

Abstract
BACKGROUND AND PURPOSE
Peroxisome proliferator-activated receptor (PPAR) agonists exert anti-albuminuric effects. However, the nephroprotective effects of these drugs remain to be fully understood. We have investigated whether gemfibrozil, GW0742 and pioglitazone protect human podocytes against nutrient deprivation (ND)-induced cell death and the role of mitochondrial biogenesis as a cytoprotective process.
EXPERIMENTAL APPROACH
Immortalized human podocytes were pre-treated with the PPAR agonists and exposed to ND (5 h) under normoxia, hypoxia or in the presence of pyruvate. Cell death was measured at the end of the ND and of the recovery phase (24 h). Mitochondrial mass, cytochrome c oxidase (COX) subunits 1 and 4 were measured as markers of mitochondrial cell content, while membrane potential as an index of mitochondrial function. PGC-1α, NRF1 and Tfam expression was studied, as crucial regulators of mitochondrial biogenesis.
KEY RESULTS
Cell pre-treatment with gemfibrozil, GW0742, or pioglitazone significantly decreased the ND-induced cell loss, necrosis and apoptosis. These effects were attenuated by hypoxia and potentiated by pyruvate. Pre-treatment with these drugs significantly increased mitochondrial cell content, while it did not affect mitochondrial function. In all these experiments pioglitazone exerted significantly larger effects than gemfibrozil or GW0742.
CONCLUSIONS AND IMPLICATIONS
Gemfibrozil, GW0742 and pioglitazone may exert direct protective effects on human podocytes. Mitochondrial biogenesis is a cell response to the PPAR agonists related to their cytoprotective activity. These results provide a mechanistic support to the clinical evidence indicating PPAR agonists as disease-modifying agents for glomerular diseases.
Keywords: podocytes, PPAR, mitochondrial biogenesis, glomerular diseases, cell death
Introduction
An anti-albuminuric effect has been shown in a number of clinical trials with PPARα and γ agonists (receptor nomenclature follows Alexander et al., 2011). The three clinically evaluated thiazolidinediones (troglitazone, rosiglitazone and pioglitazone) have been demonstrated to significantly decrease urinary albumin excretion in patients with type-2 diabetes mellitus (Sarafidis and Bakris, 2006; Mao and Ong, 2009; Sarafidis et al., 2010). Likewise, rosiglitazone has been reported to exert an anti-albuminuric effect in patients with non-diabetic glomerular diseases, including primary focal segmental glomerulosclerosis (Kincaid-Smith et al., 2008). Besides, the fibrate gemfibrozil has been shown to decrease urinary albumin excretion in patients with type-2 diabetes mellitus, hypertriglyceridaemia and microalbuminuria (Smulders et al., 1997). Together, such clinical evidence highlights the value of PPAR agonists for the treatment of glomerular diseases, although the underlying protective mechanisms remain to be fully understood.
Systemic actions (i.e. improvement of glycaemic control and dyslipidaemia) together with renal actions contribute to the therapeutic effects of PPAR agonists (Kiss-Tóth and Rőszer, 2008; Ruan et al., 2008; Mao and Ong, 2009; Yang et al., 2012). The reported anti-albuminuric effect suggests that PPAR agonists may interfere with the pathological processes impairing the integrity of the glomerular filtration barrier (GFB) and leading to the loss of the selective permeability. Podocytes are highly differentiated long-living cells involved in the formation and maintenance of the GFB (Pavenstädt et al., 2003), while their dysfunction and loss have been implicated in the onset and progression of both albuminuria and glomerulosclerosis of different aetiologies (Shankland, 2006; Wiggins, 2007). Compelling experimental results suggest that PPAR agonists may directly improve podocyte response to injury and exert cytoprotective effects. Expression of all three PPAR subtypes has been reported in rodent and human podocytes (Ren et al., 2005; Yang et al., 2006; Kanjanabuch et al., 2007; Miglio et al., 2011). Moreover, data from in vitro experiments have shown that, by interfering with the apoptotic cascades, PPAR agonists diminish cell loss induced by puromycin aminoglucoside (Kanjanabuch et al., 2007), doxorubicin (Mori et al., 2011), oxygen and glucose deprivation–reoxygenation and serum deprivation (Miglio et al., 2011). Apoptosis is known to cause podocyte depletion in many glomerulopathies (Shankland, 2006). However, additional cell death processes could contribute to the podocyte depletion, especially when specific severe insults are involved. Indeed, podocyte cytoplasmic oedema (a hallmark of necrosis) and cell detachment have been shown in rodent models of renal ischaemia and organ transplantation (Lambert et al., 1986; Wagner et al., 2008; Pippin et al., 2009). Moreover, a significant increase in the percentage of necrotic podocytes has been reported in in vitro models of ischaemia (Brukamp et al., 2007; Miglio et al., 2011). Hence, the effects of PPAR agonists on podocyte death need to be further investigated.
In order to evaluate whether preservation of podocyte survival could contribute to the anti-albuminuric effect of PPAR agonists, here the effects of gemfibrozil and pioglitazone on the nutrient deprivation (ND)-induced cell death were studied. In addition, to explore the pharmacological role of the less investigated PPARβ, the effects of the selective agonist GW0742 were also evaluated. ND is an ischaemia-related stimulus able to severely affect cell integrity. Ischaemia is known to compromise the integrity of the glomerular cells under different pathological conditions shared by a decreased glomerular blood flow (i.e. renal ischaemia, thrombotic microangiopathy, capillary loss due to glomerulosclerosis or mesangial proliferation). Hence, the protective effects exerted by PPAR agonists against the ND-induced podocyte death might be useful in understanding their therapeutic potential.
By regulating the expression of many mitochondrial proteins, PPARs are known to cause remodelling of the mitochondrial proteome and mitochondrial biogenesis (Scarpulla, 2008; Hock and Kralli, 2009). Because previous findings suggest that mitochondrial biogenesis could be a cytoprotective process (Jo et al., 2006; Yang et al., 2007a; Fujisawa et al., 2009; Miglio et al., 2009; Funk et al., 2010; Rasbach et al., 2010), here the effects of gemfibrozil, GW0742 and pioglitazone on mitochondrial mass and function were evaluated as potential cytoprotective responses to these drugs.
Methods
Cultures of human glomerular cells
In this study, we have used lines of human primary glomerular endothelial cells, immortalized mesangial cells and immortalized podocytes. Immortalized cells were obtained from primary mesangial cells and podocytes by infection with a hybrid Adeno5/SV40 virus. Glomerular cells were cultured and characterized as previously reported (Conaldi et al., 1998; Doublier et al., 2001; Collino et al., 2008). The day before the experiment, cells were plated on six-well culture plates (300 × 103 cells·per well).
In vitro models of podocyte injury
ND was achieved by replacing the culture medium with 1 mL of a bicarbonate-buffered balanced salt solution (in mM: 134, NaCl; 15.7, NaHCO3; 3.1, KCl; 1.2, CaCl2; 1.2, MgSO4; 0.25, KH2PO4; pH 7.2). Cell cultures were maintained at 37°C in a fully humidified air (95%)/CO2 (5%) incubator. Recovery was started at the designated time point by returning cell cultures to standard culture conditions. Some experiments were performed under either hypoxia alone or hypoxic nutrient deprivation (HND). Hypoxic conditions were achieved as previously reported (Miglio et al., 2011) by placing the cell cultures in an anaerobic chamber (Oxoid, Hampshire, UK) filled with a hypoxic gas mixture N2 (95%)/CO2 (5%).
Evaluation of cell loss, necrotic and apoptotic cell death
Cell loss, necrotic cell death and apoptotic cell death were determined as previously reported (Miglio et al., 2011). Briefly, cell loss was evaluated by counting the number of viable cells in a haemacytometer by the Trypan blue exclusion test, by an observer, unaware of the treatments. Necrotic cells were detected after staining with fluorescein diacetate (FDA) and propidium iodide (PI). Cells were washed twice with PBS and stained with a mixture of FDA (4.0 µM) and PI (0.4 µM). After washing, cells were examined using a fluorescence microscope (Leica Microsystems Wetzlar, Germany). Green and red cells (PI+ cells) were scored as healthy/viable and dead respectively. Apoptotic cells were detected after cell staining with DAPI. Cells were washed twice with ice-cold PBS, fixed and permeabilized with methanol (4°C; 5 min) then stained with DAPI (0.3 µM). After washing, cells were examined using a fluorescence microscope. Pyknotic and/or fragmented nuclei were scored as apoptotic nuclei. Between 80 and 120 cells or nuclei per field were counted by an observer, unaware of the treatments and, for each experimental condition, ∼5000 cells or nuclei were examined.
