

Abstract
BACKGROUND AND PURPOSE
The roles played by endothelium-derived NO and prostacyclin and by endothelial cell hyperpolarization in ACh-induced relaxation have been well characterized in arteries. However, the mechanisms underlying ACh-induced relaxation in veins remain to be fully clarified.
EXPERIMENTAL APPROACH
ACh-induced smooth muscle cell (SMC) hyperpolarization and relaxation were measured in endothelium-intact and -denuded preparations of rabbit jugular vein.
KEY RESULTS
In endothelium-intact preparations, ACh (≤10−8 M) marginally increased the intracellular concentration of Ca2+ ([Ca2+]i) in endothelial cells but did not alter the SMC membrane potential. However, ACh (10−10–10−8 M) induced a concentration-dependent relaxation during the contraction induced by PGF2α and this relaxation was blocked by the NO synthase inhibitor Nω-nitro-l-arginine. ACh (10−8–10−6 M) concentration-dependently increased endothelial [Ca2+]i and induced SMC hyperpolarization and relaxation. These SMC responses were blocked in the combined presence of apamin [blocker of small-conductance Ca2+-activated K+ (SKCa, KCa2.3) channel], TRAM 34 [blocker of intermediate-conductance Ca2+-activated K+ (IKCa, KCa3.1) channel] and margatoxin [blocker of subfamily of voltage-gated K+ (KV) channel, KV1].
CONCLUSIONS AND IMPLICATIONS
In rabbit jugular vein, NO plays a primary role in endothelium-dependent relaxation at very low concentrations of ACh (10−10–10−8 M). At higher concentrations, ACh (10−8−3 × 10−6 M) induces SMC hyperpolarization through activation of endothelial IKCa, KV1 and (possibly) SKCa channels and produces relaxation. These results imply that ACh regulates rabbit jugular vein tonus through activation of two endothelium-dependent regulatory mechanisms.
Keywords: ACh, basal NO release, calcium-activated K+ channels, endothelial cell hyperpolarization, EDHF, endothelium, NO, rabbit jugular vein, smooth muscle cell hyperpolarization, voltage-dependent K+ channels
Introduction
On the arterial side of the circulation, stimulation of endothelial cells by shear stress or agonists induces release of endothelium-derived relaxing factors such as NO, prostacyclin and endothelium-dependent hyperpolarizing factor (EDHF), which play pivotal roles in the regulation of arterial tone. For instance, endothelium-derived NO is thought to play a pivotal role in tonus regulation in large arteries (Martin et al., 1986; Vallance et al., 1989; Moore et al., 1990). Moreover, in resistance arteries and arterioles, shear stress and agonists each increase the intracellular concentration of Ca2+ ([Ca2+]i) in endothelial cells and thereby induce cell hyperpolarization through activation of two types of Ca2+-activated K+ (KCa) channels [small-conductance KCa (SKCa, KCa2.3) channels and intermediate-conductance KCa (IKCa, KCa3.1) channels], leading to endothelium-dependent smooth muscle cell (SMC) hyperpolarization and relaxation (Nagao et al., 1992; Taylor et al., 2003; Si et al., 2006; Félétou, 2009; Köhler and Ruth, 2010; Alexander et al., 2011).
In veins, as in arteries, endothelial cell stimulation by agonists induces endothelium-dependent relaxation through actions mediated by NO, prostacyclin and EDHF, although the factors responsible for this relaxation may differ among regions and among species. For example, ACh induces relaxation via both endothelium-derived NO and EDHF in human forearm veins (Martinez-León et al., 2003) but via endothelium-derived NO alone in rabbit intrapulmonary small veins (Kusama et al., 2005a). Furthermore, it has been reported that both the amount of NO released from the endothelium and the magnitude of endothelium-dependent SMC hyperpolarization are smaller in veins than in arteries, for example, in porcine pulmonary vein vs. porcine pulmonary artery (Zhang et al., 2006), human saphenous vein vs. human internal mammary artery (Liu et al., 2000) and porcine coronary artery vs. porcine cardiac vein (Zhang et al., 2004). However, it is uncertain whether this is true in other types of veins. More importantly, the mechanism underlying endothelium-stimulant-induced, endothelium-dependent relaxation in veins remains to be fully clarified.
To address the above deficits, we examined the characteristics of the action(s) by which ACh alters electrical and mechanical activities in rabbit jugular vein. To that end, we examined ACh-induced responses in endothelium-denuded preparations and in endothelium-intact preparations treated or not treated with the NO synthase inhibitor Nω-nitro-l-arginine (l -NNA) and the COX inhibitor diclofenac. We also examined the effects of blockers of SKCa, IKCa and voltage-gated K+ (KV)1 channels, as well as of blockers of other K+ channels, on ACh-induced SMC hyperpolarization and relaxation in endothelium-intact preparations.
Methods
Animals
All experiments performed in this study conformed to Guidelines on the Conduct of Animal Experiments issued by the Graduate School of Medical Sciences in Nagoya City University and were approved by the Committee on the Ethics of Animal Experiments in that institution. Male Japanese albino rabbits (supplied by Kitayama Labes, Ina, Japan), weighing 2.3–3.0 kg (12–15 weeks of age), were used in this study (total number used 35). The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Tissue preparation
Rabbits were anaesthetized by injection of pentobarbitone sodium (50 mg kg−1, i.v.), then killed by exsanguination. The external jugular vein was immediately excised and placed in Krebs solution. Once connective tissue had been carefully removed, each segment was either cut so as to make ring preparations for measuring isometric tension or cut open along its long axis to make circularly cut preparations for electrophysiological studies, as described previously (Itoh and Kajikuri, 2011). In some preparations, the endothelium was removed by gently rubbing the intimal surface with small pieces of razor blade (Itoh et al., 1992). Successful removal of the endothelium was confirmed by the absence of a relaxation or hyperpolarization response to 1 µM ACh. Guanethidine (5 µM, to prevent effects due to release of sympathetic transmitters) was present throughout the experiments.
Electrophysiological study
Membrane potentials were measured in SMCs using a conventional microelectrode technique, as described previously (Kusama et al., 2005a; Itoh and Kajikuri, 2011). ACh (3 µM) was applied for 90 s at 30 min intervals in endothelium-intact preparations. To observe the concentration-dependent effects of ACh on the membrane potential, ACh (10−10–3 × 10−6 M) was applied for 90 s at 20–30 min intervals (depending on the concentration) in a given preparation. After the control ACh response had been recorded, l -NNA (0.1 mM) was applied for 60 min and was present thereafter (Yamamoto et al., 2005; Kusama et al., 2005a). The COX inhibitor diclofenac (3 µM) was added in the presence of l -NNA for 60 min (Yamashita et al., 1999) and the final ACh response was then obtained. When the effect of a given blocker of K+ channels was to be observed, the blocker was applied as a pretreatment for over 5 min and was thereafter present during the applications of ACh (Itoh et al., 2003; Kusama et al., 2005a, b; Itoh and Kajikuri, 2011).
Isometric tension measurement
Rings (0.6–0.7 mm long) cut from the isolated vessels were suspended for measurement of isometric tension (calculated per mm length of ring) in an organ chamber between two stainless steel wires inserted into the lumen of the ring, as described previously (Kodama et al., 2009; Itoh and Kajikuri, 2011; Maekawa et al., 2012). The organ chamber was filled with 3 mL Krebs solution at 37°C and this was gassed with 95% oxygen and 5% carbon dioxide. Resting tension was adjusted to obtain maximum contraction in high K+ solution (128 mM). PGF2α was used to induce contraction since it has been shown to induce stable contraction in the rabbit jugular vein (Kodama et al., 2009; Itoh and Kajikuri, 2011; Maekawa et al., 2012).
To obtain concentration-dependent effects, ACh (10−10–10−6 M) was cumulatively applied during the contraction induced by PGF2α in endothelium-intact or -denuded strips. The concentration of PGF2α used was 3 µM in the presence of endothelium and 1 µM in its absence. To examine the roles played by endothelium-derived NO, the effect of ACh was examined both in the absence and in the presence of l -NNA (0.1 mM), which was applied as pretreatment for 60 min and was present thereafter. Since l -NNA enhanced the contraction induced by PGF2α, the concentration of PGF2α applied in the presence of l -NNA was reduced to 1 µM so as to obtain matched amplitudes of contraction. To examine the roles played by endothelium-derived PGs, the effect of ACh was examined in the absence and in the presence of diclofenac (3 µM) in endothelium-intact strips treated with l -NNA. When the effect of a given blocker of K+ channels was to be observed, the blocker was applied as a pretreatment for over 5 min during a PGF2α contraction and was present during subsequent applications of ACh.
Measurement of intracellular concentration of Ca2+ ([Ca2+]i)
The endothelial cell [Ca2+]i in rabbit jugular vein was measured by the use of Fura-2 (Kusama et al., 2005a). In brief, 3 µM Fura-2 acetoxymethyl ester (Fura-2 AM) was applied together with 0.001% Pluronic F-127 to endothelium-intact strips for 3 h at room temperature (20–23°C) under dark conditions. After loading, the strip was mounted on a fluorescence microscope and superfused with warmed (37°C) Krebs solution. The focus was adjusted to reveal individual endothelial cells. ACh (0.03–3 µM) was then applied for 90 s with a 10 min interval to obtain concentration–response curves. Fura-2 was excited by dual wavelengths [340 nm (F340) and 380 nm (F380)] for 1 s and the fluorescence collected through a 510 nm emission filter (half-width, 20 nm) with a 15 s interval. The images were captured and analysed using commercial software (AquaCosmos; Hamamatsu Photonics, Hamamatsu, Japan). Changes in [Ca2+]i are expressed as the change in the fluorescence ratio F340/F380. The mean fluorescence intensities obtained from 10 endothelial cells in a given preparation were averaged, and this average (one value for each strip) was used for the later analysis.
