

Abstract
BACKGROUND AND PURPOSE
Voltage-gated sodium channels (NaV channels) are key players in the generation and propagation of action potentials, and selective blockade of these channels is a promising strategy for clinically useful suppression of electrical activity. The conotoxin µ-CnIIIC from the cone snail Conus consors exhibits myorelaxing activity in rodents through specific blockade of skeletal muscle (NaV1.4) NaV channels.
EXPERIMENTAL APPROACH
We investigated the activity of µ-CnIIIC on human NaV channels and characterized its inhibitory mechanism, as well as the molecular basis, for its channel specificity.
KEY RESULTS
Similar to rat paralogs, human NaV1.4 and NaV1.2 were potently blocked by µ-CnIIIC, the sensitivity of NaV1.7 was intermediate, and NaV1.5 and NaV1.8 were insensitive. Half-channel chimeras revealed that determinants for the insensitivity of NaV1.8 must reside in both the first and second halves of the channel, while those for NaV1.5 are restricted to domains I and II. Furthermore, domain I pore loop affected the total block and therefore harbours the major determinants for the subtype specificity. Domain II pore loop only affected the kinetics of toxin binding and dissociation. Blockade by µ-CnIIIC of NaV1.4 was virtually irreversible but left a residual current of about 5%, reflecting a ‘leaky’ block; therefore, Na+ ions still passed through µ-CnIIIC-occupied NaV1.4 to some extent. TTX was excluded from this binding site but was trapped inside the pore by µ-CnIIIC.
CONCLUSION AND IMPLICATIONS
Of clinical significance, µ-CnIIIC is a potent and persistent blocker of human skeletal muscle NaV1.4 that does not affect activity of cardiac NaV1.5.
Keywords: sodium channel, muscle relaxant, analgesia, conotoxin, patch-clamp
Introduction
Voltage-gated sodium channels (NaV channels) are essential for rapid electrical signalling in neuronal and muscle cells. This important role is manifested by an array of muscular and neuronal diseases associated with either NaV channel inhibition or hyperactivity (e.g. George, 2005; Andavan and Lemmens-Gruber, 2011). Numerous drugs bind to NaV channels and are used to interfere with neuronal signalling, mainly to dampen electrical firing to achieve analgesia (e.g. Priest, 2009; Zuliani et al., 2010). However, since in humans there are nine genes coding for prototypical NaV channels, not all of which are implicated in pain signalling (Goldin, 2002; Wood et al., 2004); safe and effective pharmacological interference with NaV channels requires subtype-specific drugs. For example, NaV channel inhibitors used as analgesics should not affect cardiac NaV channels (NaV1.5) or those expressed in the CNS (NaV1.1, NaV1.2). Hence, there is a significant demand for new drugs with high specificity for NaV channel subtypes that are strongly expressed in the peripheral nervous system, for example, NaV1.3, NaV1.6 or NaV1.7. The strong structural conservation across the NaV channel subtypes makes this a difficult challenge for standard medicinal chemistry approaches.
Peptides from marine cone snails, referred to as conotoxins, may provide specific NaV channel inhibitors. Among them, µ-conotoxins are particularly promising candidates because they block the pore of NaV channels from the extracellular side. With a size of 16–26 amino acid residues and three disulfide bridges, µ-conotoxins are large enough to make contact with all four domains of a NaV channel α-subunit (Dudley et al., 2000; Li et al., 2001; Choudhary et al., 2003). Therefore, in contrast to low molecular weight compounds, µ-conotoxins have the potential to be tailored so that they ‘fit’ perfectly to one specific NaV subtype only. A detailed understanding of how µ-conotoxins interact with and block NaV channel subtypes is therefore likely to be the key to future drug development (for review, see Norton, 2010).
One candidate holding such promise is µ-CnIIIC, a 22-residue conopeptide from Conus consors, which displays long-lasting myorelaxing effects in rodents (Favreau et al., 2012). This activity is due to its ability to potently block rat skeletal muscle NaV1.4 and rat brain NaV1.2 channels. µ-CnIIIC does not block cardiac mouse NaV1.5 channels and rat dorsal root ganglia NaV1.8 channels and shows intermediate inhibitory activity on neuronal mouse NaV1.6 and NaV1.7 channels. In order to further the development of µ-CnIIIC as a highly selective NaV-targeting drug or lead structure, we set out to determine whether µ-CnIIIC shows a similar specificity pattern for human NaV channel subtypes. In addition, we used NaV channel chimeras to elucidate the location of molecular determinants for sensitivity and insensitivity to µ-CnIIIC and to investigate the mechanism by which it blocks the target channels.
Methods
Production of µ-CnIIIC
µ-CnIIIC, with the sequence of ZGCCNGPKGCSSKWCRDHARCC-NH2 (Z = pyroglutamate), was synthesized and folded as described in Favreau et al. (2012).
Generation of channel mutants
Wild-type NaV channels used in this study were the rat (r) isoforms of NaV1.4 (SCN4A, P15390) and NaV1.8 (SCN10A, Q62968) and the human (h) isoforms of NaV1.2 (SCN2A, Q99250), NaV1.4 (SCN4A, P35499), NaV1.5 (SCN5A, Q14524), NaV1.7 (SCN9A, Q15858) and NaV1.8 (SCN10A, Q9Y5Y9.2). Accession numbers refer to the UniProt database, and nomenclature is according to Alexander et al. (2011).
Construction of domain chimeras between rNaV1.4 and hNaV1.5 was as described previously (Leipold et al., 2011); we either introduced single domains of hNaV1.5 into the background of rNaV1.4 yielding 5444, 4544, 4454 and 4445 or replaced the first or second half of the channel protein for constructs 5544 and 4455. In addition, pore loops of hNaV1.5 were inserted into the background of rNaV1.4: domain I, from I239 to A443, termed Ip5; domain II, from G692 to V766, termed IIp5 (numbers refer to rNaV1.4; see supporting information Figure S2). The combination is termed Ip5-IIp5. Similarly, pore loops of NaV1.7 were introduced into NaV1.4 (Ip7, IIp7 and Ip7-IIp7). Further site-directed mutagenesis was performed following PCR strategies. Half-channel chimeras between rNaV1.4 and rNaV1.8 were constructed according to an equivalent strategy yielding 4488 and 8844; the boundary was between residues A1040 and L1041 in rNaV1.4 and A1169 and L1170 in rNaV1.8 respectively. Constructs based on NaV1.8 had to be expressed in Neuro-2A cells, and current recording was done in the presence of 1 µM TTX to completely block endogenous NaV channels. Chimera 4488 was thus mutated in the pore loop of domain I (Y401S) to render this channel insensitive to TTX and, hence, to make it accessible for functional assays in Neuro-2A cells; this construct was termed 4*488. Mutant rNaV1.4-Y401S served as a control.
All mutant channel constructs were verified by DNA sequencing. Plasmid DNA was isolated from E. coli using the PureYield plasmid purification kit (Promega GmbH, Mannheim, Germany).