Evaluation of caspase 3 activation
Increase in caspase 3 activity was evaluated as an index of apoptotic cell death. Using a commercial kit (Biovision Research Products, Mountain View, CA), caspase 3 activity was determined by measuring the ability of cell lysates to cleave the p-nitroaniline coupled caspase 3 substrate DEVD, according to the manufacturer's instructions.
Confocal microscopy
Indirect immunofluorescence was performed on podocytes cultured on chamber slides (Nalgen Nunc International, Rochester, NY) fixed in 4% paraformaldehyde containing 2% sucrose. Subconfluent cells were stained with a goat polyclonal anti-synaptopodin or a rabbit polyclonal anti-podocin (both from Santa Cruz Biotechnology, Santa Cruz, CA) antibodies. An immunologically irrelevant guinea pig serum was used as a control where appropriate. Alexa Fluor-488 anti-goat or Alexa Fluor-488 anti-rabbit polyclonal antibodies (Molecular Probes, Leiden, the Netherlands) was used as a secondary antibody. Confocal microscopy analysis was performed using a Zeiss LSM 5 Pascal Model Confocal Microscope (Carl Zeiss International, Jena, Germany). Hoechst 33258 was added for nuclear staining.
Quantitative RT-PCR analysis
Total RNA was extracted with OMNIzol reagent (Euroclone, Milan, Italy) according to the manufacturer's instructions. First-strand cDNA was synthesized from 0.5 µg of total RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR experiments were performed in 25 µL reaction mixtures containing 10 ng of cDNA template, the Power SYBR® Green PCR Master Mix and the AmpliTaq Gold® DNA Polymerase LD (Applied Biosystems). Relative quantization of the products was performed using a 48-well StepOne™ Real Time System (Applied Biosystems). For all real-time PCR analyses, β-actin mRNA was used to normalize RNA inputs.
Western blot analysis
Western blot analyses were performed as previously described (Miglio et al., 2009). Bcl-2, Bax, and PPARγ co-activator (PGC)-1α, were detected following incubation with polyclonal antibodies (Santa Cruz Biotechnology). The mitochondrial DNA-encoded cytochrome c oxidase (COX) subunit 1 was detected with a monoclonal antibody (Santa Cruz Biotechnology). The nuclear DNA-encoded COX4, nuclear respiratory factor (NRF)1 and the mitochondrial transcription factor A (Tfam) were detected with monoclonal antibodies (Abcam plc Cambridge Science Park, Cambridge, UK). To confirm the homogeneity of the proteins loaded, the membranes were stripped and incubated with an anti-β-actin monoclonal antibody (Sigma-Aldrich, Milan, Italy). The membranes were overlaid with Western Lightning Chemiluminescence Reagent Plus (Perkin-Elmer Life Science, Norwalk, CT) and exposed to Hyperfilm ECL film (Amersham Biosciences, Piscataway, NJ). Protein bands were quantified on film by densitometry using the software Image J 1.41 (US National Institutes of Health, Bethesda, MD, USA).
Evaluation of mitochondrial mass and membrane potential
Mitochondrial mass and membrane potential of the cells were determined with the aid of fluorescent dyes, MitoTracker Green FM and tetramethylrhodamine ethyl ester (TMRE; Molecular Probes, Eugene, OR, USA), respectively, as described by Mitsuishi et al. (2008); Hoechst 33342 (Molecular Probes) was used for nuclear staining. The value for mitochondrial mass was normalized to that for nuclei, and membrane potential was normalized to that for mitochondrial density. Cells were cultured with or without the drug treatements, then treated with the dyes for 10 min and washed twice with warm PBS. The fluorescent intensity was measured with a Victor X4 multiplate reader (Perkin-Elmer).
Data analysis
In some cases (see Results), data were fitted as sigmoidal concentration–response curves, and analysed with a four-parameter logistic equation by using the software Origin version 6.0 (Microcal Software, Northampton, MA). Statistical significance was evaluated by one-way anova followed by the post hoc Dunnett's test; differences were considered statistically significant when P < 0.05.
Materials
Pioglitazone was from Alexis (Vinci, Italy). GW0742, gemfibrozil, BADGE and all other reagents were from Sigma-Aldrich. PPAR ligands were dissolved in dimethyl sulfoxide, and the final drug concentrations were obtained by dilution of stock solutions in the experimental buffers. The final concentration of the organic solvent was less than 0.1%, which had no effect on cell viability.
Results
PPAR expression by human glomerular cells
To study whether PPAR agonists could exert protective effects on human glomerular cells, first we studied whether our cell lines express functional PPARs. Human glomerular endothelial cells, immortalized mesangial cells and podocytes were untreated or treated (72 h, as a repeated treatment; drugs and medium were replaced every 24 h) with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM). The levels of the mRNA encoding for PPARα, PPARβ or PPARγ were measured by quantitative PCR. As shown in Figure 1, PPAR genes were constitutively expressed by the three glomerular cell types. In addition, PPAR expression was significantly (P < 0.01 vs. basal level) induced by the three PPAR agonists. These results indicate that our lines of human glomerular cells express functional PPARs, and among the three cell types, podocytes may express higher PPAR levels, thus, together with the above-mentioned issues (see Introduction), strongly indicating the opportunity to study the effects exerted by PPAR agonists on this cells type.
Figure 1.
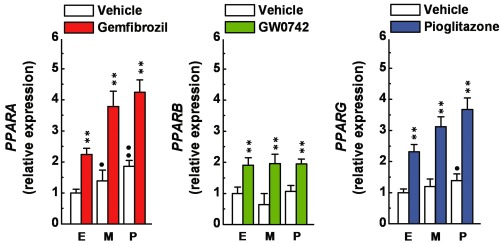
Expression of PPARA, PPARB and PPARG by human glomerular cells. Primary human endothelial cells (E), immortalized human mesangial cells (M) and immortalized human podocytes (P) were untreated or treated with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) for 72 h (drugs and medium were replaced every 24 h), then processed to determine PPARA, PPARB or PPARG expression by quantitative PCR analysis. mRNA levels were normalized to those of untreated endothelial cells. **P < 0.01 versus vehicle alone. •P < 0.05; ••P < 0.01 versus endothelial cells.
Effects of gemfibrozil, GW0742 and pioglitazone on the ND-induced cell loss
First, we evaluated the time course of the protective effects exerted by the three PPAR agonists. Cells were pre-treated with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) for 24 h (a single treatment) to 120 h (as repeated treatments), then exposed to ND (5 h). Alternatively, they were treated with these PPAR agonists at the same time as ND or throughout the course of the recovery phase (24 h; due to cell proliferation, the effects of longer cell treatments cannot be accurately measured). The overall cell loss was determined by counting the number of viable cells at the end of the recovery phase. In addition, the insult-associated and the recovery-associated cell loss were determined by counting the number of viable cells at the end of the ND and of the recovery (24 h) phase respectively. As shown in Figure 2A, the overall cell loss, the insult-associated cell loss and the recovery-associated cell loss were significantly decreased (P < 0.01 vs. vehicle alone at ≥72 h pre-treatment) in cultures pre-treated with the three PPAR agonists. Notably, significantly larger protective effects (P < 0.05 vs. gemfibrozil or GW0742) were exerted by pioglitazone on both the overall cell loss and the ND-associated cell loss, while equivalent effects were exerted by these drugs on the recovery-associated cell loss. The overall cell loss was also significantly (P < 0.05 vs. vehicle alone) decreased in cell cultures either treated with the three drugs throughout the course of the recovery phase (P < 0.05 vs. vehicle alone; Figure 2A), or pre-treated (24 h) plus post-treated (24 h; P < 0.05 vs. vehicle alone; P < 0.01 vs. pre- or post-treatment alone; Figure S1), thereby indicating that these protective effects depend on the length of cell treatment and are unaffected by the insult. Notably, the overall cell loss was decreased by the three PPAR agonists in a concentration-dependent manner; and interestingly, when compared with the single pre-treatment (24 h), a leftward shift of the concentration–response curves and a higher drug efficacy were caused by the repeated pre-treatments (72 h), while no significant change was observed on the Hill coefficient values (Table 1 and Figure S2). Collectively, these results suggest that these drugs decrease podocyte loss, most likely through receptor-dependent mechanisms.
Figure 2.
