

Abstract
BACKGROUND AND PURPOSE
Acute silencing of caveolin-1 (Cav-1) modulates receptor-mediated contraction of airway smooth muscle. Moreover, COX-2- and 5-lipoxygenase (5-LO)-derived prostaglandin and leukotriene biosynthesis can influence smooth muscle reactivity. COX-2 half-life can be prolonged through association with Cav-1. We suggested that lack of Cav-1 modulated levels of COX-2 which in turn modulated tracheal contraction, when arachidonic acid signalling was disturbed by inhibition of COX-2.
EXPERIMENTAL APPROACH
Using tracheal rings from Cav-1 knockout (KO) and wild-type mice (B6129SF2/J), we measured isometric contractions to methacholine and used PCR, immunoblotting and immunohistology to monitor expression of relevant proteins.
KEY RESULTS
Tracheal rings from Cav-1 KO and wild-type mice exhibited similar responses, but the COX-2 inhibitor, indomethacin, increased responses of tracheal rings from Cav-1 KO mice to methacholine. The phospholipase A2 inhibitor, eicosatetraynoic acid, which inhibits formation of both COX-2 and 5-LO metabolites, had no effect on wild-type or Cav-1 KO tissues. Indomethacin-mediated hyperreactivity was ablated by the LTD4 receptor antagonist (montelukast) and 5-LO inhibitor (zileuton). The potentiating effect of indomethacin on Cav-1 KO responses to methacholine was blocked by epithelial denudation. Immunoprecipitation showed that COX-2 binds Cav-1 in wild-type lungs. Immunoblotting and qPCR revealed elevated levels of COX-2 and 5-LO protein, but not COX-1, in Cav-1 KO tracheas, a feature that was prevented by removal of the epithelium.
CONCLUSION AND IMPLICATIONS
The indomethacin-induced hypercontractility observed in Cav-1 KO tracheas was linked to increased expression of COX-2 and 5-LO, which probably enhanced arachidonic acid shunting and generation of pro-contractile leukotrienes when COX-2 was inhibited.
Keywords: caveolin-1, contraction, 5-lipoxygenase, airway smooth muscle, PGE2
Introduction
Caveolae are flask-shaped invaginations of the plasma membrane found in variable numbers in different cell types, being most prominent in vascular endothelial cells, adipocytes, fibroblasts, epithelial cells and smooth muscle cells. They segregate receptors and signalling intermediates and form a micro-environment where local kinases and phosphatases can modify downstream signalling events and cell responses (Okamoto et al., 1998; Simons and Toomre, 2000; Stan, 2005). Caveolin-1 (Cav-1), the primary structural protein of caveolae, plays a key role in orchestrating activation of pathways that underpin cell proliferation, migration and contraction (Cohen et al., 2004; Halayko and Stelmack, 2005). In smooth muscle, caveolae are enriched in Ca2+-handling and binding proteins, and trimeric G-proteins (Li et al., 1995; de Weerd and Leeb-Lundberg, 1997; Darby et al., 2000; Gosens et al., 2007a) which is of functional significance, as acute disruption of plasma membrane caveolae suppresses GPCR-mediated contraction in airway smooth muscle (ASM) (Gosens et al., 2007b; Prakash et al., 2007; Sharma et al., 2010). ASM contraction in situ is regulated both by intrinsic cellular pathways and by mediators released from neighbouring cells, such as the airway epithelium. Thus, there is an intrinsic role for caveolins in regulating isolated ASM cell contraction. However, a systematic assessment of mechanisms that integrate contraction of intact multicellular airways from Cav-1 knockout (KO) & wild type mice is still lacking.
Arachidonic acid metabolites play an important role in cellular physiology, and their aberrant biosynthesis via COX-2 or 5-lipoxygenase (5-LO) pathways has been linked with allergic and inflammatory diseases (Wenzel, 1997; Wasserman, 1988; Peters-Golden and Henderson, 2007). The increased contractile function of ASM, as seen in chronic airway diseases, can be regulated by the airway epithelium which is a rich source of lipid mediators that regulate ASM tone and contractility (Barnes et al., 1988; Holtzman, 1992; Raeburn and Webber, 1994; Chung and Barnes, 1999). Epithelium-associated changes in COX-2 activity and levels of endogenous PGE2 modulated changes in airway reactivity and bronchial inflammation in experimental models of asthma (Gavett et al., 1999; Peebles et al., 2002; Torres et al., 2008). Recent research also suggests a role for Cav-1 in chronic lung diseases such as asthma, chronic obstructive pulmonary disease and pulmonary fibrosis (Gosens et al., 2010; Xia et al., 2010; Ryter et al., 2011). Moreover, allergen-naïve caveolin-1 knockout (Cav-1 KO) mice exhibit an altered lung phenotype that is likely to affect airway physiology (Murata et al., 2007). Interestingly, although reduced Cav-1 expression in whole lung has been demonstrated in allergen-challenged mice (Le Saux et al., 2008), the opposite effect has recently been reported for ASM upon allergen challenge of guinea pigs (Gosens et al., 2010). The COX-2 enzyme is co-localized with Cav-1 in caveolae, and the binding of COX-2 with Cav-1 targets the former for degradation via endoplasmic reticulum-associated mechanisms (Liou et al., 2001; Chen et al., 2010a). These studies indicate a novel function for Cav-1 in regulating both COX-2 expression and activity.
Therefore, we used Cav-1 KO mice to test the hypothesis that the lack of Cav-1 would modulate COX-2 abundance and that this modulation would affect control of tracheal contractility when signalling through the arachidonic acid cascade was disrupted by selective inhibition of the cyclooxygenase pathway. We found that responses to methacholine of tracheal preparations from Cav-1 KO mice were highly sensitive to pharmacological manipulation of COX but this was not observed in rings from wild-type mice. Notably, the effects of COX inhibition could be reversed using 5-LO pathway inhibitors, indicating shunting of arachidonic acid towards pro-contractile 5-LO metabolites in Cav-1 null airways. We also found that removal of the epithelium modified COX-2 sensitivity in Cav-1 KO preparations, and that COX-2 and 5-LO abundance was markedly increased in the airway epithelium of Cav-1 KO mice. These data suggest that, despite an increase in both COX-2 and 5-LO proteins in Cav-1 KO airways, normal functional balance was maintained because intrinsic airway contractility was not affected. However, due to the elevated activity of both pathways, when COX-2 was inhibited there was an enhanced generation of pro-contractile arachidonic acid metabolites via 5-LO, and these products maintained hypercontractility of Cav-1-deficient airways through montelukast-sensitive LT receptors.
Methods
Isolated tracheal ring preparation and epithelium removal
All animal care and experimental protocols were approved by the University of Manitoba Animal Care Committee. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). All animals were maintained on a 12 h dark and light cycle and were fed with regular laboratory chow while in-house at the university facility. Cav-1 KO Cav1tm1Mls/J and, as genetic controls, B6129SF2/J (wild-type) mice were used in this study. All mice were female, aged between 8 and 12 weeks (total = 41 Cav-1 KO mice and 41 wild-type) and were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Animals were killed with an overdose of pentobarbital (90 mg·kg−1) before tracheal isolation. For this, the chest cavity contents were removed en masse and placed in Krebs-Henseleit bicarbonate solution (K-H) of the following composition (in mM): 118 NaCl, 23.5 NaCO3, 4.69 KCl, 1.18 KH2PO4, 1.00 MgCl2, 2.50 CaCl2 and 5.55 dextrose. The K-H solution was gassed with 95% O2-5% CO2 to maintain a pH between 7.3 and 7.5. Tracheal isolations were carried out in cold K-H solution (4°C) by pinning the apex of the heart and the voice box of trachea to a dissecting dish and removing extraneous tissue. Lungs were removed and frozen for protein and RNA analysis (see below).
Each isolated trachea was cut into four segments with each segment containing three to four cartilage rings. Tracheal ring preparations were mounted between two pins: – one pin firmly fixed and the other attached to an isometric force transducer in one chamber of a Danish Myo Technology (Aarhus, Denmark) organ bath system. The paries membranaceus of the tracheal ring preparation (containing the smooth muscle layer) was placed between the two support pins. Tissue preparations were maintained in gassed K-H solution at 37°C and pH 7.3–7.5, for all subsequent studies.
For some studies, epithelium-denuded tracheal preparations were used. To remove the epithelium, tracheal segments were threaded onto silk surgical threads (Ethicon P4888C size 5); then the rings were rolled 3 revolutions on a paper towel soaked with K-H solution (Ndukwu et al., 1994). Randomly selected tracheal preparations were saved for histological evaluation of epithelial removal whilst others were used for immunohistochemical staining of Cav-1, 5-LO and COX-2.