Cell culture and transfection
HEK 293 cells (Centre for Applied Microbiology and Research, Porton Down, Salisbury, UK) were maintained in 45% Dulbecco's modified Eagles medium and 45% Ham's F12 Medium, supplemented with 10% fetal calf serum in a 5% CO2 incubator at 37°C. HEK 293 cells were trypsinized, diluted with culture medium and grown in 35 mm dishes. When grown to 30–50% confluence, the cells were transfected with a 5:1 ratio of the NaV channel expression plasmids and a vector encoding the CD8 antigen (Jurman et al., 1994) using the Rotifect transfection kit (Roth, Karlsruhe, Germany). Dynabeads (Deutsche Dynal GmbH, Hamburg, Germany) were used for visual identification of individual transfected cells. For expression of NaV1.8 channels, Neuro-2A cells [German Collection of Microorganisms and Cell Cultures (DSMZ), Braunschweig, Germany] were used. Endogenous sodium channels of Neuro-2A cells were blocked with 1 µM TTX.
Electrophysiological recordings
Sodium current was measured by applying the whole-cell configuration of the patch-clamp method to HEK 293 and Neuro-2A (for NaV1.8) cells 24–48 h after transfection as described previously (Chen et al., 2000; Schirmeyer et al., 2010). Patch pipettes were fabricated from Kimax borosilicate glass of about 1–2 MΩ resistance. EPC-9 and EPC-10 patch clamp amplifiers operated by PatchMaster software (HEKA Elektronik, Lambrecht, Germany) were used. Series resistance was corrected electronically up to 85%; recording configurations with series resistance greater than 5 MΩ were discarded.
The patch pipettes contained (in mM): 35 NaCl, 105 CsF, 10 EGTA, 10 HEPES (pH 7.4 with CsOH). The bath solution contained (in mM): 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES (pH 7.4 with NaOH). µ-CnIIIC was diluted in bath solution containing 2 mg·mL−1 BSA and stored at −20°C until use. The toxin was applied locally with a glass pipette as described previously (Chen et al., 2000). Rapid inactivation of mutant rNaV1.4-M1305C was removed by extracellular application of 100 µM 2,2′-dithiobis(5-nitropyridine) (DTNP). All experiments were performed at 20 ± 1°C.
The onset of toxin-induced current block was measured with repetitive pulses to −20 mV every 5 s. Peak currents were plotted as a function of time and fitted with a single-exponential function to estimate the time constant of block, τ, and the steady state current remaining after µ-CnIIIC equilibration (Irem). The concentration-dependence of current block was analysed with a Hill equation; the Hill coefficient, nH (h in the equation) was set to 1 assuming a first-order reaction of toxin binding:
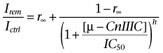 |
(1) |
IC50 is the concentration where toxin-induced current block becomes half-maximal and r∞ the remaining current fraction at limiting toxin concentration. The error of IC50 was obtained from the data fit using IgorPro software (WaveMetrics, Lake Oswego, OR, USA). The apparent on-rate kon and off-rate koff of current block were estimated by analysing the inverse of the time constant of current block, τon, as a function of µ-CnIIIC concentration:
| (2) |
The off-rate, koff, was further constrained by the inverse of the time constant of current recovery measured upon toxin removal, τoff.
Non-stationary noise analysis was performed as described earlier (Starkus et al., 2003; Heinemann and Leipold, 2011). A train off >200 10 ms test depolarizations to various voltages was applied in the absence and presence of 100 µM µ-CnIIIC. The leak-corrected mean current responses and the corresponding variances were calculated using PulseTools software (HEKA Elektronik). Variance was analysed as a function of mean current assuming a binomial distribution of channel states (Sigworth, 1977):
| (3) |
Where σ2 is the variance and I is the macroscopic mean current response of all individual test depolarizations. Nch defines the number of channels underlying the macroscopic current, and i is an estimate for the single-channel current. The maximal open probability Po,max was estimated from the variance analysis according to:
| (4) |
Data analysis and statistics
Electrophysiological data were analysed with FitMaster (HEKA Elektronik) and IgorPro (WaveMetrics) software. Data are presented as mean ± SEM with n= number of independent measurements. Significance for the difference between two data groups was tested with a Student's two-sided t-test; the resulting P-values are indicated.
Results
µ-CnIIIC blocks human NaV1.4 channels virtually irreversibly
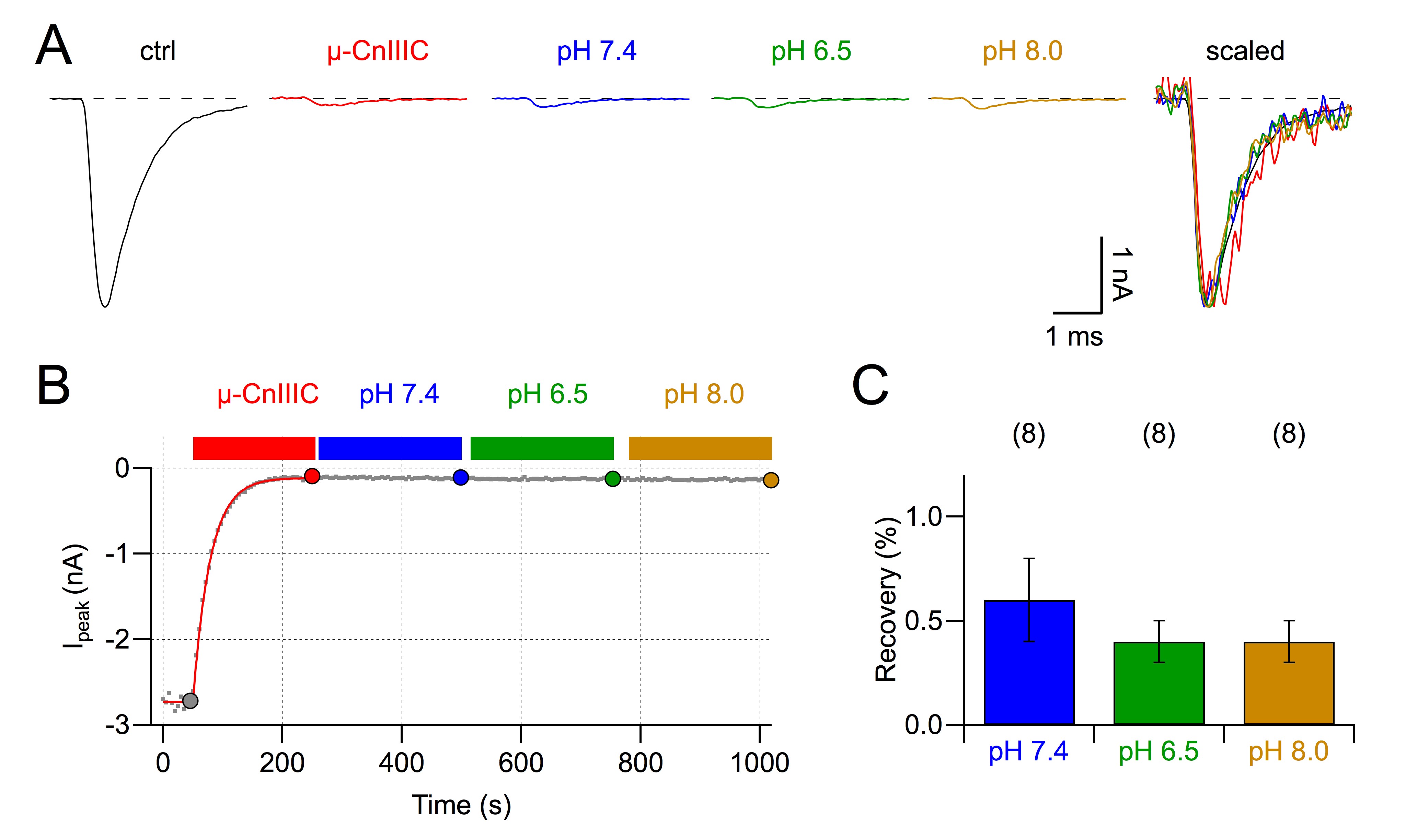
Rat skeletal muscle NaV1.4 channels are potently blocked by µ-CnIIIC, while mouse cardiac NaV1.5 and rat TTX-resistant NaV1.8 are insensitive (Favreau et al., 2012). We set out to determine whether this clear channel discrimination seen in rodents is also valid for the human paralogs. As shown in Figure 1A and B, hNaV1.4 channels, heterologously expressed in HEK 293 cells, were almost completely blocked by 1 µM µ-CnIIIC. The remaining current of about 5% did not show any obvious alterations in kinetics (Figure 1A). The onset of block, described with a single exponential function (Figure 1B), proceeded with a time constant τon of about 250 s and even after extensive washing with control saline the current did not recover. Measurements of current block at various µ-CnIIIC concentrations and simultaneous description of the steady-state block and its kinetics according to a first-order reaction (Figure 1C) revealed an apparent IC50 value of 4.95 ± 0.50 nM, an on-rate (kon) of 4.1 ± 0.5 103 M−1·s−1, a practically vanishing off-rate (koff= 2.0 ± 0.3 105 s−1) and a remaining current component at saturating toxin concentration (r∞) of 4.95 ± 0.12%. These results are very similar to those obtained for rat NaV1.4 (IC50= 1.3 ± 0.4 nM; kon= 3.6 ± 0.7 103 M−1·s−1; koff= 4.6 ± 0.7 106 s−1; r∞= 5.01 ± 0.23%). Hence, importantly, µ-CnIIIC would be expected to inhibit human skeletal muscle NaV channels in a clinical setting. This block was very slow (based on the determined kon, the time constant for the onset of µ-CnIIIC effect at the IC50 was estimated to about 13 h) and extremely long-lasting. Furthermore, the block even persisted when cells were washed with buffered salines covering pH values in the range of 6.5–8.0 (supporting information Figure S1).
