1 and 5-HT2 receptor antagonist (receptor nomenclature follows Alexander et al., 2011). Thus, we used these antagonists to examine whether the noradrenaline and 5-HT receptors in the spinal cord mediated the inhibitory effect of milnacipran on C-fibre-evoked FPs after the establishment of spinal LTP. The doses of antagonists used in the present study, (0.8 mL of 5 µM yohimbine, 30 µM idazoxan or 30 µM methysergide) were equivalent to about 1.56, 5.78 and 11.3 µg respectively. These antagonists alone at the concentrations tested were found to have no apparent influence on C-fibre-evoked FPs for at least 180 min, when applied to the recording segments at 60 min after HFS (Figure 2A). As shown in Figure 2B, the inhibitory effect of milnacipran (10 µM) on C-fibre-evoked FPs was significantly reduced when milnacipran was applied 60 min after HFS in the presence of any of these antagonists (pretreatment of 15 min). The inhibition of C-fibre-evoked FPs during 90–120 min following the administration of milnacipran (10 µM) in the presence of yohimbine, idazoxan and methysergide (150–180 min following HFS) was 10 ± 9% (n = 5), 9 ± 5% (n = 5) and 9 ± 7% (n = 6) respectively (P < 0.05 vs. 10 µM milnacipran alone; Figure 2C).
Figure 2.
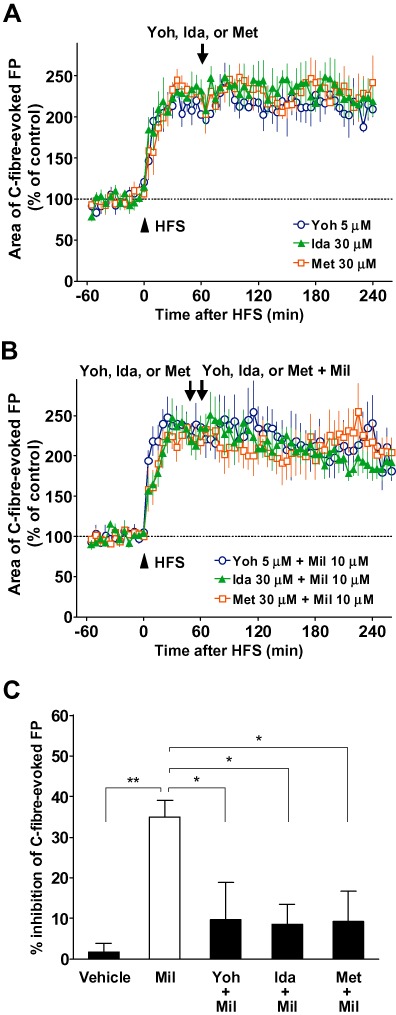
Effects of adrenoceptor or 5-HT receptor antagonists on the inhibitory effect of milnacipran (Mil) on the C-fibre-evoked field potentials (FPs) following the establishment of long-term potentiation (LTP) in naïve animals. FPs in the spinal dorsal horn were elicited by electrical stimulation of the sciatic nerve fibres at 1 min intervals in anaesthetized naïve adult rats. LTP of C-fibre-evoked FPs was induced by high-frequency stimulation (HFS; 0.5 ms duration, 40–45 V, 100 Hz, given in two trains of 1 s duration at 20 s intervals; arrowhead) of the sciatic nerve fibres. Each area of C-fibre-evoked FPs was normalized to the mean of 60 consecutive responses obtained prior to HFS (–60 to 0 min in the graph) and five consecutive responses were averaged. (A) Yohimbine (Yoh; n= 4), idazoxan (Ida; n= 3) or methysergide (Met; n= 4) was administered spinally 60 min after HFS (arrow), when the enhancement of C-fibre-evoked FPs by HFS was fully established. (B) Yohimbine (n= 5), idazoxan (n= 5) or methysergide (n= 6) was administered spinally 45 min after HFS. Yohimbine, idazoxan or methysergide plus milnacipran were administered spinally 60 min after HFS. (C) The inhibitory effects of 10 µM milnacipran on the C-fibre-evoked FPs with or without yohimbine, idazoxan or methysergide. The % inhibition of C-fibre-evoked FPs was calculated using the mean area of 30 consecutive FPs during 90–120 min following the vehicle or milnacipran administration (averaged at 150–180 min after HFS) in comparison to the pre-drug values (averaged at 30–60 min after HFS). The values in vehicle and milnacipran groups are from Figure 1. Data shown are means ± SEM. *P < 0.05; **P < 0.01, significantly different as indicated.
In addition to the inhibitory effect on early LTP demonstrated above, milnacipran (10 µM) markedly inhibited C-fibre-evoked FPs when administered spinally more than 3 h (190 min) after HFS (Figure 3). The C-fibre-evoked FPs were reduced from 218 ± 10% (averaged at 0–30 min before milnacipran administration) to 131 ± 8% (averaged 90–120 min following the application, n = 6), indicating that milnacipran can also reverse spinal LTP thought to reflect the transcriptional changes occurring in later time points (typically more than 3 h) after the induction of LTP (Sandkühler, 2009). When compared with the pre-drug values (160–190 min following HFS), the C-fibre-evoked FPs were maximally decreased by 39 ± 5% (n= 6) from 90 to 120 min following administration of 10 µM milnacipran (280–310 min following HFS), comparable to the inhibitory effect at early LTP (Figure 1). In three out of six animals here, milnacipran was removed from the chamber and replaced by yohimbine (5 µM) 120 min after the application of milnacipran, and we found that C-fibre-evoked FPs returned close to the pre-drug level (Figure 3). The mean C-fibre-evoked FPs before milnacipran administration (160–190 min after HFS) and after replacement with yohimbine (460–490 after HFS) were 210 ± 14% and 208 ± 9% (n = 3 each). By contrast, replacement of milnacipran in the chamber by vehicle (in another three animals) did not reverse the inhibitory effect of milnacipran (Figure 3). The mean C-fibre-evoked FPs before milnacipran administration (160–190 min after HFS) and after replacement with vehicle (460–490 after HFS) were 225 ± 16% and 127 ± 9% (n = 3 each, P < 0.01 vs. the yohimbine-treated group).