To examine the effect of TRAM 34 on ACh-induced endothelial cell [Ca2+]i, ACh (1 µM) was first applied (to record the control response) for 1 min, followed by a 7 min washout with Krebs solution containing TRAM-34 (10 µM). Finally, 1 µM ACh was applied in the presence of TRAM 34.
Immunohistochemical staining
Segments of external jugular vein were immersion-fixed in 4% paraformaldehyde in 10 mM phosphate buffer and embedded in optical cutting temperature (OCT) compound (Tissue Tek; SAKURA Finetechnical, Tokyo, Japan), then frozen and stored at −80°C. Sections were cut at 6 µm thickness on a cryostat, then mounted on Matsunami adhesive silane (MAS)-coated glass slides (Matsunami Glass, Kishiwada, Japan), as described previously (Kusama et al., 2005a; Itoh and Kajikuri, 2011).
The sections were incubated overnight at 4°C with anti-KV1.2 or anti-KV1.3 monoclonal antibody (each at 1:100 dilution; Abcam, Tokyo, Japan) as the primary antibody. After the sections had been rinsed with PBS, they were incubated for 1 h at room temperature with the second antibody (Alexa Fluor 488 anti-mouse IgG antibody; 1:2000 dilution; Molecular Probes, Eugene, OR, USA), followed by a wash with PBS. The fluorescence of Alexa Fluor 488 was then detected by confocal laser scanning microscopy, under identical conditions in each case.
Western blot analysis
Rabbit jugular vein was homogenized in sample buffer [62.5 mM Tris-HCl (pH 6.8), 10% glycerol and 2% SDS]. Western blotting was performed by a method described previously (Maekawa et al., 2012). A monoclonal antibody against KV1.2 or KV1.3 (each at 1:400 dilution) was used as the primary antibody. The signals from the immunoreactive bands were detected by means of an enhanced chemiluminescence-detection system (SuperSignal West Pico; Pierce, Rockford, IL, USA).
Solutions
The composition of the Krebs solution was as follows (mM): NaCl, 122; KCl, 4.7; MgCl2, 1.2; CaCl2, 2.5; NaHCO3, 15.5; KH2PO4, 1.2; glucose, 11.5. When high K+ solution was to be made, the NaCl in the Krebs solution was replaced with an equimolar amount of KCl. For example, 4.1 mM NaCl in the Krebs solution (containing 5.9 mM K+) was replaced with 4.1 mM KCl, thus making a 10 mM high K+ solution. Solutions were bubbled with 95% oxygen and 5% carbon dioxide (pH 7.3–7.4).
Drugs
The drugs used were as follows: l -NNA, charybdotoxin (ChTx), apamin, iberiotoxin (IBTx) and margatoxin (Peptides Institute Inc., Osaka, Japan); ACh-HCl (Daiichi-Seiyaku, Tokyo Japan), glibenclamide, ouabain, diclofenac sodium and TRAM 34 (Sigma Chemical Co., St. Louis, MO, USA); PGF2α (Cayman Chemical Co., Ann Arbor, MI, USA); Fura-2 AM (Molecular Probes); guanethidine (Tokyo Kasei, Tokyo, Japan).
Stock solutions were made of glibenclamide (0.1 M), Fura-2 AM (1 mM) and TRAM 34 (30 mM) (all in dimethyl sulphoxide) and of PGF2α (10 mM in ethanol). All other drugs were dissolved in ultrapure Milli-Q water (Japan Millipore Corp., Tokyo, Japan). The above stock solutions were stored at −80°C and diluted in Krebs solution just before use.
Statistical analysis
All results are expressed as mean ± SEM, with n values representing the number of rabbits used (each rabbit provided only one segment for a given experiment). The negative log of the EC50 value (pD2 value) was determined for each curve using iterative curve-fitting software fitting an asymmetric sigmoidal function (using nonlinear least square fitter supplied by Origin®, OriginLab Corporation, Northampton, MA, USA). A one-way or two-way repeated-measures anova, with post hoc comparisons made using the Scheffé procedure or Student's unpaired t-test, was used for the statistical analysis. The level of significance was set at P < 0.05.
Results
ACh-induced hyperpolarization
Each SMC in endothelium-intact preparations of rabbit jugular vein was electrically quiescent, the resting membrane potential being −50.9 ± 1.1 mV (n= 12). Application of ACh for 90 s at 20–30 min intervals induced a reproducible, concentration-dependent SMC hyperpolarization. The response consisted of two phases: a large transient, followed by a small sustained hyperpolarization (Figure 1Aa1 and B).
Figure 1.
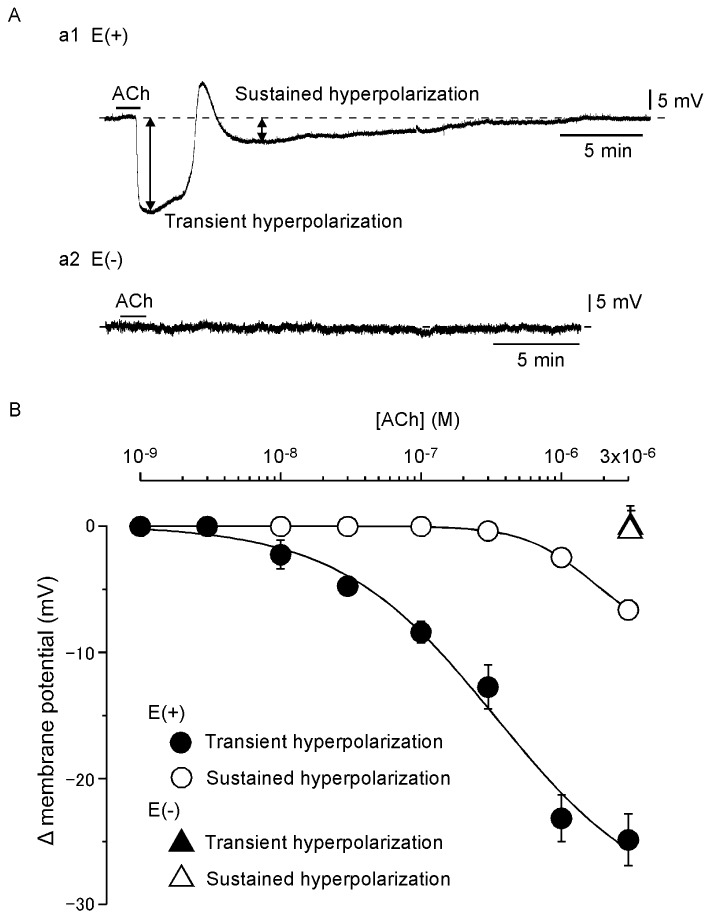
Effects of ACh on smooth muscle cell (SMC) membrane potential in endothelium-intact and -denuded rabbit jugular veins. (Aa1) Application of ACh (3 µM) for 90 s induced a large transient, followed by a small sustained SMC hyperpolarization in endothelium-intact preparation. Broken line indicates the resting membrane potential level. (Aa2) Lack of effect of ACh (3 µM) in endothelium-denuded preparation. (B) Summary of the effects of ACh in endothelium-intact preparations [E(+)] and endothelium-denuded preparations [E(–)]. Data are shown as mean ± SEM (n= 4–7).
Under resting conditions, the SMC membrane potential was −48.5 ± 2.1 mV (n= 3) in endothelium-denuded preparations, and ACh (3 µM) did not induce SMC hyperpolarization (2.3 ± 1.3 mV, n= 3; Figure 1Aa2 and B).
The NO synthase inhibitor l -NNA (0.1 mM) did not alter the resting membrane potential in SMCs in endothelium-intact preparations (n= 5; P > 0.5; Figure 2A and Bb1). Furthermore, l -NNA did not alter the ACh (3 µM)-induced transient (n= 5; P > 0.5) or sustained hyperpolarization (n= 5; P > 0.5; Figure 2Bb2). In the presence of l -NNA, the COX inhibitor diclofenac (3 µM) had no effect on the resting SMC membrane potential (n= 5; P > 0.5), but it did slightly attenuate the ACh-induced sustained hyperpolarization (n= 5; P < 0.05) with no change in the ACh-induced transient hyperpolarization (n= 5; P > 0.5). This inhibition was evident during the initial phase of the sustained hyperpolarization but not during its later phase (Figure 2A).
Figure 2.
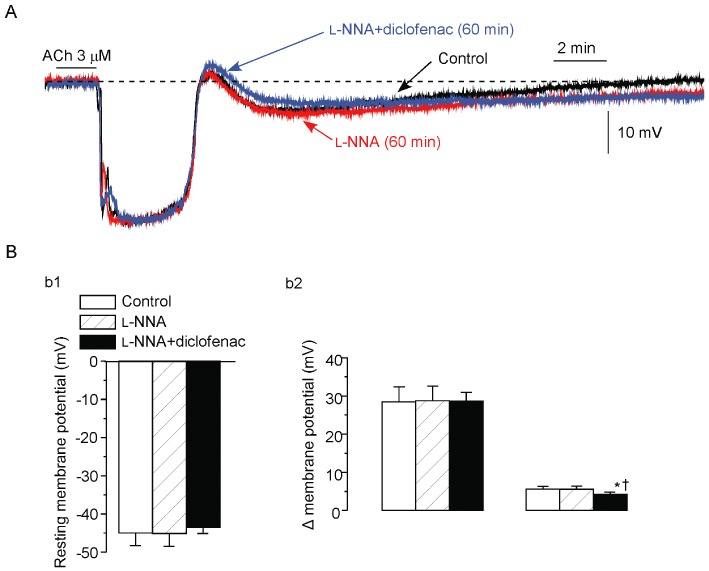
Effects of l -NNA and diclofenac on ACh-induced hyperpolarization. (A) After recording the control ACh (3 µM) response (black line), l -NNA (0.1 mM) was applied as a pretreatment for 60 min and was present thereafter. ACh was then applied in the presence of l -NNA (red line). After a 60 min pretreatment with diclofenac (3 µM) plus l -NNA, ACh was applied in their combined presence (blue line). The records shown were contiguous. (B) Effect of l -NNA with or without diclofenac on resting SMC membrane potential (b1) and the transient hyperpolarization (left panel in b2) and sustained hyperpolarization (right panel in b2) induced by ACh. Data are shown as mean ± SEM (n= 5). *P < 0.05 vs. control, †P < 0.05 vs. l -NNA.