Figure 1.
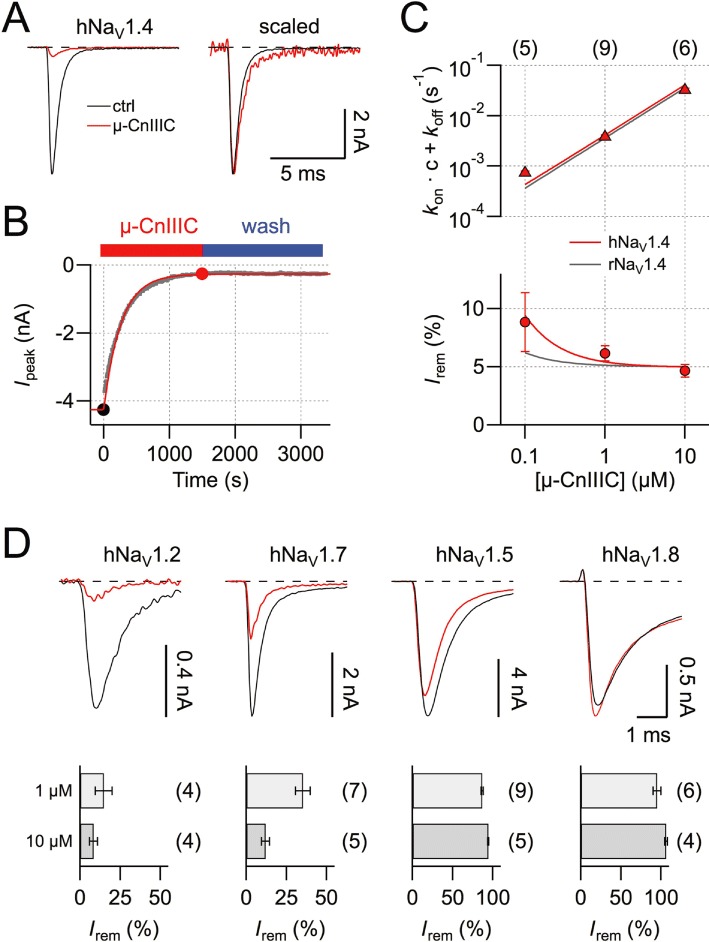
µ-CnIIIC blocks human skeletal muscle NaV1.4 channels. (A) Superposition of current traces recorded at −20 mV before (ctrl) and after application of 1 µM µ-CnIIIC (red). In the panel on the right, the red trace is scaled to match control peak currents, illustrating that the current remaining after µ-CnIIIC application does not exhibit changes in kinetics. (B) Time course of peak current reduction upon application of 1 µM µ-CnIIIC followed by wash. The continuous curve is a single-exponential data fit. Solid symbols indicate data points referring to the traces shown in (A). (C) Inverse of the mean time constant of onset of current block (top) and extrapolated remaining steady-state current (bottom) for three toxin concentrations. Red lines are global fits according to equations 1–2 for human NaV1.4; grey lines are the corresponding fit results for rat NaV1.4. (D) Superposition of current traces for hNaV1.2, hNaV1.7 and hNaV1.5 at −20 mV, as well as hNaV1.8 at 10 mV before (black) and after application of 1 µM µ-CnIIIC (red). The bar graphs show the remaining current after toxin application at the indicated concentrations. The n values in (C) and (D) are shown in parentheses.
Similar experiments were performed for human brain NaV1.2 and human peripheral nerve NaV1.7 (Figure 1D); while NaV1.2 was effectively blocked by 1 µM µ-CnIIIC (Irem= 14.7 ± 5.3%, n= 4), the sensitivity of NaV1.7 was significantly less (Irem= 35.1 ± 4.6%, n= 7, P= 0.02).
Similar to their rodent counterparts, human cardiac NaV1.5 and TTX-resistant NaV1.8 channels were only marginally affected by 10 µM µ-CnIIIC (Figure 1D).
Molecular determinants of channel specificity
Because of the similarity of human and rodent NaV channels with respect to block by µ-CnIIIC, it appeared justified to search for channel-based molecular determinants responsible for this subtype specificity using chimeras between rNaV1.4 and hNaV1.5 (Leipold et al., 2011) as well as between rNaV1.4 and rNaV1.8 (see Methods). Based on previous studies that identified important interaction sites for µ-conotoxins in the pore loops of domains I and II (Dudley et al., 1995; 2000; Chahine et al., 1998; Li et al., 2001; Cummins et al., 2002; Leipold et al., 2011), we first assayed channel chimeras in which only one half of the channel protein was replaced, that is, 4455/5544 and 4*488/8844 (each numeral indicates the origin of the respective subunit, see Figure 2A). As shown in Figure 2, all four chimeras gave rise to functional channels. However, the effect of 1 µM µ-CnIIIC differed across the panel of chimeras; while chimera 4455 showed µ-CnIIIC sensitivity comparable to wild-type NaV1.4, chimera 5544 was as insensitive as NaV1.5 (Figure 2B and C). Both chimeras based on NaV1.8 were insensitive to 10 µM µ-CnIIIC (Figure 2D and E), indicating that in NaV1.8 channels, even domains III and IV contain structural elements that prohibit channel block by this µ-conotoxin, and hence, identification of subtype-specific interaction sites would involve detailed study of the entire channel protein. We decided to focus on further NaV1.4/NaV1.5 chimeras in order to elucidate which structural differences in the first two domains are responsible for the insensitivity of NaV1.5 channels.