Figure 3.
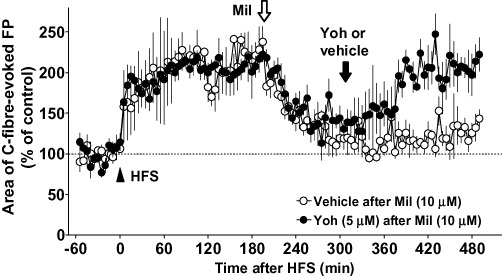
Effects of spinally administered milnacipran (Mil) on the late maintenance phase of long-term potentiation (LTP) of C-fibre-evoked field potentials (FPs) in naïve animals and its reversal by antagonizing the effect of milnacipran. FPs in the spinal dorsal horn were elicited by electrical stimulation of the sciatic nerve fibres at 1 min intervals in anaesthetized naïve adult rats. LTP of C-fibre-evoked FPs was induced by high-frequency stimulation (HFS; 0.5 ms duration, 40–45 V, 100 Hz, given in two trains of 1 s duration at 20 s intervals; arrowhead) of the sciatic nerve fibres. Each area of C-fibre-evoked FPs was normalized to the mean of 60 consecutive responses obtained prior to HFS (−60 to 0 min in the graph) and five consecutive responses were averaged. Milnacipran (10 µM) was administered spinally in the late maintenance phase of LTP (190 min after HFS; white arrow). Milnacipran was removed from the chamber and replaced by yohimbine (Yoh; n= 3) or vehicle (n= 3) 120 min after the application of milnacipran (310 min after HFS; black arrow). Data shown are means ± SEM.
Effects of milnacipran on C-fibre-evoked FPs in the nerve-injured rats
The first results suggested that C-fibre-mediated nociceptive synaptic transmission was sensitive to inhibition of 5-HT and noradrenaline reuptake after the establishment of spinal LTP, which prompted us to assess the effect of milnacipran on C-fibre-evoked FPs in a rat model of neuropathic pain (Kim and Chung, 1992; Fukuoka et al., 1998). Rats exhibiting allodynia-like behaviour to plantar stimulation 2 weeks after the modified SNL surgery were subjected to electrophysiological studies. When milnacipran was administered to the recording segments of the spinal surface of the modified SNL-model animals after ≥60 min of baseline recordings, the basal C-fibre-evoked FPs were significantly decreased, although the spinal administration of vehicle did not affect the FPs. As shown in Figure 4, 90–120 min following the spinal administration, the mean C-fibre-evoked FPs of 10, 100 and 1000 µM milnacipran groups were 63 ± 5% (n = 5), 50 ± 4% (n = 7) and 33 ± 4% (n = 4), respectively, all of which were significantly smaller (P < 0.01) than that of the vehicle group (107 ± 11%, n = 4). When compared with the pre-drug values (baseline; 0–60 min before the administration), the C-fibre-evoked FPs were maximally decreased by 37 ± 5% (n= 5), 50 ± 4% (n= 7) and 67 ± 4% (n= 4) during 90–120 min following the administration of 10, 100 and 1000 µM milnacipran respectively. Similarly, in the following pharmacological experiments to assess spinal receptors involved in the milnacipran-induced inhibition of C-fibre-evoked FPs, the lowest dose of milnacipran used in this study (10 µM) was administered to reduce any possible extraspinal or nonspecific effects. As spinal LTP of C-fibre-evoked FPs or facilitation of C-fibre-evoked action potential discharges is rarely induced by electrical stimulation of the sciatic nerve fibres after peripheral nerve injury (Rygh et al., 2000; Zhang et al., 2004; Ohnami et al., 2011), we did not apply the HFS protocol in the modified SNL-model animals.
Figure 4.
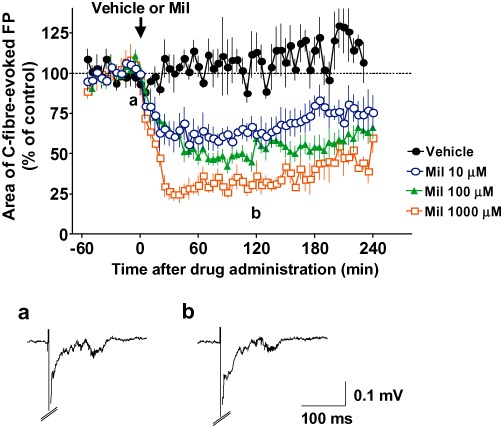
Effects of spinally administered milnacipran (Mil) on the basal C-fibre-evoked field potentials (FPs) in modified spinal nerve ligation (SNL)-model animals. FPs in the spinal dorsal horn were elicited by electrical stimulation of the sciatic nerve fibres at 1 min intervals in modified SNL-model animals. The vehicle (n= 5) or 10, 100 and 1000 µM milnacipran (n= 5, n= 7 and n= 4, respectively) was administered spinally after ≥60 min of stable baseline recordings (arrow). Each area of C-fibre-evoked FPs was normalized to the mean of 60 consecutive responses obtained prior to the administration (–60 to 0 min in the graph) and five consecutive responses were averaged. Example-averaged traces of five consecutive FPs from the 10 µM milnacipran group recorded during the times indicated on the graph (a and b) are shown. Data shown are means ± SEM.
We also examined whether the inhibitory effect of milnacipran on basal C-fibre-evoked FPs in the neuropathic pain model animals could be blocked by 5-HT or noradrenaline receptor antagonists. Because it has been shown that the analgesic effect of intrathecally administered milnacipran was blocked by yohimbine and methysergide in a rat model of neuropathic pain (Obata et al., 2005), we used these two antagonists for the electrophysiological experiments in the modified SNL-model rats. Again, yohimbine (5 µM) and methysergide (30 µM) alone were found to have no apparent influence on C-fibre-evoked FPs for at least 180 min when applied to the recording segments after ≥60 min of baseline recordings in the modified SNL-model animals (Figure 5A). As shown in Figure 5B, the inhibitory effect of milnacipran (10 µM) on C-fibre-evoked FPs was significantly reduced when milnacipran was applied in the presence of either of these antagonists (pretreatment for 15 min) in the modified SNL-model animals. The inhibition of basal C-fibre-evoked FPs during 90–120 min following the administration of milnacipran (10 µM) in the presence of yohimbine and methysergide was 9 ± 8% (n = 5) and 5 ± 3% (n = 4) respectively (P < 0.05 vs. 10 µM milnacipran alone; Figure 5C).