Smooth muscle equilibration
To establish resting tension, reference length and stable baseline, tracheal rings were equilibrated for 90–120 min with intermittent (∼20 min) instillation of 63 mM KCl-substituted K-H solution (usually three exposures) in order to contract the tissues isometrically in a receptor-independent manner. Reference length of preparations was established by stretching rings after each KCl stimulation to achieve a resting tension of 0.6 mN. The isometric force developed for each smooth muscle preparation in response to the third exposure to the KCl-substituted K-H solution was used as the reference force for subsequent contractions elicited with methacholine.
Methacholine concentration-response studies
After equilibration, tracheal rings were incubated for 30 min with either dimethyl sulfoxide (vehicle control; 1 µL·mL−1), indomethacin (non-selective COX inhibitor; 3 µM), eicosatetraynoic acid (ETYA; phospholipase A2 (PLA2) inhibitor, 10 µM) alone, or in combination with either montelukast (LTC4 and LTD4 receptor antagonist; 10 µM) or zileuton (5-LO inhibitor; 10 µM). Tracheal rings from Cav-1 KO and wild-type mice were randomly assigned to treatment groups. After incubation with inhibitors, concentration-response studies with methacholine (1.0 nM–1.0 mM) were performed.
Preparation of lung and tracheal tissue lysates from Cav-1 KO and wild-type mice
After the final administration of methacholine in myography studies, rings were washed with K-H solution and stored in protein lysis buffer at −80°C (buffer composition: 40 mM Tris, 150 mM NaCl, 1% IgepalCA-630, 1% deoxycholic acid, 1 mM NaF, 5 mM β-glycerophosphate, 1 mM Na3VO4, 10 µg·mL−1 aprotinin, 10µg·mL−1 leupeptin, 7 µg·mL−1 pepstatin A, 1 mM PMSF, pH 8.0). Entire lungs were cut into small fragments in ice cold K-H solution. For protein isolation, tissues were stored in protein lysis buffer at −80°C. To prepare homogenates, samples were thawed, homogenized using a Polytron, and supernatant collected after centrifugation (10 000×g, 5 min) and stored at −20°C for subsequent protein assay and immunoblot analyses.
Protein blot analysis
Protein content in samples was determined using the Bio-Rad protein (Bio-Rad, Hercules, CA, USA). Immunoblotting was performed using standard techniques described previously (Halayko et al., 1999; Sharma et al., 2008). Briefly, samples were size separated under reducing conditions using SDS-polyacrylamide gels, and thereafter transferred to nitrocellulose membranes. After blocking membranes using skim milk in Tris-buffered saline (TBS) with Tween-20 (0.2%), membranes were incubated with primary antibodies diluted in TBS/1% skim milk/Tween-20 overnight, then with HRP-conjugated secondary antibodies prior to visualization on film using enhanced chemiluminescence reagents (Amersham, Buckinghamshire, UK). β-actin was used to correct for equal loading of all samples.
Immunoprecipitation
Immunoprecipitation was carried out as previously described (Sharma et al., 2010). Protein-G-conjugated sepharose beads (GE Healthcare, Uppsala, Sweden) were mixed with tissue lysates and incubated overnight with Cav-1 antibody at 4°C. Beads were washed with TBS/0.1% Tween 20 and PBS, then stored at −80°C until used for protein blot analysis for COX-2 protein.
RNA isolation and real-time RT-PCR analysis
Lungs were cut into small fragments in ice cold K-H solution then stored in RNAlater* buffer at −80°C. Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, Mississauga, ON, Canada). After assessing RNA concentration and purity (Chirgwin et al., 1979), RNA (1 µg) was reverse transcribed using the Quantitect Reverse Transcription Kit (Qiagen). Real-time PCR was carried out with the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using primer pairs for COX-2, 5-LO, Cav-1 and 18S RNA [calibrator gene for qPCR using previously published method (Sharma et al., 2008)]. The primers are described in Table 1. The reaction conditions used are described previously (Sharma et al., 2008). Product specificity was determined by dissociation curve analysis. Relative quantitation of gene expression was performed using the 7500 Sequence Detection software v.1.4 (Applied Biosystems).
Table 1.
Primers used for RT-PCR analyses
| Gene | Forward primer | Reverse primer |
|---|---|---|
| COX-2 (PES-2) | TTGCTGTACAAGCAGTGGCAAAGG | ACAGGAGAGAAGGAAATGGCTGA |
| 5-LO | CCATTGCCATCCAGCTCAACCAAA | TGTCTGAGGTGTTTGGTATCGCCA |
| Cav-1 | CTCCGAGGGACATCTCTACAC | CAGCAACATCCGCATCAGCAC |
| GAPDH | AGCAATGCCTCCTGCACCACCAAC | AGACTGTGGATGGCCCCTCCGG |
| 18s RNA | CGCCGCTAGAGGTGAAATTC | TTGGCAAATGCTTTCGCTC |
Histology and immunohistochemistry
Paraffin sections (4 µm thickness) were prepared using a Shandon Finesse E Microtome Sectioner (Fisher Scientific, Ottawa, ON, Canada). After mounting on slides, sections were deparaffinized and rehydrated in xylene and graded ethanol. Heat-induced epitope retrieval was performed by placing slides in a pre-warmed steamer (60–90°C) with citrate buffer (0.1 M citric acid, 0.1 M sodium citrate, pH 6.0) for 30 min. After washing with 1X PBS, samples were incubated in blocking solution (3% BSA/PBS) for 30 min at room temperature, then washed and incubated in 3% H2O2/PBS) for 10 min, and last in Avidin and Biotin blocking solution (Vector Inc, Burlington, ON, Canada) for 15 min. Thereafter, samples were incubated in diluted primary antibodies (4°C overnight). When mouse primary antibodies were used, sections were then incubated with unconjugated AffiniPure Fab Fragment goat anti-mouse IgG (Jackson ImmunoResearch Labs, West Grove, PA, USA) for 1 h. Sections were then incubated with diluted biotinylated secondary antibody for 1 h, washed, and finally in ABC solution (Vector Inc) for 30 min. To develop slides, 3,3-diaminobenzidine substrate was added to the sections and incubated for 2–5 min, then washed in distilled water before nuclei were counter-stained with Mayer's haematoxylin and sections were mounted with Permount (Thermo Fisher Scientific, Ottawa, ON, Canada). As a negative control, primary antibodies were omitted.
Data analysis
Data are shown as means ± SEM, unless otherwise stated. Student's unpaired t-test or one-way anova was used where appropriate. When anova revealed a difference among groups, Tukey's multiple range test was used to determine significant differences between groups; P-values < 0.05 were considered significant.
Materials
Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and HRP-conjugated goat anti-rabbit IgG were obtained from Sigma, and primary antibodies were obtained from the following sources: caveolin-1 (BD Transduction Labs (Franklin Lakes, NJ, USA) and Santa Cruz Biotechnology, Santa Cruz, CA, USA), 5-LO (Santa Cruz Biotechnology), COX-2 (Calbiochem, EMD Millipore, Darmstadt, Germany). Montelukast was generously provided by Merck Frosst Canada Inc., (Kirkland Lake, PQ, Canada) and zileuton was supplied by R&D Systems, Inc., (Minneapolis, MN). Indomethacin, methacholine and ETYA were from Sigma-Aldrich, (St Louis, MO) and pentobarbital from Ceva Santé Animale (Libourne, France). All other chemicals used were of analytical grade. Receptor nomenclature follows Alexander et al., (2011).
Results
Inhibiting COX induces hyperreactivity selectively in tracheas from Cav-1 KO mice
In the absence of indomethacin, tracheal rings from Cav-1 KO mice and the wild-type controls responded similarly methacholine (Figure 1A). However, in the presence of indomethacin (3 µM), tracheal rings from Cav-1 KO mice developed hyperreactivity to methacholine whereas responses of tracheal rings from wild-type mice were unaffected (Figure 1A). In contrast to the selective effects of indomethacin on Cav-1 KO tissues, treatment with either the cysteinyl leukotriene (CysLT1) receptor antagonist montelukast (10 µM) or the 5-LO inhibitor zileuton (10 µM) was without effect on the responses of tracheal rings to methacholine, regardless of the mouse strain (Figure 1B,C). To establish the role of arachidonic acid signalling as a determinant of tracheal responses, we treated preparations with the phospholipase A2 inhibitor ETYA (10 µM) which prevents the liberation of arachidonic acid and thereby the formation of both COX and 5-LO metabolites. ETYA was without effect on basal tone or responses to methacholine in tissues from either mouse strain (data not shown), suggesting that the functional balance of COX and 5-LO metabolites produced was not changed in Cav-1 KO mice. Thus, the effects of indomethacin on Cav-1 KO tissue could be due to a disruption of the intrinsic balance between the COX and 5-LO pathways of the arachidonic acid cascade. To assess this possibility, in Cav1 KO tracheal rings treated with indomethacin, we also added montelukast (10 µM) or zileuton (10 µM). Both co-treatments reversed indomethacin-induced hyperresponsiveness (Figure 1D). These data suggest that COX inhibition in Cav-1 KO, but not in wild-type tissues, may have shunted a functionally significant fraction of arachidonic acid through the 5-LO pathway to promote contractile responses, or blocked production of COX-derived prostanoids that control contraction.