Figure 2.
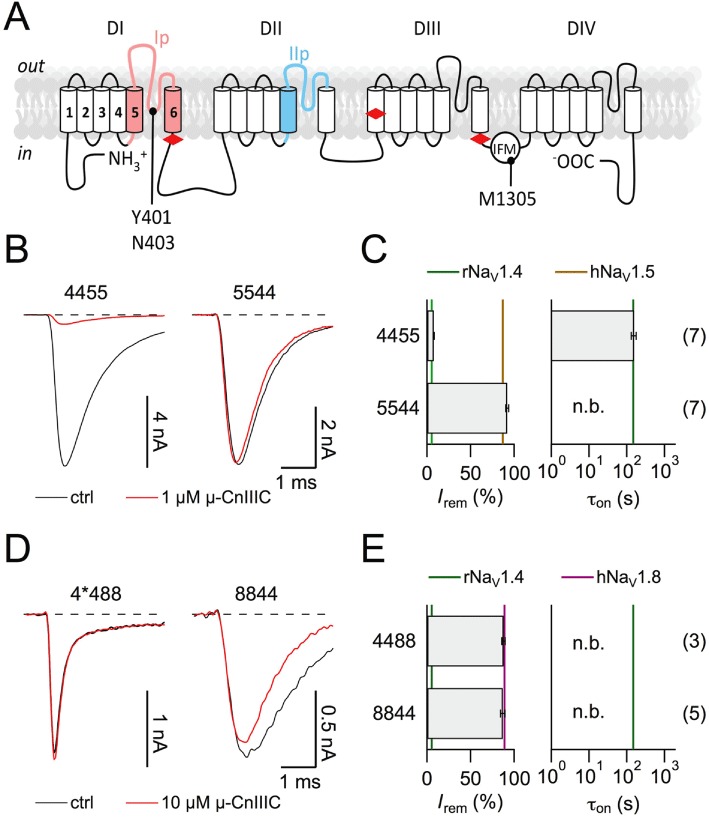
Half-channel chimeras between rNaV1.4 and hNaV1.5/rNaV1.8. (A) Topological cartoon of a NaVα-subunit with four homologous domains (DI–DIV). Boundaries used for the construction of domain chimeras (red diamonds) and pore loop chimeras are indicated. Residue numbers refer to rNaV1.4. (B, D) Superposition of current traces at −20 mV before (black) and after application of 1 µM µ-CnIIIC (red) for chimeras 4455 and 5544 in HEK 293 cells (B), as well as traces at 10 mV for 4*488 and 8844 in Neuro-2A cells with 10 µM µ-CnIIIC (D). (C, E) Mean remaining current (left) and apparent time constant of onset of block for the indicated chimeras. Vertical lines indicate the mean values of the respective wild-type channels. ‘n.b.’ refers to a situation where no onset was measured because channels were not blocked. The n values are shown in parentheses. Note that chimera 4*488 carries the additional mutation Y401S in order to make this construct resistant to TTX. In the background of rNaV1.4, this mutation does not strongly alter the ability of 1 µM µ-CnIIIC to block the channel (89.5 ± 0.4% block for Y401S, n= 6).
We initially focused on the first channel half by comparing chimeras 5444 and 4544 (Figure 3). While incorporation of domain I from NaV1.5 strongly reduced the effect of 1 µM µ-CnIIIC (Irem= 51.9 ± 3.8%, n= 7), incorporation of domain II affected µ-CnIIIC's action to a lesser extent (Irem= 8.5 ± 2.0%, n= 7), indicating that the major discriminating structural component must reside in domain I. Structures present in NaV1.5 not only affect the amount of channel blockade, but also its kinetics. For chimera 5444, the onset of block was about four times faster than for wild-type NaV1.4 channels (τon= 76.3 ± 5.4 s), and for chimera 4544, it was 20 times faster (τon= 11.6 ± 1.1 s). Hence, the structures from NaV1.5, while rendering the chimeric channel insensitive to µ-CnIIIC, actually increase the on-rate of the channel–ligand interaction.
Figure 3.
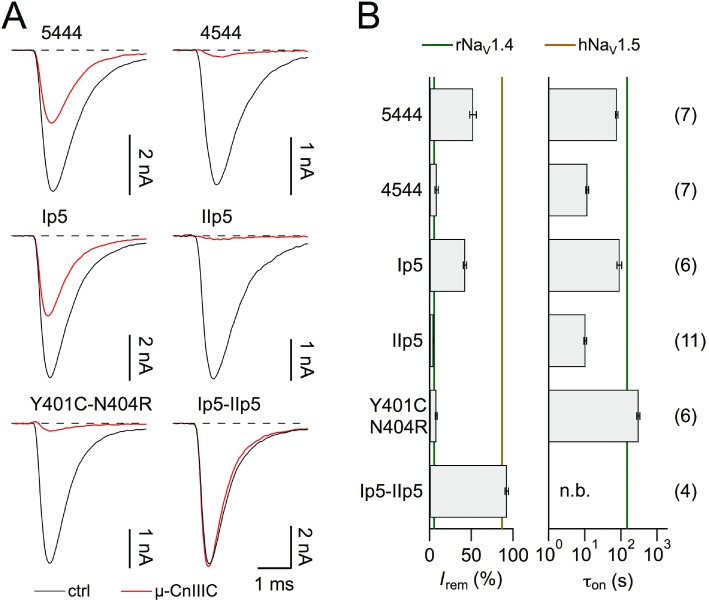
Chimeras between rNaV1.4 and hNaV1.5. (A) Superposition of current traces at −20 mV before (black) and after application of 1 µM µ-CnIIIC (red) for the indicated chimeras and the TTX-site mutant rNaV1.4-Y401C-N404R. (B) Mean remaining current (left) and apparent time constant of onset of block (right) for the indicated chimeras and mutants. Vertical lines indicate mean values for the respective wild-type channels. ‘n.b.’ refers to a situation where no onset was measured because channels were not blocked. The n values are shown in parentheses.
Insertion of just the pore loop from NaV1.5 domain I (see Figure 2A) into NaV1.4 (termed Ip5) resulted in a toxin sensitivity profile indistinguishable from that of 5444, the chimera containing the whole NaV1.5 domain I (Irem= 42.5 ± 1.9%, τon= 91.1 ± 13.6 s, n= 6; Figure 3A). For a multiple sequence alignment of pore loops from domains I and II see supporting information Figure S2. Similarly, insertion of just the pore loop from NaV1.5 domain II (IIp5) into NaV1.4 resulted in a toxin sensitivity profile resembling that of chimera 4544, the chimera containing the whole NaV1.5 domain II (Irem= 4.5 ± 0.3%, τon= 10.3 ± 0.9 s, n= 11). The combination of both pore loops (Ip5-IIp5) from NaV1.5 in NaV1.4 reproduced the µ-CnIIIC sensitivity profile of NaV1.5. We can thus conclude that the pore loops of domains I and II, which are sufficient to confer the µ-CnIIIC-related properties from NaV1.5 to NaV1.4, contain the molecular determinants for µ-CnIIIC selectivity. The pore loop of domain I appears to predominantly affect the extent of channel blockade, while the domain II pore loop has a strong impact on the on-rate of toxin binding.