The SKCa channel blocker apamin (0.1 µM) had no effect on the resting SMC membrane potential (n= 5, P > 0.1). This toxin attenuated the ACh (3 µM)-induced transient hyperpolarization (to a minor degree) but not the sustained hyperpolarization (n= 5, Figure 3A). ChTx [a blocker of IKCa, large-conductance Ca2+-activated K+ (BKCa, KCa1.1) and KV1 channels] plus apamin depolarized the SMC membrane (1.8 ± 0.4 mV, n= 5) and blocked the ACh-induced transient and sustained hyperpolarizations (n= 5; P < 0.01 in each case; Figure 3Bb2 and Dd1). In the presence of ChTx + apamin, the membrane depolarization that followed the residual ACh-induced transient hyperpolarization was greatly increased (Figure 3Dd1).
Figure 3.
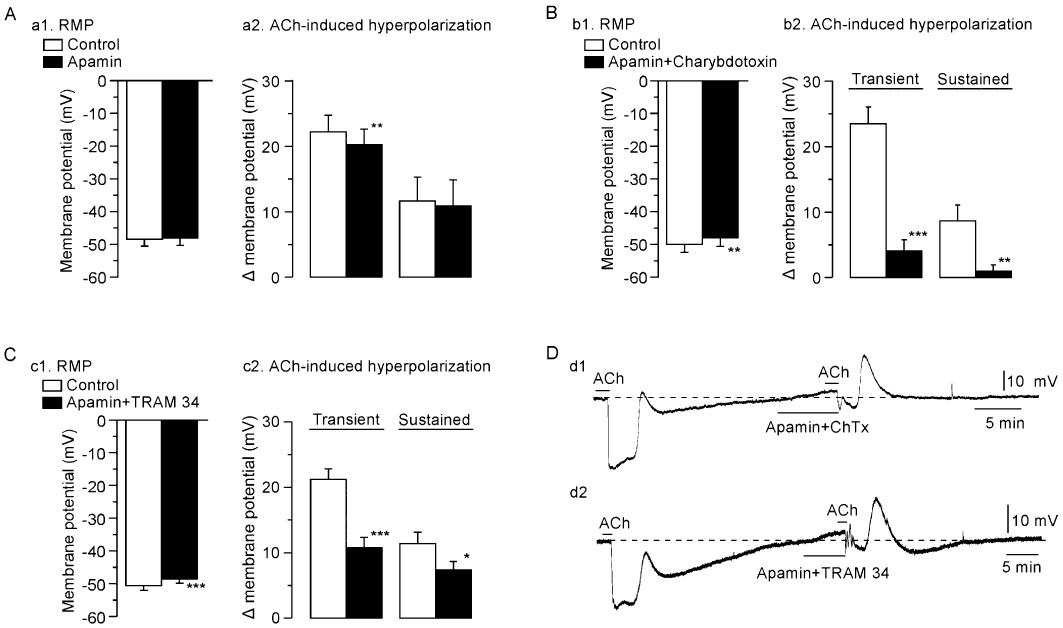
Effects of apamin alone, apamin + charybdotoxin (ChTx) and apamin + TRAM 34 on ACh-induced hyperpolarization. (A). Effect of apamin alone on resting SMC membrane potential (RMP, a1) and on ACh (3 µM)-induced hyperpolarization [transient hyperpolarization (left panel in a2) and sustained hyperpolarization (right panel in a2)]. (B). Effect of apamin + ChTx on resting SMC membrane potential (b1) and on ACh-induced hyperpolarization (b2). (C). Effect of apamin (0.1 µM) + TRAM 34 (10 µM) on resting SMC membrane potential (c1) and on ACh-induced hyperpolarization (c2). Data are shown as mean ± SEM (n= 5). *P < 0.05, **P < 0.01, ***P < 0.001 vs. ‘Control’. (D). Actual traces showing effect of apamin + ChTx (d1) or apamin + TRAM 34 (d2) on SMC membrane potential and ACh-induced hyperpolarization in endothelium-intact preparation. Broken line indicates the resting membrane potential level.
TRAM 34 (selective IKCa channel blocker; 10 µM; Harno et al., 2008; Leuranguer et al., 2008) plus apamin (0.1 µM) depolarized the resting SMC membrane (n= 5, P < 0.001) and attenuated both the transient and sustained hyperpolarizations induced by ACh (3 µM) (n= 5, Figure 3Cc2 and Dd2). In the presence of TRAM 34 + apamin, the membrane depolarization that followed the residual ACh-induced transient hyperpolarization was greatly increased (Figure 3Dd2). Figure 4 shows the concentration-dependent effects of TRAM 34 (1–30 µM) in the presence of apamin (0.1 µM) on the ACh (3 µM)-induced transient and sustained hyperpolarizations (n= 4–6).
Figure 4.
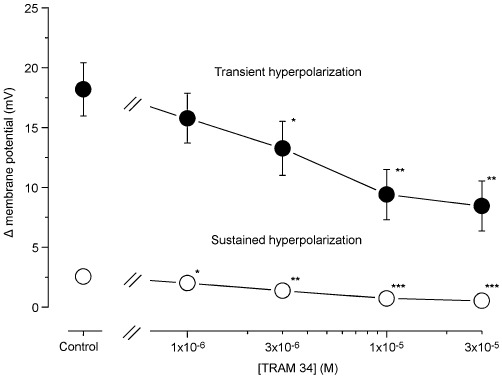
Concentration-dependent effects of TRAM 34 on ACh-induced hyperpolarization in the presence of apamin. After the control ACh (3 µM)-induced hyperpolarization had been recorded, various concentrations of TRAM 34 were applied as a pretreatment for 5 min and were present during the subsequent application of ACh. Membrane depolarizations induced by various concentrations of TRAM 34 in the presence of apamin (0.1 µM) were as follows: 0.0 ± 0.0 mV by 1 µM, 0.0 ± 0.1 mV by 3 µM, 0.7 ± 0.4 mV by 10 µM and 1.9 ± 0.6 mV by 30 µM. Data are shown as mean ± SEM (n= 4–6). *P < 0.05, **P < 0.01, ***P < 0.001 vs. ‘Control’.
The selective KV1 channel blocker margatoxin (0.3 µM) had no effect on the resting SMC membrane potential in the absence or presence of TRAM 34 (10 µM) plus apamin (0.1 µM) (n= 5–8, P > 0.1 in each case, Figure 5Aa1 and Bb1), yet it attenuated both the transient and sustained hyperpolarizations induced by 3 µM ACh in the absence and in the presence of TRAM 34 + apamin (n= 5–8, Figure 5Aa2, Bb2 and C).
Figure 5.
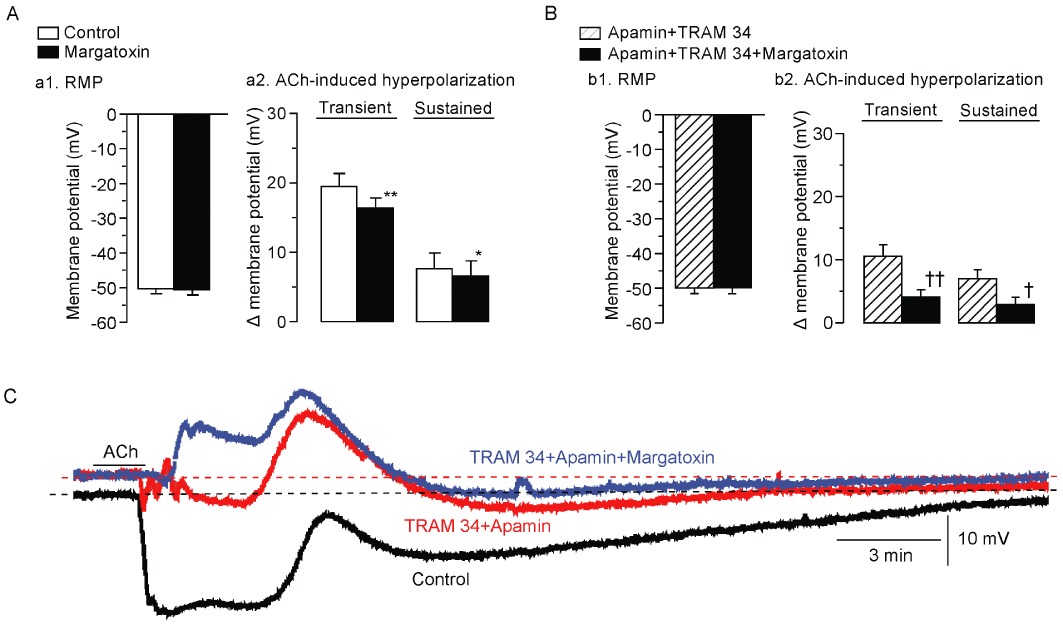
Effects of margatoxin on ACh-induced hyperpolarization in the absence and presence of TRAM 34 plus apamin. (A) Effect of margatoxin (0.3 µM) on resting SMC membrane potential (RMP, a1) and ACh (3 µM)-induced hyperpolarization in the presence of l -NNA + diclofenac (a2). Data are shown as mean ± SEM (n= 5–8). (B). Effect of margatoxin on resting SMC membrane potential (b1) and ACh-induced hyperpolarization (b2) in the presence of l -NNA + diclofenac + apamin + TRAM 34. Data are shown as mean ± SEM (n= 4). *P < 0.05, **P < 0.01 vs. ‘Control’. †P < 0.05, ††P < 0.01 vs. ‘Apamin + TRAM 34’. (C). Actual tracings showing the effect of margatoxin on ACh-induced hyperpolarization. After the control ACh (3 µM) response had been recorded (‘Control’), apamin plus TRAM 34 were applied as a pretreatment for 5 min and ACh was applied again (‘Apamin + TRAM 34’). Following a 30 min washout with Krebs solution containing l -NNA + diclofenac, apamin + TRAM 34 + margatoxin were applied as a pretreatment for 5 min and ACh was applied the final time (‘Apamin + TRAM 34 + Margatoxin’).