Figure 1.
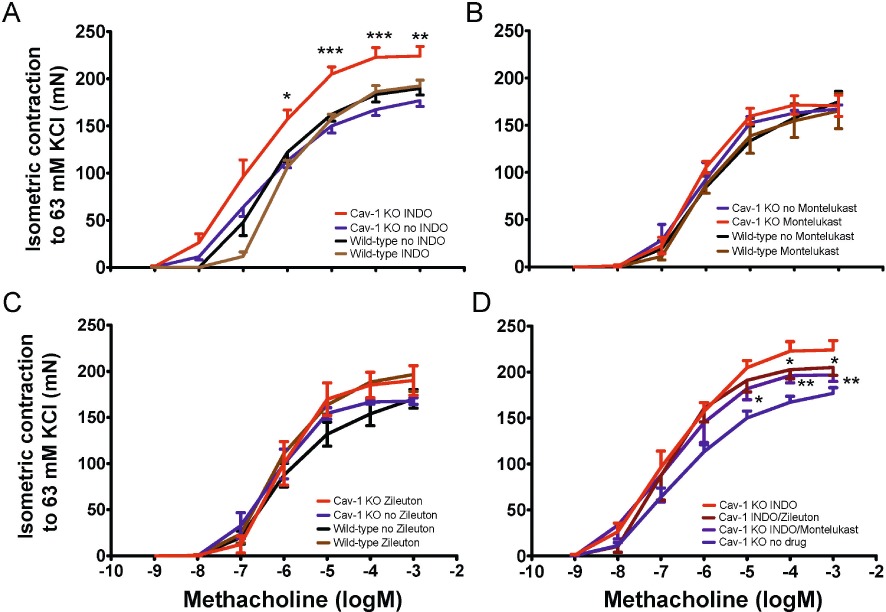
MCh-induced isometric force produced by Cav-1 KO and wild-type tracheal rings in the presence of epithelium. Trachea from Cav-1 KO and wild-type mice was isolated and sliced to obtain four equal-sized segments containing three to four cartilage rings. Tissues were equilibrated for 90–120 min with intermittent (∼20 min) instillation of 63 mM KCl-substituted K-H solution to obtain a stable resting tension at ∼0.6 mN. Isometric force was measured with increasing cumulative concentrations of methacholine (10−9–10−3 M). (A) Some tissues were incubated with indomethacin (INDO;) (n= 12 rings from 6 Cav-1 KO and n= 9 rings from 5 wild-type mice) while others received DMSO (vehicle) alone (n= 21 rings from 13 Cav-1 KO and n= 31 rings from 17 wild-type mice). With indomethacin treatment, there were larger responses in Cav-1 KO preparations, compared with indomethacin-free Cav-1 KO tissues, to methacholine between 10−6 and 10−3 M. *P < 0.05; **P < 0.01; ***P < 0.001: one-way ANOVA. (B) Tracheal rings were incubated for 30 min with montelukast (10 µM) alone before performing methacholine concentration-response studies. No significant differences were seen between Cav-1 KO and wild-type groups (n= 8 rings from 4 mice; one-way anova). (C) Tracheal rings were incubated for 30 min with zileuton (10 µM) alone before performing methacholine concentration-response studies. No significant differences were seen between Cav-1 KO and wild-type groups (n= 7 rings from 4 mice; one-way anova). (D). Tracheal rings were incubated for 30 min with indomethacin in the presence or absence of montelukast or zileuton (10 µM) before performing methacholine concentration-response studies. Compared with indomethacin-treated Cav-1 KO rings, montelukast (n= 8 rings from 8 mice, *P < 0.05, one-way ANOVA) and zileuton (n= 4 rings from 4 mice, **P < 0.01, one-way ANOVA) both significantly reversed indomethacin-induced responses in Cav-1 KO mice.
Indomethacin-induced methacholine hyperreactivity in Cav-1 KO airways is epithelium dependent
As arachidonic acid signalling is prominent in epithelial cells, tracheal rings from Cav-1 KO and wild-type mice were denuded of epithelium (Figure 2A) and the effects on responses to methacholine in the absence and presence of indomethacin measured (Figure 2B). Responses to methacholine in the absence of epithelium were indistinguishable between strains and were not different from epithelium-intact preparations. In contrast to our studies with epithelium-intact preparations, the addition of indomethacin had no effect on responses to methacholine of Cav-1 KO preparations, after removal of epithelium. These data suggest that hyperreactivity unmasked by COX inhibition in Cav-1 KO tissues was likely to have been the result of the absence of Cav-1 from epithelial cells, and not a change in contractile capacity of the ASM per se. Consistent with this conclusion, we found that KCl-induced isometric force generated by epithelium-intact or epithelium-denuded isolated tracheal rings from either strain showed no difference in maximal response to 63 mM KCl (Figure 2C). Moreover, there was no strain-dependent difference in the baseline tone of epithelium-intact or epithelium-denuded tracheal rings (Figure 2D).
Figure 2.
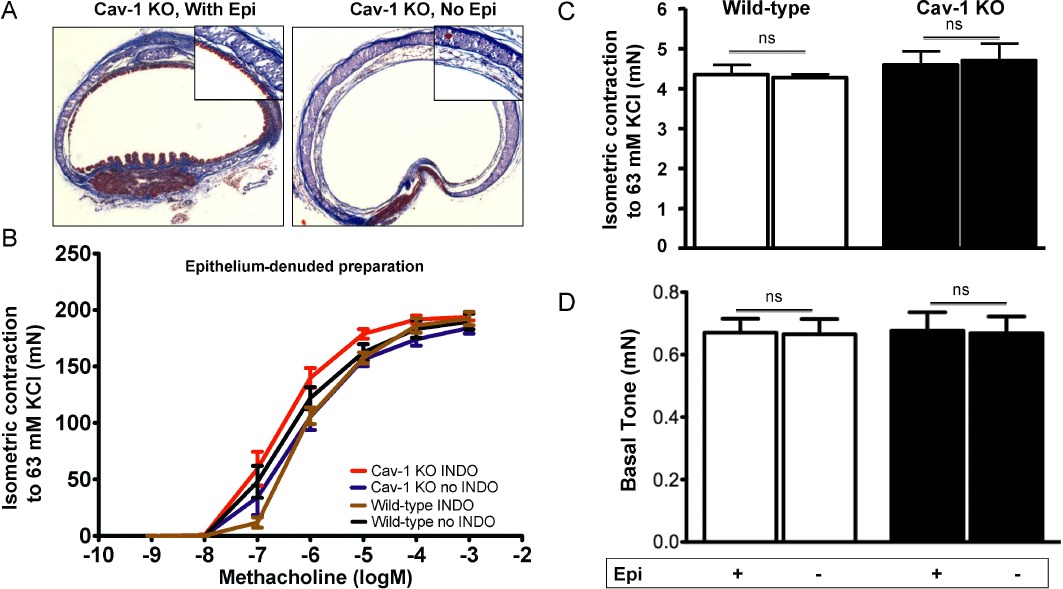
Epithelium denudation and KCl-induced isometric force in tracheal rings from Cav-1 KO and wild-type mice. The epithelium of excised tracheal segments was removed by threading with surgical silk then rolling the rings for 3 revolutions on a wetted paper towel. (A) Masson's trichrome staining of randomly selected tracheal preparations showing result of epithelial (Epi) denudation. Tracheal specimens from Cav-1 KO mice are shown. (B) Isometric methacholine concentration-response studies were performed using epithelium-denuded tracheal segments from Cav-1 KO (9 rings from 6 mice) and wild-type (9–12 rings from 5–7 animals) mice. After equilibration, rings were incubated for 30 min with either vehicle or indomethacin (INDO), then isometric force was measured with increasing concentrations of methacholine (10−9–10−3 M). Results shown are the mean ± SEM, indomethacin had minimal effects on responsiveness to methacholine (one-way anova, P > 0.05). (C) Receptor-independent force generating capacity of tracheal rings with (+) and without (−) epithelium (Epi) from wild-type and Cav-1 KO mice was assessed based on isometric force measured after 63 mM KCl substituted with K-H solution treatment. For each group, two to three tracheal rings from at least three to four mice were studied. There were no significant differences between the groups (one-way anova, P > 0.05). (D) Histogram comparing basal tone in tracheal rings from Cav-1 KO and wild-type mice with (+) and without (−) epithelium. For each group, five to seven tracheal rings from at least three to four mice were studied. There were no significant differences between the groups (one-way anova, P > 0.05). ns, not significant.