Figure 5.
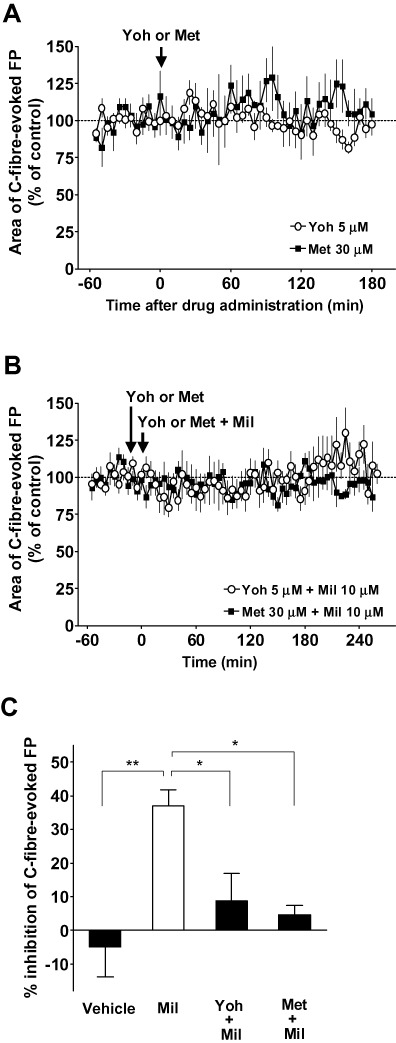
Effects of adrenoceptor or 5-HT receptor antagonists on the inhibitory effect of milnacipran (Mil) on the basal C-fibre-evoked field potentials (FPs) in modified spinal nerve ligation (SNL)-model animals. FPs in the spinal dorsal horn were elicited by electrical stimulation of the sciatic nerve fibres at 1 min intervals in modified SNL-model animals. Five consecutive responses were averaged. (A) Yohimbine (Yoh; n= 4) or methysergide (Met; n= 4) was administered spinally (arrow) after ≥60 min of stable baseline recordings. Each area of C-fibre-evoked FPs was normalized to the mean of 60 consecutive responses obtained prior to the administration (–60 to 0 min in the graph). (B) Yohimbine (n= 4) or methysergide (n= 4) was administered spinally after ≥45 min of stable baseline recordings (time at −15 min in the graph). Yohimbine or methysergide plus milnacipran were administered spinally 15 min after the administration of yohimbine or methysergide alone (time at 0 min in the graph). Each area of C-fibre-evoked FPs was normalized to the mean of 60 consecutive responses obtained prior to the administration of milnacipran (−60 to 0 min in the graph). (C) The inhibitory effects of 10 µM milnacipran on the basal C-fibre-evoked FPs with or without yohimbine or methysergide in modified SNL-model animals. The % inhibition of C-fibre-evoked FPs was calculated using the mean area of 30 consecutive FPs during 90–120 min following the vehicle or milnacipran administration in comparison to the baseline (averaged at −60 to 0 min in B or Figure 4). The values in vehicle and milnacipran groups are from Figure 4. Data shown are means ± SEM. *P < 0.05; **P < 0.01, significantly different as indicated.
Effects of milnacipran on the basal C-fibre-evoked FPs and the induction of LTP in naïve rats
To investigate whether the basal C-fibre-evoked FPs and the induction of spinal LTP in naïve animals were influenced by the spinal administration of milnacipran, the vehicle or milnacipran was applied to the recording segments following basal recordings (>60 min). In naïve animals, spinal administration of 1000 µM milnacipran (n= 5) had no significant effect on the basal C-fibre-evoked FPs compared with vehicle treatment (Figure 6A). The inhibition of C-fibre-evoked FPs at 30–60 min following the vehicle and milnacipran administration was −13 ± 3% (n = 5) and −3 ± 6% (n = 5) respectively (basal responses were averaged at 60–0 min prior to spinal administration, P > 0.05; Figure 6B). When HFS of the sciatic nerve fibres was applied 60 min following the administration, C-fibre-evoked FPs were increased equally in both vehicle- and Mil-treated rats, and the enhancement of C-fibre-evoked FPs was stably maintained for more than 240 min (Figure 6A). The mean area of C-fibre-evoked FPs at 30–60 min following HFS was 202 ± 20% (n = 5) in the vehicle-treated group and 183 ± 16% (n = 5) in the milnacipran-treated group (basal responses were averaged at 30–0 min prior to HFS, P > 0.05; Figure 6C).
Figure 6.