The selective BKCa channel blocker IBTx (0.1 µM) depolarized the SMC membrane (2.0 ± 0.3 mV, n= 4; P < 0.05) but did not alter the ACh (3 µM)-induced transient (n= 4; P > 0.1) or sustained hyperpolarization (n= 4; P > 0.1; Figure 6A and B). Both Ba2+ (blocker of the inwardly rectifying K+ channel; 30 µM) and ouabain (Na+-K+-ATPase inhibitor; 1 µM) depolarized the SMC membrane (2.7 ± 0.7 mV for Ba2+ and 2.1 ± 0.1 mV for ouabain, n= 4 in each case, P < 0.05 in each case), yet neither agent altered the ACh-induced transient (n= 4, P > 0.1) or sustained hyperpolarization (n= 4, P > 0.1; Figure 6C and D). Glibenclamide [blocker of ATP-sensitive K+ (KATP) channel; 10 µM] depolarized the SMC membrane (n= 4, P < 0.01) and attenuated the ACh-induced sustained hyperpolarization to a significant but minor degree (n= 9; P < 0.05), yet had no effect on the ACh-induced transient hyperpolarization (n= 9; P > 0.5; Figure 6E). This inhibition was evident during the initial phase of the sustained hyperpolarization but not during its later phase (Figure 6Aa2).
Figure 6.
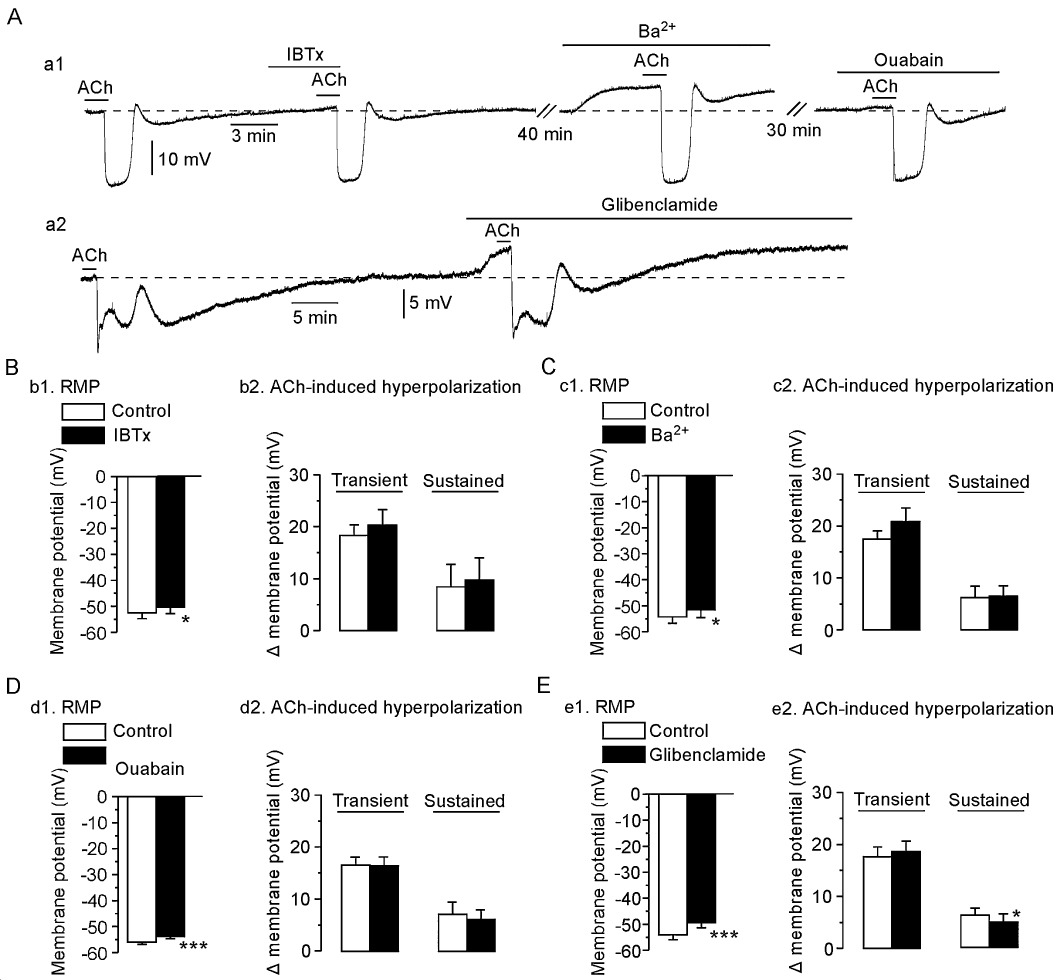
Effects of iberiotoxin (IBTx), barium chloride (Ba2+), ouabain and glibenclamide on ACh-induced hyperpolarization. (Aa1) After the control ACh (3 µM) response had been recorded, IBTx (0.1 µM) was applied as a pretreatment for 5 min and ACh was then applied in the presence of the toxin. After a 40 min washout, Ba2+ (30 µM) was applied as a pretreatment for 5 min and ACh was subsequently applied in the presence of Ba2+. Following a 30 min washout, ouabain (1 µM) was applied as a pretreatment for 5 min and ACh was applied the final time. (Aa2) After the control ACh (3 µM) response had been recorded, glibenclamide (10 µM) was applied as a pretreatment for 5 min and ACh was then applied in the presence of glibenclamide. (B) Summary of the effects of IBTx; b1, resting membrane potential (RMP) of SMC; b2, ACh-induced hyperpolarization. Summaries of the effects of Ba2+ (C), ouabain (D) and glibenclamide (E). Data are shown as mean ± SEM (n= 4–9). *P < 0.05, ***P < 0.001 vs. ‘Control’.
It has been reported that in some rat arteries an increase in the extracellular concentration of K+ ([K+]o) from 5 mM to 9.6 mM induces a SMC membrane hyperpolarization, leading to the suggestion that K+ is an endothelium-derived hyperpolarizing factor (Edwards et al., 1998). In the present endothelium-intact rabbit jugular veins, 10 mM K+ did not change the SMC membrane potential (0.2 ± 0.6 mV, n= 7), while 15 mM K+ depolarized the SMC membrane (3.1 ± 0.5 mV, n= 7).
ACh-induced increase in endothelial cell [Ca2+]i
The Fura-2 ratio was 1.05 ± 0.05 (n= 6) in endothelial cells of rabbit jugular vein under basal conditions and this was increased to 1.39 ± 0.10 after the application of 3 µM ACh (n= 6; P < 0.05; Figure 7A). The ACh-induced increase in [Ca2+]i was concentration dependent from 10−8 to 10−6 M (Figure 7B). TRAM 34 (10 µM) did not significantly modify the ACh (1 µM)-induced [Ca2+]i increase (0.44 ± 0.09 to 0.37 ± 0.09, n= 6, P= 0.545; Figure 7C).
Figure 7.
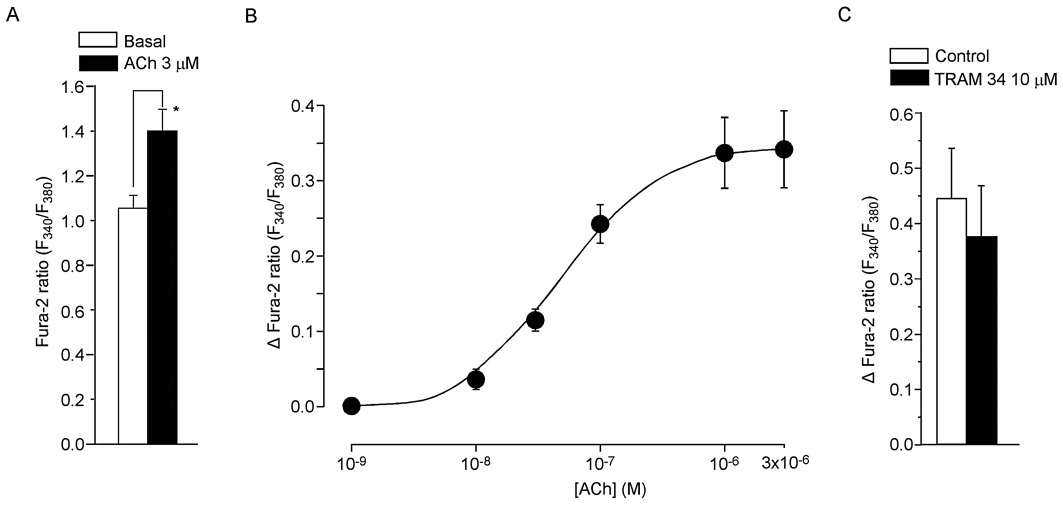
Effects of ACh on endothelial [Ca2+]i in rabbit jugular vein. (A) [Ca2+]i as estimated from the Fura-2 ratio. Application of ACh (3 µM) for 2 min significantly increased [Ca2+]i. Data are shown as mean ± SEM. *P < 0.05 vs. ‘Basal’. (B) Concentration-dependent effect of ACh (10−8–3 × 10−6 M) on [Ca2+]i[shown as Δ Fura-2 ratio (F340/F380)] in endothelial cells. Data are shown as mean ± SEM (n= 6–9). (C). Effect of TRAM 34 (10 µM) on ACh (1 µM)-induced increase in [Ca2+]i[shown as Δ Fura-2 ratio (F340/F380)] in endothelial cells. Data are shown as mean ± SEM (n= 6).