Increased COX-2 and 5-LO expression in Cav-1 KO airways and lungs
We estimated the abundance of COX-2, 5-LO and COX-1 in isolated tracheas of Cav-1 KO and wild-type mice by immunoblotting. Significantly higher levels of COX-2 and 5-LO were evident in protein lysates of whole tracheas from Cav-1 KO mice compared with those from wild type mice (Figure 3A,B), however, there was no difference in COX-1 abundance (Figure 3C). We also assessed COX-2, 5-LO and COX-1 abundance in lysates from tracheas that had been denuded of epithelium. In wild-type mouse tissues, epithelial removal had little effect on COX-2 or 5-LO abundance. However, in epithelium-denuded Cav-1 KO tracheas, we found a significantly reduced abundance of both proteins that was similar to that of epithelium-denuded or epithelium-intact wild-type tissues (Figure 3A,B). COX-1 abundance was unchanged in the epithelium-denuded tracheal rings in both strains (Figure 3C).
Figure 3.
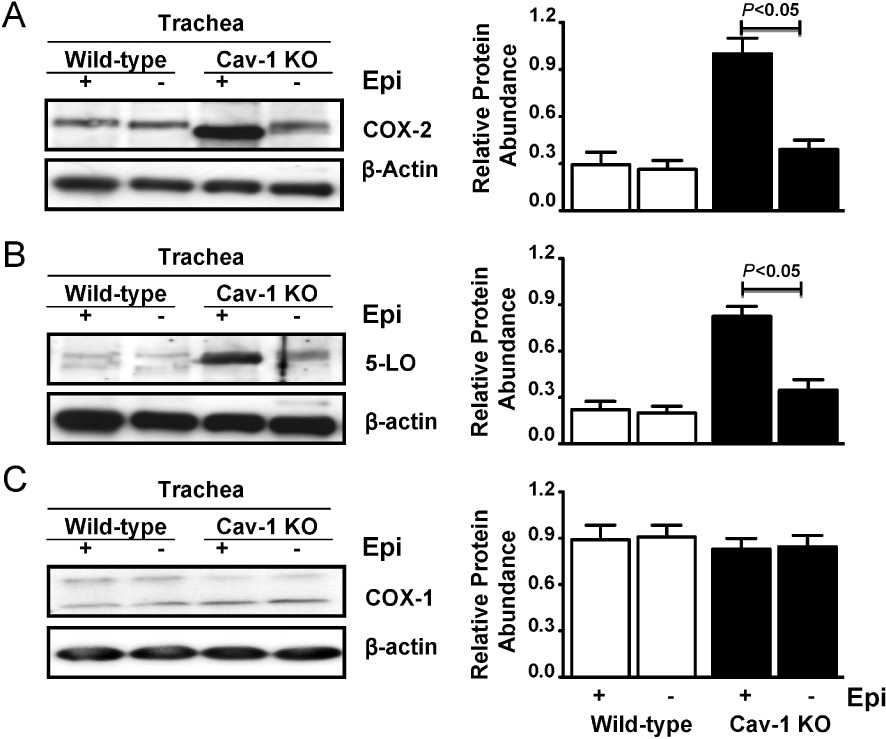
Increased expression of COX-2 and 5-LO in tracheas from Cav-1 KO mice. (A) Representative protein immunoblot and corresponding densitometry analysis (right column) for COX-2, (B) 5-LO and (C) COX-1 in lysates of tracheas with (+) and without (−) epithelium (epi) from Cav-1 KO and wild-type mice. Densitometry results represent replicates of scans of samples from 12–16 tracheal rings pooled from three to four mice from each strain. The P-values shown were obtained after one-way anova with Tukey's multiple comparison test.
In order to determine whether the differences in COX-2 and 5-LO expression that we measured in Cav-1 KO tracheas were also evident in the lungs, we performed immunoblotting using lung lysates and found that COX-2 and 5-LO protein levels to be significantly higher compared with wild-type tissue (Figure 4A,B). As Cav-1 is linked to regulation of COX-2 transcription in cancer cell lines (Rodriguez et al., 2009), we also measured COX-2 and 5-LO mRNA using qPCR. Indeed, COX-2 mRNA levels were increased by 70% and there was a threefold increase in 5-LO mRNA levels in Cav-1 KO lungs compared with wild-type tissue (Figure 4C). There is evidence for COX-2 and Cav-1 binding (Liou et al., 2001; Perrone et al., 2007) which is linked to COX-2 half-life (Rodriguez et al., 2009; Chen et al., 2010a) and so, using wild-type mouse lungs, we also performed immunoprecipitation of Cav-1 with subsequent immunoblotting for COX-2 (Figure 4D). This analysis confirmed that COX-2 did associate with Cav-1 in wild-type mouse lung cells.
Figure 4.
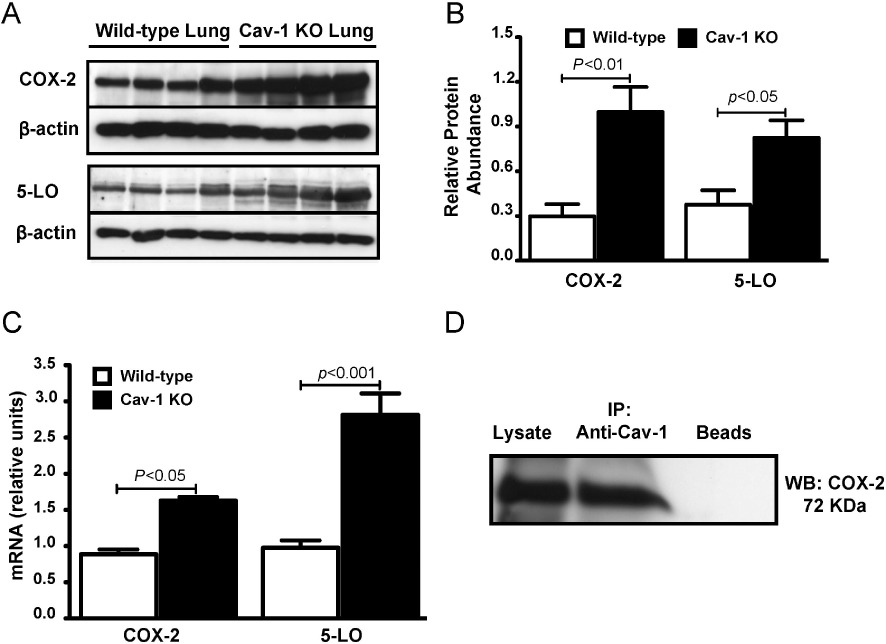
Increased expression of COX-2 and 5-LO in lungs from Cav-1 KO mice. (A) Representative Western blots and (B) corresponding densitometric analyses showing COX-2 and 5-LO abundance in whole lung lysates from Cav-1 KO and wild-type mice (n- 4–5 for each group). P-values shown were obtained after one-way anova with Tukey's multiple comparison test. (C) Histogram showing results of quantitative RT-PCR for COX-2 and 5-LO mRNA in lysates from lungs of Cav-1 KO and wild-type mice. 18S RNA was used as an internal control (n- 4–5 for each group). P-values shown were obtained after one-way anova with Tukey's multiple comparison test. (D) Western blot (WB) showing COX-2 abundance after immunoprecipitation (IP) with Cav-1 antibody and protein-G-conjugated sepharose beads from lung homogenates of wild-type mice. The lane labelled ‘Beads’ included sample but no antibody.
These data suggested that elevated COX-2 and 5-LO abundance in the respiratory system of Cav-1 deficient mice may be principally associated with accumulation in the airway epithelia, hence we next assessed the distribution of COX-2 and 5-LO by immunohistology, in resident airway cells of tracheas from Cav-1 KO and wild-type mice (Figure 5). This analysis demonstrated the epithelium to be the primary compartment in which COX-2 and 5-LO were expressed, although low levels of 5-LO and COX-2 staining were also evident in smooth muscle cells.
Figure 5.