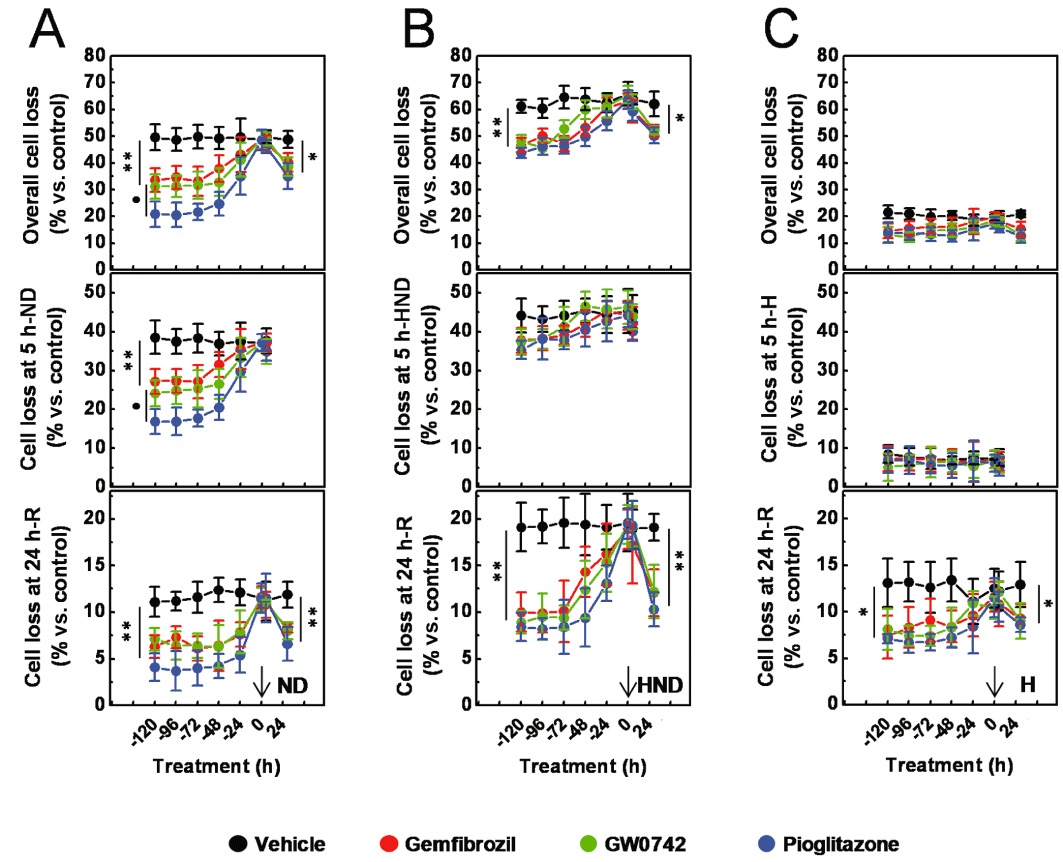
Effects of gemfibrozil, GW0742 and pioglitazone on ND-, HND-, or hypoxia (H)-induced podocyte loss. Immortalized podocytes were pre-treated with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) for 24 h (a single treatment) to 120 h (repeated treatments), then exposed (5 h) to ND (A), HND (B) or H (C). Alternatively, they were treated with the PPAR agonists either at the same time as ND (A), HND (B), H (C), or throughout the course of the recovery phase (24 h). At the end of the insult phase, the number of viable cells was assessed to determine the insult-associate cell loss; at the end of the recovery phase, the number of viable cells was assessed to determine the recovery-associated cell loss and the overall cell loss. Results are expressed as mean ± SEM. Mean of five experiments run in triplicate. *P < 0.05; **P < 0.01 versus vehicle alone. •P < 0.01 versus gemfibrozil or GW0742.
Table 1.
Estimated pEC50, Emax and Hill coefficients (nH)
| Drug | Pre-treatment (h) | pEC50 | Emax | nH |
|---|---|---|---|---|
| Gemfibrozil | 24 | 5.9 ± 1.7 | 16.9 ± 3.5 | 1.4 ± 0.4 |
| 72 | 6.2 ± 0.1 | 41.7 ± 4.5 | 1.0 ± 0.1 | |
| GW0742 | 24 | 8.0 ± 0.1 | 18.8 ± 1.4 | 1.0 ± 0.2 |
| 72 | 9.0 ± 0.1 | 36.4 ± 1.4 | 1.1 ± 0.2 | |
| Pioglitazone | 24 | 7.0 ± 0.2 | 31.3 ± 2.2 | 1.7 ± 0.1 |
| 72 | 7.9 ± 0.1 | 58.2 ± 2.8 | 1.1 ± 0.2 |
Immortalized podocytes were pre-treated with gemfibrozil (0.01–30 µM), GW0742 (0.1 nM to 0.1 µM) or pioglitazone (1 nM to 1 µM) for either 24 h (a single treatment) or 72 h (as repeated treatments), then exposed to ND (5 h). At the end of the recovery phase (24 h), the number of viable cells was assessed to determine the ND-induced cell loss. Results are expressed as mean ± SEM. Mean of four experiments run in triplicate. Data were fitted as sigmoidal concentration–response curves and analysed with a four-parameter logistic equation.
Effects of gemfibrozil, GW0742 and pioglitazone on the ND-induced podocyte injury
To better understand the nature of the protective effects, in parallel samples (72 h of repeated pre-treatments), we measured the percentage of PI+ cells, apoptotic nuclei and the caspase 3 activity, as marker of necrotic and apoptotic cell death and assessed the presence of synaptopodin and podocin, as markers of podocyte differentiation. The ND-induced rise in the percentage of PI+ cells, in apoptotic nuclei and in caspase 3 activity were significantly decreased (P < 0.05 vs. vehicle alone) by the three PPAR agonists (Figure 3A). Significantly larger effects (P < 0.05 vs. gemfibrozil or GW0742) were exerted by pioglitazone on the ND-induced increase in the percentage of PI+ (Figure 3A), while equivalent effects were exerted by these drugs on the indexes of apoptosis. As shown in Figure 4, ND decreased synaptopodin immunostaining which was preserved in cells treated with gemfibrozil, GW0742 or pioglitazone. Podocin expression was not affected by either ND or PPAR agonists (data not shown). These results indicate that ND caused an early, mainly necrotic, and a delayed, mainly apoptotic, cell death as well as loss of an essential protein. Moreover, they suggest that gemfibrozil, GW0742 and pioglitazone are endowed of both anti-apoptotic and anti-necrotic activities and preserve podocyte differentiation.
Figure 3.
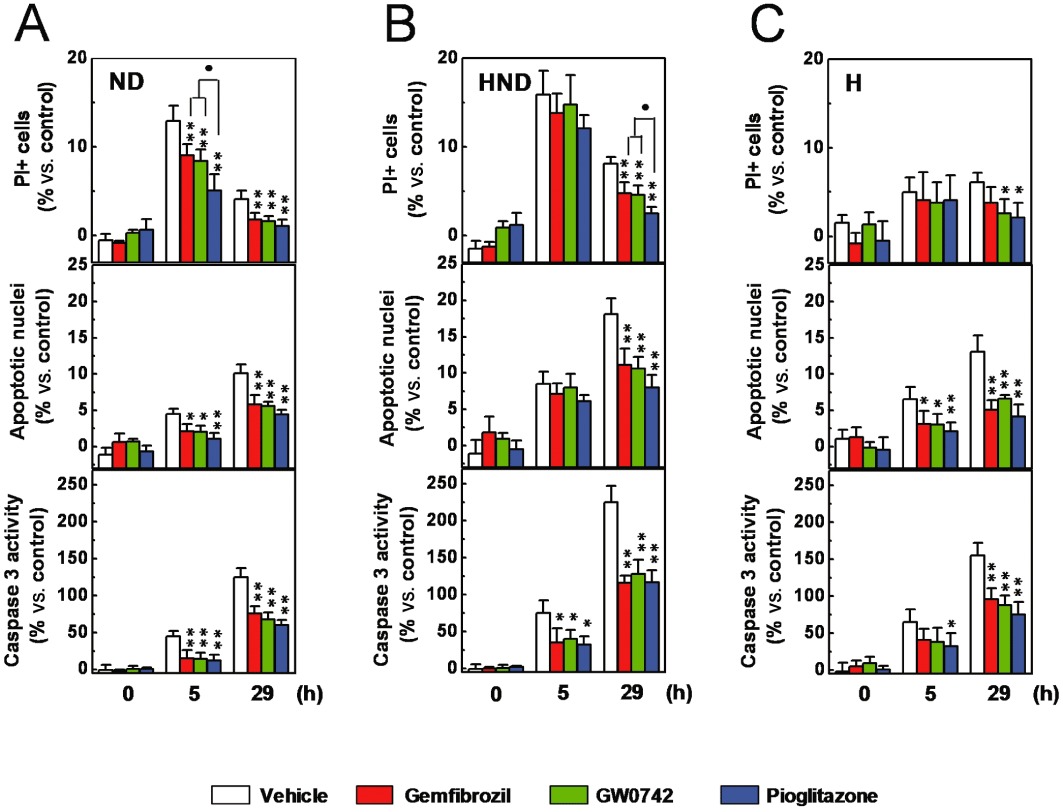
Effects of gemfibrozil, GW0742 and pioglitazone on ND-, HND- or H-induced podocyte cell death. Immortalized podocytes were pre-treated with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) for 72 h (as repeated treatments), then exposed to ND (A), HND (B), or H (C). At the end of the ND, HND or H phase and the recovery phase, the percentage of PI+ cells, apoptotic nuclei and the caspase 3 activity were measured. Results are expressed as mean ± SEM. Mean of four experiments run in triplicate. *P < 0.05; **P < 0.01 versus vehicle alone. •P < 0.05 versus gemfibrozil or GW0742.