ACh-induced relaxation during the contraction induced by PGF2α
The amplitude of contraction induced by 3 µM PGF2α in endothelium-intact preparations was similar to that induced by 1 µM PGF2α in endothelium-denuded preparations(Table 1). During the PGF2α contraction, ACh (3 × 10−10–10−6 M) concentration-dependently induced a relaxation in endothelium-intact preparations but not in endothelium-denuded preparations (Figure 8Bb1 and Table 1).
Table 1.
Absolute tension, pD2 and Emax in various conditions
| ACh | ||||
|---|---|---|---|---|
| Conditions | PGF2α (mN mm−1) | pD2 | Emax | n |
| 1 | ||||
| Endothelium (+) | 0.74 ± 0.13 | 8.6 ± 0.1 | 93.1 ± 3.7 | 7 |
| Endothelium (−) | 0.81 ± 0.15 | N.D | 2.8 ± 6.4*** | 7 |
| 2 | ||||
| None | 0.88 ± 0.10 | 8.6 ± 0.1 | 96.3 ± 1.4 | 7 |
| l -NNA | 1.09 ± 0.15 | 8.1 ± 0.1*** | 88.7 ± 3.4 | 7 |
| l -NNA + diclofenac | 1.07 ± 0.15 | 7.9 ± 0.1*** | 90.7 ± 3.9 | 7 |
| 3 | ||||
| None | 0.76 ± 0.21 | 8.9 ± 0.1 | 92.3 ± 5.1 | 7 |
| l -NNA + diclofenac | 0.80 ± 0.17 | 7.5 ± 0.3* | 87.2 ± 2.7 | 7 |
| l -NNA + diclofenac + apamin + charybdotoxin | 1.22 ± 0.32† | N.D. | −7.9 ± 17.5**†† | 7 |
| 4 | ||||
| l -NNA + diclofenac | 0.99 ± 0.10 | 7.3 ± 0.1 | 81.8 ± 0.04 | 7 |
| l -NNA + diclofenac + apamin + TRAM 34 | 1.13 ± 0.13 | 6.6 ± 0.2† | 62.9 ± 0.05†† | 7 |
| l -NNA + diclofenac + apamin + TRAM 34 + margatoxin | 1.22 ± 0.22 | 6.6 ± 0.2† | 33.0 ± 0.09†††# | 7 |
The concentration of PGF2α was 3 µM in endothelium-intact strips not treated with l -NNA but 1 µM in endothelium-intact strips treated with l -NNA and in endothelium-denuded preparations. *, **, ***P < 0.05, 0.01, 0.001 vs. ‘Endothelium (+)’ or ‘None’. †, ††, †††P < 0.05, 0.01, 0.001 vs. ‘l -NNA + diclofenac’. #P < 0.05 vs. ‘l -NNA + diclofenac + apamin + TRAM 34’.
Figure 8.
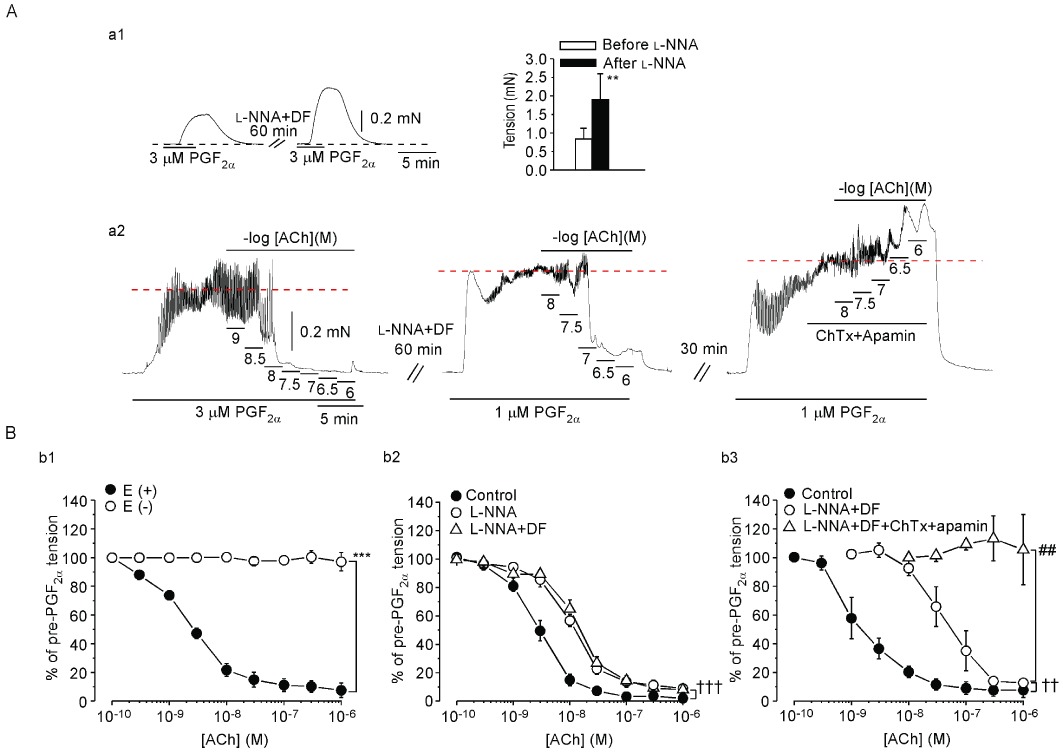
Effects of endothelium-removal, of l -NNA with or without diclofenac and of ChTx plus apamin in the presence of l -NNA + diclofenac on ACh-induced relaxation in rabbit jugular vein. (Aa1) In the presence of diclofenac (DF), PGF2α (3 µM) induced a contraction before and after a 60 min application of l -NNA (left panel). Black broken line indicates the basal tension level. Right panel shows summarized effects. Data are shown as mean ± SEM (n= 4). **P < 0.01 vs. ‘Before l -NNA’. (Aa2) First, an actual tracing was obtained of the concentration-dependent effect of ACh (left panel) in an endothelium-intact preparation. Then, l -NNA + diclofenac (DF) was applied for 60 min and was present thereafter. A 2nd tracing of the concentration-dependent effect of ACh was then obtained (middle panel). Following a 30 min washout with Krebs solution, the ACh response was observed for the last time in the presence of l -NNA + DF + ChTx + apamin (right panel). Tension levels attained after application of PGF2α are shown by red broken lines. (B). Effect of endothelium-removal (b1), of l -NNA with or without DF (b2) and of ChTx + apamin in the presence of l -NNA + DF (b3). The experiments illustrated within a given panel [i.e. (b1), (b2) or (b3)] were performed in a single rabbit, but different rabbits were used to obtain results shown in (b1), (b2) and (b3). Data are shown as mean ± SEM (n= 7). ***P < 0.001 vs. ‘E(+)’. ††P < 0.01, †††P < 0.001 vs. ‘Control’. ##P < 0.01 vs. ‘l -NNA + DF’.
l -NNA (0.1 mM) increased the contraction induced by 128 mM K+ from 0.97 ± 0.18 mN·mm−1 to 1.25 ± 0.20 mN·mm−1 in endothelium-intact preparations (n= 7; P < 0.05). l -NNA (0.1 mM) also increased the contraction induced by 3 µM PGF2α in endothelium-intact preparations (Figure 8Aa1). Since l -NNA (0.1 mM) increased the contraction induced by 3 µM PGF2α in endothelium-intact preparations, the concentration of PGF2α was reduced to 1 µM in the presence of l -NNA so as to obtain matched amplitudes of contraction (n= 7; P= 0.413; Table 1). ACh (10−10–3 × 10−9 M) did not induce a relaxation during the contraction induced by 1 µM PGF2α in the presence of l -NNA. However, at higher concentrations (10−8–10−7 M), ACh did induce such a relaxation and this relaxation was not modified by the COX inhibitor diclofenac (3 µM, n= 7, Figure 8Bb2). In the presence of l -NNA + diclofenac, combined application of ChTx (0.1 µM) and apamin (0.1 µM) blocked this ACh-induced relaxation (n= 7, right panel in Figure 8A and Figure 8Bb3, Table 1).
The KV1 channel blocker margatoxin (0.3 µM) had no effect on the relaxation induced by ACh in the presence of l -NNA + diclofenac (Figure 9A). In the presence of l -NNA + diclofenac, TRAM 34 (10 µM) plus apamin (0.1 µM) reduced the relaxation induced by ACh (10−8–3 × 10−6 M, n= 7), with the residual relaxation being partially blocked by subsequent addition of margatoxin (0.3 µM, n= 7, P < 0.05 vs. in the presence of TRAM 34 + apamin, Figure 9B and Table 1).