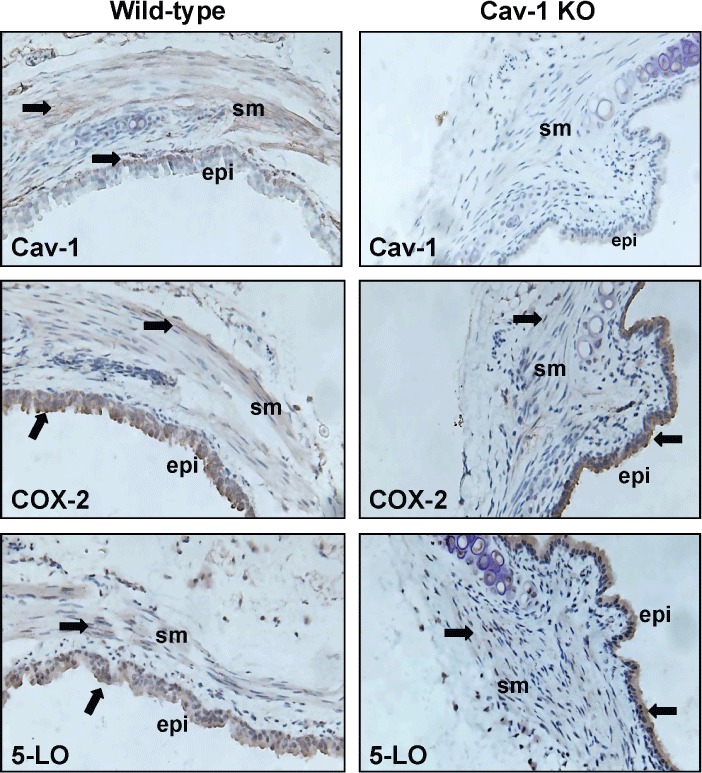
Cellular expression profile of Cav-1, COX-2 and 5-LO in airways from wild-type and Cav-1 KO mice. Representative immunohistological sections showing staining (brown colour highlighted with arrows) for Cav-1, COX-2 and 5-LO are shown. sm, airway smooth muscle layer; epi, epithelium.
Discussion
Cav-1 KO mice exhibit lung remodelling and vascular defects, suggesting a widespread effect on the physiology and function of the respiratory system (Drab et al., 2001; Jasmin et al., 2004; Murata et al., 2007). Nonetheless, before our study, the contractility of intact airways from Cav-1 KO mice had not been assessed. To our knowledge, our findings that COX-2 and 5-LO expression is increased in the lungs of Cav-1 KO mice and that in airways this increase is chiefly associated with the epithelium have not been previously reported. Our functional findings indicate that this increase in expression appears to provide a platform for modified control of airway responsiveness in situations where the COX-2 pathway of the arachidonic acid cascade is inhibited. We found that pharmacological suppression of COX with indomethacin produced hyperreactivity in Cav-1 KO airways when compared with wild type airways (despite identical intrinsic contractile capacity of tracheal smooth muscle in both types). The effects of COX-2 inhibition in Cav-1-deficient airways correlated with an increased expression of both COX-2 and 5-LO almost exclusively found in the airway epithelium. Notably, the indomethacin-induced hyperreactivity of tracheal rings was sensitive to blockade of 5-LO signalling events. This functional effect seems most likely to be the result of shunting of PLA2–derived arachidonic acid through the 5-LO pathway to produce contractile cysteinyl leukotrienes when the COX pathway was blocked (Maxis et al., 2006; Marnett, 2009). We did not detect an augmentation of contraction in airways from wild-type mice in the presence of indomethacin, indicating that the substrate shunting that occurs in airways with normal expression of COX-2 and 5-LO in the airway epithelium was not sufficient to drive a measurable change in contraction. Thus, our study indicates that Cav-1 limits the outcome of COX inhibition on the contractile response of airways, but reduction in Cav-1 abundance (in particular in the airway epithelium) facilitates COX-2 inhibition-mediated airway hyperresponsiveness.
We did not expect that tracheal smooth muscle from Cav-1 KO mice would exhibit normal contractile responses to methacholine (in the absence of indomethacin). Previous studies using in vitro and ex vivo models with acute molecular and pharmacological interventions have shown that Cav-1 plays an important role in supporting agonist-induced Ca2+ mobilization and associated contraction of isolated human ASM (Darby et al., 2000; Gherghiceanu and Popescu, 2006; Gosens et al., 2007b; Kamishima et al., 2007; Prakash et al., 2007; Sharma et al., 2010). Thus, our new findings that contractile function was not compromised in Cav-1-deficient tracheal rings were at odds with evidence showing that caveolae facilitate contraction. Significantly, earlier studies have all used molecular and pharmacological interventions to induce acute, transient inhibition or silencing of Cav-1. Thus, our current findings using tissues from mice with a constitutive Cav-1 null phenotype suggest that compensatory mechanisms, for example, augmentation of Ca2+-independent pathways involving RhoA or PKC that have been linked to caveolae (Shakirova et al., 2006; Somara et al., 2007), may be altered in ASM from Cav-1 KO mice. The data in this current study do not provide direct insight into such mechanisms but do suggest that further work is needed to dissect the interplay between Cav-1 regulated Ca2+-dependent and Ca2+-independent pathways in ASM cells.
Cyclooxygenase is the key enzyme in prostanoid biosynthesis. One isozyme, COX-1, is expressed constitutively, whereas COX-2 is induced in inflammatory conditions including lung cancer and chronic airway diseases, such as asthma and COPD (Wasserman, 1988; Walls et al., 1991; Wenzel, 1997; Gavett et al., 1999; Taha et al., 2000; Peebles et al., 2002; Khanapure et al., 2007; Torres et al., 2008; Sala et al., 2010). COX-2 is expressed in abundance by airway epithelial cells, thus allowing the synthesis of a wide profile of prostanoids (Eling et al., 1986; Churchill et al., 1989; Duniec et al., 1989; Holtzman, 1992). Prostaglandins such as PGE2 and PGI2 that are synthesized abundantly by the airway epithelium typically suppress ASM contraction or promote relaxation, though this is species dependent and related to the expression profile of prostaglandin EP receptor subtypes (Backlund et al., 2005; Ruan et al., 2008; Clarke et al., 2009; Park et al., 2010). Several studies have shown that COX-2 can form a complex with Cav-1 with subsequent localisation to caveolae (Liou et al., 2001; Perrone et al., 2007). Interestingly, association of COX-2 with Cav-1 facilitates its degradation (Chen et al., 2010a), Cav-1 can down-regulate β-catenin-Tcf/Lef-dependent transcription of COX-2 (Rodriguez et al., 2009), and it modulates post-transcriptional mechanisms such as augmented proteosomal degradation (Felley-Bosco et al., 2000; 2002) that regulate protein stability. Cav-1 can also regulate COX-2 activity, suppress PGE2 synthesis and modulate transport of PGE2-containing vesicles to the plasma membrane for release (Kojima et al., 2004; Rodriguez et al., 2009). These observations are significant because we observed that COX-2 and 5-LO abundance was markedly increased in Cav-1 KO lung and trachea, and that COX-2 was associated with Cav-1 in wild-type lungs. Thus, COX-2 expression and protein half-life are likely to be increased in Cav-1 KO mice due to the absence of Cav-1-supported degradation. This concept correlates with our data showing that indomethacin-mediated suppression of COX activity promoted hyperreactivity selectively in Cav-1 KO tracheas.
As confirmed by our immunohistochemical evidence, multiple cell types, including epithelia and smooth muscle, express COX-2 and 5-LO. However, our current study suggests that an increased expression of these proteins in the airway epithelium is the primary mechanism for the altered contractility of Cav-1 deficient airways in the presence of indomethacin. Firstly, physical removal of the airway epithelium eliminates differences in COX-2 and 5-LO abundance (as measured by immunoblotting) between Cav-1 KO and wild-type airways. This observation implies that if the proteins are increased in ASM cells, immunoblotting is not sensitive enough to detect this trend. Secondly, we show that in the absence of epithelium there is no functional consequence of indomethacin treatment on the contractility of airways from Cav-1-deficient mice. On this basis, we conclude that although 5-LO and COX-2 are expressed by ASM, any functional effect of COX inhibition of muscle-expressed protein on contraction is too small to be detected by the bioassays we employed. Thirdly, there were no differences in the depolarization- or methacholine-induced contraction of wild-type and Cav-1-deficient airways with intact epithelium. This observation suggests there was no intrinsic change in control of ASM responses. Thus, the measurable changes in airway contractility in response to COX inhibition appear to be linked to the epithelial dysfunction in Cav-1 KO tissues.