In the inner pore region of domain I, there are two important structural differences between NaV1.4 and NaV1.5 channels: residue Y401 in NaV1.4 permits TTX binding, while the homologous cysteine in NaV1.5 reduces TTX sensitivity (Backx et al., 1992). The positively charged arginine, located in three residues of the C-terminal in NaV1.5, makes the channels more resistant to extracellular divalent cations as compared to NaV1.4 with an asparagine at the homologous position (Heinemann et al., 1992). In addition, it was shown that this site affects channel block by µ-SIIIA from Conus striatus (Leipold et al., 2011). Thus, we assayed mutant NaV1.4-Y401C-N404R with 1 µM µ-CnIIIC and found that it had no effect on the remaining current and slightly slowed down the onset of block (Irem= 7.8 ± 1.0%, τon= 308 ± 31 s, n= 6, Figure 3). Thus, the effect of the NaV1.5 domain I pore loop must be accounted for by regions other than the inner pore residues C401 and R404.
Based on a first-order reaction scheme, an apparent IC50 of 1.3 nM and an almost vanishing off-rate of 4.6 10−6 s−1 was determined for wild-type rNaV1.4 (Figure 1; Favreau et al., 2012). Insertion of pore loops from NaV1.5 not only altered the degree of block and its on-rate but also accelerated the off-rate, that is, made the toxin effect reversible (Figure 4A–C). Also, if the information on the kinetics of block relief upon toxin washout is taken into account, an apparent IC50 value and the kinetic constants can be estimated more accurately (Figure 4C): Ip5 –r∞= 5.1 ± 0.1%, kon= 7.4 ± 0.1 103 M−1·s−1, koff= 5.3 ± 0.2 10−3 s−1, IC50= 708 ± 29 nM; IIp5 –r∞= 4.4 ± 0.1%, kon= 243 ± 1 103 M−1·s−1, koff= 0.7 ± 0.1 10−3·s−1, IC50= 2.7 ± 0.5 nM. Thus, the pore loop in domain I has a strong effect on the equilibrium constant, while the pore loop in domain II does not change the IC50 but strongly affects the kinetics of toxin binding and dissociation. NaV1.5 determinants do not increase the on-rate of µ-CnIIIC binding while leaving the channel largely unblocked, but the on-rate increase is accompanied by an even larger off-rate increase, resulting in lower overall affinity.
Figure 4.
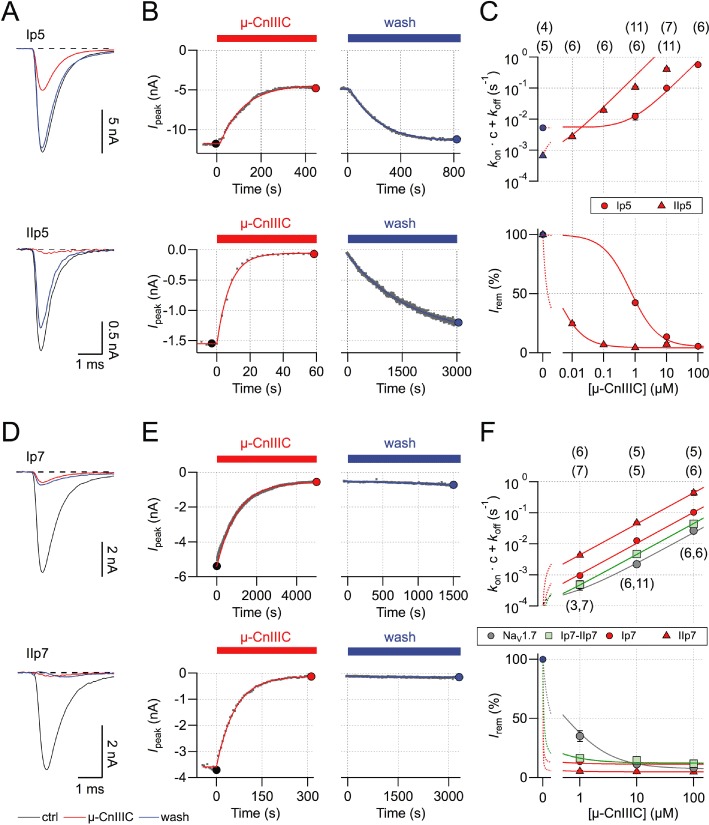
Analysis of pore-loop chimeras. (A) Superposition of current traces at −20 mV before (black) and after application of 1 µM µ-CnIIIC (red), as well as upon toxin washout (blue) for chimeras Ip5 (top) and IIp5 (bottom). (B) Time courses of toxin-induced current block and recovery kinetics upon wash with superimposed single-exponential fits. Solid symbols indicate data points from current traces shown in (A). (C) Kinetic analysis based on steady-state block and kinetics measured for various µ-CnIIIC concentrations. Lines are global data fits according to equations 1–2 for Ip5 (circles) and IIp5 (triangles). The n values are shown in parentheses. (D–F) Similar experiments as in (A–C) but for chimeras Ip7, IIp7 and Ip7-IIp7. The grey symbols in (F) indicate data for wild-type hNaV1.7.
A similar experiment was performed for the pore loops of NaV1.7. As shown in Figure 4D–F, neither of the pore loops inserted into NaV1.4 made the toxin effect clearly reversible. Similar to the NaV1.5 pore loops, toxin on-rate was faster for IIp7 than for Ip7, but neither of the pore loops substantially increased the IC50 value, which would reflect a NaV1.7 phenotype. The combination of both pore loops resembled the properties of NaV1.7 more closely. Based on the kinetics and steady-state block, the following parameters were estimated: hNaV1.7 –r∞= 7.3 ± 0.9%, kon= 0.23 ± 0.01 103 M−1·s−1, koff= 111 ± 21 10−6·s−1, IC50= 489 ± 94 nM; Ip7-IIp7 –r∞= 12.2 ± 0.4%, kon= 0.45 ± 0.02 103 M−1·s−1, koff= 23 ± 3 10−6·s−1, IC50= 50.4 ± 6.6 nM; Ip7 –r∞= 11.4 ± 0.4%, kon= 1.04 ± 0.05 103 M−1·s−1, koff= 9.1 ± 1.0 10−6 s−1, IC50= 8.7 ± 1.1 nM; IIp5 –r∞= 4.9 ± 0.1%, kon= 4.4 ± 0.1 103 M−1·s−1, koff= 21 ± 7 10−6 s−1, IC50= 4.7 ± 1.8 nM.
Persistent but incomplete channel block
An interesting feature of µ-CnIIIC is its incomplete block, leaving a remaining current of about 5% for NaV1.4 channels. To better understand this residual current component, we employed non-stationary noise analysis of current sweeps recorded in the whole-cell mode before and after toxin application. Simultaneous fit of ensemble variance versus mean current for three different voltages (−30, −10, 10 mV) under control conditions and upon saturation with 10 µM µ-CnIIIC (Figure 5A and B) yielded estimates for the maximal open probabilities and the single-channel current amplitude (Figure 5C). While Po,max did not change after toxin application (Figure 5C, left), the single-channel conductance was reduced from 23.6 ± 3.3 pS to 8.5 ± 2.9 pS (Figure 5C, right, n= 7). The total number of active channels was reduced to 14%. These results show that the residual current remaining in the presence of a saturating µ-CnIIIC concentration is carried by NaV channels with strongly reduced Na+ conductance but without marked changes in the voltage-dependence of activation. The reduced number of active channels presumably reflects channels that are either blocked completely or that have entered some kind of hibernating state not accessible to non-stationary noise analysis.