Figure 9.
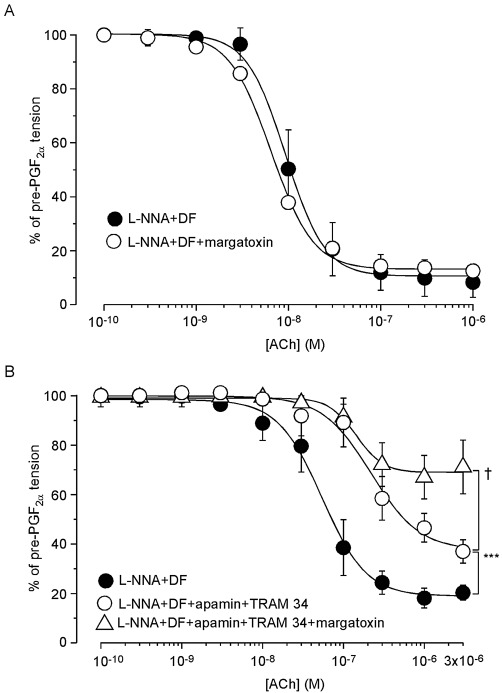
Effects of margatoxin either alone or together with TRAM 34 + apamin on ACh-induced relaxation in endothelium-intact preparations treated with l -NNA + diclofenac. (A) First, a control ACh-induced relaxation curve was recorded during a PGF2α contraction in the presence of l -NNA + diclofenac (DF) (‘l -NNA + DF’). Following a 30 min washout with Krebs solution containing l -NNA + DF, PGF2α was again applied. Margatoxin was then applied as a pretreatment for 5 min and ACh was finally cumulatively applied in the presence of l -NNA + DF + margatoxin (‘l -NNA + DF + margatoxin’). Data are shown as mean ± SEM (n= 4). (B) A control ACh-induced relaxation curve during a PGF2α contraction was recorded in the presence of l -NNA + DF (‘l -NNA + DF’). After a washout with Krebs solution for 30 min, apamin + TRAM 34 were applied as a pretreatment for 5 min during the PGF2α contraction in the presence of l -NNA + DF +apamin + TRAM 34. ACh was then cumulatively applied (‘l -NNA + DF + apamin + TRAM 34’). After a washout with Krebs solution containing l -NNA + DF for 30 min, apamin + TRAM 34 + margatoxin were applied as a pretreatment for 5 min and during the contraction induced by PGF2α in the presence of l -NNA + DF. Finally, ACh was cumulatively applied in their presence (‘l -NNA + DF + apamin + TRAM 34 + margatoxin’). Data are shown as mean ± SEM (n= 7). ***P < 0.001 vs. ‘l -NNA + DF’. †P < 0.05 vs. ‘l -NNA + DF + apamin + TRAM 34’.
Immunohistochemical staining
Localized immunoreactive responses against the KV1.2 and KV1.3 channel subtypes were identified in endothelial cells of rabbit jugular vein (Figure 10).
Figure 10.
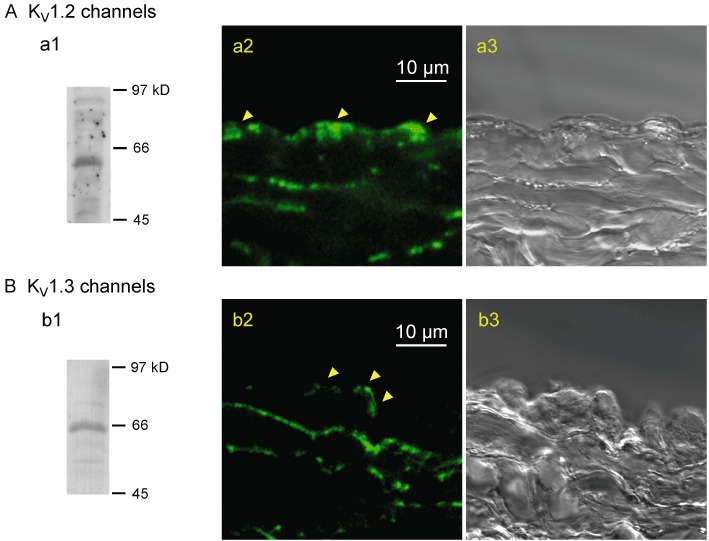
Expressions of KV1.2 and KV1.3 channels in rabbit jugular vein. (A) KV1.2, (B) KV1.3. Western blots to show the specificities of the antibodies against KV1.2 (∼60 kDa) (a1) and KV1.3 (∼65 kDa) (b1) channel proteins. Fluorescence images showing localization of antibodies against either the KV1.2 (a2) or KV1.3 channels (b2) in cross-sections of jugular vein, together with the corresponding bright-field images (a3 and b3). Arrows indicate fluorescence signals in endothelial cells. Similar observations were made in three other preparations.
Discussion and conclusions
In veins, it has been found that endothelium-derived relaxing factors such as NO, prostacyclin and EDHF play important roles in tonus regulation, although the factors responsible for these effects may differ both among regions and among species. For instance, we previously found that endothelium-derived NO is alone responsible for ACh-induced endothelium-dependent relaxation in rabbit intrapulmonary small veins (Kusama et al., 2005a). However, ACh induces relaxation via both endothelium-derived NO and EDHF in human forearm veins (Martinez-León et al., 2003) and saphenous vein (Liu et al., 2000) as well as in the porcine pulmonary vein (Zhang et al., 2006) and cardiac vein (Zhang et al., 2004). In the present experiments, we found in rabbit jugular vein that ACh (10−10–10−8 M) induced endothelium-dependent relaxation during the contraction induced by PGF2α and that the NO synthase inhibitor l -NNA significantly shifted this concentration-dependent relationship to the right, indicating that endothelium-derived NO is partly responsible for the relaxation induced by the lower concentrations (10−10–3 × 10−9) of ACh. In the presence of l -NNA, higher concentrations of ACh (10−8–10−6 M) induced endothelium-dependent SMC hyperpolarization and complete relaxation during the PGF2α contraction, both of which were blocked by ChTx + apamin. These results indicate that in this particular vein, activation of endothelial K+ channels sensitive to these two toxins is able to induce full relaxation when eNOS-function is blocked.
Role of endothelium-derived NO in ACh-induced venorelaxation
It was recently suggested that in rat aorta, asymmetric NG, NG-dimethyl-l-arginine and NG-monomethyl-l-arginine (which is endogenously produced and inhibits NOS) enhance phenylephrine-induced contraction by inhibiting the action of basally released NO (Frew et al., 1993; Al-Zobaidy et al., 2010). The phosphatidylinositol 3-kinase pathway, via activation of serine/threonine protein kinase B (PKB), causes direct phosphorylation of eNOS at Ser1177, increasing the enzyme's [Ca2+]i sensitivity and causing increased production of NO (Dimmeler et al., 1999; Fulton et al., 1999). In rabbit jugular vein we found that the eNOS inhibitor l -NNA enhanced the contraction induced by high K+ and also that induced by PGF2α (Figure 8Aa1). Furthermore, although ACh (<10−8 M) only slightly increased endothelial [Ca2+]i, at 10−10–10−8 M it induced l -NNA-sensitive relaxation. These results suggest that in rabbit jugular vein, NO is released from endothelial cells at resting [Ca2+]i levels both in the absence and in the presence of low concentrations of ACh (10−10–10−8 M) (possibly through activation of PKB/eNOS-phosphorylation), thus inhibiting contraction.
Role of endothelial cell hyperpolarization in ACh-induced relaxation in vein
It was originally found in guinea pig coronary artery that ACh induces an endothelium-dependent, two-phase SMC hyperpolarization: a large transient hyperpolarization and a subsequent slow hyperpolarization (Parkington et al., 1993; Nishiyama et al., 1998). The first phase was blocked by the non-selective IKCa channel blocker ChTx (applied either alone or together with the selective SKCa channel blocker apamin), while the second phase has been reported to be mediated either by both endothelium-derived NO and PGs or solely by PGs (both NO and PGs, Parkington et al., 1993; solely PGs, Nishiyama et al., 1998; Yamashita et al., 1999). The localization of IKCa (KCa3.1) and SKCa (KCa2.3) channels in endothelial cells has been identified in porcine coronary arteries (Burnham et al., 2002; Alexander et al., 2011) and evidence obtained using the more-selective IKCa channel blocker TRAM 34 supports IKCa channels playing a role in the ACh-induced transient hyperpolarization (Wulff et al., 2001; Gluais et al., 2005).
In rabbit jugular vein, we found that as in arteries, ACh (10−8–10−6 M) induced an endothelium-dependent two-phase SMC hyperpolarization: a large transient, followed by a small sustained hyperpolarization. l -NNA did not alter either phase of that hyperpolarization. The COX inhibitor diclofenac, applied in the presence of l -NNA, had no effect on the ACh-induced transient hyperpolarization, but it marginally attenuated the sustained hyperpolarization (the inhibition being evident during the initial phase but not during the prolonged phase of the sustained hyperpolarization; Figure 2A). Similar inhibition of the ACh-induced sustained hyperpolarization was seen when the KATP channel blocker glibenclamide was applied. Furthermore, we found that diclofenac, applied in the presence of l -NNA, did not modify the relaxation induced by ACh during the PGF2α contraction (Figure 8Bb2). These results suggest that activation of KATP channels by endothelium-derived PGs does not play a significant role in ACh-induced endothelium-dependent relaxation in rabbit jugular vein, in contrast to the situation observed in arteries.