Figure 4.
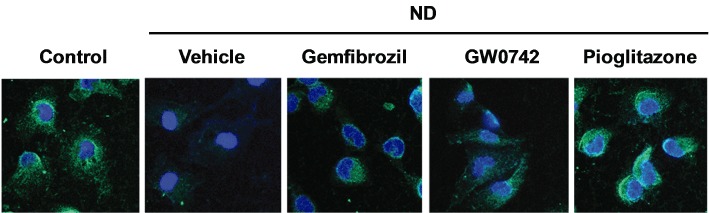
Effects of gemfibrozil, GW0742 and pioglitazone on ND-induced synaptopodin loss. Cells were treated with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) for 72 h (as repeated treatments) then exposed to ND. At the end of the recovery phase (24 h), synaptopodin expression was detected by immunofluorescence using confocal microscopy (original magnification: 630×).
Effects of hypoxia and pyruvate on the cell protection exerted by gemfibrozil, GW0742 and pioglitazone
Podocytes were pre-treated with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM), for 24 h (a single treatment) to 120 h (as repeated treatments), then exposed to ND (5 h) under hypoxia (HND) or to hypoxia alone. In comparison to the cultures exposed to ND alone (Figure 2A), higher and lower levels of cell loss were measured in HND- and hypoxia-exposed cultures respectively (Figure 2B and 2C). Consistently, higher percentages of PI+, apoptotic nuclei and levels of caspase 3 activity were detected in HND-exposed cultures, while lower rate of PI+ cells were measured in cultures exposed to hypoxia alone (Figure 3B and 3C). These results indicate that hypoxia caused a delayed, mainly apoptotic, cell death and, by promoting the recovery-associated cell death, potentiated the ND-induced lethal effects. Interestingly, the protective effects exerted by the three drugs on the ND-associated cell loss (Figure 2A) and on the increase in the percentage of PI+ cells (Figure 3C) were blunted by hypoxia (Figure 2B and 2D). On the other hand, effects on the recovery-associated cell loss and on the apoptotic indexes were not changed (Figure 2A and 2C). As hypoxia inhibits mitochondrial respiration, the above results suggest that the anti-necrotic activity of the three drugs may be related to the mitochondrial function. To explore this hypothesis, podocytes were pre-treated (72 h, as repeated treatments) with increasing concentrations of gemfibrozil (10 nM to 30 µM), GW0742 (0.1–100 nM) or pioglitazone (1 nM to 1 µM), then exposed to ND (5 h) in the presence of pyruvate (2 mM), to fuel mitochondria (Kauppinen and Nicholls, 1986; Miglio et al., 2009). The protective effects exerted by piglitazone were widely potentiated by pyruvate, whilst those of gemfibrozil or GW0742 only marginally increased (Figures 5 and S3). The interactions with hypoxia and pyruvate suggest that the protective effects exerted by the three PPAR agonists on the necrotic cell death may be related to the mitochondrial changes induced by the repeated cell treatments.
Figure 5.
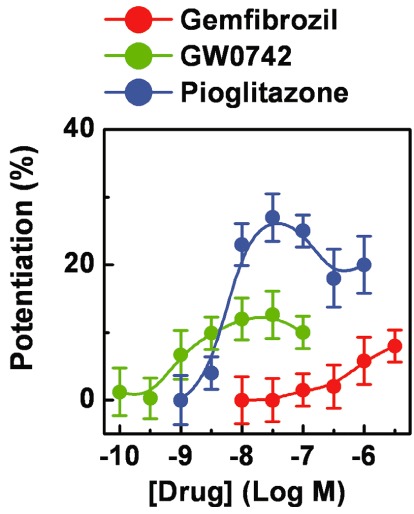
Effects of pyruvate on the PPAR agonist-induced cell protection. Immortalized podocytes were pre-treated (72 h, as repeated treatments) with gemfibrozil (0.01–30 µM), GW0742 (0.1 nM to 0.1 µM) or pioglitazone (1 nM to 1 µM), then exposed to ND (5 h) in the absence or presence of pyruvate (2 mM). At the end of the recovery phase (24 h), the number of viable cells was assessed to determine the overall cell loss. Potentiation was calculated as protective effects of the PPAR agonist in the presence of pyruvate – protective effects of PPAR agonist alone + protective effects of pyruvate alone (22 ± 5%). Results are expressed as mean ± SEM. Mean of four experiments run in triplicate.
Molecular and cellular effects of gemfibrozil, GW0742 and pioglitazone
To investigate the anti-apoptotic activity of the three PPAR agonists, podocytes were pre-treated (72 h as repeated treatments) with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM), then exposed to ND (5 h). The effects on the levels of the anti-apoptotic Bcl-2 and the pro-apoptotic Bax were measured at the end of the recovery phase (24 h). As shown in Figure 6, the Bcl-2-to-Bax ratio was markedly (43 ± 1%) decreased by ND, as a result of opposite changes in Bax and Bcl-2 levels. This effect was prevented by the three PPAR agonists that normalized Bax and Bcl-2 levels.
Figure 6.
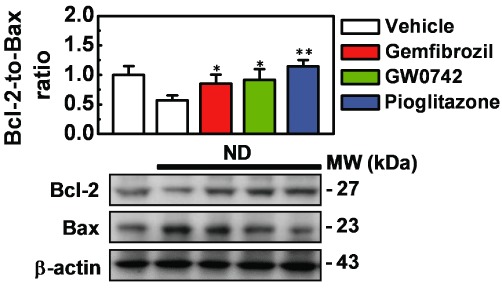
Effects of gemfibrozil, GW0742 and pioglitazone on Bcl-2 and Bax expression. Immortalized podocytes were treated (72 h, as repeated treatments) with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) then exposed to nutrient deprivation (ND; 5 h). At the end of the recovery phase (24 h), Bcl-2 and Bax expression was determined by Western blot analysis. Results are expressed as mean ± SEM. Mean of four experiments run in triplicate. *P < 0.05; **P < 0.01 versus nutrient-deprived cells treated with vehicle alone.
Then to investigate the functional linkage between anti-necrotic activity and mitochondrial function, podocytes were pre-treated with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) for 24 h (a single treatment) to 72 h (as repeated treatments). Mitochondrial mass, COX1 and COX4 levels were measured as markers of mitochondrial cell content, and mitochondrial membrane potential as a marker of mitochondrial function. Mitochondrial mass was significantly (P < 0.05 vs. vehicle alone) increased by the three PPAR agonists, and pioglitazone exerted significantly larger effects (P < 0.01 vs. gemfibrozil or GW0742; Figure 7A). Consistently, basal COX1 and COX4 levels were also significantly increased by repeated (72 h) cell treatments with the three PPAR agonists (P < 0.05 vs. vehicle alone; P < 0.05 pioglitazone vs. gemfibrozil or GW0742; Figure 7B). Basal mitochondrial membrane potential, on the other hand, was marginally affected by the three PPAR agonists (Figure 7C).
Figure 7.