Figure 5.
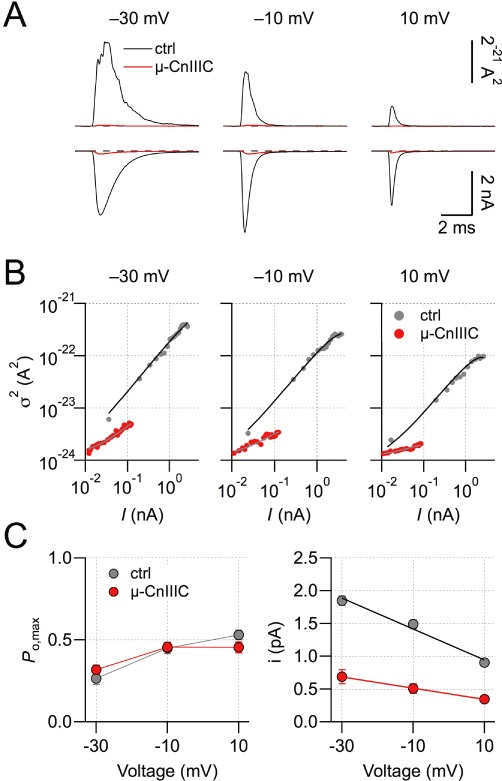
Single-channel characteristics. (A) Mean current and ensemble variance of rNaV1.4 channels at the indicated voltages based on 200 individual current recordings before (black) and after application of 10 µM µ-CnIIIC (red). (B) Ensemble variance as a function of mean current for the indicated voltages without and with toxin. Continuous lines are fits according to equation 3. (C) Voltage-dependence of the maximal open probability and the single-channel current amplitude derived from non-stationary noise analysis. Lines connect the data points in the left panel; straight lines in the right panel are linear fits for determination of the chord conductance; n= 7.
Even if channel expression in HEK 293 cells is high, the current amplitude after µ-CnIIIC application is rather small and comparable to the current sizes sometimes observed in these cells that do not express exogenous NaV channels. Thus, to eliminate the possibility that the residual current results from the activity of µ-CnIIIC-insensitive endogenous channels, we performed experiments using the channel mutant rNaV1.4-M1305C. Under control conditions, this mutant shares most of the kinetic properties of rNaV1.4 wild-type channels (not shown), and it would not be expected to influence toxin sensitivity because the mutation is located in the intracellular domain III-IV linker (Figure 2A). When treated with 100 µM of the membrane-permeable cysteine-specific agent DTNP, rapid channel inactivation was removed almost completely (Figure 6A). Upon removal of DTNP, this non-inactivating current persisted and was subsequently blocked with 10 µM µ-CnIIIC, leaving a non-inactivating residual current of 4.2 ± 0.8%. This clearly shows that the residual µ-CnIIIC-resistant current is associated with mutant NaV1.4-M1305C and not with any current component endogenous to HEK 293 cells (Figure 6A and C).
Figure 6.
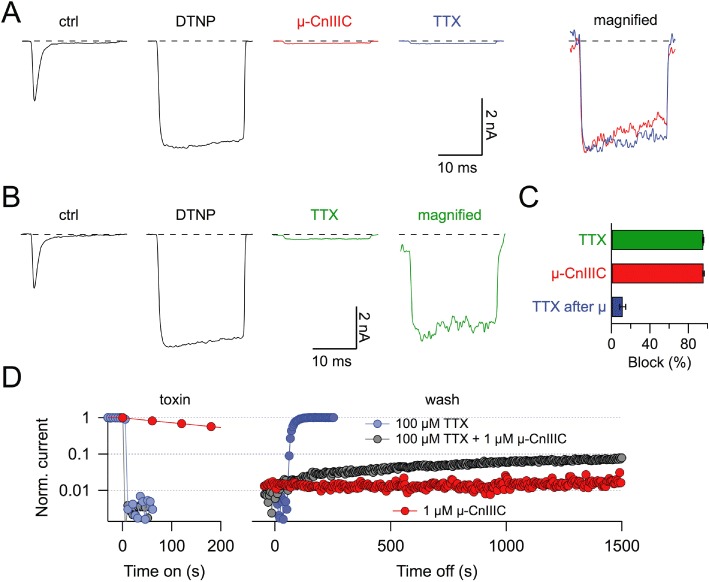
TTX block of non-inactivating, µ-CnIIIC-occupied channels. (A) Current responses at −20 mV of a HEK 293 cell expressing rNaV1.4-M1305C channels, under control conditions (ctrl), after application of 100 µM DTNP (DTNP), after subsequent application of 10 µM µ-CnIIIC without DTNP and finally after application of 1 µM TTX without DTNP or µ-CnIIIC. The panel on the right shows a magnified superposition of DTNP-modified traces with µ-CnIIIC (red) and subsequent TTX application (blue). (B) Similar experiment as in (A) but without application of µ-CnIIIC illustrating that 1 µM TTX potently blocks DTNP-modified channels. (C) Statistics of current block of non-inactivating rNaV1.4-M1305C channels induced by 1 µM TTX, 10 µM µ-CnIIIC or 1 µM TTX after equilibration in 10 µM µ-CnIIIC (n= 6), illustrating that the current remaining after µ-CnIIIC block is not readily sensitive to TTX. (D) Block kinetics and recovery kinetics of DNTP-modified rNaV1.4-M1305C channels after the application of 100 µM TTX, the same concentration of TTX together with 1 µM µ-CnIIIC or 1 µM µ-CnIIIC alone.
With this unambiguous identification of the tiny current remaining after µ-CnIIIC application, we determined whether the pore blocker TTX has an effect on this current. Rapid inactivation of NaV1.4-M1305C channels was first removed by applying DTNP. The resulting non-inactivating channels were readily blocked with 1 µM TTX (95.4 ± 0.5%; Figure 6B and C, n= 6). However, TTX only had a small effect on the residual current leaking through µ-CnIIIC-occupied channels (11.6 ± 3.0% blockade; Figure 6A and C, n= 6). This result strongly suggests that NaV1.4 channels, once occupied by µ-CnIIIC, are not readily accessible to TTX, that is, TTX cannot reach its binding site in the channel pore. When µ-CnIIIC (1 µM) and TTX (100 µM) were co-applied, the channels were blocked rapidly such as with TTX alone (Figure 6D). However, upon washing with control saline, slow recovery was observed; whereas when TTX was applied alone the block was rapidly removed and with µ-CnIIIC alone no recovery occurred. This experiment suggests that µ-CnIIIC can block the channel with TTX being located at receptor site-1.