In the present experiments, a SMC depolarization occurred after the transient hyperpolarization induced by ACh in rabbit jugular vein (Figure 1Aa1). We previously found, in rat aortic valve endothelial cells, that ACh induced a hyperpolarization followed by a depolarization, and that the latter was inhibited by Cl- channel blockers and by the Na+-K+-Cl- cotransporter inhibitor bumetanide (Ohashi et al., 1999). Furthermore, it has been reported that in the mesenteric artery of the spontaneously hypertensive rat, ACh induces a SMC depolarization through activation of Ca2+-dependent Cl- channels in the endothelial cells and thereby reduces endothelium-dependent SMC hyperpolarization (Goto et al., 2007). Thus, it is possible that the small depolarization seen here after the transient hyperpolarization may be due to activation of Cl- channels.
ChTx inhibits not only IKCa and BKCa channels but also KV1 channels (KV1.2, KV1.3 and KV1.6: Garcia et al., 1995; Alexander et al., 2011) and it is thought that KV channels are involved in endothelium-dependent hyperpolarization in both arteries and arterioles (Zygmunt et al., 1997; Eckman et al., 1998; Nishiyama et al., 1998; Quignard et al., 2000; Nilius and Droogmans, 2001; Dick and Tune, 2010). In rabbit jugular vein, we found the following: (i) ChTx + apamin blocked both phases of the ACh-induced hyperpolarization (viz. the transient and sustained phases); (ii) apamin attenuated the ACh-induced transient hyperpolarization (but only to a minor extent), although it had no significant effect on the ACh-induced sustained hyperpolarization (suggesting that KCa2.3 plays only a minor role, if any, in the ACh-induced hyperpolarization); (iii) combined application of the selective IKCa channel blocker TRAM 34 and apamin reduced both phases of the ACh-induced hyperpolarization; (iv) margatoxin (which inhibits ChTx-sensitive KV1 channels without affecting IKCa channels; Garcia et al., 1995; Alexander et al., 2011) reduced both phases of the ACh-induced hyperpolarization in the absence and in the presence of TRAM 34 + apamin; (v) the ACh-induced hyperpolarization was not modified by the BKCa channel blocker IBTx (indicating no role for BKCa channels in this hyperpolarization); (vi) the ACh-induced hyperpolarization was not altered by the blocker of the inwardly rectifying K+ channel Ba2+ or by the Na+-K+-ATPase inhibitor ouabain, while 10 mM K+ failed to modify the SMC membrane potential [suggesting that unlike in some arteries (Edwards et al., 1998), K+ may not play as significant a role as EDHF]; and (vii) while TRAM 34 + apamin reduced the ACh-induced relaxation in the presence of l -NNA + diclofenac, the residual relaxation was blocked by subsequent addition of margatoxin. These results indicate that in rabbit jugular vein, endothelial IKCa and KV1 channels are mainly responsible for the hyperpolarization and relaxation induced by ACh in the presence of l -NNA + diclofenac. However, we cannot exclude the possibility that KCa channels may contribute to the release of NO, a phenomenon previously suggested by others (Stankevicius et al., 2006; Dalsgaard et al., 2010).
Since KV channels are activated at depolarized membrane potentials, whereas SKCa and IKCa channels are each activated by a small increase in [Ca2+]i, the hyperpolarizing effect of SKCa and IKCa channel opening would be expected to prevent KV channel opening in vascular endothelial cells. We found that in the rabbit jugular vein, the KV1 channel blocker margatoxin did not significantly modify the resting SMC membrane potential in the presence or in the absence of apamin + TRAM 34. Furthermore, this toxin marginally inhibited ACh-induced endothelium-dependent SMC hyperpolarization without changing the ACh-induced relaxation. At present, however, the mechanism underlying the margatoxin-induced slight inhibition of the ACh-induced hyperpolarization remains uncertain. Most importantly, we found that margatoxin, when applied in the presence of apamin + TRAM 34, significantly inhibited both the ACh-induced SMC hyperpolarization and relaxation in rabbit jugular vein. These results suggest that in rabbit jugular vein, margatoxin-sensitive KV1 channels in endothelial cells may play a significant role in ACh-induced relaxation under conditions in which the SKCa and IKCa channels are blocked.
In arteries, the roles played by EDHF in agonist-induced endothelium-dependent relaxation have been examined under conditions in which eNOS and/or COX are blocked. Under such conditions, the function of EDHF may or may not be up-regulated, depending upon region and species. For example, NO donors inhibited the EDHF-mediated relaxation in canine coronary and rabbit carotid arteries (Olmos et al., 1995; Bauersachs et al., 1996) but had no effects in rat hepatic and mesenteric arteries (Fukao et al., 1997; Zygmunt et al., 1998) or in porcine coronary artery (Nagao and Vanhoutte, 1992). Furthermore, endothelium-derived PGs may or may not modulate the actions of EDHFs: indeed, such PGs induced inhibition in guinea pig coronary artery (Yajima et al., 1999) but had no effects in rat hepatic arteries (Zygmunt et al., 1998). Moreover, in studies on human omental resistance arteries, we previously found that in pre-eclampsia, the functions of NO and prostacyclin in endothelium-dependent relaxation are down-regulated, whereas the endothelium-dependent SMC hyperpolarization induced by bradykinin remains normal (Suzuki et al., 2000; 2002). In addition, we found here that l -NNA and diclofenac had no or only minor effects on the ACh-induced two-phase hyperpolarization (Figure 2), suggesting that the actions of EDHF in rabbit jugular vein are not modulated by endothelium-derived NO or PGs. On the basis of these results, we speculate that in the present vein, endothelial SKCa (to a minor degree), IKCa and KV1 channels may serve as a ‘second line of defence’ under conditions in which endothelial NO synthase is dysfunctional, rather than as a simple ‘back-up’ system.
We conclude that in rabbit jugular vein, endothelium-derived NO plays a primary role in endothelium-dependent regulation of contraction. In addition, we suggest that in this vein, endothelial SKCa (to a minor degree), IKCa and KV1 channels may function as a ‘second line of defence’ under conditions in which endothelial NO synthase is dysfunctional.
Acknowledgments
We thank Dr R. J. Timms for a critical reading of the manuscript. We also thank Dr J. Kajikuri and Y. Mizuno for their assistance with the present experiments. This work was partly supported by a Grant-In-Aid for Scientific Research from the Japan Society for the Promotion of Science.
Glossary
- BKCa
large-conductance KCa channel
- ChTx
charybdotoxin
- IBTx
iberiotoxin
- IKCa
intermediate-conductance KCa channel
- KATP
ATP-sensitive K+ channel
- KV
voltage-gated K+ channel
- l -NNA
Nω-nitro-l-arginine
- SKCa
small-conductance KCa channel
- TRAM
34, 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Zobaidy MJ, Craig J, Martin W. Differential sensitivity of basal and acetylcholine-induced activity of nitric oxide to blockade by asymmetric dimethylarginine in the rat aorta. Br J Pharmacol. 2010;160:1476–1483. doi: 10.1111/j.1476-5381.2010.00802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- Burnham MP, Bychkov R, Félétou M, Richards GR, Vanhoutte PM, Weston AH, et al. Characterization of an apamin-sensitive small-conductance Ca2+-activated K+ channel in porcine coronary artery endothelium: relevance to EDHF. Br J Pharmacol. 2002;135:1133–1143. doi: 10.1038/sj.bjp.0704551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard T, Kroigaard C, Misfeldt M, Bek T, Simonsen U. Openers of small conductance calcium-activated potassium channels selectively enhance NO-mediated bradykinin vasodilatation in porcine retinal arterioles. Br J Pharmacol. 2010;160:1496–1508. doi: 10.1111/j.1476-5381.2010.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick GM, Tune JD. Role of potassium channels in coronary vasodilation. Exp Biol Med. 2010;235:10–22. doi: 10.1258/ebm.2009.009201. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Eckman DM, Hopkins N, Mcbride C, Keef KD. Endothelium-dependent relaxation and hyperpolarization in guinea-pig coronary artery: role of epoxyeicosatrienoic acid. Br J Pharmacol. 1998;124:181–189. doi: 10.1038/sj.bjp.0701778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Félétou M. Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol. 2009;156:545–562. doi: 10.1111/j.1476-5381.2009.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew JD, Paisley K, Martin W. Selective inhibition of basal but not agonist-stimulated activity of nitric oxide in rat aorta by NG-monomethyl-L-arginine. Br J Pharmacol. 1993;110:1003–1008. doi: 10.1111/j.1476-5381.1993.tb13913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao M, Hattori Y, Kanno M, Sakuma I, Kitabatake A. Evidence against a role of cytochrome P450-derived arachidonic acid metabolites in endothelium-dependent hyperpolarization by acetylcholine in rat isolated mesenteric artery. Br J Pharmacol. 1997;120:439–446. doi: 10.1038/sj.bjp.0700932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia ML, Knaus H-G, Munujos P, Slaughter RS, Kaczorowski GJ. Charybdotoxin and its effects on potassium channels. Am J Physiol. 1995;269:C1–C10. doi: 10.1152/ajpcell.1995.269.1.C1. [DOI] [PubMed] [Google Scholar]