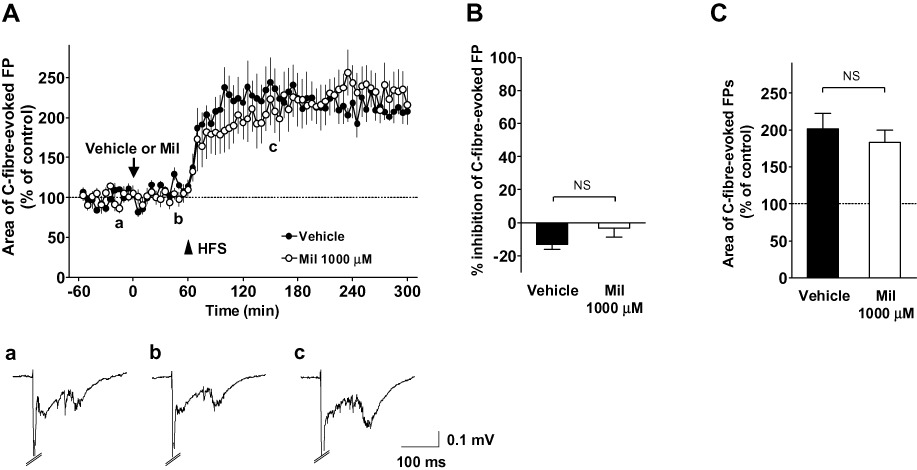
Effects of milnacipran (Mil) on the basal C-fibre-evoked field potentials (FPs) and on the induction of long-term potentiation (LTP) of C-fibre-evoked FPs in naïve animals. (A) The mean time course of C-fibre-evoked FPs prior to and after high-frequency stimulation (HFS; 0.5 ms duration, 40–45 V, 100 Hz, given in two trains of 1 s duration at 20 s intervals) following the spinal administration of the vehicle (n= 5) or 1000 µM milnacipran (n= 5). FPs were elicited by electrical stimulation of the sciatic nerve fibres at 1 min intervals in anaesthetized naïve adult rats. Each area of C-fibre-evoked FPs was normalized to the mean of 60 consecutive responses obtained prior to the administration of the vehicle or milnacipran (−60 to 0 min in the graph) and five consecutive responses were averaged. In both groups, HFS was applied 60 min after the administration (arrowhead). Example-averaged traces of five consecutive FPs from the milnacipran group recorded during the times indicated on the graph (a, b and c) are shown. (B) The inhibitory effect of vehicle or milnacipran (1000 µM) on the basal C-fibre-evoked FPs in naïve animals during 30–60 min following the administration. The area of C-fibre-evoked FPs was normalized to the mean of 60 consecutive responses obtained prior to the administration (−60 to 0 min in A). (C) Comparison of the mean C-fibre-evoked FPs during 30–60 min after HFS (time from 90 to 120 min in A) between the vehicle and milnacipran (1000 µM) groups. The area of C-fibre-evoked FPs was normalized to the mean of 30 consecutive responses obtained just prior to HFS (time from 30 to 60 min in A). Data shown are means ± SEM. NS, not significant.
Discussion and conclusions
In the present study, we investigated the effect of milnacipran, a representative SNRI, on C-fibre-mediated synaptic transmission and its plasticity in the rat spinal dorsal horn using in vivo electrophysiological studies. milnacipran significantly inhibited the basal C-fibre-evoked FPs in the modified SNL-model animals, but not in the basal condition of naïve animals. A significant inhibitory effect of milnacipran was also observed in the maintenance phase of LTP of C-fibre-evoked FPs. Thus, milnacipran inhibited C-fibre-evoked FPs in specific conditions (i.e. spinal LTP or chronic pain). We also demonstrated that the condition-dependent effect of milnacipran was mediated by spinal 5-HT1/2 receptors and α2-adrenoceptors.
SNRIs exhibit analgesic efficacy after intrathecal administration in various animal models of chronic pain (Shin and Eisenach, 2004; Obata et al., 2005; 2010; Ikeda et al., 2009); however, the mechanism has not been fully elucidated. The present study is the first to demonstrate that a monoamine reuptake inhibitor suppressed nociceptive synaptic transmission in spinal LTP and chronic pain (Figures 1, 3 and 4). Milnacipran inhibited C-fibre-evoked FPs after local spinal administration, which mostly prevents drugs from reaching other CNS regions (Beck et al., 1995; Ge et al., 2006), and the effects were blocked by spinally administered noradrenaline and/or 5-HT receptor antagonists (Figures 2, 3 and 5), all of which antagonize milnacipran-induced analgesia in chronic pain model animals (Obata et al., 2005; 2010). Milnacipran selectively inhibits both 5-HT and noradrenaline reuptake transporters, which are expressed at the bulbospinal 5-hydroxytryptaminergic and noradrenergic terminals (Moret et al., 1985; Sindrup et al., 2005), and increases the extracellular concentrations of both monoamines in the spinal cord in chronic pain conditions (Obata et al., 2010). Therefore, milnacipran could exert analgesic efficacy at the spinal level by inhibiting C-fibre-mediated nociceptive synaptic transmission by activating both the spinal 5-hydroxytryptaminergic and noradrenergic systems. The exact site of action of milnacipran in the spinal dorsal horn awaits further studies in the future. Furthermore, the basal C-fibre-evoked FPs in naïve animals were almost unchanged by milnacipran (Figure 6), and the maximal inhibitory effect of milnacipran in the maintenance phase of LTP did not exceed the pre-HFS level (Figure 1), indicating that milnacipran hardly affected the normal nociceptive transmission. These data may reflect the fact that SNRIs have limited efficacy for acute nociception (Iyengar et al., 2004; Jones et al., 2005; Suarez-Roca et al., 2006).
Antagonists of the α2-adrenoceptors mostly antagonized the effects of milnacipran in this study and the behavioural study (Obata et al., 2005). α2-adrenoceptors are expressed at the central terminals of C-fibre afferents and spinal excitatory neurons, and thought to primarily mediate analgesia by noradrenaline released from descending bulbospinal axons (Millan, 2002; Gassner et al., 2009). In fact, activation of α2-adrenoceptors suppresses excitatory neurotransmitter release from C-fibre afferents and excitatory synaptic transmission such as C-fibre-evoked FPs (Takano et al., 1993; Duflo et al., 2002; Ge et al., 2006). Although the contribution of α1-adrenoceptors, which can activate spinal inhibitory interneurons (Gassner et al., 2009), could not be completely ruled out, the spinal α2-adrenoceptors apper to play a key role in mediating the SNRI-induced inhibition of C-fibre-evoked FPs.