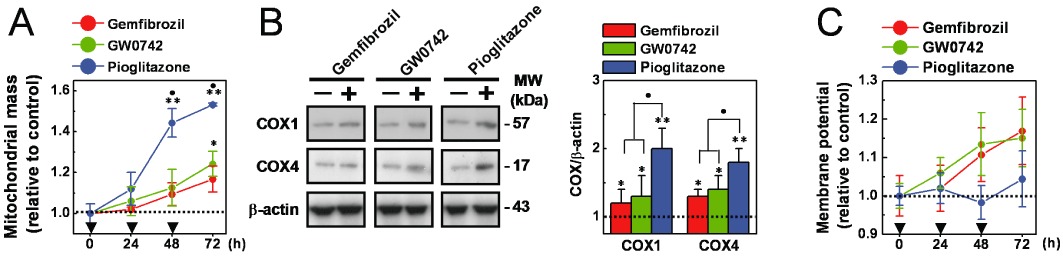
Effects of gemfibrozil, GW0742 and pioglitazone on mitochondrial cell content and function. Immortalized podocytes were pre-treated (24–72 h, as indicated in the figure) with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM). (A) Mitochondrial mass was determined by measuring fluorescence intensity after cell staining with the mitochondrial fluorescent dye MitoTracker Green FM. (C) Mitochondrial membrane potential was determined by measuring fluorescence intensity after cell staining with the potentiometric fluorescent dye TMRE. The values of mitochondrial membrane potential were normalized to those for mitochondrial mass. (B) Cells were pre-treated (72 h, as repeat treatments) with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) and COX1 and COX4 levels were determined by Western blot analyses. Results are expressed as mean ± SEM. Mean of four experiments run in triplicate. *P < 0.05; **P < 0.01 versus cells treated with vehicle alone. •P < 0.05 versus gemfibrozil or GW0742.
Finally, the effects of the three PPAR agonists on the expression of crucial regulators of the mitochondrial biogenesis processes were evaluated. As shown in Figure 8A, basal expression of PPARGC1A, which encodes for PGC-1α (the master regulator of the mitochondrial biogenesis process; Scarpulla, 2008; Hock and Kralli, 2009), was significantly increased by the three PPAR agonists (P < 0.05 vs. vehicle alone). However, a significantly higher transcriptional rate was induced by pioglitazone (P < 0.05 vs. gemfibrozil or GW0742). Western blot analyses confirmed and extended these PCR data: PGC-1α basal level was significantly increased by repeated (72 h) cell treatments with the three PPAR agonists (P < 0.01 vs. vehicle alone; P < 0.01 vs. gemfibrozil or GW0742; Figure 8C). The expression of NRF1 (a transcription factor involved in controlling the expression of nuclear genes encoding for mitochondrial proteins) and Tfam (a transcription factor involved in controlling the expression of mitochondrial genes; Scarpulla, 2008; Hock and Kralli, 2009) were also increased by the three PPAR agonists, albeit in a different manner: NRF1 was significantly (P < 0.05 vs. vehicle alone) induced by pioglitazone, and GW0742; Tfam by pioglitazone only; gemfibrozil did not exert any significant effect on these genes (Figure 8B). Similar changes were revealed by Western blot analysis (Figure 8C). Together, these results indicate that the three PPAR agonists promote several molecular and cellular changes, likely related to their protective effects on podocytes.
Figure 8.
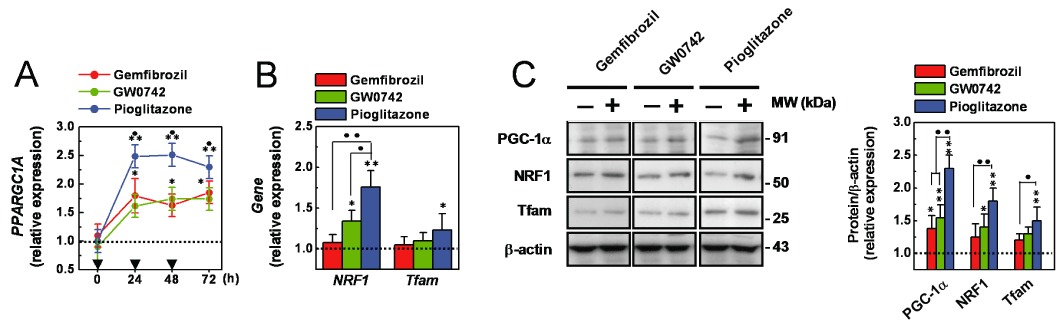
Effects of the PPAR agonists on PGC-1α, NRF1 and Tfam expression. (A) Cells were pre-treated with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) for 24–72 h and PPPARC1A expression was measured by quantitative PCR analysis. (B) Cells were treated (72 h as a repeated treatment) with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM), and NRF1 and Tfam expression was measured by quantitative PCR analysis. (C) Cells were treated (72 h as a repeated treatment) with gemfibrozil (30 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) and PGC-1α, NRF1 and Tfam expression was measured by Western blot analysis. Results are expressed as mean ± SEM. Mean of four experiments run in triplicate. *P < 0.05; **P < 0.01 versus vehicle alone. •P < 0.05; ••P < 0.01 versus gemfibrozil or GW0742.
Effects of BADGE on the pioglitazone-induced phenotypic changes
Finally, the effects of the PPARγ antagonist BADGE (Wright et al., 2000) on pioglitazone-induced responses were studied. Podocytes were treated with pioglitazone (1 µM; 72 h as repeated treatments) in the absence or presence of increasing concentrations of BADGE (0.1 nM to 10 µM). Then cells were processed to assess either the expression of PPARG and PPARGC1A, or the ND-induced cell loss. As shown in Figure 9A and 9B, the pioglitazone-induced up-regulation of PPARG and PPARGC1A expression and the decrease of ND-induced cell loss were abolished by BADGE in a concentration-dependent manner: the pIC50 values were 6.1 ± 0.2, 6.5 ± 0.2 and 6.4 ± 0.1 respectively. Interestingly, the concentration–response curves were well superimposed, thereby suggesting that these are related and PPARγ-mediated pioglitazone effects.
Figure 9.
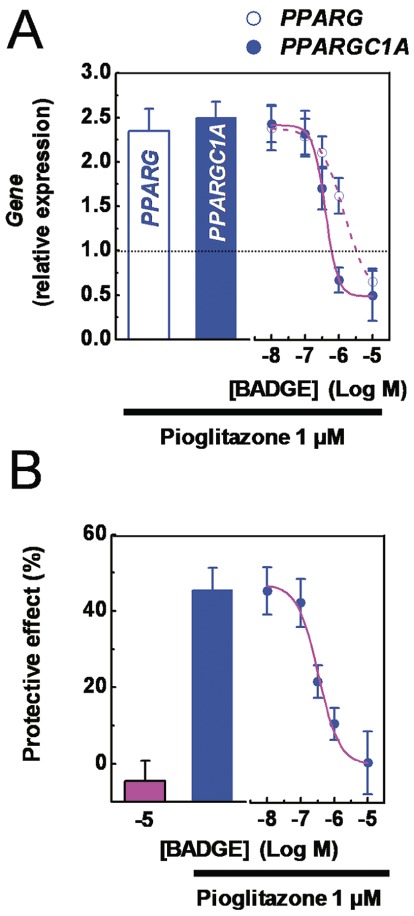
Effects of BADGE on pioglitazone-induced effects. Immortalized podocytes were treated (72 h, as a repeated treatment) with pioglitazone (1 µM) in the absence or presence of increasing concentration of BADGE (10 nM to 10 µM), and processed to determine either PPARG and PPARGC1A expression by quantitative PCR analysis. (B) Immortalized podocytes were pre-treated (72 h, as a repeated treatment) with pioglitazone (1 µM) in the absence or presence of increasing concentration of BADGE (10 nM to 10 µM), then exposed to nutrient deprivation (5 h). At the end of a recovery phase (24 h), cell number of viable cells was assessed to determine the ND-induced cell loss. Results are expressed as mean ± SEM. Mean of four experiments run in triplicate.
Discussion and conclusion
As podocyte protection is strongly related to the preservation of the integrity of the GFB, such protection is a major therapeutic goal in albuminuric glomerular diseases. Our findings, together with previous data, strongly indicate that by decreasing cell death and preserving cell differentiation, PPAR agonists could exert direct protective effects on human podocytes.
Mature podocytes are post-mitotic, highly differentiated, long-living cells and major constituents of the GFB. In our study, we have used immortalized podocytes, selected by their retention of a characteristic arborized morphology and markers (i.e. synaptopodin, podocin) of in vivo podocytes, together with the ability to proliferate, essential to carry out mechanistic studies. These characteristics were routinely evaluated to avoid any changes associated to the in vitro cultures. We showed that these cells constitutively expressed functional PPARs, which were significantly up-regulated by cell treatment with PPAR agonists. Thus, these findings indicated that these cell lines were a valid model to study not only features of podocyte biology, including cell survival and differentiation (Doublier et al., 2001; Burt et al., 2007; Collino et al., 2008; Miglio et al., 2011), but also the effects exerted by PPAR agonists on human podocytes.