- Gluais P, Edwards G, Weston AH, Falck JR, Vanhoutte PM, Félétou M. Role of SKCa and IKCa in endothelium-dependent hyperpolarizations of the guinea-pig isolated carotid artery. Br J Pharmacol. 2005;144:477–485. doi: 10.1038/sj.bjp.0706003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Edwards FR, Hill CE. Depolarization evoked by acetylcholine in mesenteric arteries of hypertensive rats attenuates endothelium-dependent hyperpolarizing factor. J Hypertens. 2007;25:345–359. doi: 10.1097/HJH.0b013e328010d616. [DOI] [PubMed] [Google Scholar]
- Harno E, Edwards G, Geraghty AR, Ward DT, Dodd RH, Dauban P, et al. Evidence for the presence of GPRC6A receptors in rat mesenteric arteries. Cell Calcium. 2008;44:210–219. doi: 10.1016/j.ceca.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Itoh T, Kajikuri J. Characteristics of the actions by which 5-HT affects electrical and mechanical activities in rabbit jugular vein. Br J Pharmacol. 2011;164:979–991. doi: 10.1111/j.1476-5381.2011.01373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Seki N, Suzuki S, Ito S, Kajikuri J, Kuriyama H. Membrane hyperpolarization inhibits agonist-induced synthesis of inositol 1,4,5-trisphosphate in rabbit mesenteric artery. J Physiol. 1992;451:307–328. doi: 10.1113/jphysiol.1992.sp019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Kajikuri J, Hattori T, Kusama N, Yamamoto T. Involvement of H2O2 in superoxide-dismutase-induced enhancement of endothelium-dependent relaxation in rabbit mesenteric resistance artery. Br J Pharmacol. 2003;139:444–456. doi: 10.1038/sj.bjp.0705255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama A, Komori K, Hattori K, Yamanouchi D, Kajikuri J, Itoh T. Sarpogrelate hydrochloride reduced intimal hyperplasia in experimental rabbit vein graft. J Vasc Surg. 2009;49:1272–1281. doi: 10.1016/j.jvs.2008.11.071. [DOI] [PubMed] [Google Scholar]
- Köhler R, Ruth P. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflügers Arch. 2010;459:969–976. doi: 10.1007/s00424-010-0819-z. [DOI] [PubMed] [Google Scholar]
- Kusama N, Kajikuri J, Watanabe Y, Suzuki Y, Katsuya H, Itoh T. Characteristics of attenuated endothelium-dependent relaxation seen in rabbit intrapulmonary vein following chronic nitroglycerine administration. Br J Pharmacol. 2005a;145:193–202. doi: 10.1038/sj.bjp.0706178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusama N, Kajikuri J, Yamamoto T, Watanabe Y, Suzuki Y, Katsuya H, et al. Reduced hyperpolarization in endothelial cells of rabbit aortic valve following chronic nitroglycerine administration. Br J Pharmacol. 2005b;146:487–497. doi: 10.1038/sj.bjp.0706363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuranguer V, Gluais P, Vanhoutte PM, Verbeuren TJ, Félétou M. Openers of calcium-activated potassium channels and endothelium-dependent hyperpolarizations in the guinea pig carotid artery. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:101–109. doi: 10.1007/s00210-008-0267-x. [DOI] [PubMed] [Google Scholar]
- Liu ZG, Ge ZD, He GW. Difference in endothelium-derived hyperpolarizing factor-mediated hyperpolarization and nitric oxide release between human internal mammary artery and saphenous vein. Circulation. 2000;102(Suppl 3):III296–III301. doi: 10.1161/01.cir.102.suppl_3.iii-296. [DOI] [PubMed] [Google Scholar]
- Maekawa T, Komori K, Kajikuri J, Itoh T. Characteristics of the actions by which 5-hydroxytryptamine affects electrical and mechanical activities in rabbit jugular-vein graft. Br J Pharmacol. 2012;166:1419–1432. doi: 10.1111/j.1476-5381.2012.01867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W, Villani GM, Jothianandan D, Furchgott RF. Depression of contractile responses in rat aorta by spontaneously released endothelium-derived relaxing factor. J Pharmacol Exp Ther. 1986;237:529–538. [PubMed] [Google Scholar]
- Martinez-León JB, Segarra G, Medina P, Vila JM, Lluch P, Peiró M, et al. Ca2+-activated K+ channels mediate relaxation of forearm veins in chronic renal failure. J Hypertens. 2003;21:1927–1934. doi: 10.1097/00004872-200310000-00021. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PK, al-Swayeh OA, Chong NWS, Evans RA, Gibson A. l -NG-nitro-arginine (L-NOARG), a novel, L-arginine-reversible inhibitor of endothelium-dependent relaxation in vitro. Br J Pharmacol. 1990;99:408–412. doi: 10.1111/j.1476-5381.1990.tb14717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao T, Vanhoutte PM. Characterization of endothelium-dependent relaxations resistant to nitro-l-arginine in the porcine coronary artery. Br J Pharmacol. 1992;107:1102–1107. doi: 10.1111/j.1476-5381.1992.tb13414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao T, Illiano S, Vanhoutte PM. Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-l-arginine in rats. Am J Physiol. 1992;263:H1090–H1094. doi: 10.1152/ajpheart.1992.263.4.H1090. [DOI] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Phys Rev. 2001;81:1416–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hashitani H, Fukuta H, Yamamoto Y, Suzuki H. Potassium channels activated in the endothelium-dependent hyperpolarization in guinea-pig coronary artery. J Physiol. 1998;510:455–465. doi: 10.1111/j.1469-7793.1998.455bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi M, Satoh K, Itoh T. Acetylcholine-induced membrane potential changes in endothelial cells of rabbit aortic valve. Br J Pharmacol. 1999;126:19–26. doi: 10.1038/sj.bjp.0702262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos L, Mombouli J-V, Illiano S, Vanhoutte PM. cGMP mediates the desensitization to bradykinin in isolated canine coronary arteries. Am J Physiol. 1995;268:H865–H870. doi: 10.1152/ajpheart.1995.268.2.H865. [DOI] [PubMed] [Google Scholar]
- Parkington HC, Tare M, Tonta MA, Coleman HA. Stretch revealed three components in the hyperpolarization of guinea-pig coronary artery in response to acetylcholine. J Physiol. 1993;465:459–476. doi: 10.1113/jphysiol.1993.sp019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignard JF, Feletou M, Edwards G, Duhault J, Weston AH, Vanhoutte PM. Role of endothelial cell hyperpolarization in EDHF-mediated responses in the guinea-pig carotid artery. Br J Pharmacol. 2000;129:1103–1112. doi: 10.1038/sj.bjp.0703175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si H, Heyken WT, Wölfle SE, Tysiac M, Schubert R, Grgic I, et al. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res. 2006;99:537–544. doi: 10.1161/01.RES.0000238377.08219.0c. [DOI] [PubMed] [Google Scholar]
- Stankevicius E, Lopez-Valverde V, Rivera L, Hughes AD, Mulvany MJ, Simonsen U. Combination of Ca2+-activated K+ channel blockers inhibits acetylcholine-evoked nitric oxide release in rat superior mesenteric artery. Br J Pharmacol. 2006;149:560–572. doi: 10.1038/sj.bjp.0706886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Kajikuri J, Suzumori K, Itoh T. Mechanisms underlying the reduced endothelium-dependent relaxation in human omental resistance artery in pre-eclampsia. J Physiol. 2000;527:163–174. doi: 10.1111/j.1469-7793.2000.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Hattori T, Kajikuri J, Yamamoto T, Suzumori K, Itoh T. Reduced function of endothelial prostacyclin in human omental resistance arteries in pre-eclampsia. J Physiol. 2002;545:269–277. doi: 10.1113/jphysiol.2002.022384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, et al. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–131. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- Wulff H, Gutman GA, Cahalan MD, Chandy KG. Delineation of the clotrimazole/TRAM-34 binding site on the intermediate conductance calcium-activated potassium channel, IKCa1. J Biol Chem. 2001;276:32040–32045. doi: 10.1074/jbc.M105231200. [DOI] [PubMed] [Google Scholar]
- Yajima K, Nishiyama M, Yamamoto Y, Suzuki H. Inhibition of endothelium-dependent hyperpolarization by endothelial prostanoids in guinea-pig coronary artery. Br J Pharmacol. 1999;126:1–10. doi: 10.1038/sj.bjp.0702254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Kajikuri J, Watanabe Y, Suzuki Y, Suzumori K, Itoh T. Chronic nitroglycerine administration reduces endothelial nitric oxide production in rabbit mesenteric resistance artery. Br J Pharmacol. 2005;146:534–542. doi: 10.1038/sj.bjp.0706365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Kajikuri J, Ohashi M, Kanmura Y, Itoh T. Inhibitory effects of propofol on acetylcholine-induced, endothelium-dependent relaxation and prostacyclin synthesis in rabbit mesenteric resistance arteries. Anesthesiology. 1999;91:1080–1089. doi: 10.1097/00000542-199910000-00029. [DOI] [PubMed] [Google Scholar]
- Zhang RZ, Yang Q, Yim AP, Huang Y, He GW. Different role of nitric oxide and endothelium-derived hyperpolarizing factor in endothelium-dependent hyperpolarization and relaxation in porcine coronary arterial and venous system. J Cardiovasc Pharmacol. 2004;43:839–850. doi: 10.1097/00005344-200406000-00014. [DOI] [PubMed] [Google Scholar]
- Zhang RZ, Yang Q, Yim AP, Huang Y, He GW. Role of NO and EDHF-mediated endothelial function in the porcine pulmonary circulation: comparison between pulmonary artery and vein. Vascul Pharmacol. 2006;44:183–191. doi: 10.1016/j.vph.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Edwards G, Weston AH, Larsson B, Höggestätt ED. Involvement of voltage-dependent potassium channels in the EDHF-mediated relaxation of rat hepatic artery. Br J Pharmacol. 1997;121:141–149. doi: 10.1038/sj.bjp.0701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Plane F, Paulsson M, Garland CJ, Högestätt ED. Interactions between endothelium-derived relaxing factors in the rat hepatic artery: focus on regulation of EDHF. Br J Pharmacol. 1998;124:992–1000. doi: 10.1038/sj.bjp.0701893. [DOI] [PMC free article] [PubMed] [Google Scholar]