One of the novel findings of our study is that 5-LO levels were elevated, chiefly in the airway epithelium, in mice lacking Cav-1. However, to our knowledge there are no prior reports that indicate Cav-1 can interact with, or is linked to, pathways that regulate the expression of 5-LO, in contrast to the interactions between Cav-1 and COX-2 described above. Thus, the mechanism for increased 5-LO in Cav-1 KO lungs is not clear and further investigation is warranted, primarily because the LTs (i.e. LTB4, LTC4, LTD4 and LTE4) are potent bronchoconstrictors, chiefly mediating contraction of airway smooth muscle via the G-protein coupled receptor, CysLT1 (Lynch et al., 1999). We have shown that inhibition of 5-LO or blockade of CysLT1 receptors was sufficient to reverse the indomethacin-induced hyperreactivity in tracheal rings with intact epithelium from Cav-1 KO mice. However, treatment of tracheal rings with montelukast or zileuton alone had no effect on the contractility of tissues from Cav-1 KO or wild-type mice. These findings suggest that 5-LO signalling only contributed to enhanced contractility of Cav-1 KO tissues when COX was inhibited. Although inhibition of 5-LO by zileuton might have caused the shunting of arachidonic acid through the COX-2 pathway, we observed no measurable effect on airway contractile responses. This observation suggests that there was insufficient shunting to produce function-altering prostanoid concentrations, or that the prostanoids generated did not augment methacholine-induced contraction, as has been reported for some exogenously added prostanoids such as PGF2α (Schaafsma et al., 2005).
As depicted schematically in Figure 6, we suggest that increased epithelial expression of both COX-2 and 5-LO in Cav-1 KO tissues was a key predisposing factor for the selective indomethacin-induced hyperreactivity. Moreover, it seems likely that the augmented generation of cysteinly LTs (and their subsequent binding to CysLT1 receptors on ASM) in Cav-1 KO airways was associated with a shunting of PLA2-derived arachidonic acid through the 5-LO pathway when COX-2 was inhibited (Maxis et al., 2006; Chen et al., 2010b). This effect may also occur in wild-type airways, but our functional evidence indicates that it is not sufficient to alter airway contractility, probably because of the much lower levels of COX-2 and 5-LO in the wild-type airway epithelium. These data suggest that Cav-1 acts as a determinant for the expression of 5-LO and COX-2, which in turn mediates the increased potential for epithelial control of airway contraction if COX-2 activity is modulated. However, in the absence of COX-2 modulation, airway contractility was unchanged in Cav-1 KO mice, this may have been because, although both COX-2 and 5-LO protein levels and activity were increased, the normal balance (ratio) of 5-LO and COX metabolites generated was maintained. We conclude that increased expression of COX-2 and 5-LO in Cav-1 KO airways predisposed these tissues to exaggerated shunting of arachidonic acid via 5-LO in the presence of indomethacin, yielding a functional enhancement of contraction. Consistent with this conclusion are our observations that epithelial denudation by itself was insufficient to produce tracheal smooth muscle hyperreactivity in Cav-1 KO preparations, thereby demonstrating that elevated epithelial COX-2 activity alone could not account for the indomethacin-induced hyperreactivity in Cav-1 deficient airways. One caveat to our interpretation is that our studies do not preclude the possibility of direct negative regulation of LT-induced contraction by PGs, a mechanism that will require further experimentation.
Figure 6.
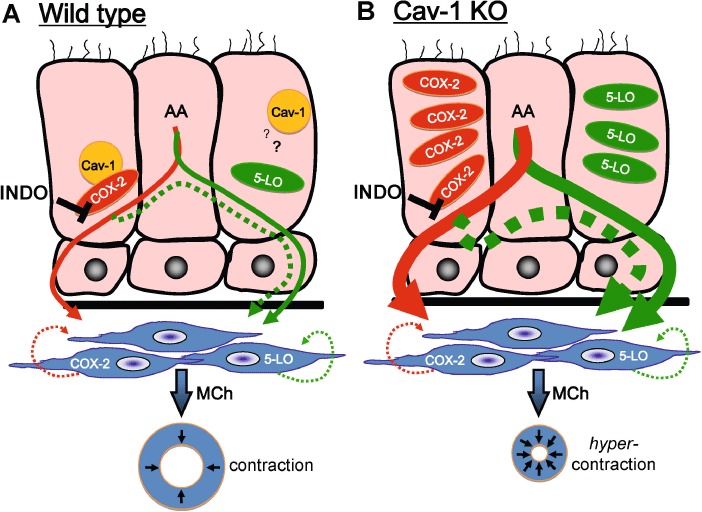
Schematic representation of the proposed effect of COX-2 inhibition on the production of arachidonic acid (AA) metabolites in epithelial cells and its association with disparate effects of airway contractility in wild-type and Cav-1 KO airways. (A) In wild-type airways, relatively low levels of COX-2 and 5-LO are expressed in the epithelium (pink cells) due to Cav-1-associated protein turnover. This is likely because of direct interaction of COX-2 with Cav-1; however, whether 5-LO interaction with Cav-1 is necessary is unknown (question mark). In wild-type tissues (in the absence of indomethacin; INDO), relatively modest basal levels of COX-2 and 5-LO metabolites are released by the epithelium (thin red and green arrows respectively), and act equally on underlying airway smooth muscle. Airway smooth muscle is also likely to produce small amounts of prostanoids (green hatched line) and leukotrienes (red hatched line) that have autocrine effects. Inhibition of the COX-2 pathway with indomethacin suppresses the generation of prostaglandins, thereby promoting shunting of AA metabolites through the 5-LO arm of the cascade (hatched green arrow); however, this imbalance is not sufficient to affect methacholine (MCh)-induced contraction of airway smooth muscle (blue cells, blue ring below represents an intact airway). (B) For Cav-1 KO airways, the lack of caveolin-1 leads to selective accumulation (and increased activity) of both COX-2 and 5-LO in airway epithelial cells, and increases basal capacity to generate prostanoids and leukotrienes (thick red and green arrows respectively); however, in the absence of indomethacin, these metabolites act equally on underlying airway smooth muscle, thus no difference in airway response from wild-type tissues is evident. As in wild-type tissue, airway smooth muscle also produces small amounts of prostanoids (green hatched line) and leukotrienes (red hatched line), but unlike Cav-1 KO epithelium COX-2 and 5-LO in smooth muscle cells is not significantly elevated. In contrast to basal conditions, inhibition of the COX-2 pathway with indomethacin leads to significant unbalancing of AA metabolite production through shunting via the 5-LO branch of the cascade (thick, hatched green arrow). Due to the elevated levels of COX-2 and 5-LO, with indomethacin treatment sufficient levels of leukotrienes are synthesized and released to promote hypercontractility of the underlying airway smooth muscle (blue), an effect that is evident by excessive contraction in response to methacholine (depicted with blue ring at the bottom of the panel).
Our data also showed that wild-type murine airways were refractory to the effects of indomethacin, indicating that Cav-1 is likely to contribute to the inhibition of pathways that can impart control of airway responsiveness to the epithelium. Our experiments do not allow precise identification of these pathways, but revealing that Cav-1 is a regulator of these pathways is of relevance. For instance, smooth muscle contractility in chronic airway diseases is regulated by the epithelium, a rich source of lipid mediators and other types of contracting and relaxing factors (Barnes et al., 1988; Fernandes et al., 1990; Holtzman, 1992; Raeburn and Webber, 1994; Chung and Barnes, 1999; Fortner et al., 2001). In asthma, aberrant biosynthesis of lipid mediators by COX-2 or 5-LO has been linked with changes in airway reactivity and bronchial inflammation (Wenzel, 1997; Wasserman, 1988; Gavett et al., 1999; Peebles et al., 2002; Peters-Golden and Henderson, 2007; Torres et al., 2008). In bovine tracheal preparations, contraction is refractory to epithelial denudation (Schaafsma et al., 2005), whereas prostanoids are released in abundance in guinea pig airways (Walls et al., 1991). Thus, our finding that Cav-1 modulates expression and activity of COX-2 and 5-LO in the airway epithelium indicates that Cav-1 should be considered as a potential regulator of mechanisms that lead to changes in bronchial constriction in pathophysiological conditions.
In summary, our findings suggest that differences in Cav-1 expression can regulate the degree to which the airway epithelium is able to control bronchoconstriction (e.g. when COX is inhibited). Cav-1 contributes in this capacity through effects on expression and activity of both epithelial COX-2, which drives the synthesis of prostaglandins such as PGE2, and 5-LO, which mediates synthesis of cysteinyl LTs. Moreover, our findings suggest that the ability of ASM to contract in response to other GPCR ligands (i.e. methacholine) is unchanged in Cav-1 KO mice, a result that differs from data showing acute transient silencing of Cav-1 suppresses Ca2+ mobilization and contraction. Our new findings suggest that other mechanisms that maintain contractility may be enhanced with long-term Cav-1 depletion. This study also reveals that Cav-1 and caveolae offer a complex control of airway responses via effects in different cell types that lead to alterations in both intrinsic cellular responses and intercellular communication.