By decreasing apoptosis and necrosis, cell treatments with gemfibrozil, GW0742 and pioglitazone diminished the ND-induced podocyte loss. In addition, by preserving the expression of synaptododin and podocin, they preserved cell differentiation. Glomerular ischaemia has been shown to cause both apoptosis and necrosis, which contribute to the podocyte depletion (Lambert et al., 1986; Brukamp et al., 2007; Wagner et al., 2008; Pippin et al., 2009; Miglio et al., 2011). Although in vitro experiments do not create an exact replica of an in vivo environment, they create an environment where the effects of specific pathological stimuli can be carefully studied. In our experiments, ND caused an early and transient increase in the percentage of necrotic cells and a delayed and progressive increase in the percentage of apoptotic cells; these effects were potentiated by hypoxia and attenuated by pyruvate. Interestingly, our data demonstrate that in addition to the lethal effect, ND blunted the expression of synaptopodin, while not affecting that of podocin (data not show). Therefore, by mimicking in vitro the lethal effects of an in vivo ischaemic insult associated to renal ischaemia and organ transplantation (Lambert et al., 1986; Wagner et al., 2008; Pippin et al., 2009), and affecting podocyte differentiation, ND (with or without hypoxia) is a valid stimulus to study podocyte responses to ischemic insults. Interestingly, because ischaemia is implicated in many glomerular disease which share the common feature of decreased blood flow (i.e. thrombotic microangiopathy, capillary loss due to glomerulosclerosis or mesangial proliferation), we could infer that the protective effects exerted by the PPAR agonists against the ND-induced podocyte injury would be of value in understanding their therapeutic potential.
Our results demonstrate that gemfibrozil, GW0742 and pioglitazone exerted anti-apoptotic and anti-necrotic effects when added as cell pre-treatments. The three PPAR agonists were also effective in decreasing cell death when added throughout the course of the recovery phase or as a pre- plus post-treatment. Therefore, these results indicate that these PPAR agonists exhibit both anti-apoptotic and anti-necrotic activities: the former seems to rely on immediate events while the latter on delayed responses. Nevertheless, by increasing the cell ability to buffer the lethal processes triggered by ND, these activities can underlie the described protective effects on human podocytes.
The anti-apoptotic effects of PPAR agonists and some related molecular events (i.e. normalization of Bcl-2 and Bax expression, here confirmed) have been previously reported both in podocytes (Kanjanabuch et al., 2007; Miglio et al., 2011), as well as in other cell types, such as neuronal and endothelial cells (Fuenzalida et al., 2007; Jung et al., 2007; Zanetti et al., 2008). By contrast, the anti-necrotic effects exerted by these drugs has never been carefully investigated. Our findings indicate that mitochondrial biogenesis is a cell response that could underlie the anti-necrotic effects exerted by gemfibrozil, GW0742 and pioglitazone on human podocytes. Indeed, in our experiments, the protective effects exerted by these drugs on the ND-induced lethality were potentiated by pyruvate, which fuels mitochondria (Kauppinen and Nicholls, 1986; Miglio et al., 2009), and markedly attenuated by hypoxia, which inhibits the oxidative processes (including mitochondrial respiration). Interestingly, when compared with those of gemfibrozil or GW0742, significantly larger effects were exerted by pioglitazone on ND-induced necrotic cell death. On the other hand, equivalent effects were exerted by these drugs on the apoptotic lethality. Noteworthy, such differences and equivalences were also observed when the relative effects on the indexes of mitochondrial biogenesis and on the Bcl-2-to-Bax ratio were compared. Hence, together, these results not only provide a mechanistic basis for the pharmacological results but also suggest that the ability to promote mitochondrial biogenesis, together with the normalization of Bcl-2 and Bax expression, could be determinants of the cytoprotective activity of PPAR agonists.
In mammalian cells, a complex transcription network controls mitochondrial biogenesis (Scarpulla, 2008; Hock and Kralli, 2009). In our experiments, gemfibrozil, GW0742 and pioglitazone significantly increased the expression of crucial regulators of this process, albeit in a different manner: the transcriptional co-activator PGC-1α (all these drugs), the transcription factors NRF1 (pioglitazone and GW0742) and Tfam (pioglitazone only). PPARGC1A has been reported to be a PPARγ and PPARβ target gene (Hondares et al., 2006; 2007). In trans-activation assays, pioglitazone and gemfibrozil activated both PPARα and PPARγ (Sauerberg et al., 2002; Kim et al., 2007; Suh et al., 2008), while GW0742 PPARβ only (Sznaidman et al., 2003). We have shown that the three receptor subtypes were induced by repeated cell treatments with these drugs (Miglio et al., 2011 and here confirmed). Thus, we may conclude that through PPARγ or PPARβ, gemfibrozil, GW0742 and pioglitazone could induce PGC-1α in human podocytes. The specific pharmacodynamic profile and the drug-induced changes in the receptor expression may explain the relative differences in the effects exerted by these drugs on the induction of PGC-1α. Due to the complexity of the transcription network, more speculative mechanisms could be proposed in the effort to explain the effects of gemfibrozil, GW0742 and pioglitazone on NRF1 and Tfam levels. Interestingly, the consistency of the effects exerted by these three drugs on the level of PGC-1α, NRF1 and Tfam on the mitochondrial cell contents and on the ND-induced necrotic cell death suggests that they may be components of the molecular/cellular pathway underlying the anti-necrotic activity of PPAR agonists. Moreover, our results suggest that PPARγ activation is the initial event of this pathway. In fact, at the same concentrations, the PPARγ antagonist BADGE abolished in a concentration-dependent manner the effects exerted by pioglitazone on both PPARG and PPARGC1A up-regulation and ND-induced cell death.
The hypothesis that mitochondrial biogenesis might be a cell response contributing to the cytoprotective effects of PPAR agonists is also indicated by previous results. Thus, thiazolidinedione-induced mitochondrial biogenesis sustains cell survival of normal and malignant T cells (Jo et al., 2006; Yang et al., 2007a), preserves the integrity of human umbilical vein endothelial cells (Fujisawa et al., 2009) and could contribute to the neuroprotection exerted by pioglitazone and rosiglitazone (Strum et al., 2007; Miglio et al., 2009; Semple and Noble-Haeusslein, 2011). Interestingly, mitochondrial biogenesis has been proposed to mediate (at least in part) the protective effects exerted by different agents shared by the ability to induce PGC-1α expression. In renal proximal tubular cells, the SIRT1 activator SRT1720 and 5-HT receptor agonists have been shown to increase PGC-1α expression, promote mitochondrial biogenesis and rescue mitochondrial function after oxidative injury (Funk et al., 2010; Rasbach et al., 2010). Hence, these findings suggest that mitochondrial biogenesis is not only a process through which cells adapt their mitochondrial apparatus to physiological cues, but also it could be a pharmacological mechanism to support cell viability. It is recognised that mitochondria influence many aspects of cell biology, including cell survival and death. As the site of oxidative phosphorylation, they provide an efficient route to generate ATP from energy-rich molecules. They possess a high capacity to buffer much of the cytosolic Ca2+ that enters the cells during the cell death processes (Bianchi et al., 2004; Dong et al., 2006). They maintain physiological levels of NAD+ during genotoxic stress (i.e. those produced by ROS) and promote cell survival by the defined ‘mitochondrial oasis effect’ (Yang et al., 2007b). These features provide the basis of understanding why an increased mitochondrial content could enhance cellular ability to buffer detrimental events affecting cellular homeostasis. For example, it has been proposed that by distributing carbohydrate and lipid substrates among more mitochondria, cells with induced mitochondrial biogenesis can more efficiently carry out oxidative processes (Weinberg, 2011). To date, mitochondrial biogenesis has never been studied in podocytes. However, convergent lines of evidence suggest that survival of this cell type critically depends on mitochondrial function. Mitochondria provide most of the cellular ATP in these cells (Abe et al., 2010). In addition, congenital and acquired mitochondrial impairment severely affects podocyte survival (Inoue et al., 2000; Hotta et al., 2001; Yamagata et al., 2002; Hagiwara et al., 2006; Diomedi-Camassei et al., 2007).
The renoprotective effects of PPAR agonists depend on a complex interaction between systemic and renal actions (Kiss-Tóth and Rőszer, 2008; Ruan et al., 2008; Mao and Ong, 2009; Yang et al., 2012). Our results indicate that, by decreasing apoptotic and necrotic cell death and by preserving cell differentiation, gemfibrozil, GW0742 and pioglitazone could exert significant direct protective effects on human podocytes. Mitochondrial biogenesis is a cell response related to the cytoprotective activity of these drugs. Compared with gemfibrozil or GW0742, pioglitazone exerted significantly larger effects, thus indicating its higher activity. These findings suggest that these drugs cause a phenotypic change that might allow to podocytes to maintain their viability and function against internal and/or external perturbations; at higher levels, these effects might contribute to the preservation of GFB integrity and therefore to the anti-albuminuric and renoprotective effects.