In animal models of neuropathic pain, spinal administration of 5-HT or SSRIs exerted analgesic effects (Bardin et al., 2000; Honda et al., 2006) and inhibited the firing of spinal dorsal horn neurons (Hains et al., 2003; Liu et al., 2010). Therefore, spinal nociceptive transmission in neuropathic conditions is likely to be suppressed when the spinal 5-HT level is elevated, which appears to overcome any pronociceptive effects of 5-HT (Millan et al., 1996; Millan, 2002; Aira et al., 2010). Previous findings suggested that the nociceptive transmission including C-fibre-evoked FPs could be significantly inhibited via spinal 5-HT1A, 5-HT1B and 5-HT2C receptor subtypes in rat models of neuropathic pain (Colpaert et al., 2002; Obata et al., 2007; Aira et al., 2010). 5-HT might not only inhibit the C-fibre terminals and spinal excitatory neurons via 5-HT1A/1B receptors (Millan, 2002; Yoshimura and Furue, 2006) but also facilitate noradrenaline release from the bulbospinal noradrenergic axons via 5-HT2C receptors (Obata et al., 2007) in the spinal dorsal horn. In the presence of methysergide, activation of spinal α2-adrenoceptors by the 5-HT-induced noradrenaline release might be also attenuated, which mostly antagonized the effects of milnacipran (Figures 2 and 5; Obata et al., 2005).
Milnacipran inhibited C-fibre-evoked FPs after the establishment of spinal LTP or abnormal pain. This indicates that monoaminergic modulation of spinal nociceptive synaptic transmission is changed after HFS and nerve injury. However, the mechanism(s) underlying the condition-dependent effect of milnacipran are unclear. Previous studies suggested that the extracellular concentrations of 5-HT and/or noradrenaline in the CNS were decreased in chronic pain conditions (Covey et al., 2000; Hains et al., 2002; Vogel et al., 2003; Liu et al., 2010). Several molecules such as calcium/calmodulin-dependent kinase II, which play critical roles in hypersensitivity and spinal LTP (Fang et al., 2002; Yang et al., 2004; Choi et al., 2005), might accelerate spinal monoamine reuptake (Ramamoorthy et al., 2011) in chronic pain and/or spinal LTP. Therefore, it is likely that blockade of monoamine receptors has no detectable influence on pain transmission, as shown by our finding that spinally administered methysergide and idazoxan (and/or yohimbine), given alone, did not change either C-fibre-evoked FPs after the establishment of spinal LTP or abnormal pain in this study, and also the paw withdrawal threshold to mechanical stimuli in a rodent post-operative pain model (Obata et al., 2010). Moreover, the decrease in monoamine levels affects the localization and/or sensitivity of their receptors (Roudet et al., 1994; Saunders and Limbird, 1999; Dziedzicka-Wasylewska et al., 2006; Omiya et al., 2008). Thus the inhibitory effect of clonidine, an α2-adrenoceptor agonist, on nociceptive transmission was potentiated after the depletion of spinal noradrenaline (Howe and Yaksh, 1982), as well as after the development of hypersensitivity (Duflo et al., 2002; Millan, 2002; Omiya et al., 2008) and LTP of C-fibre-evoked FPs (Ge et al., 2006). When monoamine concentrations are maintained at a high level by milnacipran pretreatment, the receptor function might be unaltered following HFS, and as a result, C-fibre-evoked FPs would be stable even in the presence of the drug (Figure 6). Therefore, presynaptic and presumably sequential postsynaptic functional alterations might be induced in the 5-hydroxytryptaminergic and noradrenergic systems after HFS and/or peripheral nerve injury, which may contribute to the appearance of the condition-dependent effect of milnacipran. Determination of whether chronic administration of SNRIs induces tolerance to their anti-hyperalgesic effects awaits further investigation.
In the present study, spinal LTP was induced in the presence of milnacipran (Figure 6). Recent findings suggested that several 5-HT and noradrenaline receptor subtypes have opposing roles, such as facilitation versus inhibition, for the induction of LTP or facilitation of C-fibre-evoked action potential discharges in the spinal cord (Ge et al., 2006; Rygh et al., 2006). Therefore, milnacipran-induced increases in spinal 5-HT and noradrenaline might have resulted in no net effect on the induction of spinal LTP. Further studies using subtype-specific 5-HT and noradrenaline receptor agonists and antagonists may improve our understanding of the roles of these receptor subtypes in the induction of spinal plasticity.
LTP of nociceptive synaptic transmission in the spinal dorsal horn has been proposed as a spinal mechanism of hypersensitivity (Liu and Sandkühler, 1995; Sandkühler, 2009); however, it is not fully understood whether and how LTP is related to chronic pain. Evaluation of C-fibre-evoked FPs with pharmacological treatment using in vivo preparations allowed us to compare the characteristics of spinal LTP and chronic pain, keeping in mind that acute LTP (less than 3 h after HFS) does not necessarily reflect the pathophysiological changes in neuropathic pain (e.g. transcriptional changes). Employing this approach, we previously reported that the effects of calcium channel blockers on the induction and maintenance of spinal LTP are in agreement with their effects on the development and maintenance of pathological pain respectively (Ohnami et al., 2011). As we demonstrated here, milnacipran prevented the maintenance, but not the induction, of LTP. This is compatible with previous findings showing that milnacipran potently ameliorates established chronic pain (Shin and Eisenach, 2004; Obata et al., 2005; 2010; Ikeda et al., 2009), without interfering with the development of hypersensitivity (Suarez-Roca et al., 2006). Milnacipran also inhibited C-fibre-evoked FPs in the nerve-injured animals. Most likely, peripheral nerve injury induced an LTP-like state, that is, a preexisting abnormal condition in the spinal cord, as C-fibre-evoked responses of spinal neurons were not further potentiated by HFS after nerve injury (Rygh et al., 2000; Zhang et al., 2004; Ohnami et al., 2011). Consistent with this, the basal C-fibre-evoked responses in the normal and neuropathic conditions were similar, despite the loss and/or damage of spinal nerves in neuropathic rats, as demonstrated here and previously (Matthews and Dickenson, 2001; Buesa et al., 2008; Ohnami et al., 2011), suggesting that nociceptive transmission had been already enhanced in the spinal cord by the modified SNL. The finding that milnacipran inhibited C-fibre-evoked FPs when administered spinally more than 3 h after HFS (Figure 3) may also support the importance of milnacipran for treatment of established chronic pain. However, it is most likely that the pathophysiological changes in chronic pain conditions are not completely normalized by milnacipran, considering that the continuous presence of increased monoamines by blocking their reuptake was important in the milnacipran-induced inhibition of the established spinal LTP (Figure 3).