Acknowledgments
This project is supported by an unrestricted research grant from Merck Frost Canada Inc. (MISP 34379), and in part by the Canadian Institute of Health Research (CIHR), and the Canada Research Chair Program, Canada Foundation for Innovation and the Manitoba Institute of Child Health (MICH). PS holds a CIHR-Banting & Best Canada Graduate Scholarship, and has been supported by the CIHR-National Training Program in Allergy and Asthma and by a University of Manitoba Graduate Fellowship. AJH holds a Canada Research Chair in Airway Cell and Molecular Biology.
Glossary
- 5-LO
5-lipoxygenase
- ASM
airway smooth muscle
- Cav-1
caveolin-1
Conflicts of interest
Portions of this project were supported by a competitive, unrestricted research grant from Merck Frost Canada Inc. (MISP 34379).
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backlund MG, Mann JR, Dubois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005;69(Suppl 1):28–32. doi: 10.1159/000086629. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Chung KF, Page CP. Inflammatory mediators and asthma. Pharmacol Rev. 1988;40:49–84. [PubMed] [Google Scholar]
- Chen SF, Liou JY, Huang TY, Lin YS, Yeh AL, Tam K, et al. Caveolin-1 facilitates cyclooxygenase-2 protein degradation. J Cell Biochem. 2010a;109:356–362. doi: 10.1002/jcb.22407. [DOI] [PubMed] [Google Scholar]
- Chen SH, Fahmi H, Shi Q, Benderdour M. Regulation of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase-activating protein/5-lipoxygenase by 4-hydroxynonenal in human osteoarthritic chondrocytes. Arthritis Res Ther. 2010b;12:R21. doi: 10.1186/ar2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Chung KF, Barnes PJ. Cytokines in asthma. Thorax. 1999;54:825–857. doi: 10.1136/thx.54.9.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill L, Chilton FH, Resau JH, Bascom R, Hubbard WC, Proud D. Cyclooxygenase metabolism of endogenous arachidonic acid by cultured human tracheal epithelial cells. Am Rev Respir Dis. 1989;140:449–459. doi: 10.1164/ajrccm/140.2.449. [DOI] [PubMed] [Google Scholar]
- Clarke DL, Dakshinamurti S, Larsson AK, Ward JE, Yamasaki A. Lipid metabolites as regulators of airway smooth muscle function. Pulm Pharmacol Ther. 2009;22:426–435. doi: 10.1016/j.pupt.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- Darby PJ, Kwan CY, Daniel EE. Caveolae from canine airway smooth muscle contain the necessary components for a role in Ca(2+) handling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1226–L1235. doi: 10.1152/ajplung.2000.279.6.L1226. [DOI] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Duniec ZM, Eling TE, Jetten AM, Gray TE, Nettesheim P. Arachidonic acid metabolism in normal and transformed rat tracheal epithelial cells and its possible role in the regulation of cell proliferation. Exp Lung Res. 1989;15:391–408. doi: 10.3109/01902148909087867. [DOI] [PubMed] [Google Scholar]
- Eling TE, Danilowicz RM, Henke DC, Sivarajah K, Yankaskas JR, Boucher RC. Arachidonic acid metabolism by canine tracheal epithelial cells. Product formation and relationship to chloride secretion. J Biol Chem. 1986;261:12841–12849. [PubMed] [Google Scholar]
- Felley-Bosco E, Bender FC, Courjault-Gautier F, Bron C, Quest AF. Caveolin-1 down-regulates inducible nitric oxide synthase via the proteasome pathway in human colon carcinoma cells. Proc Natl Acad Sci U S A. 2000;97:14334–14339. doi: 10.1073/pnas.250406797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felley-Bosco E, Bender F, Quest AF. Caveolin-1-mediated post-transcriptional regulation of inducible nitric oxide synthase in human colon carcinoma cells. Biol Res. 2002;35:169–176. doi: 10.4067/s0716-97602002000200007. [DOI] [PubMed] [Google Scholar]
- Fernandes LB, Preuss JM, Paterson JW, Goldie RG. Epithelium-derived inhibitory factor in human bronchus. Eur J Pharmacol. 1990;187:331–336. doi: 10.1016/0014-2999(90)90360-i. [DOI] [PubMed] [Google Scholar]
- Fortner CN, Breyer RM, Paul RJ. EP2 receptors mediate airway relaxation to substance P, ATP, and PGE2. Am J Physiol Lung Cell Mol Physiol. 2001;281:L469–L474. doi: 10.1152/ajplung.2001.281.2.L469. [DOI] [PubMed] [Google Scholar]
- Gavett SH, Madison SL, Chulada PC, Scarborough PE, Qu W, Boyle JE, et al. Allergic lung responses are increased in prostaglandin H synthase-deficient mice. J Clin Invest. 1999;104:721–732. doi: 10.1172/JCI6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherghiceanu M, Popescu LM. Caveolar nanospaces in smooth muscle cells. J Cell Mol Med. 2006;10:519–528. doi: 10.1111/j.1582-4934.2006.tb00417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosens R, Dueck G, Gerthoffer WT, Unruh H, Zaagsma J, Meurs H, et al. p42/p44 MAP kinase activation is localized to caveolae-free membrane domains in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007a;293:L1163–L1172. doi: 10.1152/ajplung.00471.2006. [DOI] [PubMed] [Google Scholar]
- Gosens R, Stelmack GL, Dueck G, Mutawe MM, Hinton M, McNeill KD, et al. Caveolae facilitate muscarinic receptor-mediated intracellular Ca2+ mobilization and contraction in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007b;293:L1406–L1418. doi: 10.1152/ajplung.00312.2007. [DOI] [PubMed] [Google Scholar]
- Gosens R, Stelmack GL, Bos ST, Dueck G, Mutawe MM, Schaafsma D, et al. Caveolin-1 is required for contractile phenotype expression by airway smooth muscle cells. J Cell Mol Med. 2010;15:2430–2442. doi: 10.1111/j.1582-4934.2010.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halayko AJ, Stelmack GL. The association of caveolae, actin, and the dystrophin-glycoprotein complex: a role in smooth muscle phenotype and function? Can J Physiol Pharmacol. 2005;83:877–891. doi: 10.1139/y05-107. [DOI] [PubMed] [Google Scholar]
- Halayko AJ, Camoretti-Mercado B, Forsythe SM, Vieira JE, Mitchell RW, Wylam ME, et al. Divergent differentiation paths in airway smooth muscle culture: induction of functionally contractile myocytes. Am J Physiol. 1999;276((1 Pt 1)):L197–L206. doi: 10.1152/ajplung.1999.276.1.L197. [DOI] [PubMed] [Google Scholar]
- Holtzman MJ. Arachidonic acid metabolism in airway epithelial cells. Annu Rev Physiol. 1992;54:303–329. doi: 10.1146/annurev.ph.54.030192.001511. [DOI] [PubMed] [Google Scholar]
- Jasmin JF, Mercier I, Hnasko R, Cheung MW, Tanowitz HB, Dupuis J, et al. Lung remodeling and pulmonary hypertension after myocardial infarction: pathogenic role of reduced caveolin expression. Cardiovasc Res. 2004;63:747–755. doi: 10.1016/j.cardiores.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Kamishima T, Burdyga T, Gallagher JA, Quayle JM. Caveolin-1 and caveolin-3 regulate Ca2+ homeostasis of single smooth muscle cells from rat cerebral resistance arteries. Am J Physiol Heart Circ Physiol. 2007;293:H204–H214. doi: 10.1152/ajpheart.00669.2006. [DOI] [PubMed] [Google Scholar]
- Khanapure SP, Garvey DS, Janero DR, Letts LG. Eicosanoids in inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr Top Med Chem. 2007;7:311–340. doi: 10.2174/156802607779941314. [DOI] [PubMed] [Google Scholar]
- Kojima F, Naraba H, Miyamoto S, Beppu M, Aoki H, Kawai S. Membrane-associated prostaglandin E synthase-1 is upregulated by proinflammatory cytokines in chondrocytes from patients with osteoarthritis. Arthritis Res Ther. 2004;6:R355–R365. doi: 10.1186/ar1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Saux CJ, Teeters K, Miyasato SK, Hoffmann PR, Bollt O, Douet V, et al. Down-regulation of caveolin-1, an inhibitor of transforming growth factor-beta signaling, in acute allergen-induced airway remodeling. J Biol Chem. 2008;283:5760–5768. doi: 10.1074/jbc.M701572200. [DOI] [PubMed] [Google Scholar]