Acknowledgments
We are grateful to Dr Valentina Sala, Dr Christian Leo (Dipartimento di Anatomia, Farmacologia e medicina Legale, Università degli Studi di Torino), Dr Francesca Lisa and Dr Maria Botto (Dipartimento di Scienza e Tecnologia del Farmaco, Università degli Studi di Torino) for technical assistance. This work was funded by the Italian Ministry of Education, University, and Research (Programmi di ricerca scientifica di rilevanza nazionale, PRIN 2007), the Regione Piemonte (2008 and 2009) and the Università degli Studi di Torino.
Glossary
- COX
cytochrome c oxidase
- FDA
fluorescein diacetate
- GFB
glomerular filtration barrier
- HND
hypoxic nutrient deprivation
- ND
nutrient deprivation
- NRF
nuclear respiratory factors
- PGC
PPARγ co-activator
- PI
propidium iodide
- Tfam
mitochondrial transcription factor A
- TMRE
tetramethylrhodamine ethyl ester
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effects of cell pre-treatment and/or post-treatment with gemfibrozil, GW0742 or pioglitazone on the ND-induced cell loss. Immortalized podocytes were pre-treated (24 h) with gemfibrozil (30 μM), GW0742 (0.1 μM) or pioglitazone (1 μM), transiently exposed to ND (5 h), and/or treated throughout the course of the recovery phase (post-treatment; 24 h). At the end of the recovery phase, the number of viable cells was assessed to determine the overall cell loss. Results are expressed as mean ± SEM. Mean of five experiments run in triplicate. *P < 0.05; **P < 0.01 versus vehicle alone. ••P < 0.01 versus pre-treatment or post-treatment alone.
Figure S2 Concentration–response curves of the effects of gemfibrozil, GW0742 and pioglitazone on ND-induced cell loss. Immortalized podocytes were pre-treated with gemfibrozil (0.01–30 μM), GW0742 (0.1 nM to 0.1 μM) or pioglitazone (1 nM to 1 μM) for either 24 h (a single treatment) or 72 h (as repeated treatments), then exposed to ND (5 h). At the end of the recovery phase (24 h), the number of viable cells was assessed to determine the overall cell loss. Results are expressedas mean ± SEM. Mean of four experiments run in triplicate. Data were fitted as sigmoidal concentration–response curves and analysed with a four-parameter logistic equation.
Figure S3 Effects of pyruvate on the protection exerted by gembibrozil, GW0742 and pioglitazone on the ND-inducedcell loss. Immortalized podocytes were pre-treated with gemfibrozil (0.01–30 μM), GW0742 (0.1 nM to 0.1 μM) or pioglitazone (1 nM to 1 μM) for either 24 h (a single treatment) or 72 h (as repeated treatments), then exposed to ND (5 h) in the absence or presence of pyruvate (2 mM). At the end of the recovery phase (24 h), the number of viable cells was assessed to determine the overall cell loss. Results are expressed as mean ± SEM. Mean of four experiments run in triplicate. Data were fitted assigmoidal concentration–response curves and analysed with a four-parameter logistic equation.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abe Y, Sakairi T, Kajiyama H, Shrivastav S, Beeson C, Kopp JB. Bioenergetic characterization of mouse podocytes. Am J Physiol Cell Physiol. 2010;299:C464–C476. doi: 10.1152/ajpcell.00563.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi K, Rimessi A, Prandini A, Szabadkai G, Rizzuto R. Calcium and mitochondria: mechanisms and functions of a troubled relationship. Biochim Biophys Acta. 2004;1742:119–131. doi: 10.1016/j.bbamcr.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Brukamp K, Jim B, Moeller MJ, Haase VH. Hypoxia and podocyte-specific Vhlh deletion confer risk of glomerular disease. Am J Physiol Renal Physiol. 2007;293:F1397–F1407. doi: 10.1152/ajprenal.00133.2007. [DOI] [PubMed] [Google Scholar]
- Burt D, Salvidio G, Tarabra E, Barutta F, Pinach S, Dentelli P, et al. The monocyte chemoattractant protein-1/cognate CC chemokine receptor 2 system affects cell motility in cultured human podocytes. Am J Pathol. 2007;171:1789–1799. doi: 10.2353/ajpath.2007.070398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collino F, Bussolati B, Gerbaudo E, Marozio L, Pelissetto S, Benedetto C, et al. Preeclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells. Am J Physiol Renal Physiol. 2008;294:F1185–F1194. doi: 10.1152/ajprenal.00442.2007. [DOI] [PubMed] [Google Scholar]
- Conaldi PG, Biancone L, Bottelli A, Wade-Evans A, Racusen LC, Boccellino M, et al. HIV-1 kills renal tubular epithelial cells in vitro by triggering an apoptotic pathway involving caspase activation and Fas upregulation. J Clin Invest. 1998;102:2041–2049. doi: 10.1172/JCI3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18:2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- Dong Z, Saikumar P, Weinberg JM, Venkatachalam MA. Calcium in cell injury and death. Annu Rev Pathol. 2006;1:405–434. doi: 10.1146/annurev.pathol.1.110304.100218. [DOI] [PubMed] [Google Scholar]
- Doublier S, Ruotsalainen V, Salvidio G, Lupia E, Biancone L, Conaldi PG, et al. Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephritic syndrome. Am J Pathol. 2001;158:1723–1731. doi: 10.1016/S0002-9440(10)64128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuenzalida K, Quintanilla R, Ramos P, Piderit D, Fuentealba RA, Martinez G, et al. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282:37006–37015. doi: 10.1074/jbc.M700447200. [DOI] [PubMed] [Google Scholar]
- Fujisawa K, Nishikawa T, Kukidome D, Imoto K, Yamashiro T, Motoshima H, et al. TZDs reduce mitochondrial ROS production and enhance mitochondrial biogenesis. Biochem Biophys Res Commun. 2009;379:43–48. doi: 10.1016/j.bbrc.2008.11.141. [DOI] [PubMed] [Google Scholar]
- Funk JA, Odejinmi S, Schnellmann RG. SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J Pharmacol Exp Ther. 2010;333:593–601. doi: 10.1124/jpet.109.161992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara M, Yamagata K, Capaldi RA, Koyama A. Mitochondrial dysfunction in focal segmental glomerulosclerosis of puromycin aminonucleoside nephrosis. Kidney Int. 2006;69:1146–1152. doi: 10.1038/sj.ki.5000207. [DOI] [PubMed] [Google Scholar]
- Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–1203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- Hondares E, Mora O, Yubero P, Rodriguez de la Concepción M, Iglesias R, Giralt M, et al. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1alpha gene transcription: an autoregulatory loop controls PGC-1alpha expression in adipocytes via peroxisome proliferator-activated receptor-gamma coactivation. Endocrinology. 2006;147:2829–2838. doi: 10.1210/en.2006-0070. [DOI] [PubMed] [Google Scholar]
- Hondares E, Pineda-Torra I, Iglesias R, Staels B, Villarroya F, Giralt M. PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem Biophys Res Commun. 2007;354:1021–1027. doi: 10.1016/j.bbrc.2007.01.092. [DOI] [PubMed] [Google Scholar]
- Hotta O, Inoue CN, Miyabayashi S, Furuta T, Takeuchi A, Taguma Y. Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int. 2001;59:1236–1243. doi: 10.1046/j.1523-1755.2001.0590041236.x. [DOI] [PubMed] [Google Scholar]
- Inoue K, Nakada K, Ogura A, Isobe K, Goto Y, Nonaka I, et al. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat Genet. 2000;26:176–181. doi: 10.1038/82826. [DOI] [PubMed] [Google Scholar]
- Jo SH, Yang C, Miao Q, Marzec M, Wasik MA, Lu P, et al. Peroxisome proliferator-activated receptor gamma promotes lymphocyte survival through its actions on cellular metabolic activities. J Immunol. 2006;177:3737–3745. doi: 10.4049/jimmunol.177.6.3737. [DOI] [PubMed] [Google Scholar]
- Jung TW, Lee JY, Shim WS, Kang ES, Kim SK, Ahn CW, et al. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. J Neurol Sci. 2007;253:53–60. doi: 10.1016/j.jns.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Kanjanabuch T, Ma LJ, Chen J, Pozzi A, Guan Y, Mundel P, et al. PPAR-gamma agonist protects podocytes from injury. Kidney Int. 2007;71:1232–1239. doi: 10.1038/sj.ki.5002248. [DOI] [PubMed] [Google Scholar]