In conclusion, the SNRI milnacipran inhibited C-fibre-mediated nociceptive synaptic transmission in the spinal dorsal horn in the maintenance phase of LTP and neuropathic conditions, but not in normal conditions, by activating the spinal 5-hydroxytryptaminergic and noradrenergic systems. This provides novel evidence regarding the analgesic mechanisms of SNRIs. The present study supports the notion that spinal LTP is a key factor in chronic pain and that SNRIs may be a promising treatment for various pain syndromes including neuropathic pain.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research (C) (20602004, 23590720) from the Japan Society for the Promotion of Science, Tokyo, Japan (M.T.).
Glossary
- CNS
central nervous system
- FP
field potential
- HFS
high-frequency stimulation
- LTP
long-term potentiation
- SNL
spinal nerve ligation
- SNRI
5-HT and noradrenaline reuptake inhibitor
- SSRI
selective serotonin reuptake inhibitor
Conflict of interest
There are no financial or other relationships that might lead to a conflict of interest.
References
- Aira Z, Buesa I, Salgueiro M, Bilbao J, Aguilera L, Zimmermann M, et al. Subtype-specific changes in 5-HT receptor-mediated modulation of C fibre-evoked spinal field potentials are triggered by peripheral nerve injury. Neuroscience. 2010;168:831–841. doi: 10.1016/j.neuroscience.2010.04.032. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin L, Schmidt J, Alloui A, Eschalier A. Effect of intrathecal administration of serotonin in chronic pain models in rats. Eur J Pharmacol. 2000;409:37–43. doi: 10.1016/s0014-2999(00)00796-2. [DOI] [PubMed] [Google Scholar]
- Beck H, Schröck H, Sandkühler J. Controlled superfusion of the rat spinal cord for studying non-synaptic transmission: an autoradiographic analysis. J Neurosci Methods. 1995;58:193–202. doi: 10.1016/0165-0270(94)00176-h. [DOI] [PubMed] [Google Scholar]
- Benrath J, Brechtel C, Stark J, Sandkühler J. Low dose of S(+)-ketamine prevents long-term potentiation in pain pathways under strong opioid analgesia in the rat spinal cord in vivo. Br J Anaesth. 2005;95:518–523. doi: 10.1093/bja/aei215. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Buesa I, Urrutia A, Aira Z, Salgueiro M, Bilbao J, Mozas M, et al. Depression of C fibre-evoked spinal field potentials by the spinal δ opioid receptor is enhanced in the spinal nerve ligation model of neuropathic pain: involvement of the µ-subtype. Neuropharmacology. 2008;55:1376–1382. doi: 10.1016/j.neuropharm.2008.08.029. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG, Shaw JL, Thompson L, Nelson DL, et al. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25:871–880. doi: 10.1016/S0893-133X(01)00298-6. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Choi SS, Seo YJ, Kwon MS, Shim EJ, Lee JY, Ham YO, et al. Increase of phosphorylation of calcium/calmodulin-dependent protein kinase-II in several brain regions by substance P administered intrathecally in mice. Brain Res Bull. 2005;65:375–381. doi: 10.1016/j.brainresbull.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Colpaert FC, Tarayre JP, Koek W, Pauwels PJ, Bardin L, Xu XJ, et al. Large-amplitude 5-HT1A receptor activation: a new mechanism of profound, central analgesia. Neuropharmacology. 2002;43:945–958. doi: 10.1016/s0028-3908(02)00119-3. [DOI] [PubMed] [Google Scholar]
- Coluzzi F, Mattia C. Mechanism-based treatment in chronic neuropathic pain: the role of antidepressants. Curr Pharm Des. 2005;11:2945–2960. doi: 10.2174/1381612054864993. [DOI] [PubMed] [Google Scholar]
- Covey WC, Ignatowski TA, Knight PR, Spengler RN. Brain-derived TNFα: involvement in neuroplastic changes implicated in the conscious perception of persistent pain. Brain Res. 2000;859:113–122. doi: 10.1016/s0006-8993(00)01965-x. [DOI] [PubMed] [Google Scholar]
- Duflo F, Li X, Bantel C, Pancaro C, Vincler M, Eisenach JC. Peripheral nerve injury alters the α2 adrenoceptor subtype activated by clonidine for analgesia. Anesthesiology. 2002;97:636–641. doi: 10.1097/00000542-200209000-00018. [DOI] [PubMed] [Google Scholar]
- Dziedzicka-Wasylewska M, Faron-Górecka A, Kuśmider M, Drozdowska E, Rogóż Z, Siwanowicz J, et al. Effect of antidepressant drugs in mice lacking the norepinephrine transporter. Neuropsychopharmacology. 2006;31:2424–2432. doi: 10.1038/sj.npp.1301064. [DOI] [PubMed] [Google Scholar]
- Fang L, Wu J, Lin Q, Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 2002;22:4196–4204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka T, Tokunaga A, Kondo E, Miki K, Tachibana T, Noguchi K. Change in mRNAs for neuropeptides and the GABAA receptor in dorsal root ganglion neurons in a rat experimental neuropathic pain model. Pain. 1998;78:13–26. doi: 10.1016/S0304-3959(98)00111-0. [DOI] [PubMed] [Google Scholar]
- Gassner M, Ruscheweyh R, Sandkühler J. Direct excitation of spinal GABAergic interneurons by noradrenaline. Pain. 2009;145:204–210. doi: 10.1016/j.pain.2009.06.021. [DOI] [PubMed] [Google Scholar]
- Ge YX, Xin WJ, Hu NW, Zhang T, Xu JT, Liu XG. Clonidine depresses LTP of C-fiber evoked field potentials in spinal dorsal horn via NO-cGMP pathway. Brain Res. 2006;1118:58–65. doi: 10.1016/j.brainres.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Hains BC, Everhart AW, Fullwood SD, Hulsebosch CE. Changes in serotonin, serotonin transporter expression and serotonin denervation supersensitivity: involvement in chronic central pain after spinal hemisection in the rat. Exp Neurol. 2002;175:347–362. doi: 10.1006/exnr.2002.7892. [DOI] [PubMed] [Google Scholar]