- Li S, Okamoto T, Chun M, Sargiacomo M, Casanova JE, Hansen SH, et al. Evidence for a regulated interaction between heterotrimeric G proteins and caveolin. J Biol Chem. 1995;270:15693–15701. doi: 10.1074/jbc.270.26.15693. [DOI] [PubMed] [Google Scholar]
- Liou JY, Deng WG, Gilroy DW, Shyue SK, Wu KK. Colocalization and interaction of cyclooxygenase-2 with caveolin-1 in human fibroblasts. J Biol Chem. 2001;276:34975–34982. doi: 10.1074/jbc.M105946200. [DOI] [PubMed] [Google Scholar]
- Lynch KR, O'Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- Marnett LJ. Mechanisms of cyclooxygenase-2 inhibition and cardiovascular side effects: the plot thickens. Cancer Prev Res (Phila) 2009;2:288–290. doi: 10.1158/1940-6207.CAPR-09-0033. [DOI] [PubMed] [Google Scholar]
- Maxis K, Delalandre A, Martel-Pelletier J, Pelletier JP, Duval N, Lajeunesse D. The shunt from the cyclooxygenase to lipoxygenase pathway in human osteoarthritic subchondral osteoblasts is linked with a variable expression of the 5-lipoxygenase-activating protein. Arthritis Res Ther. 2006;8:R181. doi: 10.1186/ar2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ, et al. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med. 2007;204:2373–2382. doi: 10.1084/jem.20062340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndukwu IM, Solway J, Arbetter K, Uzendoski K, Leff AR, Mitchell RW. Immune sensitization augments epithelium-dependent spontaneous tone in guinea pig trachealis. Am J Physiol. 1994;266((5 Pt 1)):L485–L492. doi: 10.1152/ajplung.1994.266.5.L485. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing ‘preassembled signaling complexes’ at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- Park SW, Kim HS, Choi MS, Jeong WJ, Heo DS, Kim KH, et al. The effects of the stromal cell-derived cyclooxygenase-2 metabolite PGE2 on proliferation of colon cancer cells. J Pharmacol Exp Ther. 2010;336:516–523. doi: 10.1124/jpet.110.173278. [DOI] [PubMed] [Google Scholar]
- Peebles RS, Jr, Hashimoto K, Morrow JD, Dworski R, Collins RD, Hashimoto Y, et al. Selective cyclooxygenase-1 and -2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 2002;165:1154–1160. doi: 10.1164/ajrccm.165.8.2106025. [DOI] [PubMed] [Google Scholar]
- Perrone G, Zagami M, Altomare V, Battista C, Morini S, Rabitti C. COX-2 localization within plasma membrane caveolae-like structures in human lobular intraepithelial neoplasia of the breast. Virchows Arch. 2007;451:1039–1045. doi: 10.1007/s00428-007-0506-4. [DOI] [PubMed] [Google Scholar]
- Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- Prakash YS, Thompson MA, Vaa B, Matabdin I, Peterson TE, He T, et al. Caveolins and intracellular calcium regulation in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1118–L1126. doi: 10.1152/ajplung.00136.2007. [DOI] [PubMed] [Google Scholar]
- Raeburn D, Webber SE. Proinflammatory potential of the airway epithelium in bronchial asthma. Eur Respir J. 1994;7:2226–2233. doi: 10.1183/09031936.94.07122226. [DOI] [PubMed] [Google Scholar]
- Rodriguez DA, Tapia JC, Fernandez JG, Torres VA, Munoz N, Galleguillos D, et al. Caveolin-1-mediated suppression of cyclooxygenase-2 via a beta-catenin-Tcf/Lef-dependent transcriptional mechanism reduced prostaglandin E2 production and surviving expression. Mol Biol Cell. 2009;20:2297–2310. doi: 10.1091/mbc.E08-09-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan YC, Wang Z, Du JY, Zuo WL, Guo JH, Zhang J, et al. Regulation of smooth muscle contractility by the epithelium in rat vas deferens: role of ATP-induced release of PGE2. J Physiol. 2008;586((Pt 20)):4843–4857. doi: 10.1113/jphysiol.2008.154096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Lam HC, Chen ZH, Choi AM. Deadly triplex: smoke, autophagy, and apoptosis. Autophagy. 2011;7:436–437. doi: 10.4161/auto.7.4.14501. [DOI] [PubMed] [Google Scholar]
- Sala A, Folco G, Murphy RC. Transcellular biosynthesis of eicosanoids. Pharmacol Rep. 2010;62:503–510. doi: 10.1016/s1734-1140(10)70306-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaafsma D, Gosens R, Bos IS, Meurs H, Zaagsma J, Nelemans SA. Role of contractile prostaglandins and Rho-kinase in growth factor-induced airway smooth muscle contraction. Respir Res. 2005;6:85. doi: 10.1186/1465-9921-6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakirova Y, Bonnevier J, Albinsson S, Adner M, Rippe B, Broman J, et al. Increased Rho activation and PKC-mediated smooth muscle contractility in the absence of caveolin-1. Am J Physiol Cell Physiol. 2006;291:C1326–C1335. doi: 10.1152/ajpcell.00046.2006. [DOI] [PubMed] [Google Scholar]
- Sharma P, Tran T, Stelmack GL, McNeill K, Gosens R, Mutawe MM, et al. Expression of the dystrophin-glycoprotein complex is a marker for human airway smooth muscle phenotype maturation. Am J Physiol Lung Cell Mol Physiol. 2008;294:L57–L68. doi: 10.1152/ajplung.00378.2007. [DOI] [PubMed] [Google Scholar]
- Sharma P, Ghavami S, Stelmack GL, McNeill KD, Mutawe MM, Klonisch T, et al. Beta-dystroglycan binds caveolin-1 in smooth muscle: a functional role in caveolae distribution and Ca2+ release. J Cell Sci. 2010;123((Pt 18)):3061–3070. doi: 10.1242/jcs.066712. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Somara S, Gilmont RR, Martens JR, Bitar KN. Ectopic expression of caveolin-1 restores physiological contractile response of aged colonic smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2007;293:G240–G249. doi: 10.1152/ajpgi.00064.2007. [DOI] [PubMed] [Google Scholar]
- Stan RV. Structure of caveolae. Biochim Biophys Acta. 2005;1746:334–348. doi: 10.1016/j.bbamcr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Taha R, Olivenstein R, Utsumi T, Ernst P, Barnes PJ, Rodger IW, et al. Prostaglandin H synthase 2 expression in airway cells from patients with asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161((2 Pt 1)):636–640. doi: 10.1164/ajrccm.161.2.9811063. [DOI] [PubMed] [Google Scholar]
- Torres R, Herrerias A, Serra-Pages M, Roca-Ferrer J, Pujols L, Marco A, et al. An intranasal selective antisense oligonucleotide impairs lung cyclooxygenase-2 production and improves inflammation, but worsens airway function, in house dust mite sensitive mice. Respir Res. 2008;9:72. doi: 10.1186/1465-9921-9-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls AF, Rhee YK, Gould DJ, Walters C, Robinson C, Church MK, et al. Inflammatory mediators and cellular infiltration of the lungs in a guinea pig model of the late asthmatic reaction. Lung. 1991;169:227–240. doi: 10.1007/BF02714157. [DOI] [PubMed] [Google Scholar]
- Wasserman MA. Modulation of arachidonic acid metabolites as potential therapy of asthma. Agents Actions Suppl. 1988;23:95–111. doi: 10.1007/978-3-0348-9156-1_6. [DOI] [PubMed] [Google Scholar]
- de Weerd WF, Leeb-Lundberg LM. Bradykinin sequesters B2 bradykinin receptors and the receptor-coupled Galpha subunits Galphaq and Galphai in caveolae in DDT1 MF-2 smooth muscle cells. J Biol Chem. 1997;272:17858–17866. doi: 10.1074/jbc.272.28.17858. [DOI] [PubMed] [Google Scholar]
- Wenzel SE. Arachidonic acid metabolites: mediators of inflammation in asthma. Pharmacotherapy. 1997;17((1 Pt 2)):3S–12S. [PubMed] [Google Scholar]
- Xia H, Khalil W, Kahm J, Jessurun J, Kleidon J, Henke CA. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am J Pathol. 2010;176:2626–2637. doi: 10.2353/ajpath.2010.091117. [DOI] [PMC free article] [PubMed] [Google Scholar]