Discussion and conclusions
µ-CnIIIC is a promising candidate for further clinical development
The µ-conopeptide CnIIIC shows selectivity across NaV subtypes in rodents, displaying strong activity against rat skeletal muscle NaV1.4 and rat brain NaV1.2 channels while being inactive with respect to cardiac mouse NaV1.5 and rat NaV1.8 channels (Favreau et al., 2012). Previous work on a pair of different µ-conotoxins, µ-GIIIA and µ-GIIIB, indicated that channel selectivity observed in rodents is not necessarily observed in humans (Cummins et al., 2002). However, in the case of µ-CnIIIC, we have shown in this study that the selectivity observed for the rodent channels is fully reproduced in their human paralogs. These similarities in potency and selectivity of rodent and human channels, together with efficacy seen in vivo experiments (Favreau et al., 2012), strongly support the further clinical development of µ-CnIIIC. With an apparent IC50 value of a few nM for NaV1.4, there is a safety margin towards cardiac NaV1.5 of more than 1000-fold, thus reducing the risk of cardiac side effects to a very low level. An important feature of µ-CnIIIC is its very long-lasting block with estimated reversibility time constants in the order of many hours. This stability was not compromised in solutions of varying pH (supporting information Figure S1), thus explaining the long-lasting effects also seen in an in vivo setting.
Dissection of molecular determinants of µ-CnIIIC selectivity
A detailed understanding of how µ-conotoxins interact with and block NaV channel subtypes is therefore likely to be the key to future drug development. Using chimeras between µ-CnIIIC-sensitive (NaV1.4) and insensitive channel subtypes (NaV1.5 and NaV1.8), we attempted to localize molecular determinants for the toxin insensitivity. While replacing domains of NaV1.4 with corresponding structures from either the first and or the second half of NaV1.8 was sufficient to eliminate toxin sensitivity, substituting domains from the second half of NaV1.5 into NaV1.4 had no detectable effect. Thus, in NaV1.8, all four domains harbour sites that are incompatible with µ-conotoxin activity, while for NaV1.5, the determinants are located exclusively in domains I and II. At a higher resolution, our investigation revealed that the structural determinants for µ-CnIIIC insensitivity are located in the pore loops of these domains. However, the pore loops in domains I and II contribute to toxin insensitivity in different ways. While the pore loop of domain I strongly increased the apparent IC50 of µ-CnIIIC, the pore loop of domain II had little effect on steady-state toxin binding but accelerated toxin association and dissociation. One interpretation of this observation with respect to NaV1.4 would be that while domain I is key to high-affinity toxin binding, domain II affects access of the toxin to its binding site. However, the effect of domain I pore loop is not mediated by those residues determining the different TTX sensitivity of NaV1.4 and NaV1.5 channels, suggesting that other parts of the pore loop must play an important role.
Apparently, the effect of different channel fragments on the µ-conotoxin block strongly depends on the conotoxin investigated. For example, for µ-SIIIA, a related toxin with only a few structural differences compared with µ-CnIIIC (Leipold et al., 2011), the influence of the domain I pore loop on channel block was attributable to the TTX site, but the major functional difference between NaV1.4 and NaV1.5 channels was mediated by the pore loop in domain II. In contrast, the TTX site does not have a functional effect on µ-CnIIIC activity, although the domain I pore loop determines the sensitivity of the channel to µ-CnIIIC.
Moreover, the µ-SIIIA phenotypes of rNaV1.4 and hNaV1.7 could be bidirectionally conferred by the substitution of only a single residue in the domain II pore loops of these channels (rNaV1.4: A728, hNaV1.7: N889, supporting information Figure S2). In contrast, even the exchange of the entire pore loop II was not sufficient to confer the µ-CnIIIC phenotype of NaV1.7 to NaV1.4 (Figure 4). Similar to NaV1.5, pore loop I of NaV1.7 appeared to play an important role in determining the activity of the toxin. Given the weak sequence conservation of that pore loop among the various NaV channel isoforms (supporting information Figure S2B), it will be a huge challenge to design a toxin specific for NaV1.7, for example, as an analgesic.
Residual current upon saturation with µ-CnIIIC
An interesting feature of certain µ-conotoxins is that they apparently do not achieve complete blockade of sodium channels. Using a variant of µ-GIIIA (R13Q) that leaves about 25% of the rat NaV1.4 current unblocked, French et al. (1996) studied the properties of µ-conotoxin-occupied channels, showing that the bound toxin both reduces the single-channel current size and slightly modifies channel gating. Subsequently, it was demonstrated that TTX can enter the pore to reach toxin receptor site-1 when the channel is bound by µ-GIIIA (R13Q) (Zhang et al., 2010). Here, we showed that µ-CnIIIC, similar to µ-SIIIA (Leipold et al., 2011), is an example of a natural µ-conotoxin that fails to block NaV1.4 channels completely, albeit with a residual current of only about 5% of the total control amplitude. This illustrates that the phenomenon of ‘leaky channel block’ is not a feature restricted to µ-conotoxin mutants but is also observable for native toxins. However, owing to the small amplitude of the remaining current fraction, such current signals are not accessible to direct single-channel recordings. We thus used non-stationary noise analysis to show that the remaining current was mediated by a fraction of µ-CnIIIC-occupied channels with reduced single-channel conductance; apparently, another fraction of channels was either completely blocked or went into another non-conducting state.
Unambiguous assignment of 5% residual current to exogenously expressed channels was demonstrated using a channel mutant, which had a fast inactivation that was removed by the extracellular application of DTNP. In addition, using this method, it was shown that the current remaining after saturation with µ-CnIIIC is not readily accessible to TTX block. Thus, we conclude that µ-CnIIIC blocks access of TTX to channel receptor site-1. Likewise, other low molecular weight drugs accessing receptor site-1 might not be active on µ-CnIIIC-occupied channels. On the other hand, once TTX is bound to the channel, µ-CnIIIC is apparently still able to bind to its receptor site, as demonstrated by an accelerated recovery from µ-CnIIIC block in the presence of TTX. Thus, the channel's extracellular cavity must be wide enough to simultaneously accommodate TTX and the µ-conopeptide CnIIIC, similar to µ-KIIIA (Zhang et al., 2010). The incomplete recovery of µ-CnIIIC block in the presence of TTX (Figure 6D) and the results from non-stationary noise analysis are suggestive of more than a single configuration by which the toxin can occupy the channel's pore.
Conclusions
The µ-conopetide CnIIIC from Conus consors is a very potent, selective and durable antagonist of skeletal muscle NaV1.4 sodium channels (Favreau et al., 2012). In this study, we have shown that this promising pharmacological profile is fully reproduced in the relevant human sodium channels, providing important confirmation of the clinical potential of µ-CnIIIC as a muscle relaxant. Further development may benefit from a better understanding of the molecular basis underlying the channel selectivity of µ-CnIIIC, and in this study, we showed that the structural basis for selectivity against NaV1.8 is relatively complex, involving structures located in channel domains III and IV in addition to domains I and II. By contrast, determinants of selectivity for NaV1.5 and NaV1.7 reside in the pore loops of domains I and II. Future attempts to optimize the effects of µ-CnIIIC for potential clinical applications, by increasing its safety margin with respect to cardiac NaV1.5 or by improving its pharmacokinetic properties, should therefore focus on the interaction of this toxin with the pore loop elements of the first two channel domains.
Acknowledgments
This work was supported by the European Union FP6 (CONCO). We thank JS Trimmer for providing cDNA coding for rNaV1.4, Ch. Alzheimer for hNaV1.2, AL George for hNaV1.4, hNaV1.5, N Klugbauer for hNaV1.7 and JN Wood for rNaV1.8.