- Kauppinen RA, Nicholls DG. Synaptosomal bioenergetics. The role of glycolysis, pyruvate oxidation and responses to hypoglycemia. Eur J Biochem. 1986;158:159–165. doi: 10.1111/j.1432-1033.1986.tb09733.x. [DOI] [PubMed] [Google Scholar]
- Kim NJ, Lee KO, Koo BW, Li F, Yoo JK, Park HJ, et al. Design, synthesis, and structure-activity relationship of carbamate-tethered aryl propanoic acids as novel PPARalpha/gamma dual agonists. Bioorg Med Chem Lett. 2007;17:3595–3598. doi: 10.1016/j.bmcl.2007.04.057. [DOI] [PubMed] [Google Scholar]
- Kincaid-Smith P, Fairley KF, Farish S, Best JD, Proietto J. Reduction of proteinuria by rosiglitazone in non-diabetic renal disease. Nephrology. 2008;13:58–62. doi: 10.1111/j.1440-1797.2007.00903.x. [DOI] [PubMed] [Google Scholar]
- Kiss-Tóth E, Rőszer T. PPARgamma in kidney physiology and pathophysiology. PPAR Res. 2008;2008:1–9. doi: 10.1155/2008/183108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert R, Henry M, Howden B, Jablonski P, Rae D, Tavanlis G, et al. Glomerular damage after kidney preservation. Transplantation. 1986;42:125–130. doi: 10.1097/00007890-198608000-00004. [DOI] [PubMed] [Google Scholar]
- Mao Z, Ong AC. Peroxisome proliferator-activated receptor gamma agonists in kidney disease-future promise, present fears. Nephron Clin Pract. 2009;112:c230–c241. doi: 10.1159/000224789. [DOI] [PubMed] [Google Scholar]
- Miglio G, Rosa AC, Rattazzi L, Collino M, Lombardi G, Fantozzi R. PPARgamma stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem Int. 2009;55:496–504. doi: 10.1016/j.neuint.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Miglio G, Rosa AC, Rattazzi L, Grange C, Collino M, Camussi G, et al. The subtypes of peroxisome proliferator-activated receptors expressed by human podocytes and their role in decreasing podocyte injury. Br J Pharmacol. 2011;162:111–125. doi: 10.1111/j.1476-5381.2010.01032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuishi M, Miyashita K, Itoh H. cGMP rescues mitochondrial dysfunction induced by glucose and insulin in myocytes. Biochem Biophys Res Commun. 2008;367:840–845. doi: 10.1016/j.bbrc.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Mori K, Mukoyama M, Nakao K. PPAR-alpha transcriptional activity is required to combat doxorubicin-induced podocyte injury in mice. Kidney Int. 2011;79:1274–1276. doi: 10.1038/ki.2011.36. [DOI] [PubMed] [Google Scholar]
- Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- Pippin J, Kumar V, Stein A, Jablonski P, Shankland SJ, Davis CL. The contribution of podocytes to chronic allograft nephropathy. Nephron Exp Nephrol. 2009;111:e1–e10. doi: 10.1159/000178762. [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Funk JA, Jayavelu T, Green PT, Schnellmann RG. 5-Hydroxytryptamine receptor stimulation of mitochondrial biogenesis. J Pharmacol Exp Ther. 2010;332:632–639. doi: 10.1124/jpet.109.159947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S, Xin C, Beck KF, Saleem MA, Mathieson P, Pavenstädt H, et al. PPARalpha activation upregulates nephrin expression in human embryonic kidney epithelial cells and podocytes by a dual mechanism. Biochem Biophys Res Commun. 2005;338:1818–1824. doi: 10.1016/j.bbrc.2005.10.158. [DOI] [PubMed] [Google Scholar]
- Ruan X, Zheng F, Guan Y. PPARs and the kidney in metabolic syndrome. Am J Physiol Renal Physiol. 2008;294:F1032–F1047. doi: 10.1152/ajprenal.00152.2007. [DOI] [PubMed] [Google Scholar]
- Sarafidis PA, Bakris GL. Protection of the kidney by thiazolidinediones: an assessment from bench to bedside. Kidney Int. 2006;70:1223–1233. doi: 10.1038/sj.ki.5001620. [DOI] [PubMed] [Google Scholar]
- Sarafidis PA, Stafylas PC, Georgianos PI, Saratzis AN, Lasaridis AN. Effect of thiazolidinediones on albuminuria and proteinuria in diabetes: a meta-analysis. Am J Kidney Dis. 2010;55:835–847. doi: 10.1053/j.ajkd.2009.11.013. [DOI] [PubMed] [Google Scholar]
- Sauerberg P, Pettersson I, Jeppesen L, Bury PS, Mogensen JP, Wassermann K, et al. Novel tricyclic-alpha-alkyloxyphenylpropionic acids: dual PPARalpha/gamma agonists with hypolipidemic and antidiabetic activity. J Med Chem. 2002;45:789–804. doi: 10.1021/jm010964g. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Semple BD, Noble-Haeusslein LJ. Broad-spectrum neuroprotection against traumatic brain injury by agonism of peroxisome proliferator-activated receptors. Exp Neurol. 2011;229:195–197. doi: 10.1016/j.expneurol.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- Smulders YM, van Eeden AE, Stehouwer CD, Weijers RN, Slaats EH, Silberbusch J. Can reduction in hypertriglyceridaemia slow progression of microalbuminuria in patients with non-insulin-dependent diabetes mellitus? Eur J Clin Invest. 1997;27:997–1002. doi: 10.1046/j.1365-2362.1997.2330779.x. [DOI] [PubMed] [Google Scholar]
- Strum JC, Shehee R, Virley D, Richardson J, Mattie M, Selley P, et al. Rosiglitazone induces mitochondrial biogenesis in mouse brain. J Alzheimers Dis. 2007;11:45–51. doi: 10.3233/jad-2007-11108. [DOI] [PubMed] [Google Scholar]
- Suh YG, Kim NJ, Koo BW, Lee KO, Moon SH, Shin DH, et al. Design, synthesis, and biological evaluation of novel constrained meta-substituted phenyl propanoic acids as peroxisome proliferator-activated receptor alpha and gamma dual agonists. J Med Chem. 2008;51:6318–6333. doi: 10.1021/jm8003416. [DOI] [PubMed] [Google Scholar]
- Sznaidman ML, Haffner CD, Maloney PR, Fivush A, Chao E, Goreham D, et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor delta (PPARdelta)-synthesis and biological activity. Bioorg Med Chem Lett. 2003;13:1517–1521. doi: 10.1016/s0960-894x(03)00207-5. [DOI] [PubMed] [Google Scholar]
- Wagner MC, Rhodes G, Wang E, Pruthi V, Arif E, Saleem MA, et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J Biol Chem. 2008;283:35579–35589. doi: 10.1074/jbc.M805507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JM. Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol. 2011;22:431–436. doi: 10.1681/ASN.2010060643. [DOI] [PubMed] [Google Scholar]
- Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- Wright HM, Clish CB, Mikami T, Hauser S, Yanagi K, Hiramatsu R, et al. A synthetic antagonist for the peroxisome proliferator-activated receptor gamma inhibits adipocyte differentiation. J Biol Chem. 2000;275:1873–1877. doi: 10.1074/jbc.275.3.1873. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Muro K, Usui J, Hagiwara M, Kai H, Arakawa Y, et al. Mitochondrial DNA mutations in focal segmental glomerulosclerosis lesions. J Am Soc Nephrol. 2002;13:1816–1823. doi: 10.1097/01.asn.0000019772.17954.f8. [DOI] [PubMed] [Google Scholar]
- Yang C, Jo SH, Csernus B, Hyjek E, Liu Y, Chadburn A, et al. Activation of peroxisome proliferator-activated receptor gamma contributes to the survival of T lymphoma cells by affecting cellular metabolism. Am J Pathol. 2007a;170:722–732. doi: 10.2353/ajpath.2007.060651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007b;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HC, Ma LJ, Ma J, Fogo AB. Peroxisome proliferator-activated receptor-γ agonist is protective in podocyte injury-associated sclerosis. Kidney Int. 2006;69:1756–1764. doi: 10.1038/sj.ki.5000336. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhou Y, Guan Y. PPARγ as a therapeutic target in diabetic nephropathy and other renal diseases. Curr Opin Nephrol Hypertens. 2012;21:97–105. doi: 10.1097/MNH.0b013e32834de526. [DOI] [PubMed] [Google Scholar]
- Zanetti M, Stocca A, Dapas B, Farra R, Uxa L, Bosutti A, et al. Inhibitory effects of fenofibrate on apoptosis and cell proliferation in human endothelial cells in high glucose. J Mol Med. 2008;86:185–195. doi: 10.1007/s00109-007-0257-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.