- Hains BC, Willis WD, Hulsebosch CE. Serotonin receptors 5-HT1A and 5-HT3 reduce hyperexcitability of dorsal horn neurons after chronic spinal cord hemisection injury in rat. Exp Brain Res. 2003;149:174–186. doi: 10.1007/s00221-002-1352-x. [DOI] [PubMed] [Google Scholar]
- Honda M, Uchida K, Tanabe M, Ono H. Fluvoxamine, a selective serotonin reuptake inhibitor, exerts its antiallodynic effects on neuropathic pain in mice via 5-HT2A/2C receptors. Neuropharmacology. 2006;51:866–872. doi: 10.1016/j.neuropharm.2006.05.031. [DOI] [PubMed] [Google Scholar]
- Howe JR, Yaksh TL. Changes in sensitivity to intrathecal norepinephrine and serotonin after 6-hydroxydopamine (6-OHDA), 5,6-dihydroxytryptamine (5,6-DHT) or repeated monoamine administration. J Pharmacol Exp Ther. 1982;220:311–321. [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jager T, et al. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Ishida Y, Naono R, Takeda R, Abe H, Nakamura T, et al. Effects of intrathecal administration of newer antidepressants on mechanical allodynia in rat models of neuropathic pain. Neurosci Res. 2009;63:42–46. doi: 10.1016/j.neures.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Iyengar S, Webster AA, Hemrick-Luecke SK, Xu JY, Simmons RM. Efficacy of duloxetine, a potent and balanced serotonin-norepinephrine reuptake inhibitor in persistent pain models in rats. J Pharmacol Exp Ther. 2004;311:576–584. doi: 10.1124/jpet.104.070656. [DOI] [PubMed] [Google Scholar]
- Jones CK, Peters SC, Shannon HE. Efficacy of duloxetine, a potent and balanced serotonergic and noradrenergic reuptake inhibitor, in inflammatory and acute pain models in rodents. J Pharmacol Exp Ther. 2005;312:726–732. doi: 10.1124/jpet.104.075960. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- King T, Rao S, Vanderah T, Chen Q, Vardanyan A, Porreca F. Differential blockade of nerve injury-induced shift in weight bearing and thermal and tactile hypersensitivity by milnacipran. J Pain. 2006;7:513–520. doi: 10.1016/j.jpain.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Lee YC, Chen PP. A review of SSRIs and SNRIs in neuropathic pain. Expert Opin Pharmacother. 2010;11:2813–2825. doi: 10.1517/14656566.2010.507192. [DOI] [PubMed] [Google Scholar]
- Liu FY, Qu XX, Ding X, Cai J, Jiang H, Wan Y, et al. Decrease in the descending inhibitory 5-HT system in rats with spinal nerve ligation. Brain Res. 2010;1330:45–60. doi: 10.1016/j.brainres.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Liu XG, Sandkühler J. Long-term potentiation of C-fiber-evoked potentials in the rat spinal dorsal horn is prevented by spinal N-methyl-D-aspartic acid receptor blockage. Neurosci Lett. 1995;191:43–46. doi: 10.1016/0304-3940(95)11553-0. [DOI] [PubMed] [Google Scholar]
- Liu X, Sandkühler J. Characterization of long-term potentiation of C-fiber-evoked potentials in spinal dorsal horn of adult rat: essential role of NK1 and NK2 receptors. J Neurophysiol. 1997;78:1973–1982. doi: 10.1152/jn.1997.78.4.1973. [DOI] [PubMed] [Google Scholar]
- Mao J, Chen LL. Systemic lidocaine for neuropathic pain relief. Pain. 2000;87:7–17. doi: 10.1016/S0304-3959(00)00229-3. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Dickenson AH. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain. 2001;92:235–246. doi: 10.1016/s0304-3959(01)00255-x. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Seguin L, Honore P, Girardon S, Bervoets K. Pro- and antinociceptive actions of serotonin (5-HT)1A agonists and antagonists in rodents: relationship to algesiometric paradigm. Behav Brain Res. 1996;73:69–77. doi: 10.1016/0166-4328(96)00073-3. [DOI] [PubMed] [Google Scholar]
- Minami K, Hasegawa M, Ito H, Nakamura A, Tomii T, Matsumoto M, et al. Morphine, oxycodone, and fentanyl exhibit different analgesic profiles in mouse pain models. J Pharmacol Sci. 2009;111:60–72. doi: 10.1254/jphs.09139fp. [DOI] [PubMed] [Google Scholar]
- Moret C, Charveron M, Finberg JP, Couzinier JP, Briley M. Biochemical profile of midalcipran (F 2207), 1-phenyl-1-diethyl-aminocarbonyl-2-aminomethyl-cyclopropane (Z) hydrochloride, a potential fourth generation antidepressant drug. Neuropharmacology. 1985;24:1211–1219. doi: 10.1016/0028-3908(85)90157-1. [DOI] [PubMed] [Google Scholar]
- Obata H, Saito S, Koizuka S, Nishikawa K, Goto F. The monoamine-mediated antiallodynic effects of intrathecally administered milnacipran, a serotonin noradrenaline reuptake inhibitor, in a rat model of neuropathic pain. Anesth Analg. 2005;100:1406–1410. doi: 10.1213/01.ANE.0000149546.97299.A2. [DOI] [PubMed] [Google Scholar]
- Obata H, Ito N, Sasaki M, Saito S, Goto F. Possible involvement of spinal noradrenergic mechanisms in the antiallodynic effect of intrathecally administered 5-HT2C receptor agonists in the rats with peripheral nerve injury. Eur J Pharmacol. 2007;567:89–94. doi: 10.1016/j.ejphar.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Obata H, Kimura M, Nakajima K, Tobe M, Nishikawa K, Saito S. Monoamine-dependent, opioid-independent antihypersensitivity effects of intrathecally administered milnacipran, a serotonin noradrenaline reuptake inhibitor, in a postoperative pain model in rats. J Pharmacol Exp Ther. 2010;334:1059–1065. doi: 10.1124/jpet.110.168336. [DOI] [PubMed] [Google Scholar]