Glossary
- DTNP
2,2′-dithiobis(5-nitropyridine)
- NaV
channel, voltage-gated sodium channel
- TTX
tetrodotoxin
Conflicts of interest
None to declare.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Block of rNaV1.4 channels at various pH values. (A) Current traces of rNaV1.4 channels at −20 mV before and after application of 10µM µ-CnIIIC, as well as upon wash with control saline (pH 7.4), followed by wash with solution of pH 6.5 and pH 8.0. In the panel on the right all traces are super imposed, scaled to peak. (B) Time course of peak inward current for the indicated conditions. (C) Statistics on recovery from µ-CnIIIC block for 200-s wash each, obtained with solutions of the indicated pH values. n-values are provided in parentheses.
{kind=link}
Figure S2 Sequence alignments of NaV-channel regions particularly important for µ-conotoxin action. (A) Topological cartoon of an NaV α-subunit with four homologous domains (DI-DIV). Boundaries used for the construction of domain chimeras (red diamonds) and pore loop chimeras (colour) are indicated. Residue numbers refer to rNaV1.4. (B) Multiple sequence alignments of the indicated NaV-channel types for the pore region of domain I (pink) and of domain II (blue).
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andavan GS, Lemmens-Gruber R. Voltage-gated sodium channels: mutations, channelopathies and targets. Curr Med Chem. 2011;18:377–397. doi: 10.2174/092986711794839133. [DOI] [PubMed] [Google Scholar]
- Backx PH, Yue DT, Lawrence JH, Marban E, Tomaselli GF. Molecular localization of an ion-binding site within the pore of mammalian sodium channels. Science. 1992;257:248–251. doi: 10.1126/science.1321496. [DOI] [PubMed] [Google Scholar]
- Chahine M, Sirois J, Marcotte P, Chen L, Kallen RG. Extrapore residues of the S5–S6 loop of domain 2 of the voltage-gated skeletal muscle sodium channel (rSkM1) contribute to the µ-conotoxin GIIIA binding site. Biophys J. 1998;75:236–246. doi: 10.1016/s0006-3495(98)77510-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gordon D, Heinemann SH. Modulation of cloned skeletal muscle sodium channels by the scorpion toxins Lqh II, Lqh III, and Lqh αIT. Pflügers Arch. 2000;439:423–432. doi: 10.1007/s004249900181. [DOI] [PubMed] [Google Scholar]
- Choudhary G, Yotsu-Yamashita M, Shang L, Yasumoto T, Dudley SC., Jr Interactions of the C-11 hydroxyl of tetrodotoxin with the sodium channel outer vestibule. Biophys J. 2003;84:287–294. doi: 10.1016/S0006-3495(03)74849-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Aglieco F, Dib-Hajj SD. Critical molecular determinants of voltage-gated sodium channel sensitivity to µ-conotoxins GIIIA/B. Mol Pharmacol. 2002;61:1192–1201. doi: 10.1124/mol.61.5.1192. [DOI] [PubMed] [Google Scholar]
- Dudley SC, Jr, Todt H, Lipkind G, Fozzard HA. A µ-conotoxin-insensitive Na+ channel mutant: possible localization of a binding site at the outer vestibule. Biophys J. 1995;69:1657–1665. doi: 10.1016/S0006-3495(95)80045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley SC, Jr, Chang N, Hall J, Lipkind G, Fozzard HA, French RJ. µ-conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J Gen Physiol. 2000;116:679–690. doi: 10.1085/jgp.116.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favreau PH, Benoit E, Hocking H, Carlier L, D'hoedt D, Leipold E, et al. Pharmacological characterization of a novel µ-conopeptide, CnIIIC, indicates potent and preferential inhibition of sodium channel subtypes (NaV1.2/1.4) and reveals unusual activity on neuronal nicotinic acetylcholine receptors. Br J Pharmacol. 2012;166:1654–1668. doi: 10.1111/j.1476-5381.2012.01837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French RJ, Prusak-Sochaczewski E, Zamponi GW, Becker S, Kularatna AS, Horn R. Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron. 1996;16:407–413. doi: 10.1016/s0896-6273(00)80058-6. [DOI] [PubMed] [Google Scholar]
- George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. Evolution of voltage-gated Na+ channels. J Exp Biol. 2002;205:575–584. doi: 10.1242/jeb.205.5.575. [DOI] [PubMed] [Google Scholar]
- Heinemann SH, Leipold E. Tools for studying peptide toxin modulation of voltage-gated sodium channels. 2011. In e-book: Toxins and Ion Transfers, SFET Editions. http://www.sfet.asso.fr.
- Heinemann SH, Terlau H, Imoto K. Molecular basis for pharmacological differences between brain and cardiac sodium channels. Pflügers Arch. 1992;422:90–92. doi: 10.1007/BF00381519. [DOI] [PubMed] [Google Scholar]
- Jurman ME, Boland LM, Liu Y, Yellen G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- Leipold E, Markgraf R, Miloslavina A, Kijas M, Schirmeyer J, Imhof D, et al. Molecular determinants of the subtype specificity of µ-conotoxin SIIIA targeting neuronal voltage-gated sodium channels. Neuropharmacology. 2011;61:105–111. doi: 10.1016/j.neuropharm.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Li RA, Ennis IL, French RJ, Dudley SC, Jr, Tomaselli GF, Marban E. Clockwise domain arrangement of the sodium channel revealed by µ-conotoxin (GIIIA) docking orientation. J Biol Chem. 2001;276:11072–11077. doi: 10.1074/jbc.M010862200. [DOI] [PubMed] [Google Scholar]
- Norton RS. Mu-conotoxins as leads in the development of new analgesics. Molecules. 2010;15:2825–2844. doi: 10.3390/molecules15042825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest BT. Future potential and status of selective sodium channel blockers for the treatment of pain. Curr Opin Drug Discov Devel. 2009;12:682–692. [PubMed] [Google Scholar]
- Schirmeyer J, Szafranski K, Leipold E, Mawrin C, Platzer M, Heinemann SH. A subtle alternative splicing event of the NaV1.8 voltage-gated sodium channel is conserved in human, rat, and mouse. J Mol Neurosci. 2010;41:310–314. doi: 10.1007/s12031-009-9315-3. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ. Sodium channels in nerve apparently have two conductance states. Nature. 1977;270:265–267. doi: 10.1038/270265a0. [DOI] [PubMed] [Google Scholar]
- Starkus JG, Varga Z, Schönherr R, Heinemann SH. Mechanisms of the inhibition of Shaker potassium channels by protons. Pflügers Arch. 2003;447:44–54. doi: 10.1007/s00424-003-1121-0. [DOI] [PubMed] [Google Scholar]
- Wood JN, Boorman JP, Okuse K, Baker MD. Voltage-gated sodium channels and pain pathways. J Neurobiol. 2004;61:55–71. doi: 10.1002/neu.20094. [DOI] [PubMed] [Google Scholar]
- Zhang MM, Gruszczynski P, Walewska A, Bulaj G, Olivera BM, Yoshikami D. Cooccupancy of the outer vestibule of voltage-gated sodium channels by µ-conotoxin KIIIA and saxitoxin or tetrodotoxin. J Neurophysiol. 2010;104:88–97. doi: 10.1152/jn.00145.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuliani V, Rivara M, Fantini M, Costantino G. Sodium channel blockers for neuropathic pain. Expert Opin Ther Pat. 2010;20:755–779. doi: 10.1517/13543771003774118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.