- Ohnami S, Tanabe M, Shinohara S, Takasu K, Kato A, Ono H. Role of voltage-dependent calcium channel subtypes in spinal long-term potentiation of C-fiber-evoked field potentials. Pain. 2011;152:623–631. doi: 10.1016/j.pain.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Omiya Y, Yuzurihara M, Suzuki Y, Kase Y, Kono T. Role of α2-adrenoceptors in enhancement of antinociceptive effect in diabetic mice. Eur J Pharmacol. 2008;592:62–66. doi: 10.1016/j.ejphar.2008.06.087. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Shippenberg TS, Jayanthi LD. Regulation of monoamine transporters: role of transporter phosphorylation. Pharmacol Ther. 2011;129:220–238. doi: 10.1016/j.pharmthera.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randić M, Jiang MC, Cerne R. Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. J Neurosci. 1993;13:5228–5241. doi: 10.1523/JNEUROSCI.13-12-05228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudet C, Mouchet P, Feuerstein C, Savasta M. Normal distribution of alpha 2-adrenoceptors in the rat spinal cord and its modification after noradrenergic denervation: a quantitative autoradiographic study. J Neurosci Res. 1994;39:319–329. doi: 10.1002/jnr.490390309. [DOI] [PubMed] [Google Scholar]
- Rygh LJ, Kontinen VK, Suzuki R, Dickenson AH. Different increase in C-fibre evoked responses after nociceptive conditioning stimulation in sham-operated and neuropathic rats. Neurosci Lett. 2000;288:99–102. doi: 10.1016/s0304-3940(00)01201-5. [DOI] [PubMed] [Google Scholar]
- Rygh LJ, Suzuki R, Rahman W, Wong Y, Vonsy JL, Sandhu H, et al. Local and descending circuits regulate long-term potentiation and zif268 expression in spinal neurons. Eur J Neurosci. 2006;24:761–772. doi: 10.1111/j.1460-9568.2006.04968.x. [DOI] [PubMed] [Google Scholar]
- Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Sandkühler J, Liu X. Induction of long-term potentiation at spinal synapses by noxious stimulation or nerve injury. Eur J Neurosci. 1998;10:2476–2480. doi: 10.1046/j.1460-9568.1998.00278.x. [DOI] [PubMed] [Google Scholar]
- Saunders C, Limbird LE. Localization and trafficking of α2-adrenergic receptor subtypes in cells and tissues. Pharmacol Ther. 1999;84:193–205. doi: 10.1016/s0163-7258(99)00032-7. [DOI] [PubMed] [Google Scholar]
- Shin SW, Eisenach JC. Peripheral nerve injury sensitizes the response to visceral distension but not its inhibition by the antidepressant milnacipran. Anesthesiology. 2004;100:671–675. doi: 10.1097/00000542-200403000-00030. [DOI] [PubMed] [Google Scholar]
- Sindrup SH, Otto M, Finnerup NB, Jensen TS. Antidepressants in the treatment of neuropathic pain. Basic Clin Pharmacol Toxicol. 2005;96:399–409. doi: 10.1111/j.1742-7843.2005.pto_96696601.x. [DOI] [PubMed] [Google Scholar]
- Suarez-Roca H, Quintero L, Arcaya JL, Maixner W, Rao SG. Stress-induced muscle and cutaneous hyperalgesia: differential effect of milnacipran. Physiol Behav. 2006;88:82–87. doi: 10.1016/j.physbeh.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Ueta K, Tamagaki S, Mashimo T. Antiallodynic and antihyperalgesic effect of milnacipran in mice with spinal nerve ligation. Anesth Analg. 2008;106:1309–1315. doi: 10.1213/ane.0b013e318167889a. [DOI] [PubMed] [Google Scholar]
- Svendsen F, Tjølsen A, Hole K. LTP of spinal Aβ and C-fibre evoked responses after electrical sciatic nerve stimulation. Neuroreport. 1997;10:3427–3430. doi: 10.1097/00001756-199711100-00002. [DOI] [PubMed] [Google Scholar]
- Takano M, Takano Y, Yaksh TL. Release of calcitonin gene-related peptide (CGRP), substance P (SP), and vasoactive intestinal polypeptide (VIP) from rat spinal cord: modulation by alpha 2 agonists. Peptides. 1993;14:371–378. doi: 10.1016/0196-9781(93)90055-l. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Murakami H, Honda M, Ono H. Gabapentin depresses C-fiber-evoked field potentials in rat spinal dorsal horn only after induction of long-term potentiation. Exp Neurol. 2006;202:280–286. doi: 10.1016/j.expneurol.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Vogel C, Mössner R, Gerlach M, Heinemann T, Murphy DL, Riederer P, et al. Absence of thermal hyperalgesia in serotonin transporter-deficient mice. J Neurosci. 2003;23:708–715. doi: 10.1523/JNEUROSCI.23-02-00708.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallenstein S, Zucker CL, Fleiss JL. Some statistical methods useful in circulation research. Circ Res. 1980;47:1–9. doi: 10.1161/01.res.47.1.1. [DOI] [PubMed] [Google Scholar]
- Yang HW, Hu XD, Zhang HM, Xin WJ, Li MT, Zhang T, et al. Roles of CaMKII, PKA, and PKC in the induction and maintenance of LTP of C-fiber-evoked field potentials in rat spinal dorsal horn. J Neurophysiol. 2004;91:1122–1133. doi: 10.1152/jn.00735.2003. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Furue H. Mechanisms for the anti-nociceptive actions of the descending noradrenergic and serotonergic systems in the spinal cord. J Pharmacol Sci. 2006;101:107–117. doi: 10.1254/jphs.crj06008x. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Zhou LJ, Hu XD, Hu NW, Zhang T, Liu XG. Acute nerve injury induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn of intact rat. Sheng Li Xue Bao. 2004;56:591–596. [PubMed] [Google Scholar]
- Zhang XC, Zhang YQ, Zhao ZQ. Different roles of two nitric oxide activated pathways in spinal long-term potentiation of C-fiber-evoked field potentials. Neuropharmacology. 2006;50:748–754. doi: 10.1016/j.neuropharm.2005.11.021. [DOI] [PubMed] [Google Scholar]